DOI:
10.1039/C8CS00947C
(Tutorial Review)
Chem. Soc. Rev., 2019,
48, 4626-4638
Catalytic cascade reactions by radical relay
Received
2nd May 2019
First published on 12th July 2019
Abstract
Radical cascade reactions are an attractive tool for the rapid construction of complex molecular architectures. Although a large number of powerful radical cascades have been developed, stoichiometric amounts of reagents and/or additives are often required to mediate these processes. Radical relay strategies, in which radical character is recycled, require only a catalytic amount of reagent and are particularly attractive as they promise cascades that are high in atom economy. This tutorial review highlights recent advances in this rapidly developing area by setting out and dissecting the reaction designs underpinning state-of-the-art processes involving radical relays. Advances in the field of radical relay cascades will open the door to more efficient synthesis with far-reaching benefits for the makers and end-users of complex molecules.

Huan-Ming Huang
| Huan-Ming Huang carried out doctoral studies under the direction of Professor David J. Procter, funded by a President's Scholarship. After a short stay in Manchester as an EPSRC postdoctoral fellow in the Procter group, he recently joined the group of Professor Frank Glorius at the University of Münster (Germany) as an Alexander von Humboldt postdoctoral research fellow. His research interests include the development of sustainable catalytic reactions and novel radical chemistry. |

Monserrat H. Garduño-Castro
| Monserrat H. Garduño-Castro was born in Mexico City, Mexico. She received her BSc (Pharmaceutical and Biological Chemistry) and Master (Organic Chemistry) degrees from the National Autonomous University of Mexico (UNAM) in 2012 and 2015, respectively. In 2015, she joined the group of Professor David Procter at the University of Manchester to carry out her doctoral studies, funded by CONACyT, and is interested in the development of new radical cyclisation cascades using SmI2. |

Charlotte Morrill
| Charlotte Morrill studied for her undergraduate degree at the University of Manchester. In 2015 she joined the group of Professor David Procter for her doctoral studies, funded by the BBSRC and under the joint supervision of Professor Nicholas Turner. Her research interests include the integration of radical cyclisation cascades with biocatalysis. |

David J. Procter
| David J. Procter was born in Leyland in Lancashire, England. He obtained his BSc in Chemistry from the University of Leeds in 1992 and his PhD in 1995 working with Professor Christopher Rayner on organosulfur and selenium chemistry. He then spent two years as a Postdoctoral Research Associate with Professor Robert Holton at Florida State University in Tallahassee, USA working on the synthesis of Taxol. In late 1997 he took up a Lectureship at the University of Glasgow in Scotland and was promoted to Senior Lecturer in 2004. In 2004, he moved to a Readership at the University of Manchester. David was promoted to Professor in 2008 and is currently an EPSRC Established Career Fellow. |
Key learning points
• understand the concept of radical relays and their potential role in the future of sustainable synthesis.
• appreciate that many different metal and organic catalysts can be used to mediate radical relay reactions.
• be familiar with the most significant advancements in catalytic radical relay reactions and the product architectures accessible.
• understand the various mechanisms underpinning radical relay processes and thus be able to design new transformations.
• appreciate the challenges in the field and future trends.
|
1. Introduction
Syntheses involving the use of many reagents in many reaction steps are being superseded by the use of cascade reactions that deliver complex targets in a more efficient and economical fashion.1,2 The winning combination of high reactivity and high selectivity associated with open-shell intermediates makes radical processes ideal for use in cascade reactions and there has been a recent acceleration in the progress made in this field.3–5 Despite the emergence of many elegant radical cascade reactions, these processes often require the use of stoichiometric amounts of reagent or additive. In fact, even in state-of-the-art transition metal-catalyzed methods and emerging photoredox strategies, stoichiometric co-reductants, co-oxidants, or additives are typically required to achieve catalytic turnover.6
So-called ‘radical relays’ have been used to sidestep the use of stoichiometric reagents in important radical cascade reactions and have allowed redox-neutral processes to be developed. For the purpose of the Review, we define a radical relay as a redox-neutral process in which radical character is regenerated and thus only a catalytic amount of radical-generating reagent is required. We suggest that a radical relay process involves three key stages: (1) radical formation – radical character is generated by single-electron transfer (SET) or addition of a radical; (2) radical relocation – radical character is propagated during a bond-forming/breaking sequence; (3) radical rebound – radical character is recycled, typically by SET back to a metal catalyst or the expulsion of a radical that acts as a catalyst.
While visible-light enabled redox-neutral radical cross-coupling,7 intramolecular redox-neutral processes,8 and titanocene,9 cobalt10 and copper-catalysed11 radical reactions have been the subject of excellent reviews, herein, we aim to bring these disparate areas of radical chemistry together in a more general review on catalytic radical relay reactions by highlighting the underpinning strategies common to many of the processes and showcasing the application of radical relays in cascade approaches to important targets. The Review will embrace the use of various metals (e.g., Ti(III), Cu(I), Ru(II), Mn(III), Co(II), Rh(II), Fe(II), Ir(III), Sm(II), Ni(I)) and organic catalysts to mediate radical relay processes and is organised into intramolecular examples (Section 2), processes involving hydrogen atom transfer (Section 3), and intermolecular examples (Section 4). By highlighting a selection of the most important examples of radical relays, we aim to inspire new work in this important field. The efficiency and sustainability promised by radical relays suggests that they will play a crucial role in the future of radical chemistry12,13 and the expedient construction of molecular architectures of societal importance.
2. Intramolecular catalytic cascade reactions involving radical relay
Intramolecular radical cascade reactions have been used to quickly assemble complex polycyclic scaffolds, including those found in natural products, through the formation of multiple bonds in one operation.1–3 A variety of elegant, catalytic cyclisation cascades involving radical relays have now been reported.
2.1 Cu(I)-Catalysed cascades by radical relay
Many radical relays are initiated by cleavage of a weak bond by reductive SET from a low-valent metal. An early example of an intramolecular catalytic cascade reaction, operating by radical relay, was reported by Aubé and co-workers.14 In the presence of 5 mol% [Cu(PPh3)Cl]4, enantiomerically pure oxaziridine 1 was converted to pyrroline 6 in 66% yield and ≥95% ee. The proposed mechanism involves the generation of nitrogen-centered radical 2 by reduction of the N–O bond in oxaziridine 1, by SET reduction by Cu(I). After 5-exo-trig cyclisation, and 1,4-aryl migration in radical 3, the four-membered ring intermediate 5 is formed. In this case the radical-rebound stage is thought to involve homolytic substitution (SH2) at oxygen regenerating the Cu(I) catalyst. A retro [2+2] cyclisation then delivers the final product 6 (Scheme 1).
 |
| | Scheme 1 Cu(I)-Catalysed cascade synthesis of pyrrolines by radical relay (Aubé, 1992).14 | |
Interestingly, when the diastereoisomeric oxaziridine 1′ was submitted to the same conditions, aziridine 8 was obtained in 53% yield (Scheme 2). Nitrogen centered radical intermediate 2′ is again generated through SET reduction by Cu(I) and undergoes 5-exo-trig cyclisation to give primary radical 3′. It is thought that phenyl transfer is disfavoured in this diastereoisomer due to steric interactions between the α-methyl substituent and the phenyl substituent on the pyrrolidine ring. Thus, the aziridine motif is formed through radical addition to nitrogen and the formation of ketyl radical 7. Product 8 is then formed by back electron-transfer to Cu(II) and regeneration of the Cu(I) catalyst.
 |
| | Scheme 2 Cu(I)-Catalysed cascade synthesis of aziridines by radical relay (Aubé, 1992).14 | |
2.2 Ti(III)-Catalysed cascades by radical relay
Ti(III)-Based reductive SET reagents are widely used to mediate the cyclisations and cyclisation cascades of epoxides. Either a stoichiometric amount of Ti(III) reagent or a combination of catalytic Ti(III) with a stoichiometric amount of co-reductant is typically required. In 2003, Gansäuer and co-workers developed an example of a Ti(III)-catalysed cascade reaction that operates by radical relay (Scheme 3).15 Bicyclic tetrahydrofuran architectures 12 were obtained in moderate to good yields from unsaturated epoxides 9 using 10 mol% Cp2TiCl2. The active Ti(III) catalytic species is generated in situ by reduction of Cp2TiCl2 with Mn. β-Metalloxy radical 10 is formed by reductive SET cleavage of a C–O bond in the epoxide by Ti(III) and 5-exo-trig cyclisation generates the corresponding tertiary radical intermediate 11. As in the work of Aubé, radical rebound by homolytic substitution at oxygen forms the tetrahydrofuran ring of the product and regenerates the Ti(III) catalyst. A computational study provided support for the proposed mechanism.
 |
| | Scheme 3 Ti(III)-Catalysed cascade synthesis of tetrahydrofurans by radical relay (Gansäuer, 2003).15 | |
More recently, Gansäuer and co-workers refined their radical relay approach and reported a catalytic, intramolecular radical arylation of epoxides (Scheme 4).16 Using 1 mol% Cp2TiCl2 and catalytic amounts of Mn and Coll·HCl, a variety of five- and six-membered bicyclic and tricyclic scaffolds were obtained in excellent yields (70–96%) after short reaction times. The reaction sequence involves reductive epoxide opening by the Ti(III) catalyst and 5-exo-trig cyclisation onto the aryl ring to generate intermediates 15. Radical rebound is achieved by intramolecular electron-transfer to regenerate the Ti(III) catalyst, before rearomatisation takes place by proton transfer. Radical relay approaches sidestep the need for a stoichiometric reductant and an external proton source to quench the Ti alkoxides.
 |
| | Scheme 4 Ti(III)-Catalysed cascade arylation of epoxides by radical relay (Gansäuer, 2012).16 | |
In 2016, the Ti(III)-catalysed radical relay approach was used by Gansäuer and co-workers to prepare dihydropyrrolizine and tetrahydroindolizine scaffolds (Scheme 5).17 Following a mechanism analogous to that set out in Scheme 4, a variety of bicyclic motifs could be obtained in good to excellent yields with excellent regiocontrol. Furthermore, catalytic radical cyclisation cascades were also described: epoxide 20 underwent a 6-endo-trig/5-exo-trig cyclisation cascade to give 21 with complete diastereocontrol.
 |
| | Scheme 5 Ti(III)-Catalysed cascade synthesis of dihydropyrrolizine and tetrahydroindolizine scaffolds by radical relay (Gansäuer, 2016).17 | |
2.3 Ru(II)-Catalysed cascades by radical relay
The potential to develop redox-neutral radical reactions using visible-light and photoredox catalysts is well-appreciated7 and cascade reactions have recently been developed that operate by radical relay. While pioneering work by Yoon and co-workers on a Ru(II)-catalysed [2+2] enone cycloaddition was designed to operate by radical relay,18 the team have since shown that initiation of a radical chain process is most likely.13 In 2011, the team reported a radical relay, Ru(II)-catalysed [3+2] cycloaddition of aryl cyclopropyl ketones (Scheme 6).19 The radical fragmentation of a strained ring system is a common feature of known radical relays. In these systems, release of ring strain drives the relocation of radical character. Different alkene and alkyne motifs were used as radical acceptors and a wide range of bicyclic scaffolds was prepared in excellent yield with good diastereocontrol. In addition to 2.5 mol% of Ru(II) catalyst, a stoichiometric amount of Lewis acid was used to activate the cyclopropyl ketone substrates. Mechanistically, SET reduction of the activated ketone generates a ketyl radical intermediate. Radical fragmentation then gives a lanthanide enolate radical 23. Sequential 5-exo-trig/dig and 5-exo-trig cyclisation – including radical rebound onto a metal enolate – delivers lanthanide ketyl radical intermediate 25 that loses an electron to give product 26. Radical addition to an enolate or enolate-equivalent is a common strategy used in relays to generate α-metalloxyradicals prior to radical fragmentation and catalyst regeneration. Although the overall process is redox-neutral, a stoichiometric amount of TMEDA was required to achieve efficient catalysis (Scheme 6).
 |
| | Scheme 6 Ru(II)-Catalysed [3+2] cycloadditions by radical relay (Yoon, 2011).19 | |
2.4 Ir(III)-Catalysed cascades by radical relay
In 2014, Knowles and co-workers reported an Ir(III)-catalysed intramolecular hydroamination of aryl-substituted alkenes using a radical relay strategy promoted by visible-light (Scheme 7).20 The radical heterocyclisation was achieved in excellent yield with only 2 mol% Ir(ppy)2(dtbbpy)PF6 catalyst and without the need for additives. In this relay, radical formation is achieved by oxidative SET by Ir(III) to generate radical cations 28 from simple secondary amines 27. After 5-exo-trig cyclisation, the resultant benzylic radicals 29 are reduced by Ir(II) and the Ir(III) photocatalyst is regenerated. The product of intramolecular olefin hydroamination 31 is formed after proton transfer in intermediate 30.
 |
| | Scheme 7 Ir(III)-Catalysed intramolecular alkene hydroamination by radical relay (Knowles, 2014).20 | |
In a recent study, the Stephenson group has developed an Ir(III)-catalysed radical cascade approach to a library of medicinally-relevant 1-aminonorbornanes (Scheme 8).21 The radical relay is again driven by radical fragmentation of a strained ring system and uses 2 mol% [Ir(dF[CF3]ppy)2(dtbbpy)](PF6) and catalytic ZnCl2. Furthermore, the procedure was adapted to continuous flow processing and could be used to prepare 1-aminonorbornanes on gram scale. The proposed mechanism also involves radical formation by SET oxidation of an amine: starting amine 32a is oxidised by the photoexcited Ir(III) catalyst and the resultant radical cation intermediate 33 undergoes fragmentation. β-Iminium radical 34 then undergoes a 6-exo-trig/5-exo-trig cyclisation cascade that assembles the norbornane scaffold. Finally, radical cation 36 can undergo SET reduction by Ir(II) to regenerate the Ir(III) photocatalyst. The authors highlight that an alternative endgame in which 36 oxidises the starting material 32a may be more favourable (Scheme 8).
 |
| | Scheme 8 Ir(III)-Catalysed radical relay approach to 1-aminonorbornanes reactions by radical relay (Stephenson, 2019).21 | |
2.5 Co(II)-Catalysed cascades by radical relay
Several radical relays utilize radical formation by capture of a carbene by a low valent metal catalyst. In 2011, Zhang and co-workers reported the Co(II)-catalysed enantioselective cyclisation of unsaturated allylic diazoacetates to give bicyclic cyclopropanes in excellent yield and with high enantioselectivity (Scheme 9).22 Co(III)-Bound alkyl radical 40 is thought to form by decomposition of the diazoacetate in the presence of an enantiopure Co(II)–porphyrin catalyst. After 5-exo-trig cyclisation, resultant radical 41 undergoes cyclisation with concomitant regeneration of the Co(II) catalyst.
 |
| | Scheme 9 Allylic diazoacetates in Co(II)-catalysed enantioselective cascade reactions by radical relay (Zhang, 2011).22 | |
Zhang's team has also described Co(II)-catalysed intramolecular radical aziridinations involving allylsulfamoyl and allyloxycarbonyl azides that operate by similar radical relays (Scheme 10).23,24 The method shows wide substrate scope and excellent yields and stereoselectivities. Furthermore, the newly developed method was used in the synthesis of a NK1 antagonist.23
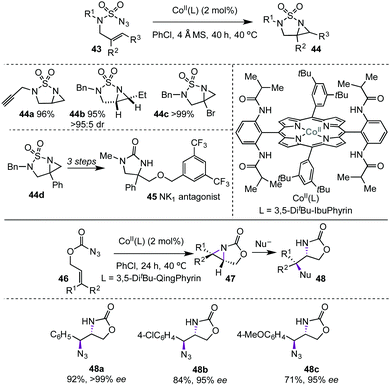 |
| | Scheme 10 Co(II)-Catalysed cascade synthesis of bicyclic aziridines by radical relay (Zhang, 2016 and 2017).23,24 See Scheme 9 for the structure of 3,5-di-tBu-QingPhyrin. | |
In 2016, the de Bruin group described a cascade synthesis of substituted 1H-indenes that uses a Co(II)-catalyst in a radical relay approach (Scheme 11).25 In an approach related to that of Zhang,23,24 base-mediated decomposition of unsaturated N-tosyl hydrazones 49 in the presence of a Co(II) catalyst gave Co(III)-bound alkyl radicals 50. 5-endo-Trig cyclisation of 50 generated benzylic radicals 51 that undergo fragmentation to regenerate the Co(II) catalyst. Rearomatization of 52 generates the indene products 53.
 |
| | Scheme 11 Co(II)-Catalysed synthesis of substituted 1H-indenes by radical relay (de Bruin, 2016).25 | |
2.6 Sm(II)-Catalysed cascades by radical relay
SmI2 is arguably the most well-known reductive SET reagent. In most examples of its use, SmI2 must be used in stoichiometric amounts. We have recently reported a SmI2-catalysed radical cyclisation cascade that operates by radical relay (Scheme 12).26 Using 5 mol% SmI2, aryl cyclopropyl ketones containing alkene or alkyne motifs undergo cascade cyclisation and deliver bicyclic products containing up to four contiguous stereocenters in high yield and with good diastereocontrol. Crucially, stoichiometric additives and co-reductants are not required. Catalytic dearomatising radical cyclisation cascades are also possible.
 |
| | Scheme 12 Sm(II)-Catalysed cyclisation cascades by radical relay (Procter, 2019).26 | |
Inspired by the work of Yoon,19 the radical relay is based on the radical fragmentation of a strained ring system and a rebound involving radical addition to a metal enolate. The proposed mechanism involves generation of ketyl radical intermediate 55 and radical fragmentation to give samarium(III)-enolate radical 56. Subsequent 5-exo-dig/5-exo-trig radical cyclisation cascade generates ketyl radical intermediate 58 and back electron transfer to Sm(III) regenerates the Sm(II) catalyst and delivers bicyclic product 59a. Radical inhibitors, radical probes, computational studies, and EPR experiments were used to gain support for the radical relay mechanism.
3. Catalytic radical relay cyclisation cascades involving hydrogen atom transfer
Hydrogen atom transfer (HAT) processes constitute a powerful tool for the selective functionalisation of remote and unreactive C–H bonds.27 HAT is also a popular strategy for the relocation of radical character in radical relays. Selected examples of catalytic radical relays that depend on HAT processes are described in the following section.
3.1 Sn-Catalysed cascades by radical relay involving HAT
In 1992, Kim reported an early example of a catalytic radical relay process.28 The relay is built around a central HAT process and also involves fragmentation of an epoxide to generate the reactive radical that undergoes HAT. The process is catalysed by tributyltin radical, formed in situ from n-Bu3SnH (30 mol%) and AIBN, and converts epoxyketones 60 to carbobicyclic tertiary alcohols 64 (Scheme 13). The proposed mechanism begins with addition of tributyltin radical to epoxyketones 60 to give Sn(IV) ketyl radicals 61 that undergo fragmentation to form the oxygen-centered radicals 62. After 1,5-HAT, secondary radicals 63 undergo 5-exo-trig cyclisation to form new Sn(IV) ketyl radicals, and fragmentation regenerates the tributyltin radical and delivers the products 64 (Scheme 13). Again, the rebound stage of the relay involves radical addition to a metal enolate.
 |
| | Scheme 13 Sn-Catalysed cascade synthesis of bicyclic scaffolds by radical relay (Kim, 1992).28 | |
 |
| | Scheme 14 Sulfur-catalysed cascade reactions by radical relay involving HAT (Rawal, 1992).29 | |
3.2 Sulfur-catalysed cascades by radical relay involving HAT
Also in 1992, Rawal and co-workers described a similar radical relay approach to carbobicyclic architectures that is catalysed by thiyl radical, generated in situ from AIBN and (PhS)2 under visible light irradiation (Scheme 14).29 The thiyl radical is thought to add to the enol acetate moiety to generate α-acetoxy radicals 66 that then fragment to give oxygen-centered radicals 67. After 1,5-HAT and 5-exo-trig cyclisation, elimination regenerates the thiyl radical and delivers the bicyclic tertiary alcohol products 69.
3.3 Cu(I)-Catalysed cascades by radical relay involving HAT
In 2018, Zhu and co-workers reported a process for the arylation of remote C(sp3)–H bonds that operates by a Cu(I)-catalysed radical relay involving 1,5-HAT (Scheme 15).30 The scope of the process is broad and proved amenable for the late stage functionalisation of natural product derivatives. Again, radical formation by SET reduction of a weak bond is used to trigger the relay. The proposed mechanism involves generation of amidyl radical 72 by SET reduction of the N–F bond in 70 by the Cu(I) catalyst. After 1,5-HAT, and base-promoted transmetallation of ArB(OH)2 with Cu(II), Cu(III) intermediate 74 is formed. Reductive elimination regenerates the Cu(I) catalyst and delivers the arylation product 75. This report highlights an alternative approach for the regeneration of low-valent metal catalysts: access to higher oxidation states of a metal can be exploited in reductive elimination-type processes.
 |
| | Scheme 15 Cu(I)-Catalysed arylation of remote C(sp3)–H bonds by radical relay involving HAT (Zhu, 2018).30 | |
Nagib and co-workers independently developed a related Cu(I)-catalysed radical relay strategy for remote C–C bond formation that harnesses 1,5-HAT (Scheme 16).31 It is proposed that a CuIAr species, formed by transmetallation from ArB(OH)2, reduces the N–F bond in 76. The resultant nitrogen-centered radical undergoes 1,5-HAT, and formation of a CuIIIAr intermediate. Reductive elimination forms the δC–H arylation product 77 and the Cu(I) catalyst is regenerated. Notably, the process is not limited to the activation and coupling of benzylic positions (Scheme 16).
 |
| | Scheme 16 Cu(I)-Catalysed arylation of remote C(sp3)–H by radical relay (Nagib, 2019).31 | |
3.4 Co(II)-Catalysed cascades by radical relay involving HAT
Recently, the group of Zhang developed an enantioselective Co(II)-catalysed radical relay cyclisation that involves activation of remote C(sp3)–H bonds in aliphatic diazo compounds (Scheme 17).32 As in the group's previous approaches, carbene capture by a low-valent metal was used for radical formation. The approach allows the construction of 5-membered saturated heterocycles with high enantioselectivity. Co(III)-Bound alkyl radical 79 is initially thought to arise from reaction of diazo compounds (derived from 78) with the Co(II) catalyst. 1,5-HAT then generates a new Co(III)-bound alkyl radical 80 and radical cyclisation forms the 5-membered products 81 and regenerates the Co(II) catalyst. Impressively, the enantioselectivity of the process can be controlled by a D2-symmetric chiral amidoporphyrin ligand. The process shows broad scope and good functional group tolerance.
 |
| | Scheme 17 Co(II)-Catalysed, enantioselective cyclisations by radical relay involving HAT (Zhang, 2018).32 | |
3.5 Ir(III)-Catalysed cascades by radical relay involving HAT
In 2016, the groups of Knowles33 and Rovis34 independently reported photoredox-catalysed processes for C–C bond formation at the expense of unactivated C(sp3)–H bonds in simple, N–H amides. Both strategies operate by radical relays featuring 1,5-HAT, triggered by oxidative SET by Ir(III) (Scheme 18). Amidyl radicals 86 are generated by proton-coupled electron transfer (PCET) involving base and the Ir(III) photocatalyst. Carbon-centred radicals 87, formed by 1,5-HAT, are then trapped by electron-deficient olefins. The resultant electrophilic radicals 88 are reduced by Ir(II), regenerating the Ir(III) photocatalyst, and protonation of the resultant anion delivers the final product.
 |
| | Scheme 18 Ir(III)-Catalysed cascades by radical relay involving HAT (Knowles and Rovis, 2016).33,34 | |
3.6 Pd(0)-Catalysed cascades by radical relay involving HAT
Gevorgyan and co-workers have recently used a photoactivated Pd(0) catalyst in a radical relay strategy involving HAT. The Heck-type reaction proceeds by activation of a remote C(sp3)–H bond (Scheme 19).35 Of note, radical relays were reported in which 1,6-HAT and 1,7-HAT, in addition to more common 1,5-HAT processes, were used in the radical translocation stage. The proposed mechanism involves excitation of Pd(0) and SET reduction of iodides 89. The resultant primary radical intermediates 91 undergo HAT to generate more stable radical intermediates 92. Intermolecular coupling with the alkene partner then generates the secondary alkyl radical 94. After elimination to give the alkenylation products 95 and Pd(II) species, the Pd(0) catalyst is regenerated by reductive elimination (Scheme 19).
 |
| | Scheme 19 Pd(0)-Catalysed cascades by radical relay involving HAT (Gevorgyan, 2019).35 | |
4. Intermolecular catalytic cascade reactions involving radical relay
The development of efficient radical relays that operate in an intermolecular setting is arguably even more challenging as fast, intramolecular radical processes – e.g., initial cyclisation or HAT – can not be relied upon to underpin selective cascade reactions. Despite this challenge, powerful radical relays have been used to drive catalytic intermolecular cascades that deliver important carbocyclic and heterocyclic products.
4.1 Intermolecular [3+2] reactions by radical relay
In recent years, several research groups have reported catalytic intermolecular couplings that can be considered as formal [3+2] cycloadditions. The approaches employ different transition metal catalysts but have in common the use of radical relays (Scheme 20). For example, in 2016, Yoon and co-workers reported an enantioselective [3+2] reaction mediated by a Ru(II) photocatalyst and visible light.36 In 2017, the Lin group described a Ti(III)-catalysed radical [3+2] coupling of N-acylaziridines and alkenes enabled by radical relay.37 Soon after, the same group reported an enantioselective [3+2] reaction of cyclopropyl ketones and alkenes.38 Finally, more recently, Meggers and co-workers reported an enantioselective intermolecular Rh(III)-catalysed [3+2] process promoted by visible light.39 A common strategy underpins the reports. Radicals are formed by SET reduction; radicals are relocated by fragmentation of a strained ring system; and, radicals rebound by addition to an enolate-type intermediate. Metal-mediated carbonyl reduction generates a ketyl radical 110 that undergoes fragmentation. The resulting radical 111 is then trapped by the alkene/alkyne coupling partner to form a new radical intermediate 112. Finally, cyclisation constructs a 5-membered ring and back electron-transfer regenerates the metal catalyst (Scheme 20).
 |
| | Scheme 20 Catalytic intermolecular [3+2] couplings by radical relay. | |
 |
| | Scheme 21 Co(II)-Catalysed furan synthesis by intermolecular radical relay (Zhang, 2012).40 See Scheme 10 for the structure of 3,5-ditbu-IbuPhyrin. | |
4.2 Co(II)-Catalysed intermolecular couplings by radical relay
Zhang's team has adapted the use of their Co(II)–porphyrin catalysts for use in intermolecular radical relays: complex furans can be constructed from α-diazocarbonyl compounds and alkynes (Scheme 21).40 The coupling utilises 2 mol% of the Co(II) catalyst and exhibits good functional group tolerance. In common with their previous relays, carbene capture by Co(II) is used for radical formation. Co(III)-Bound carbon-centred radical 115 then adds to the alkyne to give alkenyl radical 116. Addition to the carbonyl is then proposed to form radical 117 and subsequent fragmentation delivers the furan product 118 and the regenerated Co(II) catalyst.
 |
| | Scheme 22 Mn(III)-Catalysed pyridine synthesis by intermolecular radical relay (Chiba, 2011).41 | |
4.3 Mn(III)-Catalysed intermolecular reactions by radical relay
In 2011, Chiba and co-workers described a Mn(III)-catalysed union of vinyl azides and cyclopropanols in a formal [3+3] approach to azaheterocycles (Scheme 22).41 This method was also used in the synthesis of (±)-Melinonine-E. The radical relay again takes advantage of a facile radical fragmentation of a strained ring system. Oxygen-centred radicals 121 are generated by oxidative SET by Mn(III). After radical fragmentation, the resultant primary radicals 122 couple with the vinyl azide partner to form iminyl radicals that are trapped by Mn(II) to generate Mn(III)-intermediates 123. After cyclisation and protonation, the Mn(III)-catalyst is regenerated and the initial products 126 are oxidised in situ to form pyridines 127.
4.4 Iron-catalysed intermolecular reactions by radical relay
An Fe(II)-catalysed intermolecular coupling of 2H-azirines and enamides has been used in a synthesis of pyrroles by Guan and co-workers (Scheme 23).42 Reminiscent of the reductive opening of epoxides by Gansäuer's team, the radical relay is thought to involve opening of 2H-azirines 128 by reductive SET from Fe(II) to give the secondary radical intermediates 130. Subsequent coupling with enamides 129 gives radical intermediates 131 that cyclise to form the heterocyclic products and regenerate Fe(II).
 |
| | Scheme 23 Fe(II)-Catalysed synthesis of pyrroles by intermolecular radical relay (Guan, 2018).42 | |
4.5 Ir(III)-Catalysed intermolecular reactions by radical relay
In addition to Ru(II), Ir(III) photocatalysts have also been used in intermolecular couplings that operate by radical relay. In this case, oxidative SET by Ir(III) is used for radical formation. For example, the group of Zheng has developed a [4+2] annulation reaction that uses an Ir(III)–photoredox catalyst and visible light.43 This method allows cyclohexenes to be constructed by the coupling of cyclobutylanilines and alkynes (Scheme 24). Once again, radical fragmentation of a strained ring system is used to drive radical relocation in the relay. The reaction is proposed to proceed by SET oxidation of cyclobutylanilines 134 by the photoexcited Ir(III)-catalyst. The resultant amine radical cation 136 undergoes ring opening to give radical iminium intermediate 137. Subsequent intermolecular radical addition to the alkyne partners gives alkenyl radical species 138 that cyclise on to the iminium to give new amine radical cations 139. The relay is closed by SET reduction of the amine radical cation by Ir(II) to deliver the desired cyclohexenyl amines 140 and to regenerate the Ir(II) photocatalyst.
 |
| | Scheme 24 Ir(III)-Catalysed construction of cyclohexenyl amines by intermolecular radical relay (Zheng, 2015).43 | |
4.6 Cu(I)-Catalysed intermolecular reactions by radical relay
A three-component coupling of alkyl halides, olefins and trifluoromethylthiolate induced by visible-light and a Cu(I) catalyst has been developed by Peters, Fu and co-workers (Scheme 25).44 Again, radical formation involves reduction of a weak bond – a carbon-halogen bond in this case – by reductive SET. Visible light excitation of a Cu(I)SCF3 species triggers reduction of the alkyl halide coupling partner to form radical intermediates 144 and a Cu(II)SCF3 species. Subsequent intermolecular radical coupling with an alkene partner generates secondary alkyl radicals 145 that are oxidised by the Cu(II) species. Subsequent coupling with trifluoromethylthiolate forms the products 146 with concomitant regeneration of the Cu(I) catalyst.
 |
| | Scheme 25 Cu(I)-Catalysed three-component coupling by intermolecular radical relay (Peters and Fu, 2018).44 | |
Liu, Lin and co-workers have reported a related Cu(I)-catalysed three component, radical relay approach to CF3-substituted allenyl nitriles that involves the coupling of 1,3-enynes, Togni's reagent and TMSCN.45 Products of 1,2-addition or 1,4-addition could be obtained depending on the choice of ligand for copper (Scheme 26). The proposed mechanism involves generation of the CF3 radical by SET reduction of Togni's reagent by the Cu(I) catalyst. Subsequent addition to the 1,3-enynes generates propargylic radicals 149. With bisoxazoline ligand (L1), the 1,2-addition product 152 was obtained after reductive elimination from a Cu(III) species 150 and isomerisation (Path A). Interestingly, the 1,4-addition product 153 could be accessed, presumably via reductive elimination from an allenyl Cu(III) species (Path B), by switching to the use of phenanthroline-type ligand (L2).
 |
| | Scheme 26 Cu(I)-Catalysed three-component coupling to give allenyl nitriles by intermolecular radical relay (Liu and Lin, 2018).45 | |
Very recently, a Cu(I)-catalysed radical relay involving redox-active oxime esters, styrenes and boronic acids, mediated by visible-light, has been described by the group of Xiao and Chen (Scheme 27).46 Iminyl radical 157 was generated from oxime derivative 154 by reduction with a Cu(I) species. After β-scission, primary radical 158 was trapped by alkene to form the more stable secondary radical intermediate 159 which was trapped by Cu(II). The resultant Cu(III) intermediate 160 undergoes reductive elimination to regenerate the Cu(I) catalyst and deliver the product 161.
 |
| | Scheme 27 Cu(I)-Catalysed multicomponent coupling of redox-active oxime esters, styrenes and boronic acids by intermolecular radical relay (Xiao and Chen, 2018).46 | |
4.7 Nickel-catalysed intermolecular reactions by radical relay
Molander and co-workers have recently reported a radical relay that employs two metal catalysts working synergistically. The approach involves the amidoarylation of unactivated olefins and combines the use of an Ir(III)-photocatalyst and a Ni catalyst in a radical relay (Scheme 28).47 Strategically, oxidative SET by Ir(III) triggers the relay while the Ni catalyst facilitates intermolecular coupling and rebound via a reductive elimination-type process. It is proposed that amidyl radical 164 is generated from amide 162 by proton-coupled electron transfer (PCET) involving the excited Ir(III) photocatalyst. After 5-exo-trig cyclisation, radical intermediate 165 is captured by the Ni(0) catalyst to form Ni(I) species 166. Oxidative addition of an aryl halide followed by reductive elimination generates the product 168 and Ni(I). Reduction of Ni(I) by Ir(II) regenerates both the Ni(0) and the Ir(III) catalysts. A related transformation has been independently described by Leonori and co-workers (Scheme 28)48 and involves a radical relay catalysed by Ni(I). After transmetalation with, for example, phenylboronic acid, the resultant Ni(I) species generates amidyl radical intermediate 171 by SET reduction of the N-aryloxy amide. After 5-exo-trig cyclisation, the resultant radical intermediate is trapped by Ni(II) to form Ni(III) intermediate 172. Subsequent reductive elimination delivers product 173 and regenerates the Ni(I) catalyst. In addition to the use of arylboronic acids, dialkylzinc partners could also be used as the organometallic partners in the intermolecular radical relay.
 |
| | Scheme 28 Ni/Ir-Catalysed amidoarylation of unactivated olefins by intermolecular radical relay (Molander, 2019);47 Ni-catalysed amidoarylation of unactivated olefins by intermolecular radical relay (Leonori, 2019).48 | |
4.8 Metal-free, catalytic intermolecular reactions by radical relay
The group of Melchiorre has made significant advances in enantioselective synthesis by combining the use of photoredox catalysts with enantiopure organocatalysts in processes involving radical relay.49 For example, a recent report describes the construction of quaternary carbon stereocentres by enantioselective radical conjugate addition using a dual catalytic approach that employs a chiral amine organocatalyst and tetrabutylammonium decatungstate (TBADT) as a photocatalyst (PC) (Scheme 29). Key to their approach is the use of a carbazole-containing organocatalyst that opens up a radical rebound pathway. The reaction begins with condensation of the amine organocatalyst and ketone to generate iminium ion 176. Nucleophilic alkyl radicals 177, generated by hydrogen atom abstraction from a methylene group in coupling partner 175 by the excited photocatalyst, undergo radical conjugate addition to 176 to give iminium radical 178. Crucially, the amine catalyst possesses an in-built, electron-rich carbazole unit that can quench the unstable radical cation 178 by fast intramolecular SET. Subsequently tautomerisation gives the imine-containing radical-cation 180. SET oxidation by the carbazoliumyl radical cation 180 then regenerates the photocatalyst and hydrolysis of the resultant imine 181 liberates the product 182 and regenerates the organocatalyst.
 |
| | Scheme 29 Merging organocatalysis and photoredox catalysis in enantioselective conjugate additions by radical relay (Melchiorre, 2016).49 | |
Finally, the group of Maruoka developed an enantiopure thiol catalyst for an enantioselective, [3+2] coupling of vinyl cyclopropanes 183 and enol ethers 184 (Scheme 30).50 Again, the relay is driven by radical fragmentation of a strained ring system. The mechanism proceeds by addition of thiyl radical – formed by reaction of the enantiopure thiol catalyst with benzoyl peroxide under irradiating conditions – to 183 with ring-opening of the cyclopropane motif. The resultant electron-deficient radical 185 then undergoes addition to the electron-rich alkene acceptor. Subsequent radical cyclisation constructs the 5-membered ring of the products 187 and radical elimination regenerates the thiyl catalyst. The stereochemical course of the radical cyclisation is controlled by the carefully-designed substituents on sulfur.
 |
| | Scheme 30 Sulfur-catalysed, enantioselective intermolecular couplings by radical relay (Maruoka, 2014).50 | |
5. Summary and outlook
Radical sequences, including cyclisation cascades, are widely recognised as versatile tools for the construction of high-value, and often complex, molecular architectures. Radical relay strategies are playing an increasingly important role in radical chemistry and will be key to a future in which the sustainability of synthetic processes will be of paramount importance. This review has showcased some of the most important advances in both intramolecular and intermolecular radical processes employing radical relays, including rare examples of enantioselective transformations. An attempt has also been made to define the term ‘radical relay’ and to classify processes according to the catalysts used and the strategies employed. Furthermore, the Review has served to highlight where significant challenges lie. For example, as many radical relays employ strained-ring starting materials or diazocompounds, can more general starting materials serve as inputs for radical relays? Radical additions and HAT are predominantly used to relocate radical character in relays but can alternative radical processes be used? Can further progress be made towards the development of enantioselective radical relays? The design of dual catalytic radical relays is particularly challenging: Can further breakthroughs be made in this area? Finally, and perhaps most important of all, can general design principles be developed that allow any synthetic radical process to be upgraded to a catalytic relay process? We hope that this Review will stir interest, and encourage further exploration, in this exciting, emerging field.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We gratefully acknowledge funding from the UK Engineering and Physical Sciences Research Council (EPSRC; EP/M005062/01 – Postdoctoral Fellowship to H.-M. H. and EPSRC Established Career Fellowship to D. J. P.), CONACyT, México (PhD Scholarship No. 510789 to M. H. G.-C.) and the BBSRC DTP (Studentship to C. M.).
Notes and references
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed
 .
.
- R. Ardkhean, D. F. J. Caputo, S. M. Morrow, H. Shi, Y. Xiong and E. A. Anderson, Chem. Soc. Rev., 2016, 45, 1557–1569 RSC .
- K. Hung, X. Hu and T. J. Maimone, Nat. Prod. Rep., 2018, 35, 174–202 RSC .
- L. J. Sebren, J. J. Devery and C. R. J. Stephenson, ACS Catal., 2014, 4, 703–716 CrossRef CAS .
- M. P. Plesniak, H.-M. Huang and D. J. Procter, Nat. Rev. Chem., 2017, 1, 0077 CrossRef .
- M. D. Kärkäs, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef .
- J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193–5203 RSC .
- Y. Okada and K. Chiba, Chem. Rev., 2018, 118, 4592–4630 CrossRef CAS .
- A. Gansäuer, S. Hildebrandt, E. Vogelsang and R. A. Flowers II, Dalton Trans., 2016, 45, 448–452 RSC .
- C. te Grotenhuis and B. de Bruin, Synlett, 2018, 29, 2238–2250 CrossRef CAS .
- F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS .
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS .
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS .
- J. Aubé, X. Peng, Y. Wang and F. Takusagawa, J. Am. Chem. Soc., 1992, 114, 5466–5467 CrossRef .
- A. Gansäuer, B. Rinker, M. Pierobon, S. Grimme, M. Gerenkamp and C. Mück-Lichtenfeld, Angew. Chem., Int. Ed., 2003, 42, 3687–3690 CrossRef .
- A. Gansäuer, M. Behlendorf, D. von Laufenberg, A. Fleckhaus, C. Kube, D. V. Sadasivam and R. A. Flowers II, Angew. Chem., Int. Ed., 2012, 51, 4739–4742 CrossRef .
- S. Hildebrandt and A. Gansäuer, Angew. Chem., Int. Ed., 2016, 55, 9719–9722 CrossRef CAS .
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS .
- Z. Lu, M. Shen and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 1162–1164 CrossRef CAS .
- A. J. Musacchio, L. Q. Nguyen, G. Hudson Beard and R. R. Knowles, J. Am. Chem. Soc., 2014, 136, 12217–12220 CrossRef CAS .
- D. Staveness, T. M. Sodano, K. Li, E. A. Burnham, K. D. Jackson and C. R. J. Stephenson, Chem, 2019, 5, 215–226 CAS .
- X. Xu, H. Lu, J. V. Ruppel, X. Cui, S. Lopez de Mesa, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 15292–15295 CrossRef CAS .
- H. Jiang, K. Lang, H. Lu, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2016, 55, 11604–11608 CrossRef CAS .
- H. Jiang, K. Lang, H. Lu, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2017, 139, 9164–9167 CrossRef CAS .
- B. G. Das, A. Chirila, M. Tromp, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2016, 138, 8968–8975 CrossRef CAS .
- H.-M. Huang, J. J. W. McDouall and D. J. Procter, Nat. Catal., 2019, 2, 211–218 CrossRef CAS .
- L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569–1586 CrossRef CAS .
- S. Kim and J. S. Koh, J. Chem. Soc., Chem. Commun., 1992, 1377–1378 RSC .
- V. H. Rawal and V. Krishnamurthy, Tetrahedron Lett., 1992, 33, 3439–3442 CrossRef CAS .
- Z. Li, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2018, 57, 13288–13292 CrossRef CAS .
- Z. Zhang, L. M. Stateman and D. A. Nagib, Chem. Sci., 2019, 10, 1207–1211 RSC .
- Y. Wang, X. Wen, X. Cui and X. P. Zhang, J. Am. Chem. Soc., 2018, 140, 4792–4796 CrossRef CAS .
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS .
- J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef .
- P. Chuentragool, D. Yadagiri, T. Morita, S. Sarkar, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 1794–1798 CrossRef CAS .
- A. G. Amador, E. M. Sherbrook and T. P. Yoon, J. Am. Chem. Soc., 2016, 138, 4722–4725 CrossRef CAS .
- W. Hao, X. Wu, J. Z. Sun, J. C. Siu, S. N. MacMillan and S. Lin, J. Am. Chem. Soc., 2017, 139, 12141–12144 CrossRef CAS .
- W. Hao, J. H. Harenberg, X. Wu, S. N. MacMillan and S. Lin, J. Am. Chem. Soc., 2018, 140, 3514–3517 CrossRef CAS .
- X. Huang, J. Lin, T. Shen, K. Harms, M. Marchini, P. Ceroni and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 5454–5458 CrossRef CAS .
- X. Cui, X. Xu, L. Wojtas, M. M. Kim and X. P. Zhang, J. Am. Chem. Soc., 2012, 134, 19981–19984 CrossRef CAS .
- Y.-F. Wang, K. K. Toh, E. P. J. Ng and S. Chiba, J. Am. Chem. Soc., 2011, 133, 6411–6421 CrossRef CAS .
- M. N. Zhao, Z. H. Ren, D. S. Yang and Z. H. Guan, Org. Lett., 2018, 20, 1287–1290 CrossRef CAS .
- J. Wang and N. Zheng, Angew. Chem., Int. Ed., 2015, 54, 11424–11427 CrossRef CAS .
- J. He, C. Chen, G. C. Fu and J. C. Peters, ACS Catal., 2018, 8, 11741–11748 CrossRef CAS .
- F. Wang, D. Wang, Y. Zhou, L. Liang, R. Lu, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2018, 57, 7140–7145 CrossRef CAS .
- X. Y. Yu, Q. Q. Zhao, J. Chen, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 15505–15509 CrossRef CAS .
- S. Zheng, Á. Gutiérrez-Bonet and G. A. Molander, Chem, 2019, 5, 339–352 CAS .
- L. Angelini, J. Davies, M. Simonetti, L. Malet Sanz, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2019, 5003–5007 CrossRef CAS .
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS .
- T. Hashimoto, Y. Kawamata and K. Maruoka, Nat. Chem., 2014, 6, 702–705 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Monserrat H.
Garduño-Castro
,
Monserrat H.
Garduño-Castro

































