
Photoluminescent metal–organic frameworks and their application for sensing biomolecules
Jing
Dong†
ab,
Dan
Zhao†
a,
Yi
Lu
 *a and
Wei-Yin
Sun
*a
*a and
Wei-Yin
Sun
*a
aCoordination Chemistry Institute, State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing National Laboratory of Microstructures, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210023, China. E-mail: luyi@nju.edu.cn; sunwy@nju.edu.cn; Tel: +86 25 89683485
bNanjing Tech University, Nanjing 211816, China
First published on 10th September 2019
Abstract
Photoluminescence of metal–organic frameworks (MOFs) is sensitive to the structure and concentration of chemical species in the surroundings since MOFs combine the advantages of highly ordered porous structures, varied luminescence origins and diversified host–guest interactions. The diversity and combination flexibility of the organic and inorganic components together with the voids within MOFs offer ample possibilities for tuning their luminescence properties. On the basis of their intrinsic framework structures and biocompatible building blocks, MOFs have stimulated great interest in the area of biosensors. By elaborating on these points, this review will provide up-to-date developments in luminescent MOFs (LMOFs) with emphasis on synthetic approaches and their application in sensing biomolecules. The design outline of LMOFs including functionalization with fluorescent linkers and metal centers and incorporating fluorescent guest molecules within MOFs is presented, and the sensing properties of LMOFs for biomolecules such as DNA/RNA, enzymes/proteins, amino acids, glucose, ascorbic acid, and antibiotics are summarized.
 Jing Dong | Jing Dong received her master’s degree in engineering from Northeast Agricultural University in 2013. At present, she is a PhD student and pursuing her doctorate course in inorganic chemistry in Nanjing University. Her research focuses on the synthesis of metal–organic framework materials and their biosensor properties. |
 Dan Zhao | Dan Zhao obtained her master’s degree in inorganic chemistry from Bohai University, China, in 2013. Then she joined Nanjing University, China, where she obtained her PhD degree in chemistry and worked in the field of functional coordination polymer materials. Her current research interests cover the design and synthesis of functional metal–organic framework (MOF) hybrid materials and the investigation of their physical chemical properties. |
 Yi Lu | Yi Lu is currently an associate professor at Nanjing University (NJU). She received her PhD degree from NJU under the supervision of Prof. Wei-Yin Sun in 2011, studying the synthesis and catalytic properties of MOFs. Sponsored by the Chinese Scholarship Council (CSC) from 2008 to 2010, she studied in the Scripps Research Institute (TSRI) as a visiting student under the supervision of Prof. Jin-Quan Yu, working on Pd-catalyzed C–H activation reactions. She joined the School of Chemistry and Chemical Engineering at NJU as an assistant professor in 2011. Her research interest focuses on selectively Rh(III)-catalyzed C–H functionalization and small molecule catalysis by MOF materials. |
 Wei-Yin Sun | Wei-Yin Sun graduated from East China Normal University, China, in 1986 and received his PhD degree from Osaka University, Japan, in 1993. Since 1996 he has been a Professor at Nanjing University, where he pursues his research on coordination chemistry with interests in the design and synthesis of metal–organic architectures with specific structures and properties. He has published more than 350 research papers. He is a member of the Advisory Board of CrystEngComm and the Editorial Board of J. Coord. Chem., and an Associate Editor of Chinese Chem. Lett., an Executive Associate Editor of Chinese J. Inorg. Chem. and has been a Fellow of the Royal Society of Chemistry (FRSC) since 2014. |
1. Introduction
Photoluminescence includes fluorescence and phosphorescence depending on multiple spin states during the radiative relaxation process.1,2 Such emission phenomena are quite fascinating and the materials emitting light can be employed for different applications such as lighting,3–5 imaging,6 sensing,7 or solar light harvesting.8 Recently, more and more attention has been focused on exploring the applications of such light-emitting phenomena of a relatively new and unique class of materials: metal–organic frameworks (MOFs).9–12MOFs, as a class of hybrid materials, assembled by combining metal ions or clusters and organic bridging ligands via coordination linkages, have rapidly developed in the past few decades as one of the most active fields in coordination chemistry.13–19 Their successful applications in varied fields, including gas storage and separation,20 heterogeneous catalysis,21 light harvesting and chemical sensing,22 as well as drug delivery and bioimaging have been demonstrated.23,24 The hybrid nature of MOFs makes them promising candidates for fabricating luminescent sensors to detect a definite species.25 The photoluminescence of MOFs can originate from the metal centers or ligand units, and be tuned by interplay/interactions within the building components:26 (1) the organic linkers with aromatic moieties or extended π systems give rise to optical emission or photoluminescence upon excitation; (2) the metal components including lanthanides or various inorganic clusters can also contribute to luminescence. In addition, some guest molecules within MOFs can also emit or induce luminescence.
Importantly, the luminescence of MOFs is sensitive to the interactions between the guest species and framework, which makes it possible for MOFs to be utilized as sensors. When a guest species interacts with the framework, the photoluminescence of MOFs can respond quickly by exhibiting varying degrees of fluorescence enhancement, attenuation or even quenching. The advantage of MOFs as luminescent probes is that the framework structure can provide a number of interaction sites for the analyte, hence enhancing the detection sensitivity. The tunability of MOFs allows them to interact with the target species selectively, which can improve the specificity and selectivity of the detection. In addition, MOFs tend to allow ratiometric detection, increasing the detection accuracy by reducing the impact of the environment, interference and other factors.12 Over the past few years, a great deal of effort has been focused on designing MOF-based probes for sensing gases,27,28 explosives,29 solvents and biomolecules.30,31 Of particular interest are the potential chemical sensors in the environmental and biological systems.32 Accordingly, this review is designed to focus on providing an updated critical overview of the synthetic approaches and applications of luminescent MOFs for sensing biomolecules. The design outline of luminescent MOFs and the sensing properties of such MOFs for biomolecules including DNA/RNA, enzymes/proteins, amino acids, glucose, ascorbic acid, antibiotics and so on are summarized. Such biomolecules are either essential or harmful for human health and/or the living environment, and consequently, rapid and efficient detection of these biomolecules is a great issue. Herein, we particularly pay attention to recently reported studies on fabrication of luminescent MOFs with respect to sensing biomolecular species.
2. Luminescent metal–organic frameworks (LMOFs)
Luminescence can be described as the transformation of energy absorbed by materials into light energy and is generally determined by the structures and interatomic/intermolecular attraction forces of the materials.33 The most common source of energy in the absorption process is light and the pathway of relaxation from the excited states accompanying the emission of photons is classified into two basic types: fluorescence and phosphorescence. Fluorescence, a spin-allowed transition, which occurs from the lowest singlet excited electronic state (S1) to the ground state (S0) with typical lifetimes in the order of nanoseconds (ns). Emission accompanying the transition from an excited triplet electronic state (T1) to the ground singlet state is defined as phosphorescence.The luminescence characteristics of MOFs give rise to an important sub-category of MOFs with photon emission initiated by the absorption of excitation energy.34 Due to the structural diversity of MOFs, including multiple types of ligand molecules, inorganic ions or clusters and guest molecular or ionic species, their photoluminescence can originate in several different ways:19 metal center/ligand-based emission, ligand-to-ligand charge transfer (LLCT), ligand-to-metal charge transfer (LMCT), metal-to-ligand charge transfer (MLCT), metal-to-metal charge transfer (MMCT) and processes that involve guest molecules in the LMOF pores, such as guest centered or sensitized emission (Fig. 1a). Organic linkers, typically those with π-conjugated backbones, have little spin–orbit coupling, so selection rules are determined by the symmetry of the singlet ground and excited states. Hence, the strongest emission is usually from the lowest excited singlet state to the singlet ground state (fluorescence). Such transitions are either π–π* or n–π* in nature. By Hund's rule a lower-lying triplet state must exist, which can be accessed by non-radiative energy transfer. However, in the case of organic species, the triplet state is non-emissive as the spin selection rules cannot be relaxed in the absence of strong spin–orbit coupling. The electronic states involved in the luminescence phenomena of a complex are briefly summarized in the energy spectrum (Fig. 1b). Here, it is worth noting that small organic molecules in LMOFs usually suffer from self-quenching resulting in a low quantum yield of their luminescence. On the other hand, metal-center emitted luminescence is basically observed in cases where f-orbital metal ions or metal clusters serve as nodes. Moreover, some LMOFs are found to have high absorption struts coordinated to metal centers to facilitate intersystem crossing from singlet to triplet excited states to amplify the emission, which is called the antenna effect. Such characteristics of LMOFs enable them to be potential luminescent probe materials. The inherent crystallinity of MOFs allows for precise structural determination by X-ray diffraction, providing exact knowledge of atomic positions and the interaction site that may be involved in the detection of analytes.35–39 In addition, the porosity of LMOFs provides multiple advantages to the related sensors. First and foremost, the porosity constrains analyte–MOF distance, providing possible close-interactions between the framework and analyte species. Second, alterations in the size and chemical environment such as hydrophilicity/hydrophobicity, polarizability etc. of the pores can be used to control the selectivity towards the sensed species. Last but not least, the porosity of the LMOFs can enhance the adsorption of the analyte into the pores, resulting in accumulated concentration of the analyte in the sensor and improving sensing performance.
 | ||
| Fig. 1 (a) Schematic representation of the varied origins of photoluminescence in a porous MOF wherein metal centers (pale blue) are linked by organic linkers (canary yellow) with an incorporated guest (orange). (b) Schematic illustration of the electronic states involved in the luminescence of an octahedral coordination complex. | ||
To realize sensing of biomolecules, luminescence design is crucial for achieving desired LMOFs. For example, the pore size of MOFs is important and should be tunable to encapsulate a definite luminescent guest species. It has been demonstrated that ligand exchange is useful for tuning the pore size of MOFs by using organic ligands with varied lengths. In addition, post-synthetic modification (PSM) is powerful to introduce functional groups into frameworks; for instance, excitation wavelength can be changed from the ultraviolet (UV) to visible region by introducing an amino group in the organic ligand of the framework. Therefore, in the following section on the design outline of LMOFs, we will summarize the design and fabrication strategies of LMOFs according to the building units of MOFs, namely linkers, nodes and guest species.
2.1 Design outline of LMOFs
Development of MOFs as solid-state luminescent materials takes advantage of their structural diversity, functionality, crystallinity, porosity and reusability compared to traditional luminescent materials.40 LMOFs can be designed by tailoring their structures with fluorescent linkers such as organic components and metal centers, and/or by introducing guest molecules such as organic dyes, transition metal complexes, and quantum dots (QDs) into the frameworks (Fig. 2). Such synthetic flexibility of the organic and inorganic components together with the voids within MOFs offers ample possibilities for tuning their photophysical properties. | ||
| Fig. 2 Approaches described in this article for designing LMOFs. | ||
 | ||
| Fig. 3 (a) Preparation of bio-MOF-101-BCPPE from bio-MOF-101 via ligand exchange. (b) Photographs of bio-MOF-101, H2BCPPE and bio-MOF-101-BCPPE showing different colors upon UV irradiation. (c) Emission spectra of H2BCPPE, bio-MOF-101 and bio-MOF-101-BCPPE. Adapted from ref. 51. | ||
The ligand exchange process can be applied to adjust the pore size of MOFs and systematically changing the length of the ligands can precisely control the pore size of MOFs, which will enable the MOFs to encapsulate specific guest species to improve the properties of the MOFs.55–58 Furthermore, functional groups such as carboxylate, pyridyl, amino, amide, hydroxyl, azide and conjugated aromatic groups can be introduced into organic ligands to tune the coordination ability of the ligand, stability, photoluminescence and sensing properties of the resulting MOFs. For example, Eddaoudi and coworkers reported a 12-connected rare-earth secondary-building unit (SBU) based fcu-MOF, linked by 1,4-naphthalenedicarboxylate (1,4-NDC2−).59 Afterwards, a series of fcu-MOFs were obtained by replacing the ligand 1,4-NDC2− with NH2-BDC2−, 2-fluoroterephthalate (F-BDC2−) or 2-nitroterephthalate (NO2-BDC2−) to tune their luminescence properties and detection capacity.60 A UiO-66 analogue MOF UiO-66-(COOH)2 with uncoordinated carboxylate groups was obtained by using 1,2,4,5-benzenetetracarboxylic acid (H4BTEC) instead of terephthalic acid, and luminescent ligand 1,4-H2NDC was incorporated into UiO-66-(COOH)2via ligand exchange to generate UiO-66-Hybrid with mixed ligands of BTEC and 1,4-NDC. The uncoordinated carboxylate groups in UiO-66-Hybrid can further bind metal ions, and Eu@UiO-66-Hybrid with the emitting center Eu(III) was isolated by reaction of UiO-66-Hybrid with Eu(NO3)3·6H2O (Fig. 4a).61 As expected, UiO-66-Hybrid exhibits distinct emission compared with its parent MOF UiO-66-(COOH)2, and Eu@UiO-66-Hybrid gives characteristic emission of Eu(III) (Fig. 4b and c).
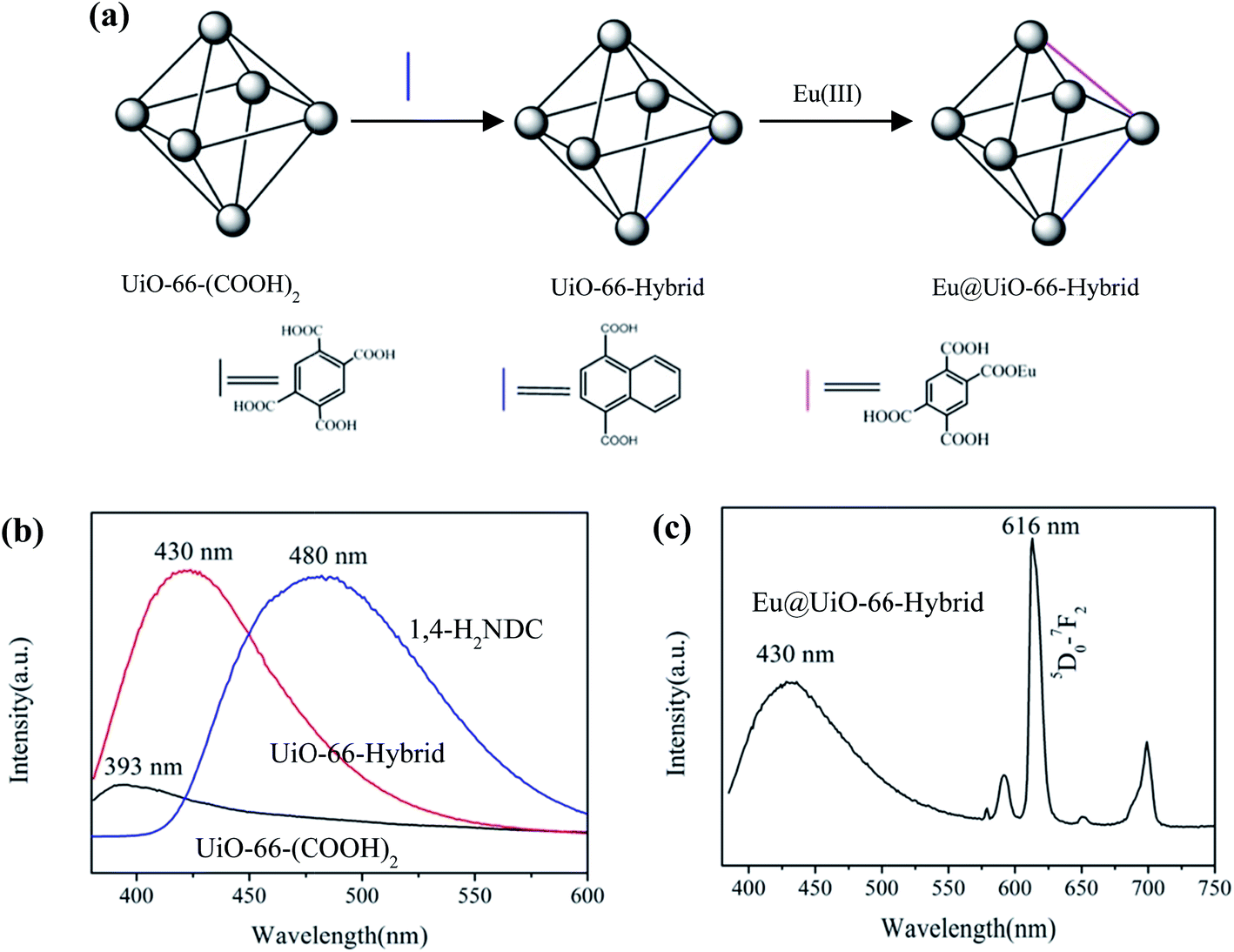 | ||
| Fig. 4 (a) Preparation of Eu@UiO-66-Hybrid through ligand exchange. Emission spectra of UiO-66-(COOH)2, UiO-66-Hybrid, 1,4-H2NDC (b) and Eu@UiO-66-Hybrid (c). Adapted from ref. 61. | ||
In addition to ligand exchange, metal ions as nodes in MOFs can also be exchanged to give opportunities for tuning the properties of MOFs. The analogue MOFs [M3(L)2(DABCO)(H2O)]·solvent (Zn3-MOF, M = Zn; Cu3-MOF, M = Cu) with the same framework structures were synthesized by solvothermal reactions of corresponding metal salts with mixed organic ligands [1,1′:3′,1′′-terphenyl]-4,4′′,5′-tricarboxylic acid (H3L) and 1,4-diazabicyclo[2.2.2]octane (DABCO). The Zn(II) ions in the Zn3-MOF can be exchanged with Cu(II) to give [Zn3Cu(L)2(DABCO)(H2O)]·solvent (Zn2Cu-MOF) (Fig. 5a and b), and the Cu2Zn-MOF could be obtained in the same way.62 It is interesting to find that these MOFs show distinct photoluminescence performance; particularly, in contrast to the non-emissive Cu3-MOF, the Cu2Zn-MOF displays strong emission (Fig. 5c). More importantly, the Zn2Cu-MOF and Cu2Zn-MOF with heterometallic centers could not be obtained by direct synthesis using mixed metal salts.62
 | ||
| Fig. 5 Photographs of metal-ion exchange from the crystals of the Zn3-MOF (a) to Zn2Cu-MOF (b), and emission spectra of the Zn3-MOF, Zn2Cu-MOF, Cu3-MOF and Cu2Zn-MOF (c). Adapted from ref. 62. | ||
PSM is another widely applied approach to functionalize organic linkers.63–67 Generally, an active site on a ligand is a prerequisite for transforming into another functional group. Qian et al. functionalized the pyridyl sites of MOF-867 with methyl groups, affording a cationic framework ZJU-101, which can be used for the removal of anionic Cr2O72−.64 Through a diazotization route, the amino group in IRMOF-3 was successfully converted into azide, which can be reduced into amine by H2S, leading to a turn-on fluorescence probe for H2S.65 A series of porous pcu MOFs based on rare earth metal (RE) clusters [RE4(μ3-OH)4(COO)6]2+ (RE3+ = Y3+, Sm3+, Eu3+, Gd3+, Tb3+, Dy3+, Ho3+, Er3+, Tm3+, and Yb3+) with amino functional groups were reported.66 An Yb3+-based MOF exhibits near-infrared emission and the excitation energy for Yb3+ sensitization can be carefully adjusted to lower energy, thus successfully shifting the excitation wavelength into the range suitable for biological imaging application (Fig. 6).66 However, the understanding of the effect of ligand functionalization on the framework is still limited. A study of the effect of polar functional groups on the dynamics of the frameworks based on the isostructural ZIF-8, ZIF-90 and ZIF-65 reveals that high pressure can convert ZIF-90 to a “gate open” phase, while ZIF-65 undergoes a transition to a “gate closed” structure as demonstrated by Moggach et al.67
 | ||
| Fig. 6 Porous pcu MOFs with varied functional amino groups. Adapted from ref. 66. | ||
Another important component of MOFs is metal centers acting as nodes in the formation of MOFs through coordination with organic ligands.68 Transition-metal ions with unpaired electrons can be efficient quenchers, while MOFs with d10 metal centers usually show luminescence because of their unique electronic configuration.69 It is noteworthy that lanthanide ions (Ln3+) can emit sharp, but weak luminescence since the transitions are forbidden by electric dipole selection rules.70
The fluorescence light-emitting mechanism is usually the charge transfer luminescence generated from an allowed charge-transfer from the excited state to the ground state. MLCT and LMCT are typical charge transfers found in MOFs. MLCT corresponds to the electronic transition from a metal-centered orbital to an organic linker-localized orbital, while LMCT involves the electronic transition from an organic linker-localized orbital to a metal-centered orbital. For instance, the MOF Cu3(C7H2NO5)2·3H2O (C7H2NO5 = 2,6-dicarboxy-4-hydroxypyridine) shows blue fluorescence at 398 and 478 nm upon excitation at 333 nm, while CuAg2(C7H3NO5)2 and the free organic linker 4-hydroxypyridine-2,6-dicarboxylic acid display green fluorescence at 515 and 526 nm upon excitation at 358 and 365 nm, respectively.71 Compared to the free organic linker, the MOF Cu3(C7H2NO5)2·3H2O displays two large blue shifts of 128 and 48 nm, while CuAg2(C7H3NO5)2 shows one small blue shift of 11 nm, suggesting that the luminescence emission of Cu3(C7H2NO5)2·3H2O may be originated from MLCT. The calculated energy bands support that the luminescence of the MOF Cu3(C7H2NO5)2·3H2O is ascribed to MLCT from the Cu 3d to the O 2p and N 2p orbital, while the luminescence of CuAg2(C7H3NO5)2 is originated from the π–π* transition of the organic linker. On the other hand, Cu(II) can also quench metal-based emission. For example, the uranyl complex UO2(C5H2N2O4)·H2O (C5H2N2O4 = 3,5-pyrazole dicarboxylate) exhibits characteristic UO22+ emission when either the linker or uranyl unit is excited; however, the Cu(II)-doped analogue (UO2)Cu(C5H2N2O4)2(H2O)2 displays no emission, regardless of the excitation wavelength.72 A similar phenomenon also occurred in the Cu(II) and Nd(III) heterometallic MOF Cu3(trans-3-(3-pyridyl)acrylate)6Nd2(NO3)6 and the homometallic one Nd(C8H6NO2)·3H2O; the presence of Cu(II) in the former one quenches the luminescence.73 MLCT luminescence has been observed in the Mn(II) MOF Mn(Hbidc) (H3bidc = 1H-benzimidazole-5,6-dicarboxylic acid).74 This Mn(II) MOF exhibits a strong red emission in the range of 625–850 nm. The strongest emission in Mn(Hbidc) is located at 726 nm, which is significantly red-shifted from the original emission at 440 nm observed in the free organic linker H3bidc. The strong emission of Mn(Hbidc) is attributed to MLCT luminescence, in which the relatively large π-conjugated system of the benzimidazole ring within Hbidc2− enforces the charge transfer from the Mn2+ ion to the organic linker. A few cases of luminescent silver MOFs have been reported in which charge-transfer character of the emission was observed. Excitation of the two-dimensional (2D) layered structure Ag(4-(2-pyrimidylthiomethyl)benzoate) at λ = 370 nm produces an intense green emission with a peak maximum at 530 nm, and the origin of the emission was assigned to LMCT and/or MLCT modified by metal-centered (ds/dp) states having Ag–Ag interactions.75 The complex [Ag(bpy)]n[Ag(1,2,4-HBTC)]n (bpy = 4,4′-bipyridine, 1,2,4-H3BTC = 1,2,4-benzenetricarboxylic acid) exhibits an intense fluorescent emission with a maximum at 502 nm (λex = 410 nm) due to MLCT.76 Wang and coworkers utilized the 4-cyanobenzoate (L) ligand to construct a 2D network, [AgL]n·nH2O, which exhibits tunable yellow-to-white photoluminescence by variation of excitation light (Fig. 7a).77 The solid-state sample of [AgL]n·nH2O displays broad emissions centered at 427 and 566 nm when excited under 355 and 330 nm UV radiation. While the high energy emission has a life time of 0.87 ns typical for fluorescence and is assigned to the π–π* intra-ligand transition, the low energy emission exhibits a long lifetime of 2.60 ms suggesting a phosphorescence characteristic. This emission was explained by the presence of an MLCT transition between the Ag 4d electrons and the π* orbital of the ligand. When the excitation light was adjusted to 349–350 nm, the emission peaks at 427 and 566 nm are comparable in intensity, which results in a white-light emission with a quantum yield of 10.86% (Fig. 7b). The Commission International de l'Eclairage (CIE) chromaticity coordinates of the white-light emissions excited by 350 and 349 nm light are approximately (0.31, 0.33) and (0.33, 0.34), respectively, close to that of pure white light (Fig. 7c).
 | ||
| Fig. 7 (a) Structure of the Ag-MOF. (b) Emission spectra of the Ag-MOF excited by 355 and 330 nm UV radiation. (c) CIE chromaticity coordinates of the white-light emissions excited by 350 and 349 nm light. Adapted from ref. 77. | ||
LMCT was reported in a wide range of Zn(II) and Cd(II) MOFs, in which the d electrons are in the core-like orbital, primarily in structures containing benzene derivatives, wherein a green colored emission was observed. For example, MOFs [Zn(2,3-pydc)(bpp)]3·2.5H2O and [Cd(2,3-pydc)(bpp)(H2O)]·3H2O (2,3-H2pydc = pyridine-2,3-dicarboxylic acid, bpp = 1,3-bis(4-pyridyl)propane) display intense fluorescent emissions at 436 and 438 nm upon excitation at 372 and 370 nm, respectively, although the organic linker 2,3-pydcH2 only gives very weak photoluminescence upon excitation at 370 nm.78 The homochiral Cd-MOF Cd3(dtba)3(bpp)3 (H2dtba = 2,2′-dithiobisbenzoic acid) exhibits temperature dependent luminescence. The room temperature blue emission of Cd3(dtba)3(bpp)3 at 434 nm with a shoulder peak at 482 nm is attributed to the metal perturbed intra-ligand emission of dtba or bpp when excited at 355 nm, while the emission at 10 K at about 507 nm originates LMCT (Fig. 8).79
 | ||
| Fig. 8 Fluorescence emission spectra of the Cd-MOF at different temperatures (blue: 10 K; red: 298 K). Adapted from ref. 79. | ||
Lanthanide ions (Ln3+) are characterized by a gradual filling of the 4f orbital, from 4f0 (for La3+) to 4f14 (for Lu3+). These electronic [Xe]4fn configurations (n = 0–14) generate a variety of electronic energy levels, resulting in intricate optical properties (Fig. 9a).80–83 Such electronic energy levels are well defined due to the shielding of the 4f orbital by the filled 5s25p6 sub-shells, and they are less sensitive to the chemical environments around lanthanide ions. Consequently, each lanthanide ion exhibits narrow and characteristic 4f–4f transitions. Ln3+ ions except La3+ and Lu3+ can generate luminescent f–f emissions from the UV to visible or near-infrared (NIR) range. Eu3+, Tb3+, Sm3+ and Tm3+ emit red, green, orange and blue light, respectively, while Yb3+, Nd3+ and Er3+ display the well-known near-infrared luminescence (Fig. 9b).84
 | ||
| Fig. 9 (a) Electronic excited-state energy levels for the Ln3+ series. (b) Luminescence spectra of some lanthanide ions. Adapted from ref. 81 and 84. | ||
Unfortunately, lanthanide ions suffer from weak light absorption and low quantum yields due to the forbidden f–f transitions, making the direct excitation of lanthanides very difficult unless high-power laser excitation is employed. This problem can be overcome by complexation of the desired lanthanide with a strongly absorbing linker, which can sensitize the lanthanide ions by the so-called “antenna effect”: light is absorbed by organic ligands around the lanthanide ions, energy is transferred to the lanthanide ions from organic ligands, and then luminescence is generated from the lanthanide ions.85 For example, Ng and coworkers reported a series of Ln-MOFs: [Ln(ADA)1.5(phen)]n (Ln3+ = Eu3+, Gd3+, Tb3+, La3+, Ce3+, Pr3+, Nd3+, and Y3+; phen = 1,10-phenanthroline) for sensing Ni2+ by introducing the ligand 1,3-adamantanediacetic acid (H2ADA). Further investigation found that [Eu(ADA)1.5(phen)]n is a bifunctional chemosensor, which can be used to sense Ni2+ through luminescence turn-off and to detect the amino acid valine (Val) by luminescence turn-on.86
Because of the similar chemical properties of lanthanide ions, the Ln-SBUs can be easily replaced by each other, affording mixed-Ln-MOFs, which may show the characteristic emissions of each lanthanide ion. For example, by doping Eu3+ into an isostructural Tb-MOF to produce a mixed-Ln-MOF EuxTb1−x-DMBDC (DMBDC = 2,5-dimethoxy-1,4-benzenedicarboxylate), a targeted self-referencing luminescent thermometer based on the emissions of Tb3+ and Eu3+ was realized in a wide temperature range from 10 to 300 K (Fig. 10a).87 Different from the traditional solid-solution type mixed-Ln-MOF mentioned above, core–shell type mixed-Ln-MOF hierarchical single crystals were also achieved via a solution-mediated epitaxial growth method.88 In the resultant MOF crystals, each component can maintain its own luminescence, and multicolor to white light emissions are achieved through the color mixing technique by controlling the domain and orientation of the hierarchical crystals (Fig. 10b).
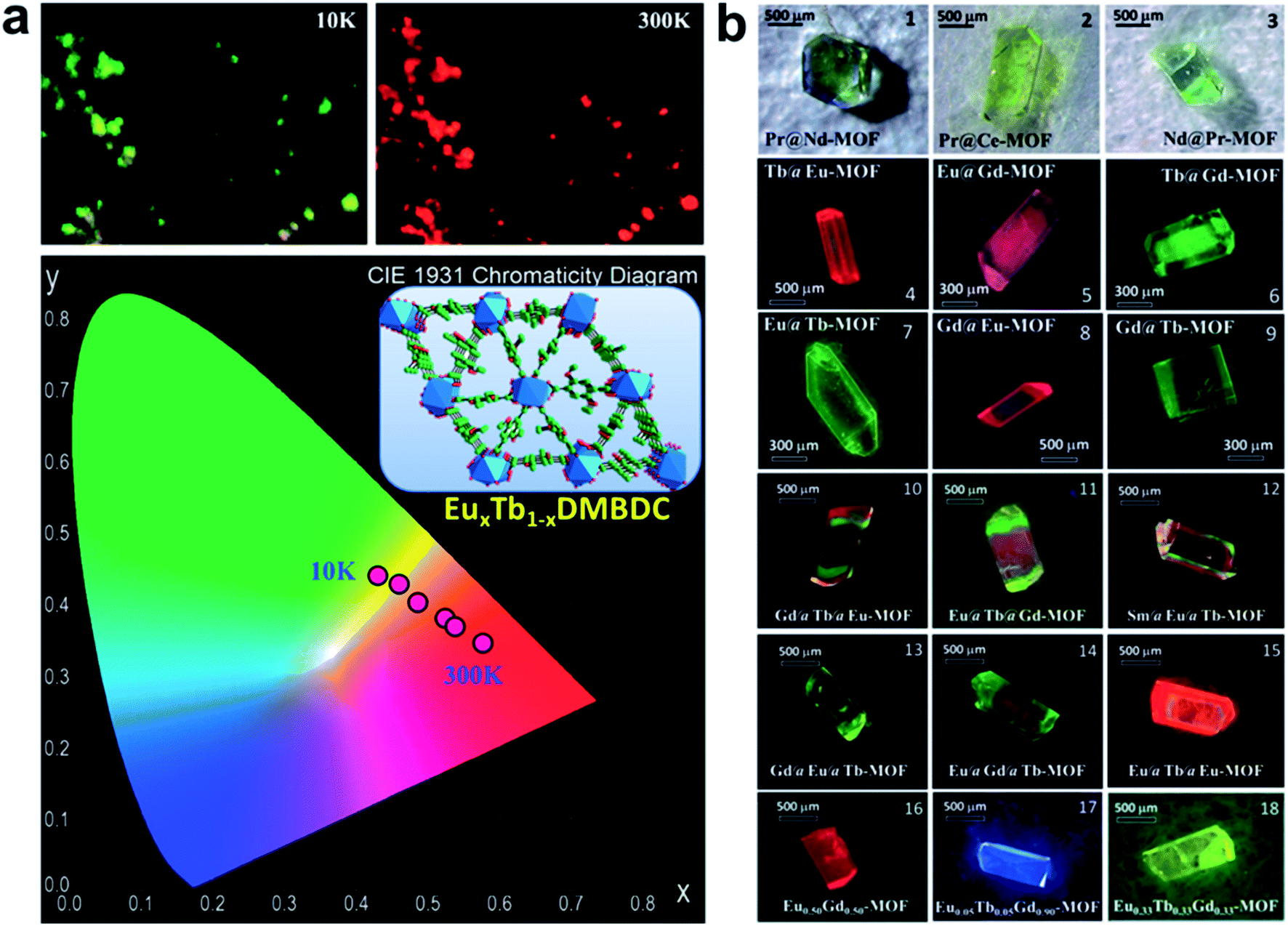 | ||
| Fig. 10 (a) Photograph and CIE chromaticity diagram of the LMOF EuxTb1−xDMBDC at 10 and 300 K, excited at 312 nm. (b) Photographs of bimetallic and trimetallic hierarchical single crystals showing visually distinguishable colors. Adapted from ref. 87 and 88. | ||
 | ||
| Fig. 11 Schematic illustration of Ln3+ incorporation into bio-MOF-1 and the corresponding excitation and emission spectra. Adapted from ref. 90. | ||
Yan and coworkers prepared MOF-76(Y):Ln hybrids via ionic doping of MOF-76 developed by Yaghi's group.91,92 The photoluminescence color of MOF-76(Y):10 mol% Eu3+/10 mol% Tb3+ could be tuned from yellow to yellow-green, warm white and orange by changing the excitation wavelength in the range of 300–380 nm. Later, a series of MOF-76(Ln) compounds were reported by using the ionic doping approach and their adsorption ability for different dyes was evaluated. The samples soaked in cationic dyes were dyed blue, red, violet and pink, but the samples soaked in anionic dyes remained colorless.93 Furthermore, lanthanide ions can be incorporated into MOFs to realize color tuning and up-conversion materials.94–97
Fluorescent dyes are also fascinating chromophore guests due to their high photoluminescence quantum yield and excellent optical and electronic properties. Incorporating organic dyes into the porous structure of MOFs can also tune the photophysical properties of MOFs. For instance, 2-thenoyltrifluoroacetone (TTA) was implanted into the Eu3+ (or Eu3+/Tb3+)-exchanged bio-MOF-1 via the “ship-in-bottle” method, and it not only can improve the luminescence intensity of Eu3+ but also can slightly change the emission intensity of the bio-MOF.98 The emission spectrum of double lanthanide ion (Eu3+/Tb3+)-functionalized bio-MOF-1 hybrids showed CIE coordinates close to those of the white region. Yan's group reported DCM@MOFs (DCM = 4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran) with well-defined blue/red two-color emission, exhibiting selective luminescent response to volatile organic compounds.99 Wu's group established a luminescent dye@MOF platform for probing volatile organics based on two emissions of the host and guest.100 By treatment of 4,8-disulfonyl-2,6-naphthalenedicarboxylic acid (H4L) with InCl3 under solvothermal conditions, the anionic MOF {[Me2NH2]0.125[In0.125(H2L)0.25]·xDMF}n with a three-dimensional (3D) framework structure and large one-dimensional (1D) channels was obtained. This In(III) MOF can serve as a host for adsorbing fluorescent dyes effectively to generate luminescent dye-loaded MOFs, and the dye-loaded MOFs realized the probing of different organic solvent molecules by tuning the energy transfer efficiency between two different emissions, especially for sensing DMF. The luminescence response of the dye-loaded MOFs is sensitive to organic solvent molecules, in which the intensity ratio of blue-to-red emission varies after exposure to specific molecules, showing that the materials have potential for application in the fabrication of ratiometric luminescent sensors (Fig. 12).101
 | ||
| Fig. 12 3D framework of {[Me2NH2]0.125[In0.125(H2L)0.25]·xDMF}n and photographs of the corresponding crystals after immersion in solution of dyes. Adapted from ref. 101. | ||
Encapsulation of luminescent nanoparticles within nonluminescent MOFs is another strategy to give a material that can be used for luminescence sensing.102–104 For example, by encapsulating branched poly-(ethylenimine)-capped carbon quantum dots (BPEI-CQDs) with a high fluorescence quantum yield into a zeolitic imidazolate framework material (ZIF-8), a fluorescent-functionalized MOF, BPEI-CQDs/ZIF-8, was obtained. The fluorescent-functionalized MOF not only maintains the fluorescence activity and sensing selectivity derived from BPEI-CQDs but also can strongly and selectively accumulate target analytes due to the adsorption property of the MOF. The obtained BPEI-CQDs/ZIF-8 composite has been used to develop an ultrasensitive and highly selective sensor for Cu2+ ions, with a wide response range (2 to 1000 nM) and a very low detection limit of 80 pM, and has been successfully applied in the detection of Cu2+ ions in environmental water samples.105 Besides CQDs, CdTe/CdS/ZnS QDs can also be encapsulated into ZIF-8 to form a QDs/CDs@ZIF-8 composite for detection of Cu2+. The dispersed sample of the QDs/CDs@ZIF-8 composite in water exhibits orange luminescence, with emission peaks at 430 and 620 nm upon excitation at 370 nm. When Cu2+ was added to the suspension of the QDs/CDs@ZIF-8 composite, the luminescence changed from orange to purple due to the quenching of the 620 nm emission while the luminescence at 430 nm remained unchanged. The limit of detection was calculated to be 1.53 nM (Fig. 13).106
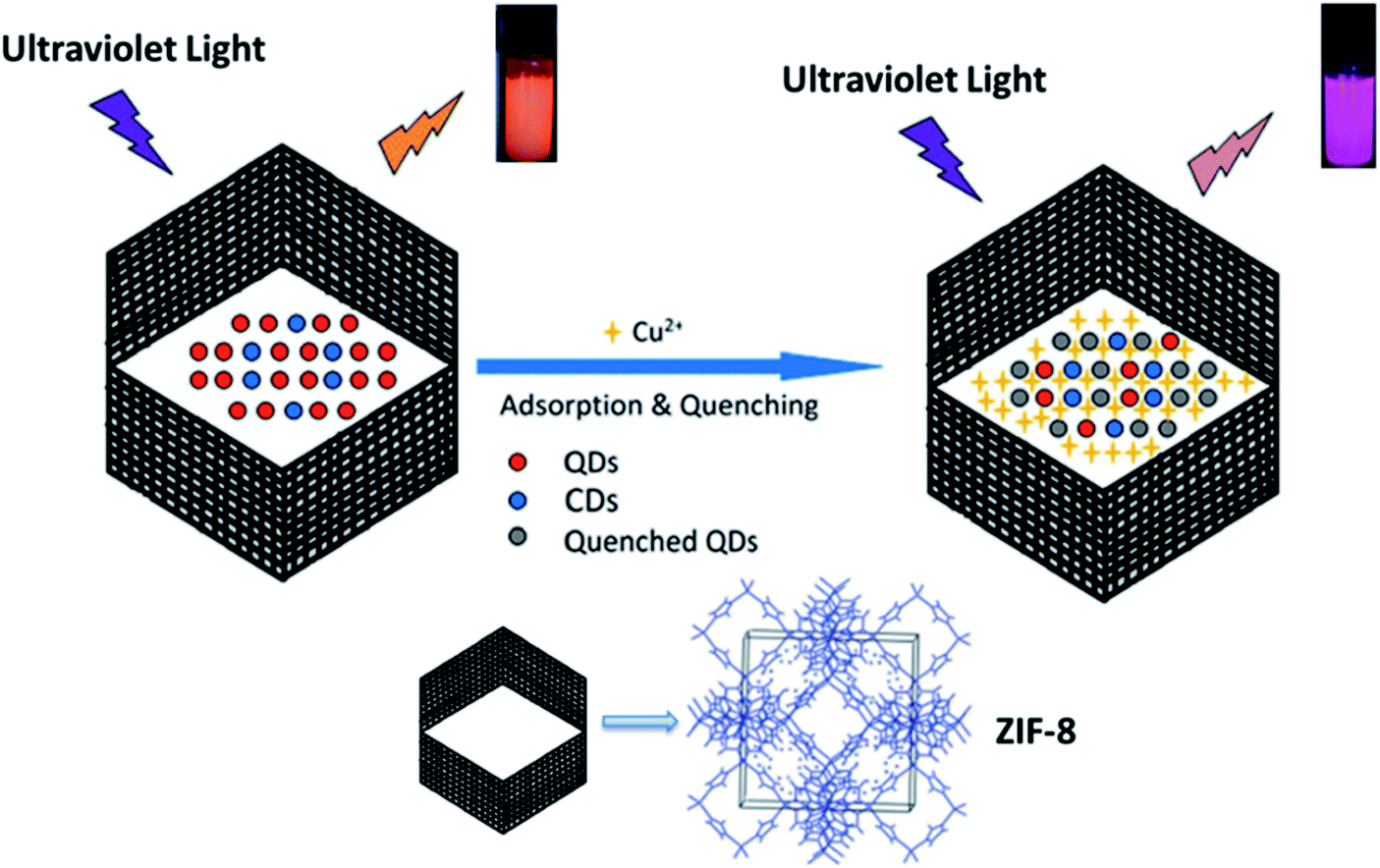 | ||
| Fig. 13 Process for sensing Cu2+ based on the fluorescent QDs/CDs@ZIF-8 composite. Adapted from ref. 106. | ||
2.2 Application of LMOFs for sensing biomolecules
Biosensing has been proved to be a promising tool to detect a wide range of compounds rapidly and selectively, with applications in security,107 healthcare for point-of-care diagnostics108 and environmental safety.109 Development of chemical approaches for the detection and screening of biological molecules is an important and highly active field of research because of the scientific and practical significance of biosensors. The increasing demand for accurate sensing has significantly promoted the design and fabrication of functional materials. Varied organic or inorganic materials have been investigated and employed as platforms for designing biosensors. Some of the unique properties of MOFs (dispensability, luminescence, non-toxicity, and biodegradability), which are essential for in vivo applications, especially in the case of nanocrystal MOFs (NMOFs), appear to be attractive but remain largely unexplored. Of particular interest are chemical sensors owing to their potential application in environmental and biological systems.110–118The biosensing approaches using LMOF-based biosensors can be summarized in the following ways: (i) LMOFs are used as fluorescence quenchers toward the fluorophores of analytes based on fluorescence resonance energy transfer (FRET),119 photoinduced electron-transfer (PET)120 or charge transfer;121 (ii) intentional preparation of MOFs combining fluorescence or luminescence properties, which can sensitively respond to their local environment or guest species.25 Although the chemosensing applications of LMOFs have been abundantly reported in the literature, biological applications have been limited due to the challenging issues described below. Because of their highly tunable characteristics, LMOFs are attractive candidates as biosensors for certain analytes.11,122,123
 | ||
| Fig. 14 (a) The principle of the sensing platform. (b) Fluorescence spectra of the FAM-labeled DNA–Cu(H2dtoa) in the presence of different concentrations of target DNA. (c) Fluorescence spectra of the FAM-labeled probe DNA 2–Cu(H2dtoa) in the presence of different concentrations of thrombin. Adapted from ref. 124. | ||
The same quenching mechanism also existed in the detection of HIV-1 double-stranded DNA (dsDNA) and Sudan virus (SUDV) RNA sequences using [Cu3(Cmdcp)2(dps)4·(H2O)4(SO4)]n (H3CmdcpBr = N-carboxymethyl-3,5-dicarboxylpyridinium bromide; dps = 4,4′-dipyridyl sulphide). The MOF [Cu3(Cmdcp)2(dps)4·(H2O)4(SO4)]n exhibited strong quenching properties through π–π stacking, and electrostatic and hydrogen bonding interactions with nucleic acids, which were ascribed to the aromatic moieties, positively charged pyridinium and coordination unsaturated Cu(II) centers.120 Transient complexes were formed by two FAM-labeled probe DNA sequences with [Cu3(Cmdcp)2(dps)4·(H2O)4(SO4)]n referred to as P-DNA-1 and P-DNA-2, and were specific for HIV DNA and SUDV RNA molecules, respectively. The formation of an MOF–probe complex leads to a decrease in the photoluminescence intensity of the MOF. Then, this group using the same ligand H3CmdcpBr and lanthanide metals (Dy and La) constructed two water-stable MOFs {[Dy(Cmdcp)(H2O)3](NO3)·2H2O}n (Fig. 15a) and {[La4(Cmdcp)6(H2O)9]}n.125 The MOF {[Dy(Cmdcp)(H2O)3](NO3)·2H2O}n exhibits a chair-type pore shape with tessellating H2O and free NO3− anions on the pore surface, which can form electrostatic, π-stacking and/or hydrogen bonding interactions with P-DNA, and this transient complex can be used as an effective, selective and fluorescent sensing platform for the detection of Ebola virus RNA sequences. {[La4(Cmdcp)6(H2O)9]}n can adsorb carboxyfluorescein (FAM)-tagged probe DNA (P-DNA) and quench the fluorescence of FAM by a PET process (Fig. 15b and c). Besides the ligand H3CmdcpBr discussed above, ligands H3CbdcpBr (N-(4-carboxybenzyl)-3,5-dicarboxylpyridinium bromide) and H2dcbbBr (1-(3,5-dicarboxybenzyl)-4,4′-bipyridinium bromide) were also used to prepare water-stable MOFs such as {[La2(Cbdcp)3(H2O)10]}n, {[Zn(HCbdcp)2]H2O}n, [Cu(dcbb)2]n and {[Cu(dcbb)2(H2O)2]·10H2O}n for sensing virus DNA or RNA by a PET process.126–128
 | ||
| Fig. 15 (a) Mechanism of fluorescent biosensors based on {[Dy(Cmdcp)(H2O)3](NO3)·2H2O}n (Dy-MOF). (b) Fluorescence spectra of P-DNA (50 nM) incubated with different concentrations of Dy-MOF. (c) Fluorescence spectra of the P-DNA@Dy-MOF system (50 nM/11 μM) incubated with different concentrations of T0. Adapted from ref. 125. | ||
Several other functional MOFs, including UiO-66-NH2,129 UiO-66,130 MIL-101,131,132 MIL-88A,119,133,134 [Gd(TIA)(HCOO)]n (TIA = 5-triazole isophthalate), {[La2(TDA)3]·2H2O}n (TDA = 2,2′-thiodiacetic acid), Cd(L)·(HDMA)2(DMF)(H2O)3 (H4L = bis-(3,5-dicarboxy-phenyl)terephthalamide, DMA = N,N-dimethylacetamide), Zn(L)·(HDMA)2(DMF)(H2O)6 and [Cu3(BTC)2(H2O)3]n (BTC = 1,3,5-benzenetricarboxylate),135–137 have also been employed as fluorescent sensing platforms for DNA/RNA detection with good sensitivity and selectivity. Such MOFs mentioned above can be applied as fluorescence quenchers to recognize DNA/RNA through reasonable fluorescence changes. For instance, UiO-66-NH2 can act as a fluorescent sensing platform for DNA detection (Fig. 16).129 The fluorophore (FAM) labeled free ssDNA shows strong fluorescence emission. When UiO-66-NH2 is introduced, the PET process may occur due to the π–π stacking and/or hydrogen bonding interactions between the ssDNA and UiO-66-NH2, which leads to the fluorescence quenching of FAM (off-state). When the target ssDNA is added to the system, the dsDNA separates from UiO-66-NH2, and results in the recovery of the fluorescence of ssDNA (turn-on state). In addition, such biomolecule sensing platforms can also be used to distinguish complementary and mismatched target sequences. An oligonucleotide sequence associated with HIV was employed for this purpose. The fluorescence of a FAM labeled ssDNA probe (PHIV) is strong, and the addition of UiO-66-NH2 resulted in a significant decrease in fluorescence intensity. After the complementary target T1, single-base mismatched target T2 and mismatched target T3 to the ssDNA were added into the UiO-66-NH2-PHIV composite, the fluorescence intensity increased by 67%, 26% and 14%, respectively. The results demonstrate that this sensing platform can identify the complementary and mismatched target sequences. In general, it can be seen that the design principle for such DNA sensing platforms is the use of three components, i.e., a probe ssDNA, MOF and target ssDNA, based on a “mix-and-detect” strategy. Namely, the fluorophore labeled probe ssDNA as an illuminant mixes with the MOF as a fluorescence quencher (turn off), and then the target ssDNA binds with the probe ssDNA to generate dsDNA and recovers the fluorescence (turn on).
 | ||
| Fig. 16 3D structure of UiO-66-NH2 and the proposed principle for detection of fluorophore-labeled DNA by UiO-66-NH2. Adapted from ref. 129. | ||
 | ||
| Fig. 17 (a) 3D structure of [La2(atp)3(H2O)2]·DMF·4H2O. (b) Change of the fluorescence spectra of [La2(atp)3(H2O)2]·DMF·4H2O with the enzymatic oxidation of catechol. Inset: photographs of fluorescence change of [La2(atp)3(H2O)2]·DMF·4H2O with the enzymatic reaction under UV irradiation from a handheld UV lamp (centered at 365 nm). (c) Response mechanism of [La2(atp)3(H2O)2]·DMF·4H2O to o-quinone. Adapted from ref. 138. | ||
Besides enzymes, proteins can also be identified using functionalized LMOFs. Recently, a fluorescence proximity assay (FPA) based on the MOF UiO-66-NH2 was developed for sensing target proteins sensitively and selectively.140 Moreover, a simple luminescence approach based upon MOF-5 conjugated with anti-BSA was also reported for BSA sensing.141 The authors demonstrated the covalent conjugation of anti-BSA on a carboxyl-functionalized MOF-5 surface. The photoluminescence properties of the bioconjugate were then investigated in the presence of the target analyte (BSA). The immunocomplex formation showed considerable quenching of the bioconjugate without affecting the structural stability of the MOF. In addition to the MOFs discussed above, the fluorescent composite material of up conversion nanoparticles (UCNPs) and HKUST-1 was also prepared for sensing proteins.142 The composite UCNPs/HKUST-1 was prepared by the reaction of copper nitrate with H3BTC after dispersing the polyacrylic acid modified UCNPs in ethanol through a solvothermal method (Fig. 18a). The fluorescence intensity of the composite reduced gradually with the increase of bovine hemoglobin (BHB) concentration (Fig. 18b). The detection limit is 0.062 mg mL−1, which is higher than that of traditional molecularly imprinted polymers (MIPs).
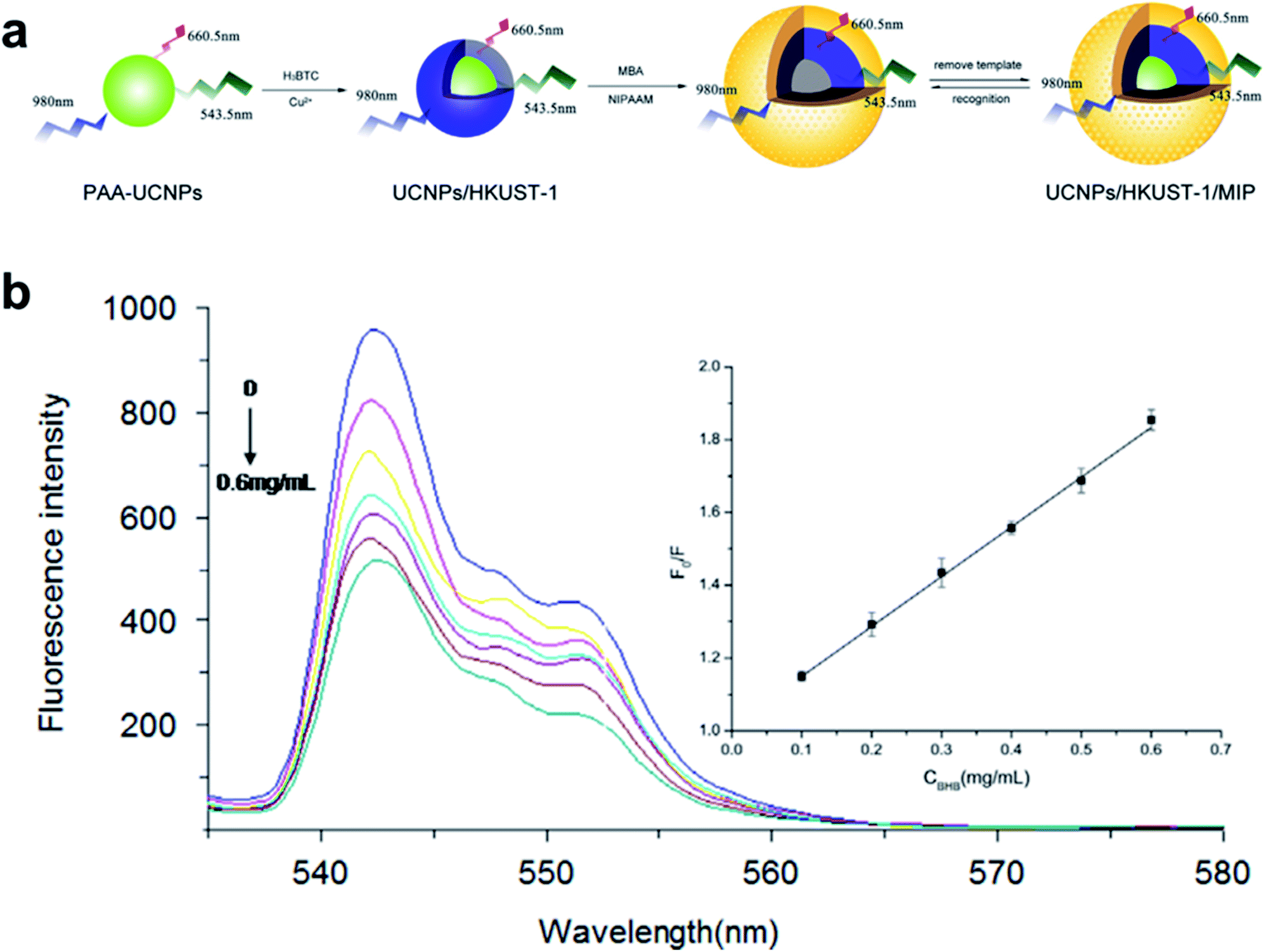 | ||
| Fig. 18 (a) Synthesis of fluorescent materials based on UCNPs and MOFs. (b) Fluorescence emission spectra of fluorescent materials with the addition of a certain concentration of BHB protein solution. The inset is the Stern–Volmer curve. F and F0 are the fluorescence intensities of the composite materials in the presence and absence of BHB. Adapted from ref. 142. | ||
 | ||
| Fig. 19 Proposed mechanism for sensing small-sized amino acids Gly and Ser via destruction of the MOF. Adapted from ref. 144. | ||
Moorthy and co-workers recently synthesized a homochiral and water-stable pyrene-tetralactic acid ligand (H4PLA) based Zn-MOF, Zn-PLA, with strong fluorescence.145 The fluorescence of Zn-PLA can be quenched specifically by histidine (His) among the tested amino acids. Both D-(+)- and L-(−)-His exhibit the fluorescence quenching effect. The reason was proposed to be based on the exchange of the cationic dimethylammonium species in the MOF by protonated His in water. Furthermore, the fluorescence quenching of Zn-PLA by D-(+)-His was thermodynamically more favorable than that by L-(−)-His. Therefore, the homochiral Zn-PLA can be used as an enantioselective fluorescent sensor for His. More recently, Zhang et al. successfully prepared a water-stable 3D homochiral MOF, {[Cd(L)(bpy)]·DMA·5H2O}n, from achiral ligands 4,4′-((naphthalene-1,4-dicarbonyl)bis(azanediyl))dibenzoic acid (H2L) and bpy. The π-conjugated naphthyl moiety acts as a fluorescence emission center, and the LMOF can be a fluorescence sensor for enantioselective detection of penicillamine (PEN) in water, a sulfur-containing amino acid.146 The fluorescence of {[Cd(L)(bpy)]·DMA·5H2O}n in aqueous solution is strong under 340 nm excitation. When D- or L-enantiomers were added to the system, the fluorescence intensity obviously decreased; however, the change caused by D-PEN was more significant than that caused by L-PEN. This is mainly because the formation of intermolecular hydrogen bonds between D-PEN and the Cd-MOF is easier than that between L-PEN and the Cd-MOF.
It has been reported that the Eu3+-functionalized MOF Eu3+@MIL-121 with high water tolerance can be employed as a highly selective and sensitive luminescent probe for hippuric acid (HA) in urine, i.e. benzoylglycine, which is a derivative of Gly and is considered to be the final and main metabolite of toluene. Eu3+@MIL-121 was achieved by encapsulating Eu3+ ions into the channels of MIL-121 (Fig. 20a).148 The shortened emission lifetime and decreased luminescence intensity of Eu3+ within Eu3+@MIL-121 upon addition of HA imply that HA may weakly coordinate to Eu3+ resulting in the quenching of luminescence (Fig. 20b and c). Another reason is that the energy absorbed by the ligand could be transferred to the HA molecules due to the presence of intermolecular π–π stacking and/or hydrogen bonding interactions between the framework and HA, which reduced the efficiency of the ligand to Eu energy transfer and resulted in the quenching effect on the luminescence intensity of Eu3+.
 | ||
| Fig. 20 (a) Schematic representation of the fabricated Eu3+@MIL-121 as a fluorescent sensor for HA. (b) Suspension-state photoluminescence spectra. (c) The relative intensities of 5D0 → 7F2 at 614 nm for Eu3+@1 dispersed in various aqueous solutions. Adapted from ref. 148. | ||
 | ||
| Fig. 21 (a) Principle of fluorescence detection of glucose and H2O2 based on the MIL-53(Fe) nanozyme. (b) Fluorescence spectral changes in response to glucose concentration based on MIL-53(Fe). Adapted from ref. 149. | ||
More recently, a similar fluorescence sensor has been developed for the detection of hydrogen peroxide by using a 2D nanosheet MOF, In-aip (H2aip = 5-aminoisophthalic acid), HRP as a catalyst and catechol as a reaction substrate.150 In this enzyme-assisted system, the fluorescence quenching of the In-aip MOF is realized by o-benzoquinone, the oxidation product of catechol catalyzed by HRP, due to the interactions between the o-benzoquinone and amino group of the aip2− ligand in the In-aip MOF. Furthermore, glucose can be oxidized to glucose acid together with the generation of H2O2 in the presence of O2 catalyzed by GOx as mentioned above, and thus this H2O2 sensing system can also be used to detect glucose with the assistance of GOx. This In-aip glucose sensing system has high selectivity and accuracy.150
In contrast to the complicated glucose sensing systems with the assistance of enzymes as described above, an amine functionalized isoreticular MOF, IRMOF-3, was utilized as a fluorescent sensor for selective detection of glucose without any modification.151 When IRMOF-3 was added to the analyte solution of glucose, the fluorescence intensity of IRMOF-3 decreased obviously due to the interactions occurring at the MOF surface between the diol groups of the glucose and –NH2/–COO− ones in the ligands of the MOF IRMOF-3. It was found that the fluorescence intensity decreased linearly with the logarithm concentration of glucose in the range of 1 to 225 μM and the detection limit of glucose is 0.56 μM.
In addition, a paper-based biosensor was designed for the detection of glucose in human serum and urine using the Ag@Au nanoprism-Ir-Zne MOF.152 The phosphorescent Ir-Zne was obtained by reaction of Zn(II) with [Ir(ppy)2(H2dcbpy)]PF6 (ppy = 2-phenylpyridine, H2dcbpy = 4,4′-dicarboxy-2,2′-bipyridine). Zhao et al. reported a metal–organic gel (MOG) obtained by combining Cu(II) and Co(II) with the organic ligand 2,4,6-tri(4-carboxyphenyl)-1,3,5-triazine as a fluorescence sensing system for detecting H2O2 and glucose.153 The detection limit of glucose is 0.33 μM. The nanocomposite g-C3N4@MOF obtained using graphitic C3N4 nanosheets (g-C3N4) and the Cu(II)-TA MOF (CuMOF) was developed for the fluorescence sensing of H2O2 and glucose.154 The determination concentration of glucose ranges from 0.1 to 22 μM, and the detection limit is 59 nM.
 | ||
| Fig. 22 Schematic drawing of Ce-MOFs for the detection of AA (left and right) and change of emission spectra of ZJU-136-Ce upon addition of AA (center). Adapted from ref. 158. | ||
We found that a series of Cd(II)-based LMOFs [Cd2Na(L)(BDC)2.5]·9H2O, [Cd2(L)(2,6-NDC)2]·DMF·5H2O and [Cd2(L)(BPDC)2]·DMF·9H2O (BDC2− = 1,4-benzenedicarboxylate, 2,6-NDC2− = 2,6-naphthalenedicarboxylate) can act as potential luminescence sensors for detecting antibiotics upon introducing the amino triazole ligand N1-(4-(1H-1,2,4-triazole-1-yl)benzyl)-N1-(2-aminoethyl)ethane-1,2-diamine (L).160 The strong emissions of these MOFs can be quenched efficiently by trace amounts of NF (NZF; NFT; furazolidone, FZD) antibiotics. The detection limits of these MOF sensors toward NZF are about 162, 75 and 60 ppb, respectively. The results illustrate that these LMOFs show fast response and high sensitivity for trace amounts of antibiotics (Fig. 23).160
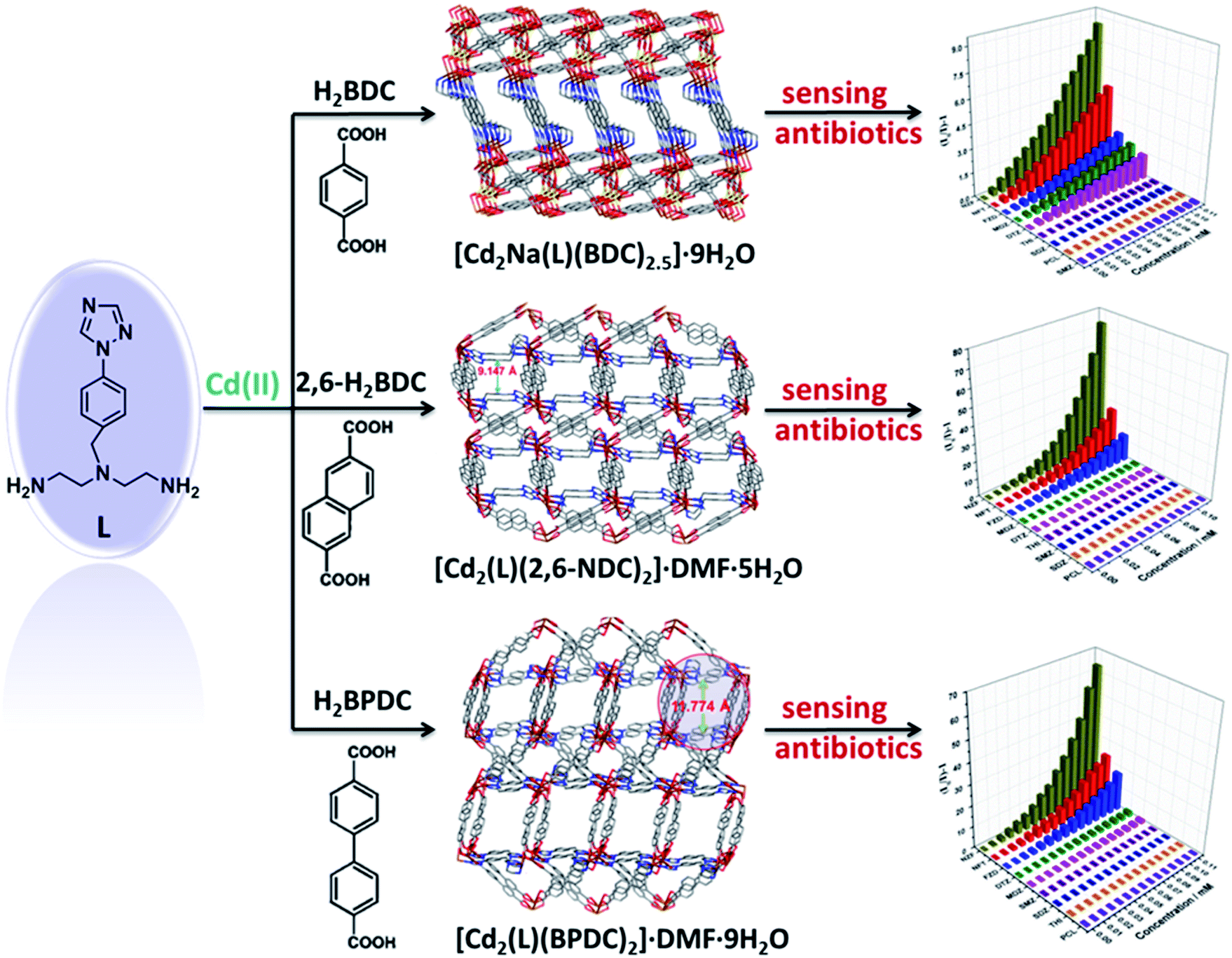 | ||
| Fig. 23 Synthetic procedure for the preparation of luminescent Cd(II)-MOFs and their Stern–Volmer plots for sensing antibiotics. Adapted from ref. 160. | ||
A lanthanide LMOF [Eu2(BCA)3(H2O)(DMF)3]·0.5DMF·H2O (Eu-BCA, BCA2− = 2,2′-biquinoline-4,4′-dicarboxylate) thin-film coated on a SSWM (stainless steel wire mesh) was synthesized and it was found that the Eu-BCA thin-film shows good luminescence properties and recyclability for selectively sensing NFT and NZF antibiotics.161 Similarly, some other examples of LMOF-based chemical sensors for the detection of definite antibiotics have been reported. For instance, a Mg-LMOF, [Mg2(APDA)2(H2O)3]·5DMA·5H2O, achieved by reacting Mg2+ salt with the amino-functionalized ligand 4,4′-(4-aminopyridine-3,5-diyl)dibenzoic acid (H2APDA) can be used as a luminescent sensor for the effective detection of antibiotics with high selectivity and sensitivity.162 The detection limits of the Mg-LMOF toward NFT and NZF are about 126 and 108 ppb, respectively. LMOFs [Zn2(Py2TTz)2(BDC)2]·2DMF·0.5H2O and [Cd2(Py2TTz)2(BDC)2]·2DMF (Py2TTz = 2,5-bis(4-pyridyl)thiazolo[5,4-d]thiazole) with 2-fold interpenetrated 3D structures are able to detect antibiotics, particularly NZF in water.163 A carbazole-functionalized Cd-LMOF [Cd3(CBCD)2(DMA)4(H2O)2]·10DMA (H3CBCD = 4,4′-(9-(4′-carboxy-[1,1′-biphenyl]-4-yl)-9H-carbazole-3,6-diyl)dibenzoic acid) was designed and employed as a fluorescent sensor for the detection of nitrofuran antibiotics NFT and NZF with high sensitivity and selectivity due to the coaction mechanism of electron and energy transfers.164
In addition to the nitrofuran antibiotics, LMOFs have been successfully applied to the detection of sulfonamide and tetracycline antibiotics.165–167 Zhu et al. developed a Zn-LMOF, [Zn3(μ3-OH)(HL)L(H2O)3]·H2O (H3L = 5-(4-carboxy-phenoxymethyl)-isophthalic acid), with a highly ordered structure.165 It was found that this Zn-LMOF has good stability in simulated wastewater with high sensitivity and fast response as a biosensor for a series of sulfonamide antibiotics through fluorescence quenching.165 The In-based MOF [In2(sbdc)3(H2O)4]·(H2O)8 (sbdc2− = trans-4,4′-stilbenedicarboxylate) and the Zr-based MOF PCN-128Y have been demonstrated to be useful for detecting tetracycline antibiotics.166,167
In addition, lanthanide-functionalized MOFs can also be utilized for detecting dipicolinic acid (DPA), a unique and major constituent of bacterial spores, including Bacillus anthracis (anthrax).171 For example, Tb3+/Eu3+ co-doped bio-MOF-1 (Tb/Eu@bio-MOF-1) obtained by encapsulating Tb3+ and Eu3+ cations into bio-MOF-1 has been reported for ratiometric detection of DPA.172,173 Tb/Eu@bio-MOF-1 exhibits an orange-red emission color (615 nm) due to the efficient energy transfer from Tb3+ to Eu3+. In the presence of DPA, the fluorescence color of Tb/Eu@bio-MOF-1 changed to green (545 nm) gradually due to the chelation of DPA with Tb enabling ligand-to-metal energy transfer.
The luminescence intensity of Ln-MOFs can be tuned by host–guest interactions, thereby providing opportunities for chemical sensing of molecules such as lysophosphatidic acid (LPA), a biomarker of ovarian cancer. For example, a series of lanthanide zeolite-like isostructural mixed-crystal ZMOFs Eu0.2206Tb0.7794-ZMOF (MZMOF-1), Eu0.3525Tb0.6415-ZMOF (MZMOF-2) and Eu0.6059Tb0.3941-ZMOF (MZMOF-3) were achieved by varying the molar ratios of Tb3+ to Eu3+ and were found to exhibit the characteristic emission bands of both Tb3+ and Eu3+ (Fig. 24a).174 Particularly, MZMOF-3 was found to exhibit the ability to selectively detect LPA, even in the presence of major components of blood plasma. The fluorescence emission properties of MZMOF-3 with respect to LPA suggest an energy-transfer mechanism. The increased intensity of Eu3+ transitions and the decreased intensity of Tb3+ transitions induced by energy transfer from Tb3+ to Eu3+ were significantly enhanced by the presence of LPA (Fig. 24b and c).174
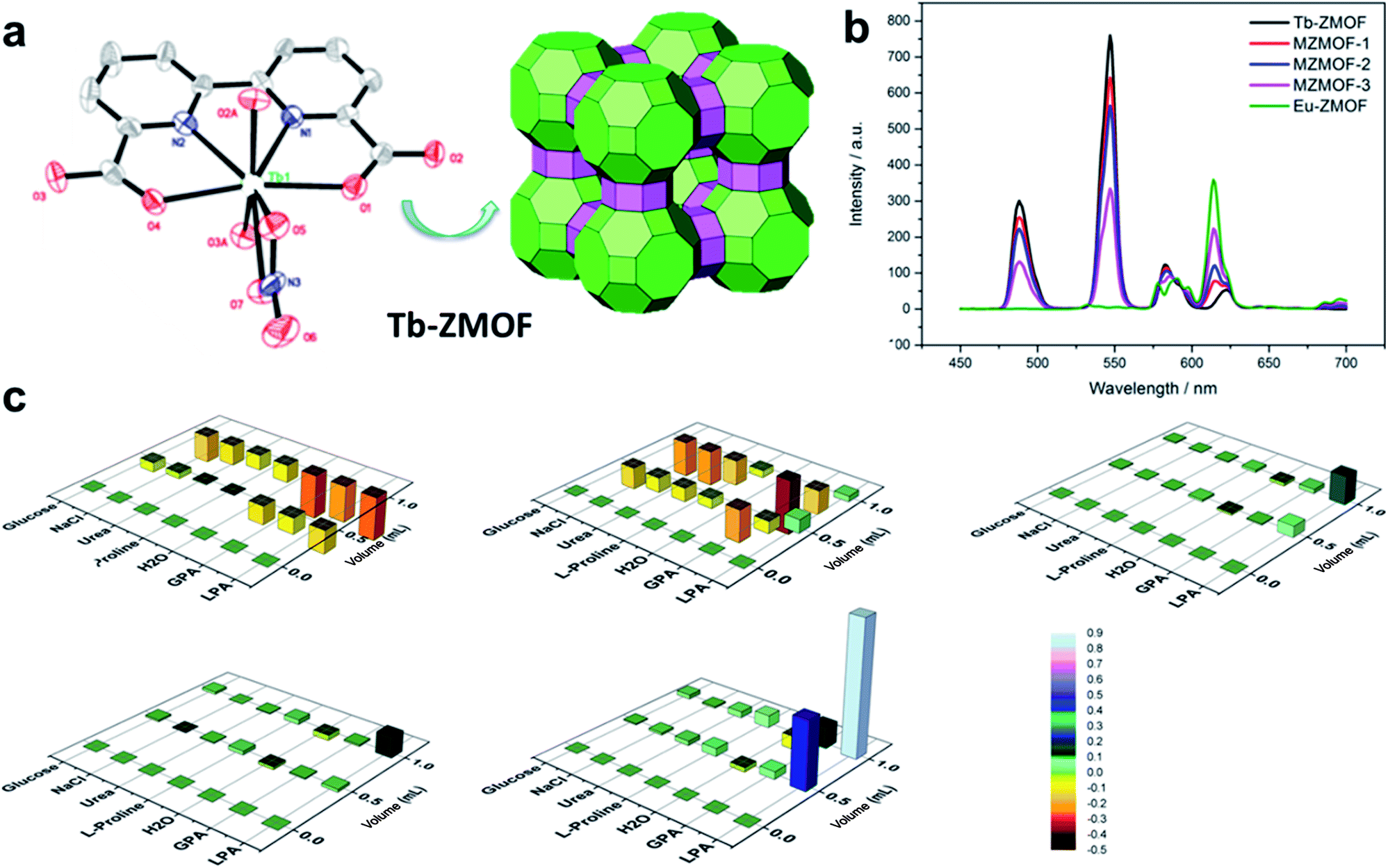 | ||
| Fig. 24 (a) Tiling representation of the rho topology of the Tb-ZMOF. (b) Relative luminescence intensity changes of mixed-crystal ZMOFs. (c) Relative luminescence intensity changes of mixed-crystal ZMOFs in the presence of different analytes. Adapted from ref. 174. | ||
Recently, composites of QDs and MOFs have been designed for enzyme activity sensing.175 For example, Lin and co-workers reported a fluorescence biosensor for gelatinase A (also known as MMP-2, a crucial member of matrix metalloproteinases) based on FRET between poly(9,9-dioctyl fluorenyl-2,7-diyl) dots (Pdots) and the MOF [Cu(H2dtoa)]n, linked by a polypeptide chain (COOH-GHHYYGPLGVRGCNH2).176 The fluorescence of Pdots was quenched by [Cu(H2dtoa)]n with the assistance of the polypeptide chain linker through the FRET process. The polypeptide chain comprises the specific MMP-2 substrate. Therefore, the addition of MMP-2 can result in the release of Pdots from the MOF and restoration of fluorescence. Therefore, MMP-2 can be quantitatively detected with high sensitivity.
3. Conclusion and outlook
In the past few years, the application of LMOFs as probes or chemical sensors has been developed rapidly. Various LMOF based sensors have been judiciously designed and demonstrated to be efficient and powerful for detecting varied species. In this review, we have comprehensively discussed the typical LMOFs, bio-sensing properties of LMOFs, well-established synthetic approaches and PSM strategies for enhancing their functional capabilities and expanding their potential bio-sensing applications. Generally, it can be seen that the LMOF-based sensing system has the advantages of rapid response, high selectivity, sensitivity with a low detection limit and so on, which may be ascribed to the tunable, functionalizable and diverse MOF structures. At the same time, such a sensing system has limitations including a complicated system for sensing large biomolecules such as DNA/RNA and enzymes/proteins, and difficulty in fabricating sensor devices. In addition, there are still challenges in this research area and further studies are required.177 One of the biggest challenges faced in the application of LMOFs in the biological field is stability as well as biocompatibility of MOFs, since most of the MOFs are sensitive to moisture, let alone to the aquatic biological environment. In order to prevent water molecules from attacking coordination bonds, it is necessary to introduce strong metal–ligand coordination interactions to enhance their thermodynamic stability and/or to increase the steric resistance of the ligand to avoid water substitution reactions and improve their dynamic stability. Fortunately, there are some strategies to achieve water-stable MOFs by using high valent metal centers (for example Zr4+) and carboxylate ligands, metal–polyazolate interactions or creating hydrophobic structures.178 Such progress will inspire researchers to make much more efforts and carry out systematic studies in this field, and great achievements can be expected in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Basic Research Program of China (grant no. 2017YFA0303504) and the National Natural Science Foundation of China (grant no. 21573106 and 21671097) for financial support of this work. This work was also supported by a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.References
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC
.
- T. N. Nguyen, F. M. Ebrahim and K. C. Stylianou, Coord. Chem. Rev., 2018, 377, 259–306 CrossRef CAS
- C.-H. Huang, T.-W. Kuo and T.-M. Chen, ACS Appl. Mater. Interfaces, 2010, 2, 1395–1399 CrossRef CAS PubMed
- S. Banerjee, C. D. Malliakas, J. I. Jang, J. B. Ketterson and M. G. Kanatzidis, J. Am. Chem. Soc., 2008, 130, 12270–12272 CrossRef CAS PubMed
- J. Chen, F. Zhao and D. Ma, Mater. Today, 2014, 17, 175–183 CrossRef CAS
- K. Y. Choi, G. Liu, S. Lee and X. Chen, Nanoscale, 2012, 4, 330–342 RSC
- R. Thakar, Y. Chen and P. T. Snee, Nano Lett., 2007, 7, 3429–3432 CrossRef CAS PubMed
- C. A. Kent, B. P. Mehl, L. Ma, J. M. Papanikolas, T. J. Meyer and W. Lin, J. Am. Chem. Soc., 2010, 132, 12767–12769 CrossRef CAS PubMed
- P. Kumar, A. Deep and K.-H. Kim, TrAC, Trends Anal. Chem., 2015, 73, 39–53 CrossRef CAS
- D. Kukkar, K. Vellingiri, V. Kumar, A. Deep and K.-H. Kim, TrAC, Trends Anal. Chem., 2018, 109, 227–246 CrossRef CAS
- D. Kukkar, K. Vellingiri, K.-H. Kim and A. Deep, Sens. Actuators, B, 2018, 273, 1346–1370 CrossRef CAS
- Y. Zhang, S. Yuan, G. Day, X. Wang, X. Yang and H.-C. Zhou, Coord. Chem. Rev., 2018, 354, 28–45 CrossRef CAS
- J. J. I. V. Perry, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC
- M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef PubMed
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC
- K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Chem. Soc. Rev., 2017, 46, 3402–3430 RSC
- Y.-S. Kang, Y. Lu, K. Chen, Y. Zhao, P. Wang and W.-Y. Sun, Coord. Chem. Rev., 2019, 378, 262–280 CrossRef CAS
- Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS PubMed
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS PubMed
- C. He, D. Liu and W. Lin, Chem. Rev., 2015, 115, 11079–11108 CrossRef CAS PubMed
- J. Rocha, C. D. S. Brites and L. D. Carlos, Chem.–Eur. J., 2016, 22, 14782–14795 CrossRef CAS PubMed
- Y. Cui, B. Chen and G. Qian, Coord. Chem. Rev., 2014, 273, 76–86 CrossRef
- G. Lu and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 7832–7833 CrossRef CAS PubMed
- R.-B. Lin, S.-Y. Liu, J.-W. Ye, X.-Y. Li and J.-P. Zhang, Adv. Sci., 2016, 3, 1500434 CrossRef PubMed
- B. Wang, X.-L. Lv, D. Feng, L.-H. Xie, J. Zhang, M. Li, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS PubMed
- Y. Deng, Z.-Y. Yao, P. Wang, Y. Zhao, Y.-S. Kang and W.-Y. Sun, Sens. Actuators, B, 2017, 244, 114–123 CrossRef CAS
- Z. Q. Liu, Y. Zhao, P. Wang, Y. S. Kang, M. Azam, S. I. Al-Resayes, X. H. Liu, Q. Y. Lu and W.-Y. Sun, Dalton Trans., 2017, 46, 9022–9029 RSC
- D. Liu, K. Lu, C. Poon and W. Lin, Inorg. Chem., 2014, 53, 1916–1924 CrossRef CAS PubMed
- D. Maspoch, D. Ruiz-Molina and J. Veciana, Chem. Soc. Rev., 2007, 36, 770–818 RSC
- M. P. Suh, Y. E. Cheon and E. Y. Lee, Coord. Chem. Rev., 2008, 252, 1007–1026 CrossRef CAS
- Z.-Q. Liu, Y. Zhao, Y. Deng, X.-D. Zhang, Y.-S. Kang, Q.-Y. Lu and W.-Y. Sun, Sens. Actuators, B, 2017, 250, 179–188 CrossRef CAS
- Z.-Q. Liu, K. Chen, Y. Zhao, Y.-S. Kang, X.-H. Liu, Q.-Y. Lu, M. Azam, S. I. Al-Resayes and W.-Y. Sun, Cryst. Growth Des., 2018, 18, 1136–1146 CrossRef CAS
- Y. Liu, Y. Zhao, X.-H. Liu, Y.-S. Kang, P. Wang and W.-Y. Sun, Dalton Trans., 2018, 47, 15399–15404 RSC
- X.-D. Zhang, Y. Zhao, K. Chen, P. Wang, Y.-S. Kang, H. Wu and W.-Y. Sun, Dalton Trans., 2018, 47, 3958–3964 RSC
- X.-D. Zhang, Y. Zhao, K. Chen, J.-H. Guo, P. Wang, H. Wu and W.-Y. Sun, Sens. Actuators, B, 2019, 282, 844–853 CrossRef CAS
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS
- A. Olaya-Castro and G. D. Scholes, Int. Rev. Phys. Chem., 2011, 30, 49–77 Search PubMed
- P. P. Cui, P. Wang, Y. Zhao and W. Y. Sun, Cryst. Growth Des., 2019, 19, 1454–1470 CrossRef CAS
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Chem. Sci., 2012, 3, 126–130 RSC
- K. Y. Cho, H. An, X. H. Do, K. Choi, H. G. Yoon, H.-K. Jeong, J. S. Lee and K.-Y. Baek, J. Mater. Chem. A, 2018, 6, 18912–18919 RSC
- O. Karagiaridi, W. Bury, A. A. Sarjeant, C. L. Stern, O. K. Farha and J. T. Hupp, Chem. Sci., 2012, 3, 3256–3260 RSC
- M. Erkartal, U. Erkilic, B. Tam, H. Usta, O. Yazaydin, J. T. Hupp, O. K. Farha and U. Sen, Chem. Commun., 2017, 53, 2028–2031 RSC
- C.-W. Tsai, J. W. Niemantsverdriet and E. H. G. Langner, Microporous Mesoporous Mater., 2018, 262, 98–105 CrossRef CAS
- H.-F. Zhang, M. Li, X.-Z. Wang, D. Luo, Y.-F. Zhao, X.-P. Zhou and D. Li, J. Mater. Chem. A, 2018, 6, 4260–4265 RSC
- J.-Q. Jiang, C.-X. Yang and X.-P. Yan, Chem. Commun., 2015, 51, 6540–6543 RSC
- P. Wang, K. Chen, Q. Liu, H.-W. Wang, M. Azam, S. I. Al-Resayes, Y. Lu and W.-Y. Sun, Dalton Trans., 2017, 46, 11425–11430 RSC
- S. S. Zhao, H. Zhang, L. Wang, L. Chen and Z. G. Xie, J. Mater. Chem. C, 2018, 6, 11701–11706 RSC
- X. Z. Song, S. Y. Song, S. N. Zhao, Z. M. Hao, M. Zhu, X. Meng, L. L. Wu and H. J. Zhang, Adv. Funct. Mater., 2014, 24, 4034–4041 CrossRef CAS
- H. Li, Y. J. Li, Z. Zhang, X. L. Pang and X. D. Yu, Mater. Des., 2019, 172, 107712 CrossRef CAS
- D. Ghosh, A. Pal, S. Ghosh, A. Gayen, M. M. Seikh and P. Mahata, Eur. J. Inorg. Chem., 2019, 3076–3083 CrossRef CAS
- C. Liu, T.-Y. Luo, E. S. Feura, C. Zhang and N. L. Rosi, J. Am. Chem. Soc., 2015, 137, 10508–10511 CrossRef CAS PubMed
- C. Liu, C. Zeng, T.-Y. Luo, A. D. Merg, R. Jin and N. L. Rosi, J. Am. Chem. Soc., 2016, 138, 12045–12048 CrossRef CAS PubMed
- O. Karagiaridi, W. Bury, E. Tylianakis, A. A. Sarjeant, J. T. Hupp and O. K. Farha, Chem. Mater., 2013, 25, 3499–3503 CrossRef CAS
- D.-X. Xue, A. J. Cairns, Y. Belmabkhout, L. Wojtas, Y. Liu, M. H. Alkordi and M. Eddaoudi, J. Am. Chem. Soc., 2013, 135, 7660–7667 CrossRef CAS PubMed
- D.-X. Xue, Y. Belmabkhout, O. Shekhah, H. Jiang, K. Adil, A. J. Cairns and M. Eddaoudi, J. Am. Chem. Soc., 2015, 137, 5034–5040 CrossRef CAS PubMed
- P. Yi, H. Huang, Y. Peng, D. Liu and C. Zhong, RSC Adv., 2016, 6, 111934–111941 RSC
- J. F. Feng, S. Y. Gao, T. F. Liu, J. L. Shi and R. Cao, ACS Appl. Mater. Interfaces, 2018, 10, 6014–6023 CrossRef CAS PubMed
- P. P. Cui, X. D. Zhang, P. Wang, Y. Zhao, M. Azam, S. I. Al-Resayes and W. Y. Sun, Inorg. Chem., 2017, 56, 14157–14163 CrossRef CAS PubMed
- W. X. Li, J. H. Gu, H. X. Li, M. Dai, D. J. Young, H. Y. Li and J. P. Lang, Inorg. Chem., 2018, 57, 13453–13460 CrossRef CAS PubMed
- Q. Zhang, J. Yu, J. Cai, L. Zhang, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2015, 51, 14732–14734 RSC
- X. Zhang, J. Zhang, Q. Hu, Y. Cui, Y. Yang and G. Qian, Appl. Surf. Sci., 2015, 355, 814–819 CrossRef CAS
- T.-Y. Luo, C. Liu, S. V. Eliseeva, P. F. Muldoon, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2017, 139, 9333–9340 CrossRef CAS PubMed
- C. L. Hobday, T. D. Bennett, D. Fairen-Jimenez, A. J. Graham, C. A. Morrison, D. R. Allan, T. Duren and S. A. Moggach, J. Am. Chem. Soc., 2018, 140, 382–387 CrossRef CAS PubMed
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed
- Y.-L. Li, Y. Zhao, P. Wang, Y.-S. Kang, Q. Liu, X.-D. Zhang and W.-Y. Sun, Inorg. Chem., 2016, 55, 11821–11830 CrossRef CAS PubMed
- Q. Zhang, J. Wang, A. M. Kirillov, W. Dou, C. Xu, C. Xu, L. Yang, R. Fang and W. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 23976–23986 CrossRef CAS PubMed
- J.-P. Zou, Q. Peng, Z. Wen, G.-S. Zeng, Q.-J. Xing and G.-C. Guo, Cryst. Growth Des., 2010, 10, 2613–2619 CrossRef CAS
- M. Frisch and C. L. Cahill, Dalton Trans., 2005, 1518–1523 RSC
- N. S. Gunning and C. L. Cahill, Dalton Trans., 2005, 2788–2792 RSC
- Y. Wei, Y. Yu and K. Wu, Cryst. Growth Des., 2008, 8, 2087–2089 CrossRef CAS
- L. Han, D. Q. Yuan, B. L. Wu, C. P. Liu and M. C. Hong, Inorg. Chim. Acta, 2006, 359, 2232–2240 CrossRef CAS
- S. Zhang, Z. Wang, H. Zhang, Y. Cao, Y. Sun, Y. Chen, C. Huang and X. Yu, Inorg. Chim. Acta, 2007, 360, 2704–2710 CrossRef CAS
- M.-S. Wang, S.-P. Guo, Y. Li, L.-Z. Cai, J.-P. Zou, G. Xu, W.-W. Zhou, F.-K. Zheng and G.-C. Guo, J. Am. Chem. Soc., 2009, 131, 13572–13573 CrossRef CAS PubMed
- G.-H. Wang, Z.-G. Li, H.-Q. Jia, N.-H. Hu and J.-W. Xu, CrystEngComm, 2009, 11, 292–297 RSC
- R. Feng, F.-L. Jiang, L. Chen, C.-F. Yan, M.-Y. Wu and M.-C. Hong, Chem. Commun., 2009, 5296–5298 RSC
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed
- P. Escribano, B. Julian-Lopez, J. Planelles-Arago, E. Cordoncillo, B. Viana and C. Sanchez, J. Mater. Chem., 2008, 18, 23–40 RSC
- S. V. Eliseeva and J.-C. G. Buenzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC
- J.-C. G. Buenzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef CAS PubMed
- N. Sabbatini, M. Guardigli and J. M. Lehn, Coord. Chem. Rev., 1993, 123, 201–228 CrossRef CAS
- K. Zheng, Z.-Q. Liu, Y. Huang, F. Chen, C.-H. Zeng, S. Zhong and S. W. Ng, Sens. Actuators, B, 2018, 257, 705–713 CrossRef CAS
- Y. Cui, H. Xu, Y. Yue, Z. Guo, J. Yu, Z. Chen, J. Gao, Y. Yang, G. Qian and B. Chen, J. Am. Chem. Soc., 2012, 134, 3979–3982 CrossRef CAS PubMed
- M. Pan, Y.-X. Zhu, K. Wu, L. Chen, Y.-J. Hou, S.-Y. Yin, H.-P. Wang, Y.-N. Fan and C.-Y. Su, Angew. Chem., Int. Ed., 2017, 56, 14582–14586 CrossRef CAS PubMed
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–8377 CrossRef CAS PubMed
- J. An, C. M. Shade, D. A. Chengelis-Czegan, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2011, 133, 1220–1223 CrossRef CAS PubMed
- T.-W. Duan and B. Yan, J. Mater. Chem. C, 2014, 2, 5098–5104 RSC
- N. L. Rosi, J. Kim, M. Eddaoudi, B. L. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed
- X. Lian and B. Yan, RSC Adv., 2016, 6, 11570–11576 RSC
- C. Li and J. Lin, J. Mater. Chem., 2010, 20, 6831–6847 RSC
- J. Yang, Q. Yuo, G. D. Li, J. J. Cao, G. H. Li and J. S. Chen, Inorg. Chem., 2006, 45, 2857–2865 CrossRef CAS PubMed
- D. Weng, X. Zheng and L. Jin, Eur. J. Inorg. Chem., 2006, 4184–4190 CrossRef CAS
- X. Zhang, B. Li, H. Ma, L. Zhang and H. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 17389–17394 CrossRef CAS PubMed
- X. Shen and B. Yan, J. Colloid Interface Sci., 2015, 451, 63–68 CrossRef CAS PubMed
- D. Yan, Y. Tang, H. Lin and D. Wang, Sci. Rep., 2014, 4, 4337 CrossRef PubMed
- M.-J. Dong, M. Zhao, S. Ou, C. Zou and C.-D. Wu, Angew. Chem., Int. Ed., 2014, 53, 1575–1579 CrossRef CAS PubMed
- S.-N. Zhao, X.-Z. Song, M. Zhu, X. Meng, L.-L. Wu, J. Feng, S.-Y. Song and H.-J. Zhang, Chem.–Eur. J., 2015, 21, 9748–9752 CrossRef CAS PubMed
- S. Saha, G. Das, J. Thote and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 14845–14851 CrossRef CAS PubMed
- S. Jin, H.-J. Son, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 955–958 CrossRef CAS PubMed
- J. Aguilera-Sigalat and D. Bradshaw, Coord. Chem. Rev., 2016, 307, 267–291 CrossRef CAS
- X. Lin, G. Gao, L. Zheng, Y. Chi and G. Chen, Anal. Chem., 2014, 86, 1223–1228 CrossRef CAS PubMed
- Y. Ma, G. Xu, F. Wei, Y. Cen, Y. Ma, Y. Song, X. Xu, M. Shi, S. Muhammad and Q. Hu, J. Mater. Chem. C, 2017, 5, 8566–8571 RSC
- D. Banerjee, Z. Hu and J. Li, Dalton Trans., 2014, 43, 10668–10685 RSC
- G.-Y. Wang, C. Song, D.-M. Kong, W.-J. Ruan, Z. Chang and Y. Li, J. Mater. Chem. A, 2014, 2, 2213–2220 RSC
- S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881–2885 CrossRef CAS PubMed
- K. Li, H. G. Nguyen, X. Lu and Q. Wang, Analyst, 2010, 135, 21–27 RSC
- Y. Wang, R. Hu, G. Lin, I. Roy and K.-T. Yong, ACS Appl. Mater. Interfaces, 2013, 5, 2786–2799 CrossRef CAS PubMed
- H. Zhang, H. Zhang, A. Aldalbahi, X. Zuo, C. Fan and X. Mi, Biosens. Bioelectron., 2017, 89, 96–106 CrossRef CAS PubMed
- R. Ranjan, E. N. Esimbekova and V. A. Kratasyuk, Biosens. Bioelectron., 2017, 87, 918–930 CrossRef CAS PubMed
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed
- M. L. Foo, R. Matsuda and S. Kitagawa, Chem. Mater., 2014, 26, 310–322 CrossRef CAS
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC
- H. Tan, G. Tang, Z. Wang, Q. Li, J. Gao and S. Wu, Anal. Chim. Acta, 2016, 940, 136–142 CrossRef CAS PubMed
- S.-P. Yang, S.-R. Chen, S.-W. Liu, X.-Y. Tang, L. Qin, G.-H. Qiu, J.-X. Chen and W.-H. Chen, Anal. Chem., 2015, 87, 12206–12214 CrossRef CAS PubMed
- H.-S. Wang, J. Li, J.-Y. Li, K. Wang, Y. Ding and X.-H. Xia, NPG Asia Mater., 2017, 9, e354 CrossRef CAS
- Z. Q. Liu, Y. Zhao, X. D. Zhang, Y. S. Kang, Q. Y. Lu, M. Azam, S. I. Al-Resayes and W. Y. Sun, Dalton Trans., 2017, 46, 13943–13951 RSC
- H.-S. Wang, Coord. Chem. Rev., 2017, 349, 139–155 CrossRef CAS
- X. Zhu, H. Zheng, X. Wei, Z. Lin, L. Guo, B. Qiu and G. Chen, Chem. Commun., 2013, 49, 1276–1278 RSC
- L. Qin, L.-X. Lin, Z.-P. Fang, S.-P. Yang, G.-H. Qiu, J.-X. Chen and W.-H. Chen, Chem. Commun., 2016, 52, 132–135 RSC
- S.-P. Yang, W. Zhao, P.-P. Hu, K.-Y. Wu, Z.-H. Jiang, L.-P. Bai, M.-M. Li and J.-X. Chen, Inorg. Chem., 2017, 56, 14880–14887 CrossRef CAS PubMed
- H.-Q. Zhao, G.-H. Qiu, Z. Liang, M.-M. Li, B. Sun, L. Qin, S.-P. Yang, W.-H. Chen and J.-X. Chen, Anal. Chim. Acta, 2016, 922, 55–63 CrossRef CAS PubMed
- H.-Q. Zhao, S.-P. Yang, N.-N. Ding, L. Qin, G.-H. Qiu, J.-X. Chen, W.-H. Zhang, W.-H. Chen and T. S. A. Hor, Dalton Trans., 2016, 45, 5092–5100 RSC
- H.-T. Zhang, J.-W. Zhang, G. Huang, Z.-Y. Du and H.-L. Jiang, Chem. Commun., 2014, 50, 12069–12072 RSC
- Y. Wu, J. Han, P. Xue, R. Xu and Y. Kang, Nanoscale, 2015, 7, 1753–1759 RSC
- J. M. Fang, F. Leng, X. J. Zhao, X. L. Hu and Y. F. Li, Analyst, 2014, 139, 801–806 RSC
- G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef CAS PubMed
- R. Mejia-Ariza, J. Rosselli, C. Breukers, A. Manicardi, L. Terstappen, R. Corradini and J. Huskens, Chem.–Eur. J., 2017, 23, 4180–4186 CrossRef CAS PubMed
- C. Serre, C. Mellot-Draznieks, S. Surble, N. Audebrand, Y. Filinchuk and G. Ferey, Science, 2007, 315, 1828–1831 CrossRef CAS PubMed
- S.-N. Zhao, L.-L. Wu, J. Feng, S.-Y. Song and H.-J. Zhang, Inorg. Chem. Front., 2016, 3, 376–380 RSC
- P. Ling, J. Lei, L. Zhang and H. Ju, Anal. Chem., 2015, 87, 3957–3963 CrossRef CAS PubMed
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed
- Y. Li, A. Guo, L. Chang, W.-J. Li and W.-J. Ruan, Chem.–Eur. J., 2017, 23, 6562–6569 CrossRef CAS PubMed
- M. Xu, S. Yuan, X.-Y. Chen, Y.-J. Chang, G. Day, Z.-Y. Gu and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 8312–8319 CrossRef CAS PubMed
- G. Zhang, H. Dong and X. Zhang, Chem. Commun., 2019, 55, 8158–8161 RSC
- L. M. Bharadwaj, J. Porous Mater., 2014, 21, 99–104 CrossRef
- T. Guo, Q. Deng, G. Fang, D. Gu, Y. Yang and S. Wang, Biosens. Bioelectron., 2016, 79, 341–346 CrossRef CAS PubMed
- H.-S. Wang and J.-P. Wei, Nanoscale, 2015, 7, 11815–11832 RSC
- W. Li, X. Qi, C.-Y. Zhao, X.-F. Xu, A.-N. Tang and D.-M. Kong, ACS Appl. Mater. Interfaces, 2017, 9, 236–243 CrossRef CAS PubMed
- P. Chandrasekhar, A. Mukhopadhyay, G. Savitha and J. N. Moorthy, Chem. Sci., 2016, 7, 3085–3091 RSC
- Q. F. Zhang, M. Y. Lei, F. Kong and Y. Yang, Chem. Commun., 2018, 54, 10901–10904 RSC
- H. Li, X. Feng, Y. Guo, D. Chen, R. Li, X. Ren, X. Jiang, Y. Dong and B. Wang, Sci. Rep., 2014, 4, 4366 CrossRef PubMed
- J.-N. Hao and B. Yan, Chem. Commun., 2015, 51, 14509–14512 RSC
- T. R. Lin, Y. M. Qin, Y. L. Huang, R. T. Yang, L. Hou, F. G. Ye and S. L. Zhao, Chem. Commun., 2018, 54, 1762–1765 RSC
- D. Ning, Q. Liu, Q. Wang, X.-M. Du, W.-J. Ruan and Y. Li, Sens. Actuators, B, 2019, 282, 443–448 CrossRef CAS
- A. Kumar, A. R. Chowdhuri, A. Kumari and S. K. Sahu, Mater. Sci. Eng., C, 2018, 92, 913–921 CrossRef CAS PubMed
- P. H. Huang, C. P. Hong, J. F. Zhu, T. T. Chen, C. T. Chan, Y. C. Ko, T. L. Lin, Z. B. Pan, N. K. Sun, Y. C. Wang, J. J. Luo, T. C. Lin, C. C. Kang, J. J. Shyue and M. L. Ho, Dalton Trans., 2017, 46, 6985–6993 RSC
- T. T. Zhao, Z. W. Jiang, S. J. Zhen, C. Z. Huang and Y. F. Li, Microchim. Acta, 2019, 186, 168 CrossRef PubMed
- N. Bagheri, M. Dastborhan, A. Khataee, J. Hassanzadeh and M. Kobya, Spectrochim. Acta, Part A, 2019, 213, 28–36 CrossRef CAS PubMed
- Y. Y. Yuan, S. L. Yang, C. X. Zhang and Q. L. Wang, CrystEngComm, 2018, 20, 6989–6994 RSC
- C. Guo, Q. Jin, Y. Wang, B. Ding, Y. Li, J. Huo and X. Zhao, Sens. Actuators, B, 2016, 234, 184–191 CrossRef CAS
- D. Yue, Y. Huang, J. Zhang, X. Zhang, Y. Cui, Y. Yang and G. Qian, Eur. J. Inorg. Chem., 2018, 173–177 CrossRef CAS
- D. Yue, D. Zhao, J. Zhang, L. Zhang, K. Jiang, X. Zhang, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2017, 53, 11221–11224 RSC
- B. Wang, X. L. Lv, D. W. Feng, L. H. Xie, J. Zhang, M. Li, Y. B. Xie, J. R. Li and H. C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS PubMed
- D. Zhao, X.-H. Liu, Y. Zhao, P. Wang, Y. Liu, M. Azam, S. I. Al-Resayes, Y. Lu and W.-Y. Sun, J. Mater. Chem. A, 2017, 5, 15797–15807 RSC
- F. Zhang, H. Yao, T. S. Chu, G. W. Zhang, Y. Wang and Y. Y. Yang, Chem.–Eur. J., 2017, 23, 10293–10300 CrossRef CAS PubMed
- N. Xu, Q. H. Zhang, B. S. Hou, Q. Cheng and G. A. Zhang, Inorg. Chem., 2018, 57, 13330–13340 CrossRef CAS PubMed
- Z. W. Zhai, S. H. Yang, M. Cao, L. K. Li, C. X. Du and S. Q. Zang, Cryst. Growth Des., 2018, 18, 7173–7182 CrossRef CAS
- N. Xu, Q. H. Zhang and G. A. Zhang, Dalton Trans., 2019, 48, 2683–2691 RSC
- X. D. Zhu, K. Zhang, Y. Wang, W. W. Long, R. J. Sa, T. F. Liu and J. Lu, Inorg. Chem., 2018, 57, 1060–1065 CrossRef CAS PubMed
- Q. Liu, D. Ning, W. J. Li, X. M. Du, Q. Wang, Y. Li and W. J. Ruan, Analyst, 2019, 144, 1916–1922 RSC
- Y. Zhou, Q. Yang, D. N. Zhang, N. Gan, Q. P. Li and J. Cuan, Sens. Actuators, B, 2018, 262, 137–143 CrossRef CAS
- Y. L. Xu, Y. Liu, X. H. Liu, Y. Zhao, P. Wang, Z. L. Wang and W. Y. Sun, Polyhedron, 2018, 154, 350–356 CrossRef CAS
- Y. L. Xu, Y. Liu, X. H. Liu, Y. Zhao, Z. L. Wang and W. Y. Sun, Isr. J. Chem., 2019, 59, 267–272 CrossRef CAS
- H.-S. Wang, W.-J. Bao, S.-B. Ren, M. Chen, K. Wang and X.-H. Xia, Anal. Chem., 2015, 87, 6828–6833 CrossRef CAS PubMed
- H. Tan, C. Ma, L. Chen, F. Xu, S. Chen and L. Wang, Sens. Actuators, B, 2014, 190, 621–626 CrossRef CAS
- Y. Zhang, B. Li, H. Ma, L. Zhang and Y. Zheng, Biosens. Bioelectron., 2016, 85, 287–293 CrossRef CAS PubMed
- Y. Zhang, B. Li, H. Ma, L. Zhang, H. Jiang, H. Song, L. Zhang and Y. Luo, J. Mater. Chem. C, 2016, 4, 7294–7301 RSC
- S.-Y. Zhang, W. Shi, P. Cheng and M. J. Zaworotko, J. Am. Chem. Soc., 2015, 137, 12203–12206 CrossRef CAS PubMed
- K. Wang, N. Li, J. Zhang, Z. Zhang and F. Dang, Biosens. Bioelectron., 2017, 87, 339–344 CrossRef CAS PubMed
- W. Yang, G. Zhang, W. Weng, B. Qiu, L. Guo, Z. Lin and G. Chen, RSC Adv., 2014, 4, 58852–58857 RSC
- Z. Q. Liu, Y. Q. Huang and W. Y. Sun, Chin. J. Inorg. Chem., 2017, 33, 1959–1969 CAS
- C. H. Wang, X. L. Liu, N. K. Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |