
Visualization of carboxylesterase 2 with a near-infrared two-photon fluorescent probe and potential evaluation of its anticancer drug effects in an orthotopic colon carcinoma mice model†
Yan
Wang
ab,
Feifei
Yu
b,
Xianzhu
Luo
ab,
Mingshun
Li
ab,
Linlu
Zhao
*b and
Fabiao
Yu
 *ab
*ab
aThe Key Laboratory of Life-Organic Analysis, Key Laboratory of Pharmaceutical Intermediates and Analysis of Natural Medicine, College of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, China
bInstitute of Functional Materials and Molecular Imaging, Key Laboratory of Emergency and Trauma, Ministry of Education, College of Pharmacy, Key Laboratory of Hainan Trauma and Disaster Rescue, College of Clinical Medicine, College of Emergency and Trauma, Hainan Medical University, Haikou 571199, China. E-mail: fbyu@yic.ac.cn; zhaolinlu@hainmc.edu.cn
First published on 9th March 2020
Abstract
We establish a near-infrared two-photon fluorescent probe for the detection of CE2 with high selectivity and sensitivity. This probe exhibits low cytotoxicity and superior tissue penetration ability for evaluating the real-time activity of CE2 in living cells, in cancer tissues, and in a colon carcinoma mice model.
Carboxylesterases (CEs), a component of α/β hydrolase fold protein, play pivotal roles in the hydrolysis of substances containing esters, amides, carbamates and thioesters.1–4 CE2, a major isoform of CEs, is found to be highly expressed in many cancer cells and mediates the activation process of many prodrugs such as gemcitabine, irinotecan (CPT-11) and capecitabine (CAPE).5,6 Therefore, the individual differences in the CE2 activity have been closely associated with the cytotoxicity and efficacy of the relevant clinical anticancer drugs.7 Furthermore, CE2 is considered to be the essential determinant in intestinal first-pass metabolism, especially for oral anticancer prodrugs.8 In view of the critical roles of CE2 in metabolism of various anticancer prodrugs, it is quite necessary to develop suitable techniques for accurate and sensitive detection of CE2 under complex physiological conditions.
Several effective methods for evaluating the CE2 activity have been established.9,10 However, these technical methods usually involve time-consuming and complex procedures, high cost, and skilled operators, which may largely restrict their application in rapid detection. As attractive alternatives, fluorescent probes have become indispensable tools toward exploring bioactive molecules.11–13 Although small-molecule based fluorescent probes have been synthesized and utilized for the detection of CE2 in vitro and in vivo,14–16 few probes are competent for practical applications mainly due to their relatively short excitation/emission wavelength at which there is strong intrinsic autofluorescence background from living tissues. This issue greatly restricts the deep-tissue penetration and measurement accuracy under physiological conditions. Compared to the single-photon confocal imaging, the developed two-photon (TP) microscopy features the ability to avoid biological autofluorescence and to reduce photodamage, because the near-infrared (NIR) wavelength can achieve a deeper penetration and can exhibit a higher 3D temporal spatial resolution.17–20 Hence, a two-photon fluorescent probe has attracted extensive research interest in sensing of bioactive macromolecule CE2 in sophisticated living systems. However, there still exist obstacles of the TP probes, which mainly lie in the deficiency with emission wavelength since most of these TP probes only can emit fluorescence at short wavelength (<550 nm).21 This issue may fail the practical bio-imaging especially in deep tissues as the collection efficiency of fluorescence signals will be greatly decreased, further affecting the sensing accuracy and sensitivity of TP probes.22,23 Therefore, it is of great significance to develop TP probes with excitation/emission wavelength in the NIR region21 which can not only minimize background fluorescence interference owing to reducing absorption by bio-molecules but also achieve deeper penetration to facilitate the precise detection of CE2 in living systems.
As illustrated in Scheme 1, the overall design strategy of the probe DCM-CES2 was based on the attachment of an enzyme-active moiety L-leucine to an excellent NIR TP chromophore dicyanomethylene-4H-pyran (DCM),11,13,24 because it showed large Stokes shifts and NIR emission (>650 nm). Since the CE2 could specifically hydrolyze the compound with large alcohol and small acyl group, our probe DCM-CES2 was able to serve as a highly selective substrate for CE2. The detailed synthetic route is shown in the ESI.† The compounds were characterized by 1H NMR, 13C NMR and HR-MS.
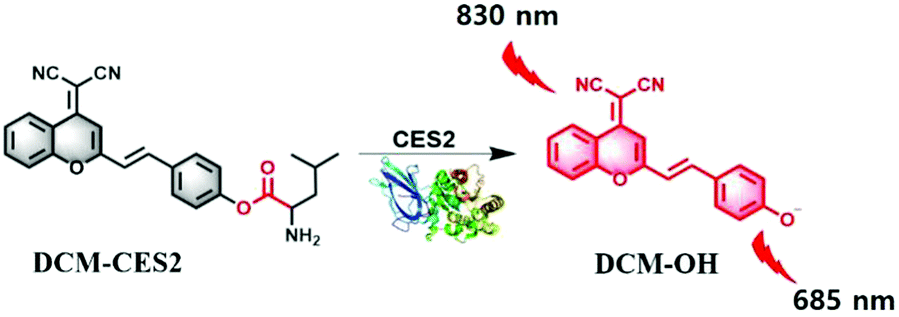 | ||
| Scheme 1 Illustration of the molecular structure of DCM-CES2 and its proposed fluorescence response mechanism toward CE2. | ||
We inspected the spectral characteristics of the probe DCM-CES2 under simulated physiological conditions (PBS buffer pH = 7.4, 10 mM at 37 °C). As shown in Fig. 1a, the probe DCM-CES2 provided characteristic absorption centred at 445 nm. With the treatment of CE2, a new maximum absorption peak appeared at 560 nm, which belonged to the released fluorophore DCM. The color changed from light yellow to rose red allowing the colorimetric detection of CE2 via the naked eye, accompanied by a large Stokes shift of 125 nm in the fluorescence spectra. The reaction with CE2 would switch-on the fluorescence emission of DCM-CES2 by restoring the intramolecular charge transfer (ICT) process, thereby offered a characteristic fluorescence emission peak centred at 685 nm in the NIR region (Fig. 1b). The fluorescence intensity displayed a good linear relationship. The regression equation was F560nm = 10.27[CE2] + 6.963 with a linear regression constant r = 0.9963 (Fig. 1c). Based on the standard method of 3σ/k, the limit of detection for CEs was 0.087 μg mL−1, which indicated favourable sensitivity for the CE2 detection in vivo.25 As indicated in Fig. 1d, there appeared a quick response within 5 min. Continuous monitoring demonstrated that the reaction completed within 10 min. The maximal TP absorption cross section (δ) was measured to be 26 GM at 830 nm (Fig. S2, ESI†), implying that DCM-CES2 was able to track CE2 via a TP microscope. These results validated that the designed probe could serve as a near-infrared off–on fluorescent chemical tool for detecting CE2 under physiological conditions.
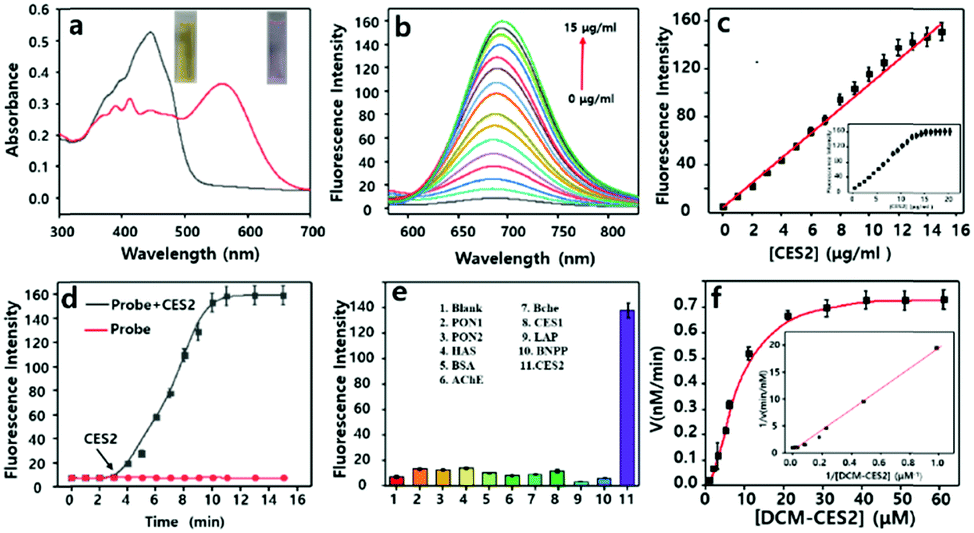 | ||
| Fig. 1 Spectral characterization of DCM-CES2 (10 μM). (a) The UV-Vis absorption spectrum with CE2 (0–10 μg mL−1) for 10 min. (b) The fluorescence spectrum with CE2 (0–15 μg mL−1) for 10 min. (c) Linear relationship between the fluorescence intensity and the activity of CE2. (d) Time-dependent kinetic measurement of the fluorescent response to CE2 (5 μg mL−1). (e) Selectivity towards various species. Blank, paraoxonase (PON1 and PON2, 10 μg mL−1), human serum albumin (HSA, 0.5 mg mL−1), bovine serum albumin (BSA, 0.5 mg mL−1), acetylcholinesterase (AChE, 0.1 μg L−1), butyrylcholinesterase (BChE, 20 U L−1), CES1 (10 μg mL−1), loperamide (LPA, 100 μg mL−1), bis(4-nitrophenyl)phosphate (BNPP, 100 μg mL−1), and CE2 (10 μg mL−1). (f) Lineweaver–Burk plot for the enzymatic reaction. The Michaelis–Menten equation was expressed as: V = Vmax[probe]/(Km + [probe]), where V is the reaction rate, [probe] refers to the probe concentration (substrate), and Km is the Michaelis constant. λex = 560 nm. | ||
A series of reconstructed human enzymes with hydrolytic activity were selected to assess their possible interactions with DCM-CES2 (Fig. 1e). DCM-CES2 was incubated with the selected enzymes, respectively, only CE2 led to a remarkable increase in fluorescence intensity at 685 nm, while the other hydrolases exhibited negligible fluorescence changes. To further confirm the CE2-dependent fluorescence changes, we chose two representative esterase inhibitors to perform the chemical inhibition assays. CE2 was pre-incubated with the non-covalent inhibitor loperamide (LPA) and covalent inhibitor bis(4-nitrophenyl)phosphate (BNPP) followed by the addition of the probe DCM-CES2, respectively (Fig. 1e). Both inhibitors restricted fluorescence emission compared with the CE2 group, which confirmed that the fluorescence change was dependent on CE2. The results indicated that DCM-CES2 could serve as an effective chemical tool to detect CE2 with high selectivity in cells. We also determined the Michaelis constant (Km). The value of Km was 33.43 μM, and the corresponding catalytic constant (Kcat) and catalytic efficiency (Kcat/Km) were 3.75 s−1 and 0.17 μM−1 s−1, respectively, demonstrating that CE2-mediated hydrolysis of DCM-CES2 possessed good kinetic property, high catalytic activity, and powerful affinity (Fig. 1f).
We next utilized the probe DCM-CES2 to detect the level of CE2 in living cells. The cytotoxicity of the probe was evaluated by the Cell Counting Kit-8 assay (Fig. S4, ESI†) with human hepatocellular liver carcinoma cell line (HepG2) and human colorectal cancer cell line (HT-29). The viability rates of the selected model cell lines indicated the relatively low cytotoxicity. To test the capability of DCM-CES2 responding to the CE2 activity in living cells, the HepG2 cells were treated under different treatment conditions. The addition of DCM-CES2 could generate strong red fluorescence, which revealed the reactivity to the CE2-overexpressed system (Fig. S6h, ESI†). Then, two representative inhibitors, BNPP for CEs and LPA for CE2, were adopted to validate the specific detection of CE2 in cells. As expected, there appeared severe fluorescence suppression in Fig. S6i and j (ESI†), respectively. The faint fluorescence was attributed to the inhibition of intracellular CE2 activity, but the CE2-selective inhibitor LPA could restrain almost all fluorescence emissions, demonstrating the specific detecting ability of our designed probe. We also pretreated the cells with 5-fluorouracil (5-FU, 70 μM, a CE2 inducer26) for 48 h at 37 °C. Interestingly, a stronger fluorescence increase was observed in Fig. S6k (ESI†), which confirmed the specific response to endogenous CE2 in living cells. The results obtained by flow cytometry assays (Fig. S6m–q, ESI†) were in well accordance with the aforementioned imaging outcomes.
CE2 offered a high activation effect on metabolism of anticancer prodrug irinotecan (CPT-11) in HT-29 cells.27 At the same situation, anticancer prodrug capecitabine (CAPE) held a stronger tendency to be first activated by CE2 and finally metabolized to drug 5-FU that could inhibit the growth of cancer cells.28 In view of the crucial role of CE2 involved in the activation process of various anticancer prodrugs, it was quite necessary to visualize CE2 for better understanding of its metabolic mechanism in drug metabolism. We next attempted to utilize the probe DCM-CES2 to detect the CE2 activity change during the activation procedure in living cells. The HT-29 cells were parallelly divided into seven groups and treated with CPT-11 (20 μM) for 0, 2, 4, 8, 12, 24, 48 h, respectively, then incubated with the probe for 30 min (Fig. 2a and b). Upon treatment with CPT-11 for the initial 4 h, there appeared a fluorescence enhancement in the testing system, while continuous effect of CPT-11 gradually weakened the fluorescence signal as the incubation time extended to 24 h. When the examination was continuous for 48 h, we noticed that the activity of CE2 gradually returned to the original level, which was in well agreement with the metabolic cycle of CPT-11.29 We speculated that the HT-29 cells were initially suffering from a stress response to metabolize the prodrug, which would promote CE2 activate stronger, but then occupied by amount of prodrug molecules. Once the prodrug was decomposed, it would lead to the decreased vitality of CE2, thus reducing the obtained fluorescence intensity as the control group. Further evaluation was carried out to detect the CE2 activation response to CAPE in the HT-29 cells. As shown in Fig. 2b, the overall fluorescence trend was almost the same as that in Fig. 2a, except for acquiring a stronger fluorescence increase at 48 h. This phenomenon is probably caused by the presence of 5-FU which not only served as a metabolite from CAPE through multiple enzymes including CE2 but also could be an inducer to stimulate the activity increase in CE2.26 The results demonstrated that different anticancer drugs offered diverse activation processes of CE2, which might instruct various cancer cell resistance mechanisms to different drugs. Flow analysis is shown in Fig. 2c and d. In addition, the PE Annexin V/7-AAD assay of HT-29 cells was also studied to illustrate cell apoptosis caused by both anticancer drugs (Fig. 2e and f). All these results suggested that DCM-CES2 could be utilized to detect the crucial role of CE2 during the anti-cancer drug activation process.
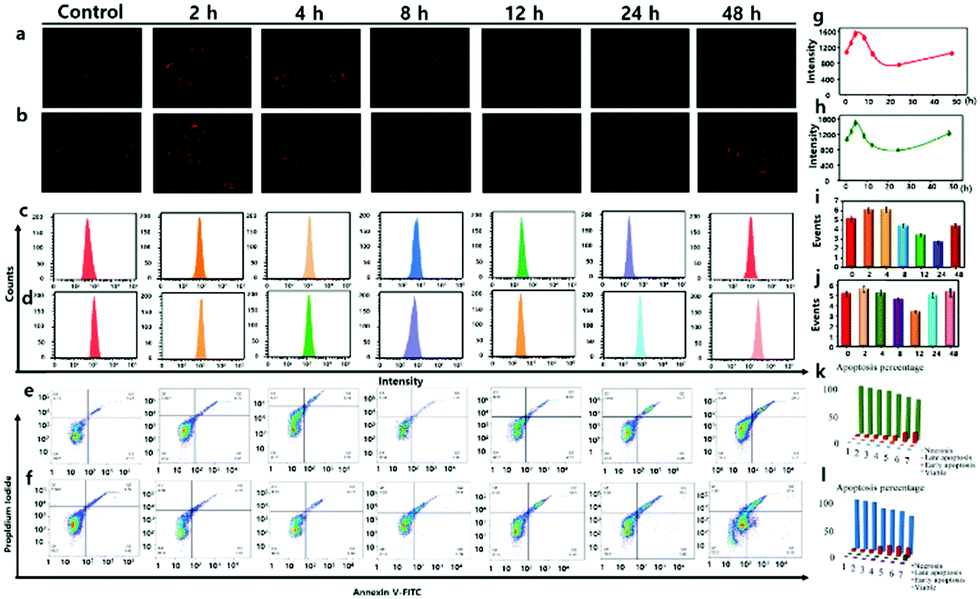 | ||
| Fig. 2 Two-photon confocal fluorescence imaging (λex = 830 nm, λem = 650–750 nm) and flow cytometry assay (λex = 560 nm, λem = 650–750 nm) for the detection of CE2 in HT-29 cells. (a and b) HT-29 cells pretreated with CPT-11 and CAPE, respectively, and then incubated with DCM-CES2. (c and d) Corresponding flow-cytometric analysis to (a) and (b), respectively. (e and f) PE annexin V/7-AAD assay of HT-29 cells in (a) and (b), respectively, using flow cytometry, Q1: necrosis cells, Q2: late apoptotic cells, Q3: early apoptotic cells, and Q4: survival cells. (g and h) Mean fluorescence intensities of the images in (a) and (b), respectively. (i and j) Mean values of (c) and (d), respectively. (k and l) Apoptosis percentages of (e) and (f), respectively. The data are shown as mean (±SD). | ||
We further evaluated the in vivo applicability of our NIR-emissive TP probe. We performed TP imaging of CE2 in subcutaneous intestinal cancer slices (100 μm) to validate the deep imaging capability of our probe. The tissue slice was incubated with DCM-CES2 (20 μM) for 60 min, then washed with PBS three times before confocal fluorescence imaging (Fig. 3a). The cancer tissues displayed strong fluorescence response with a collection window from 650 nm to 750 nm (λex = 830 nm). We utilized the z-scan mode of the TP microscope to visualize the fluorescence signals at different depths of mice colon cancer slices (0–160 μm), and then reconstructed its 3D fluorescence image (Fig. 3b). These assays indicated that the probe DCM-CES2 could measure CE2 at a penetration depth of up to 160 μm in tissues by TP microscopy, which could be attributed to its superior NIR-emissive TP fluorescence properties for in vivo sensing of CE2. Many of the reported fluorescent probes for imaging of CE2 activity were obstructed to achieve applications for in vivo experiments since their emissions were not in the NIR region.30 Motivated by the predominant NIR-emissive properties of the designed probe, we then explored its ability in real-time in vivo visualization of the CE2 activity in tumour lesions. To optimize the imaging time in mice, the probe DCM-CES2 (100 μM, 30 μL in PBS) was administered to the human colorectal xenograft tumour mice model by intra-tumoral injection and then scanned at time points 10, 20, 30, 60, 90, 120, 150 and 180 min using an IVIS Lumina Kinetic Series III imaging system. Images were taken with an excitation filter of 560 nm and emission windows from 650 nm to 750 nm. As displayed in Fig. S7 (ESI†), the control group was injected with PBS. As expected, the in situ injection of DCM-CES2 resulted in gradual fluorescence increase, indicating its rapid activation by CE2 in vivo. The intensity reached maximum at 150 min after injection and remained stable up to 180 min.
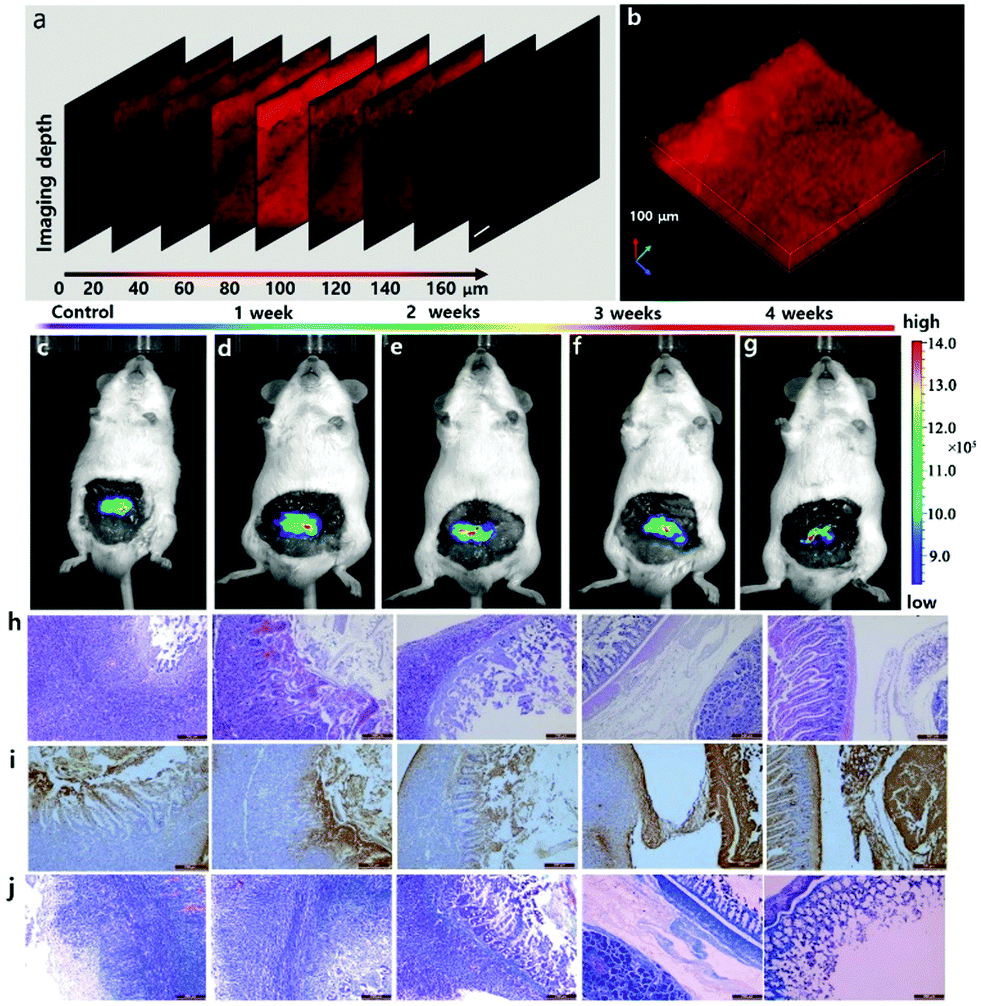 | ||
| Fig. 3 (a) The confocal z-scan TP imaging at different depths. (b) 3D fluorescence image of (a). Scale bar: 100 μm. (c–g) Evaluation of CE2 in the orthotopic tumor-bearing mice model treated with CPT-11 by tail intravenous injection (20 mg kg−1) for 0–4 weeks and fluorescence images of CE2 with DCM-CES2. (h) H&E stained colonic cancer tissue histopathology images. (i) Regional TUNEL staining in the colonic cancer tissues. (j) Masson's stained slice of colonic cancer tissues. | ||
After successfully validating the availability of DCM-CES2 to achieve real-time monitoring of CE2 in mice, we further investigated the therapeutic effect of the CE2-related anticancer prodrugs. Herein, human colonic cancer cell MC38 was inoculated in colon to construct the orthotopic tumour-bearing mice model, which was then treated with CPT-11 by tail intravenous injection for 0, 1, 2, 3 and 4 weeks. We recorded fluorescence images of these mice in five parallel groups after 150 min post abdominal intra-tumoral injection of DCM-CES2 (100 μM, 30 μL in PBS), respectively. It was worth noting that the “probe light-up” region of tumour lesions gradually narrowed, and in the 4th week pronounced improvement of tumour was exhibited (Fig. 3c–g). The experimental results demonstrated that the endogenous CE2 in tumour lesions was always activated during the treatment of colon cancer with CPT-11. The results in H&E, TUNEL and Masson's stained slices (Fig. 3h–j) illustrated the degree of tumour treatment, which provided the satisfactory treatment outcomes with anticancer prodrug CPT-11. Hence, this promising probe could give convincing assessment to the anticancer efficacy of CES-2-related prodrugs.
In conclusion, we have successfully designed and synthesized a NIR-emissive two-photon fluorescent probe DCM-CES2 for endogenous CE2 detection in living cells and in vivo. The probe offers a switch-on behavior for the detection of CE2 activity under simulated physiological systems with a large Stokes shift of 125 nm and a low LOD of 0.087 μg mL−1. Cell experiments illustrate that DCM-CES2 can realize specific detection of endogenous CE2 with low cytotoxicity and satisfactory cell membrane permeability. Tissue fluorescence imaging exhibits high resolution at a penetration depth of up to 160 μm due to its excellent NIR-emissive TP properties. DCM-CES2 has been applied for real-time monitoring of CE2 activity during the activation process of anticancer prodrugs CPT-11 and CAPE in cells. The probe also achieves in situ and in vivo evaluation of therapeutic efficacy of anticancer prodrug CPT-11 at tumour lesions in the orthotopic colonic mice model. The results of DCM-CES2 reveal that this NIR two-photon probe holds potential advantages for the further study of the biological function of CE2 in complex systems.
This work was supported by the Hainan Provincial Natural Science Foundation of China (819QN225 and 2019RC210), the National Nature Science Foundation of China (No. 21775162), the Hainan High Education Research Project (Hnky2019ZD-30 and Hnky2019ZD-52), the Talent Program of Hainan Medical University (HYPY201905, XRC180006, and XRC180010), and the Hundred-Talent Program (Hainan 2018).
Conflicts of interest
The authors declare no conflicts of interest.Notes and references
- S. C. Laizure, V. Herring, Z. Hu, K. Witbrodt and R. B. Parker, Pharmacotherapy, 2013, 33, 210–222 CrossRef CAS PubMed
.
- L. Feng, Z.-M. Liu, J. Hou, X. Lv, J. Ning, G.-B. Ge, J.-N. Cui and L. Yang, Biosens. Bioelectron., 2015, 65, 9–15 CrossRef CAS PubMed
- M. Hosokawa, Molecules, 2008, 13, 412–431 CrossRef CAS PubMed
- M. Li, C. Zhai, S. Wang, W. Huang, Y. Liu and Z. Li, RSC Adv., 2019, 9, 40689–40693 RSC
- K. Kailass, O. Sadovski, M. Capello, Y. A. Kang, J. B. Fleming, S. M. Hanash and A. A. Beharry, Chem. Sci., 2019, 10, 8428–8437 RSC
- S. P. Sanghani, S. K. Quinney, T. B. Fredenburg, Z. Sun, W. I. Davis, D. J. Murry, O. W. Cummings, D. E. Seitz and W. F. Bosron, Clin. Cancer Res., 2003, 9, 4983–4991 CAS
- Q. Jin, L. Feng, D.-D. Wang, Z.-R. Dai, P. Wang, L.-W. Zou, Z.-H. Liu, J.-Y. Wang, Y. Yu and G.-B. Ge, ACS Appl. Mater. Interfaces, 2015, 7, 28474–28481 CrossRef CAS PubMed
- D. Wang, L. Zou, Q. Jin, J. Hou, G. Ge and L. Yang, Acta Pharm. Sin. B, 2018, 8, 699–712 CrossRef PubMed
- H. Zhou, J. Tang, J. Zhang, B. Chen, J. Kan, W. Zhang, J. Zhou and H. Ma, J. Mater. Chem. B, 2019, 7, 2989–2996 RSC
- X. Chen, D. Lee, S. Yu, G. Kim, S. Lee, Y. Cho, H. Jeong, K. T. Nam and J. Yoon, Biomaterials, 2017, 122, 130–140 CrossRef CAS PubMed
- X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–659 CrossRef CAS PubMed
- M. Gao, F. Yu, C. Lv, J. Choo and L. Chen, Chem. Soc. Rev., 2017, 46, 2237–2271 RSC
- X. Wu, W. Shi, X. Li and H. Ma, Acc. Chem. Res., 2019, 52, 1892–1904 CrossRef CAS PubMed
- Z. Yan, J. Wang, Y. Zhang, S. Zhang, J. Qiao and X. Zhang, Chem. Commun., 2018, 54, 9027–9030 RSC
- X. Tian, F. Yan, J. Zheng, X. Cui, L. Feng, S. Li, L. Jin, T. D. James and X. Ma, Anal. Chem., 2019, 91, 15840–15845 CrossRef CAS PubMed
- D. Li, Z. Li, W. Chen and X. Yang, J. Agric. Food Chem., 2017, 65, 4209–4215 CrossRef CAS PubMed
- W. Qu, X. Zhang, Y. Ma, F. Yu and H. Liu, Spectrochim. Acta, Part A, 2019, 222, 117240 CrossRef CAS PubMed
- J. Wang, Q. Chen, N. Tian, W. Zhu, H. Zou, X. Wang, X. Li, X. Fan, G. Jiang and B. Z. Tang, J. Mater. Chem. B, 2018, 6, 1595–1599 RSC
- Y. Lou, C. Wang, S. Chi, S. Li, Z. Mao and Z. Liu, Chem. Commun., 2019, 55, 12912–12915 RSC
- M. Gao, R. Wang, F. Yu and L. Chen, Biomaterials, 2018, 160, 1–14 CrossRef CAS PubMed
- H. M. Kim and B. R. Cho, Chem. Rev., 2015, 115, 5014–5055 CrossRef CAS PubMed
- D. Kim, H. Moon, S. H. Baik, S. Singha, Y. W. Jun, T. Wang, K. H. Kim, B. S. Park, J. Jung and I. Mook-Jung, J. Am. Chem. Soc., 2015, 137, 6781–6789 CrossRef CAS PubMed
- W. R. Zipfel, R. M. Williams, R. Christie, A. Y. Nikitin, B. T. Hyman and W. W. Webb, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7075–7080 CrossRef CAS PubMed
- A. Feng, Y. Jia, L. Huang, L. Wang, G. Zhou, S. Wang and P. Liu, Spectrochim. Acta, Part A, 2019, 220, 117108 CrossRef CAS PubMed
- G. Xu, W. Zhang, M. K. Ma and H. L. McLeod, Clin. Cancer Res., 2002, 8, 2605–2611 CAS
- Q. Jin, L. Feng, D.-D. Wang, J.-J. Wu, J. Hou, Z.-R. Dai, S.-G. Sun, J.-Y. Wang, G.-B. Ge, J.-N. Cui and L. Yang, Biosens. Bioelectron., 2016, 83, 193–199 CrossRef CAS PubMed
- M. H. Wu, B. Yan, R. Humerickhouse and M. E. Dolan, Clin. Cancer Res., 2002, 8, 2696–2700 CAS
- S. K. Quinney, S. P. Sanghani, W. I. Davis, T. D. Hurley, Z. Sun, D. J. Murry and W. F. Bosron, J. Pharmacol. Exp. Ther., 2005, 313, 1011–1016 CrossRef CAS PubMed
- T. Hu, C. Liu, Q. Li, J. Xiong, Y. Ma, G. Wu and Y. Zhao, Medicine, 2018, 97, e0349 CrossRef PubMed
- Z.-M. Liu, L. Feng, J. Hou, X. Lv, J. Ning, G.-B. Ge, K.-W. Wang, J.-N. Cui and L. Yang, Sens. Actuators, B, 2014, 205, 151–157 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Probe synthesis and characterization, cell imaging, mice imaging, and supplementary figures. See DOI: 10.1039/d0cc00297f |
| This journal is © The Royal Society of Chemistry 2020 |